

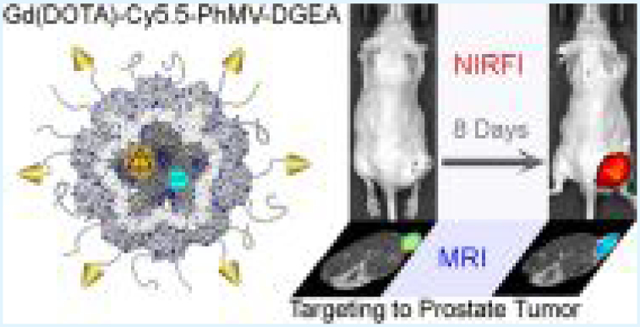
Abstract
One of the greatest challenges in nanomedicine is the low efficiency with which nanoparticles are delivered to lesions such as tumors in vivo. Here, we show that Physalis mottle virus (PhMV)-like nanoparticles can be developed as bimodal contrast agents to achieve long circulation, specific targeting capability, and efficient delivery to tumors in vivo. The self-assembling coat protein nanostructure offers various opportunities to modify the internal and external surfaces separately. After loading the internal cavity of the particles with the fluorescent dye Cy5.5 and paramagnetic Gd(III) complexes, we modified the outer surface by PEGylation and conjugation with targeting peptides. Using this combined approach, we were able to monitor a human prostate tumor model for up to 10 days by near-infrared fluorescence and magnetic resonance imaging, with up to 6% of the injection dose remaining. Our results show that PhMV-like nanoparticles provide a promising and innovative platform for the development of next-generation diagnostic and therapeutic agents.
Keywords: ultrahigh field magnetic resonance imaging, near-infrared fluorescence imaging, prostate cancer, long circulation, plant virus-like particles
Graphical Abstract
INTRODUCTION
The targeted delivery of nanoparticles (NPs) to malignant tissues can improve cancer diagnosis and therapy and is therefore a key goal in the field of cancer nanomedicine.1 However, only 0.7% (median) of the total NP injection dose (ID) is delivered to solid tumors because the particles are rapidly cleared from the circulation.2 This poor efficiency of delivery is a major challenge in the translation of nanotechnology innovations into clinical applications, requiring the development of a targeted nanoparticle delivery system with long circulation in vivo.3 To achieve this goal, the cellular internalization of NPs and tumor tissue accumulation/retention must be optimized. Hence, optimizations of NP properties such as size, uniformity, shape, surface charge, and surface modifications are critically important to improving the targeting efficiency.3
Synthetic nanoparticles have been tested in several clinical trials, but biological materials offer particular advantages for biomedical applications.4 For example, virus-like particles (VLPs) are very similar to natural viruses, in that they assemble into discrete nanoparticles, but they do not contain a viral genome.5 VLPs have many advantageous properties such as, easy to manufacture within precise quality control tolerances, the coat protein (CP) monomers can be edited by genetic engineering and modified by selective chemical conjugation, and are highly biocompatible and biodegradable.5 Physalis mottle virus (PhMV) is a +ssRNA virus from the family Tymoviridae that forms ~30 nm icosahedral capsids with T = 3 symmetry.6 VLPs can be expressed in Escherichia coli.7 They are monodisperse, symmetrical, polyvalent, and stable in a range of aqueous conditions. Importantly, lysine residues are exposed on the exterior and cysteine residues on the interior, providing labeling handles suitable for the development of multifunctional molecular-imaging agents.7,8 Other VLPs, such as those based on cowpea chlorotic mottle virus, cowpea mosaic virus (CPMV), tobacco mosaic virus (TMV), hepatitis B virus (HBV), and bacteriophage MS2, have been developed as delivery vehicles for drugs and vaccines.4,5,9-12 We recently reported that PhMV can be functionalized with imaging reagents and drugs.13 However, it is unclear whether such VLPs could be used as long-circulating, targeted contrast agents for molecular imaging, and the in vivo fate of these particles is still unpredictable. While many different synthetic scaffolds are being under exploration for cancer nanomedicine, only a few biological materials and a few plant viral species have been investigated to date. It is currently not possible to predict in vivo fates of new materials based on structural information, and in vivo studies are required to gain insights into the in vivo properties to tailor new materials, such as novel plant VLPs, for biomedical applications.
Recent reports highlighted the application of plant virus nanoparticles and virus-like nanoparticles for the design of high-relaxivity magnetic resonance imaging (MRI) contrast agents. This includes the development of Gd-hydroxypyridonate-modified MS2 nanoparticles,14,15 Gd-1,4,7,10-tetraaza-cyclododecane-1,4,7,10-tetraacetic acid (DOTA)-loaded cowpea mosaic virus (CPMV) nanoparticles,16 as well as metal-free tobacco mosaic virus (TMV) with paramagnetic organic radical compounds.17 Ideally, multifunctional contrast agents should be suitable for both near-infrared fluorescence (NIRF) imaging and ultrahigh field magnetic resonance imaging (UHFMRI) because these techniques work well in combination.12,18 The imaging reagents in each case are nonionizing, and a single nanoparticle featuring such contrast agents would achieve high sensitivity, deep tissue penetration, and high spatial resolution, making them suitable for multiscale scanning from the cellular level to the organ.12
Here, we prepared bimodal contrast agents by conjugating the NIRF dye Cy5.5 and the MRI reagent Gd3+(DOTA) to the internal surface of PhMV NPs, as well as labeling the external surface with DGEA peptides (Asp-Gly-Glu-Ala) targeting PC-3 prostate cancer cells. Prostate cancer was chosen as a tumor model to demonstrate proof of principle and because of the clinical significance.19,20 As a model system, we chose the human prostate cancer cell lines, PC-3, which expresses high levels of integrin α2β1.21 Expression of integrin α2β1 has been linked to a worse prognosis, thus making it an important diagnostic marker and therapeutic target. Here, we tested the ability of our PhMV-based contrast agent to image PC-3 cells in vitro and in vivo. We also investigated the pharmacokinetics (PK) and protein corona of the contrast agent in human blood to determine its stability and targeting specificity.
RESULTS AND DISCUSSION
Preparation and Characterization of Gd-Cy5.5-PhMV-Methoxypoly(ethylene glycol) (mPEG)/DGEA Particles.
VLPs of PhMV are composed of 180 identical 21 kDa coat protein subunits that self-assemble to form a ~30 nm icosahedral capsid (Figure 1A). The structural data indicate four lysine residues on the solvent-exposed exterior surface, and additional five lysine side chains are found on the interior surface. In contrast, only a single cysteine residue is presented on the internal surface.8,23 Accordingly, multifunctional PhMV NPs can be developed to increase potency, reduce immunogenicity, increase cellular uptake, and allow specific cell targeting.23
Figure 1.

(A) Structure of the Physalis mottle virus (PhMV) coat protein (CP) consisting of a (yellow), b (magenta), and c (green) subunits; the protein subunits are identical; a–c denotes their position within the icosahedral T = 3 lattice. The surface-exposed residues highlighted as external lysine (blue) and internal cysteine residues (red). Representation of interior and exterior (with 180° rotation) of the asymmetric PhMV CP subunit. The structure of the assembled hollow capsid shown as outer and inner surfaces as whole sphere and half-cut view, respectively. Methodology for (B) internal and (C) external modifications, respectively. Images were created using UCSF Chimera software (PDB: 1E57) and ChemDraw v15.0.
We expressed the PhMV coat protein in E. coli.7 The internal surface was modified with the macrocyclic MRI contrast agent DOTA-maleimide (Figure S1) and the NIRF dye Cy5.5-maleimide using thiol-maleimide chemistry (Figure 1B), whereas the external surface was conjugated using amine-N-hydroxysuccinimide (NHS) ester chemistry, either with mPEG-NHS as control NPs (Gd-Cy5.5-PhMV-mPEG) or with the DGEA peptide (via a heterofunctional NHS-PEG-maleimide linker) as integrin α2β1-targeted NPs (Gd-Cy5.5-PhMV-DGEA) (Figure 1C). The loading can be controlled by adjusting the reaction time, pH, and excess of reagents used. Conditions were optimized to load a smaller quantity of dye to avoid fluorescence quenching while maximizing Gd(DOTA) loading to achieve a better MRI signal. UV/visible spectrophotometry and inductively coupled plasma atomic emission spectroscopy (ICP-AES), respectively, were used to determine Cy5.5 and Dy(DOTA) loading, and determined ~20 Cy5.5 molecules and ~130 chelated Gd3+ ions, covering ~12 and ~73% of the available cysteine side chains, are available, respectively. Transmission electron microscopy (TEM) was used to confirm the structural integrity of the nanoparticles (Figure 2A-C). The typical monodisperse, spherical, ~30 nm nanostructures of the native PhMV NPs (Figure 2A1) were not significantly altered by the internal and external modifications to generate Gd-Cy5.5-PhMV-mPEG (Figure 2B1) or Gd-Cy5.5-PhMV-DGEA (Figure 2C1). The stability and dispersion of the particle in aqueous solution were monitored by size exclusion chromatography (SEC) and dynamic light scattering (DLS). The particles eluted from the Superose 6 column as a single peak at ~7.5 mL (Figure 2A2-C2). The hydrodynamic diameters of the native PhMV, Gd-Cy5.5-PhMV-mPEG, and Gd-Cy5.5-PhMV-DGEA particles were ~30.7, ~33.6, and ~35.7, respectively, with narrow size distribution (Figure S2). We attributed the slight increase in particle size to the protruding PEG and peptide. These results indicated that both Gd-Cy5.5-PhMV-mPEG and Gd-Cy5.5-PhMV-DGEA remained stable and monodisperse and retained the ~30 nm diameter of the native virus. As a general rule, large nanoparticles are preferentially filtered out by reticuloendothelial system (RES), whereas tiny nanoparticles tend to be cleared via the urinary system.24 Nanoparticles in the range of 10–50 nm diameter are therefore desirable for in vivo applications.25
Figure 2.

(A1) Transmission electron micrograph (TEM) image of native PhMV. Higher-magnification TEM image of (B1) Gd-Cy5.5-PhMV-mPEG and (C1) Gd-Cy5.5-PhMV-DGEA, and the corresponding size exclusion chromatography (SEC) profile of (A2) PhMV, (B2) Gd-Cy5.5-PhMV-mPEG, and (C2) Gd-Cy5.5-PhMV-DGEA. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the NPs with (D1) white light and (D2) fluorescence imaging. The agarose gel electrophoresis of the NPs with (E1) white light and (E2) fluorescence imaging.
Denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 2D1,D2) and native agarose gel electrophoresis (Figure 2E1,E2) were used to investigate the surface modifications. All samples except native PhMV showed bright Cy5.5 fluorescence at the same position of Coomassie-stained bands, showing that Cy5.5 was covalently attached to the PhMV coat protein (Mr = ~26 kDa), which was also present in all samples (Figure 2D1,D2). The increased PhMV CP molecular weight (MW) as compared to the wild type CP (21 kDa) is attributed to the cloning strategy; the CP was inserted between the Xho I and Hind III sites of the pRSET-A vector (Invitrogen), which leads to an addition of 39 amino acids (5 kDa) from the vector backbone at the N-terminus of the CP. The addition of amino acids include a hexa-histidine tag used for purification; however, the additional sequence does not alter the morphology or stability of the particles.26 The Gd-Cy5.5-PhMV-mPEG particles were represented by an additional band at ~29 kDa after conjugation with Cy5.5 (Mr = 767.44 Da), Gd(DOTA) (Mr = 646.16 Da), and PEG (Mr = ~2k Da), which corresponds to the anticipated increase in the mass of the coat protein monomer. Subsequent conjugation of Gd-Cy5.5-PhMV-PEG-Mal with the DGEA peptide (Mr = 493.4 Da) led to the appearance of a ~30 kDa band matching the anticipated mass of the coat protein–PEG-DGEA monomer. Both Gd-Cy5.5-PhMV-mPEG and Gd-Cy5.5-PhMV-DGEA generated two bands (52–58 kDa) that attributed to the dimer of CP. Densitometric analysis of the particles by AlphaImager gel documentation systems demonstrated that ~46% of the surface was labeled with mPEG (~331 mPEG per particle) and ~25% coverage with the DGEA peptide (~180 DGEA per particle). The surface charge of particles was investigated by agarose gel electrophoresis and ζ potential measurement (Figure 2E1,E2). The mobility shift toward the anode of the Gd-Cy5.5-PhMV particles was almost the same as native PhMV because all of the Gd(DOTA) and Cy5.5 cargo were loaded inside the particles, which minimized the effect on surface charge. PEGylation of the external surface altered the surface charge because the noncharged PEG chain removes positive amines from the particle surface. Accordingly, the ζ potential of Gd-Cy5.5-PhMV-mPEG (+2.13) was lower than the native PhMV (+4.42). Labeling with the DGEA peptide reduced the positive charge further (+1.07). According to recent opinion, NPs with a near-neutral surface charge may be taken up more efficiently and would avoid charge-induced toxicity in vivo.3,27
T1 of gadolinium(III) ion-based contrast agents are effective in shortening the longitudinal relaxation time (T1) at low field-strength magnetic (≤1.5 T). However, the longitudinal relaxivity (r1) rapidly decreases with increasing field strength and so does their effectiveness during MRI.28 Therefore, the development of contrast agents compatible with higher-field MRI scanners is always an urgent task. The increase in the relaxivity of Gd-based contrast agents can be achieved, e.g., by including more inner-sphere water molecules and ensuring a relatively fast exchange rate (τM in the order of nanoseconds) for the metal-coordinated water molecules and medium rotational correlation times (τR) for the metal complex.28 These strategies could be realized by immobilizing the Gd-complex on nanostructured materials, thus slowing down their molecular tumbling while maintaining the water residence time.29-31 We therefore conjugated Gd(DOTA)-maleimide onto the internal surface of PhMV NPs to increase the relaxivity. Figure 3A,B shows the transverse relaxivity (r2) and longitudinal relaxivity (r1) of the Gd-Cy5.5-PhMV-mPEG NPs in different magnetic fields of 1.5 and 7 T, respectively. The r1 were measured as 31.0 and 8.2 mM−1 s−1 at 1.5 and 7 T, respectively, which is about 7-fold of commercially available T1 MRI contrast agents (3–5 mM−1 s−1).28 The r1-relaxivity decreased more than the r2-relaxivity as the magnetic field became stronger, increasing the ratio of r2/r1. The r2/r1 of Gd-Cy5.5-PhMV-mPEG was 1.4 in a medium-strength magnetic field, increasing to 2.5 in an ultrahigh-strength magnetic field. This phenomenon is consistent with the relaxivity theory.32 The longitudinal relaxivities of recently reported functionalized NPs are summarized in Table S1. Our Gd-Cy5.5-PhMV-mPEG particles are among the most efficient, with a higher longitudinal relaxivity than Gd-P22 (21.7 mM−1 s−1) at 28 MHz, almost double that of TMV (14.6 mM−1 s−1) and CPMV (15.5 mM−1 s−1) at ~60 MHz, and double that of Gd-MS2 in both mild (16.9 mM−1 s−1) and ultrahigh-strength (4.3 mM−1 s−1) magnetic fields. To investigate the MRI contrast capability of the functionalized VLPs at ultrahigh magnetic field, the T1-mapping phantom images of the Gd-Cy5.5-PhMV-mPEG with different concentrations in phosphate-buffered saline (PBS) buffer (10 mM, pH 7.4) were scanned at 7 T (Figure 3C). The T1-mapping images showed a positive contrast trend from 0 to 43 μM with an increase of Gd3+ concentration, confirming the suitability of the NPs for T1-UHFMRI applications.
Figure 3.

r1/2-Relaxivity of Gd-Cy5.5-PhMV-mPEG measured at 37 °C in magnetic fields of (A) 1.5 and (B) 7 T. (C) T1-mapping phantoms of Gd-Cy5.5-PhMV-mPEG at different concentrations of Gd3+ obtained in a 7 T scanner.
Cancer-Cell-Targeting Properties of Gd-Cy5.5-PhMV-DGEA.
Next, we tested the in vitro cytotoxicity of the targeted NPs (Gd-Cy5.5-PhMV-DGEA) and their untargeted counterparts (Gd-Cy5.5-PhMV-mPEG) using the methyl thiazolyl tetrazolium assay in PC-3 cell line (Figure S4). No significant cytotoxicity was observed. This indicates that the PhMV nanoparticles are not cytolytic, as reported for other plant virus particles.12,33
The specificity of the DGEA peptide to integrin α2β1 has been systematically studied in various prostate cancer cells.34 These studies demonstrated that α2β1 integrin is overexpressed in PC-3, while the LNCaP and CWR-22 cells express androgen receptors and prostate-specific antigens. Expression was also assessed in mesenchymal stem cells, and low expression levels of α2β1 integrins were reported.35 Based on these studies, we chose the integrin α2β1+ PC-3 cell line for our studies. The in vitro-targeting ability of our NPs was determined by incubating integrin α2β1+ PC-3 cells with 2 μg/mL of Gd-Cy5.5-PhMV-DGEA (targeting group) or Gd-Cy5.5-PhMV-mPEG (control group) at 37 °C for 3 h. The cells were then qualitatively evaluated by confocal laser scanning microscopy (CLSM) (Figure 4A), and the quantity of NPs was measured by fluorescence-activated cell sorting (Figure 4B,C). Intense Cy5.5 fluorescence (pseudocolored in magenta) was observed in the targeting group (Figure 4A). The nuclei and membranes of the PC-3 cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) and A555-labeled wheat germ agglutinin (green), respectively, revealing that most of the Gd-Cy5.5-PhMV-DGEA NPs were inside the cells and probably localized in endosomes, presumably following receptor-mediated endocytosis.12,22 In contrast, only very weak signals were detected in the control group reflecting the lack of integrin α2β1-targeting ligands. The uptake of the NPs into PC-3 cells was blocked by the addition of 2 μM free DGEA 30 min before Gd-Cy5.5-PhMV-DGEA. Only weak fluorescence was detected, caused by the passive uptake of NPs. The mean fluorescence intensities (MFIs) of the cells (Figure 4B,C) confirm that DGEA-decorated NPs were taken up efficiently by the PC-3 cells, with the MFI increasing by ~10-fold, ~3-fold, and ~2-fold compared to the blank, nontargeted control, and DGEA-blocked groups, respectively. These results supported our confocal microscopy data, although the control particles were nevertheless taken up efficiently by the cancer cells via passive internalization, consistent with earlier reports.12,13 The efficient internalization of these particles is probably due to the near-neutral surface charge (Gd-Cy5.5-PhMV-mPEG: +2.13, Gd-Cy5.5-PhMV-DGEA: +1.07) in agreement with the so-called hierarchical-targeting strategy, in which near-neutral NPs have more affinity for the cell surface while reducing charge-induced toxicity.3,18,36 Our contrast agents are therefore promising candidates for future in vivo imaging and tumor-homing applications.
Figure 4.

In vitro-targeting uptake experiments were conducted by incubating PC-3 cells with 2 μg/mL Gd-Cy5.5-PhMV-DGEA (targeted group) and Gd-Cy5.5-PhMV-mPEG (control group) at 37 °C for 3 h. The blocking experiment was carried out by incubating PC-3 cells with Gd-Cy5.5-PhMV-DGEA in the presence of an excess of free DGEA (2 μM). The results were first observed by (A) confocal laser scanning microscopy (CLSM) (scale bar = 30 μm) and then (B, C) quantitative analysis by flow cytometry. The inset shows mean fluorescence intensities (MFIs) of cells in each sample quantified from three replicates with standard deviations (±) in the corresponding cell panels (p < 0.05). All samples were measured in triplicates and analyzed using FlowJo v10 software.
Targeted in Vivo NIRF and MR Imaging and ex Vivo Biodistribution Studies.
We tested the ability of Gd-Cy5.5-PhMV-DGEA to target PC-3 tumors in mouse models by bimodal NIRF imaging and T1-mapping MRI. PC-3 xenografts using athymic nude mice (n = 6) were scanned before and tracked from 4 to 240 h (10 days) after the intravenous injection of 200 μg per mouse of our contrast agent. The choice of the dose was based on our previous experience with cowpea mosaic virus (CPMV), which has similar morphology and particle size with the PhMV.37
Figure 5A shows whole-body, quantitative NIRF imaging of the VLPs in a PC-3 tumor mouse model. When the main organs were visualized in vivo, the NIRF signal clearly highlighted the PC-3 tumor and distinguished it from the surrounding tissues after 4 h postinjection (more NIRF imaging before 8 h postinjection was shown in Figure S5). Figure 5B demonstrates the visualization of the main organs in vivo. Tumor uptake increased over time and reached a maximum of 144 h (6 days) postinjection. This was followed by gradual washing-out, but high tumor retention was still evident at 240 h (10 days) postinjection. The target-binding specificity was confirmed by the significantly lower tumor uptake in the control group at the same time points. The PhMV NPs were cleared by deposition in the liver and spleen, which began ~4 h postinjection. The fluorescence signal in the lymph nodes (32–144 h postinjection) declined slightly by 144 h postinjection and was extinguished by 192 h postinjection, indicating long circulation time of the NPs.
Figure 5.

In vivo near-infrared fluorescence (NIRF) imaging. (A) NIRF images of subcutaneous PC-3 (α2β1 positive) prostate tumors in athymic nude mice (n = 6) before and 4–240 h after the intravenous injection of Gd-Cy5.5-PhMV-DGEA (targeted group) or Gd-Cy5.5-PhMV-mPEG (control group). (B) Visualization of the main organs in vivo. (C) Quantitative analysis of fluorescence intensity in tumors (p < 0.01). (D) Ex vivo NIRF images of main organs (brain, lung, heart, liver, spleen, kidneys, and tumor) after bimodal scanning (240 h postinjection) and (E) quantitative analysis of fluorescence intensity in each organ based on ex vivo NIRF imaging (p < 0.01 in the tumor). Noninjected mice were used as the blank control group.
The enhanced signal in the tumor was quantified by comparing the fluorescence intensity in the region-of-interest (ROI) within the tumor to preinjection mice as the background, normalized to 1 (Figure 5C). For the targeted group, the signal intensity in the PC-3 tumor region increased from ~2.8-fold (*p < 0.01) at 4 h postinjection to ~7-fold (*p < 0.01) at 144 h postinjection and declined slightly thereafter but still remained 5.9-fold (*p < 0.01) 240 h postinjection. The ex vivo NIRF images (Figure 5D) clearly show differences in fluorescence intensity between the dissected tumor and other tissues. Compared to the blank (noninjected mice) and nontargeted control NPs, the targeted NPs showed much more intense fluorescence within the tumor (as well as in the liver and spleen). The semiquantitative biodistribution data agreed with our ex vivo and in vivo NIRF images (Figure 5E). Notably, the DGEA-targeted NPs achieved a ~3-fold (**p < 0.01) increase in tumor-targeting efficiency compared to the nontargeted control group, although the latter also achieved significant tumor accumulation.
Our NIRF imaging showed that NPs were initially detected in the liver and spleen and kept accumulating until 144 h postinjection when tumor uptake reached its peak. We observed similar behavior in the lymph nodes. Clearance by the liver and spleen followed the trend previously reported for other plant viral nanoparticles and many synthetic nanoparticles.38 PEGylated NPs with a diameter of less than 50 nm and a neutral surface charge are taken up efficiently into draining lymph nodes, which appears to be the preferred form of lymphatic transport.25,39 Most NP contrast agents are filtered predominantly by the RES in the liver and/or spleen, but even though the same was true of our NPs, they showed much more efficient tumor homing.2,24 The nontargeting NPs were internalized passively due to the enhanced permeability and retention (EPR) effect. Washout effects were less apparent for the targeted nanoparticle, and this may be explained by the base level expression of α2β1 integrin in these organs.40 The tumor-targeting ability of Gd-Cy5.5-PhMV-DGEA is therefore likely to reflect a combination of its nanoparticle properties (favorable surface and uniform small size), passive homing based on EPR,41 lymphatic transport,25,39 cellular transport,24 and active receptor-mediated interactions via the targeting of α2β1 integrins.42
After NIRF imaging at a certain selected time point, we immediately transferred the mice to a 7 T MRI scanner. T1-mapping MR imaging of deep cross sections revealed the accumulation and retention of the NPs in tumors, with the signal strength mirroring the trends observed in the NIRF images (Figure 6). The NPs highlighted the tumor for at least 10 days, unlike small molecular contrast agents, which usually only achieve image retention for a few minutes in lesions.43 As the T1 relaxation time decreased, the color of the T1 maps changed from red to blue, as shown in the scale bar (Figure 6A), reflecting the accumulation of the NPs. Quantitative analysis of T1 relaxation times in the tumor ROI (Figure 6B) revealed that positive contrast in the tumor region appeared at the first scan time point of 6 h postinjection as ~15% shortening of T1 and increased thereafter. The average T1 value in the whole tumor 144 h postinjection declined by ~25% in the targeted group (compared with the prescan tumor) but by only ~17% in the control group (i.e., nontargeted VLPs). Indeed, the T1 value declined by ~41% in some regions of the tumor (blue color) due to the heterogeneous structure of the tumor tissue. In both treatment groups, there was a slow recovery in the T1 relaxation times 240 h postinjection. The trend in the T1 values was consistent with the NIRF signals. These results also showed that the NPs were distributed in specific tumor regions, indicating that they could be used for long-term monitoring of tumor microenvironments.44 It should be noted that imaging was achieved at significantly lower doses per the contrast agents: here, we demonstrate tumor imaging at 0.23 μmol/kg Gd, compared to 0.1 mmol/kg for Prohance45 per 0.01 mmol/kg for Feridex.46 Even so, we achieved significant NIRF and MRI contrast enhancements at these low doses, reflecting two key properties of our nanoparticles: the prolonged circulation time and the specific tumor homing and accumulation conferred by the DGEA peptide.
Figure 6.

(A) In vivo T1-map of subcutaneous PC-3 (α2β1 overexpression) prostate tumors in athymic nude mice (n = 6) obtained before and 6–240 h after the intravenous injection of Gd-Cy5.5-PhMV-DGEA (targeted group) or Gd-Cy5.5-PhMV-mPEG (control group). (B) Quantitative analysis of T1 reduction in tumors. (C) Biodistribution of Gd3+ in the main organs of mice after bimodal scanning (240 h postinjection). Noninjected mice were used as the blank control group.
Biodistribution of our NPs was determined using ICP-AES to measure the Gd3+ content per organ (Figure 6C). More Gd3+ accumulated in the tumors of mice treated with the targeted NPs (137.8 ng/g organ) than those treated with the control NPs (77.5 ng/g organ)—this agrees with the imaging data. Gd3+ was initially detected in the spleen, followed by the liver and kidneys, in agreement with the ex vivo NIRF data. The Gd3+ levels allowed us to determine that the quantity of NPs remaining in the mouse as a proportion of the initial dose (ID%) was 38.8 ID% for the targeted NPs (5.5 ID% in the tumor) and 24.7 ID% for the untargeted NPs (1.8 ID% in the tumor), even 10 days postinjection. This is a vast improvement of the median value of 0.7 ID% reported for NPs in tumors in studies published over the last decade.2 The washout rate of targeted NPs was slower than that of the control NPs, reflecting the expression of α2β1 integrin in some normal organs (e.g., liver and spleen).22,47 Furthermore, the spleen accumulated more NPs than the liver, which is consistent with previous reports that uncoated nanoparticles concentrated in the liver and spleen, but with the addition of PEGylation, this biodistribution was shifted toward the spleen.48
Finally, the mice were euthanized, and organs (brain, lung, heart, liver, spleen, kidney, and tumor) were sectioned for immunofluorescence imaging and colocalization studies (Figure 7). Based on previous studies showing the accumulation of NPs in immune system cells,49 we stained the sections with A488-labeled antimouse antibodies specific for CD45 for immune system cells, CD68 for macrophages, and F4/80 for Kuffer cells (all shown in green). The PhMV particles were colocalized by scanning for Cy5.5 fluorescence (pseudocolored white), and nuclei were counterstained with DAPI (blue). We identified some immune system cells (CD45+ and CD68+) in the tumor, colocalized with aggregated PhMV particles (Figures 7 and S6). In addition to the tumor localization, we also detected particles in the spleen and liver colocalized with CD68+ and F4/80+ cells (macrophages and Kuffer cells, respectively). A very weak signal in the kidney indicated that a small quantity of particles is cleared by the renal system, but there was almost no fluorescence in the brain, lung, or heart (Figure S7). The uptake of NPs by phagocytes is well documented.2 NPs can be digested by tumor-resident mononuclear cells or transported to tumors via monocytes.2 Our results revealed that the immune cells could deliver NPs to the tumor after some have been captured by immune cells in the bloodstream, which result in prolonged (up to 10 days) intratumor signal enhancement.To determine whether cellular uptake occurs while particles are in blood circulation, we performed intravital imaging by a two-photon laser scanning microscopy (2PLSM), which allows the real-time monitoring of the vasculature and circulation and is an excellent tool for in situ quantitative pharmacokinetics analysis of fluorescent NPs.50 The central nervous system (CNS) vasculature of nude mice (n = 3) was imaged through cranial windows using a stereotactic holder to stabilize the cranium.51 As shown in Figure 7B, there was no signal before the injection of NPs, whereas the dye-tagged NPs immediately produced homogeneous fluorescence within the vasculature after NP administration. The bright signal lasted for at least 2 h with only minor fading, indicating a prolonged half-life in the circulation. Notably, we observed some bright red spots (green arrow) flowing in the circulation, reflecting the uptake and transportation of NPs (Video 1, Supporting Information). These results show that immune cells in the bloodstream contribute to the prolonged half-life and enhanced tumor accumulation of our NPs by cell transportation.
Figure 7.

(A) Histological analysis of cryosectioned tissue slices from mice (n = 6) by confocal microscopy after bimodal scanning (240 h postinjection). The scale bar in the tumor and spleen is 30 μm and in the liver is 100 μm. Sections were stained with A488-labeled antimouse CD45 for immune cells, CD68 for macrophage cells, and F4/80 for Kuffer cells (green). The PhMV particles were colocalized by scanning for Cy5.5 (pseudocolored white). Nuclei were stained with DAPI (blue). (B) Intravital imaging for real-time monitoring of A647-PhMV-mPEG (pseudocolored red) in the circulation of nude mice (n = 3) up to 120 min. The green arrow indicates cell uptake and transport of A647-PhMV-mPEG in the blood (scale bar = 150 μm).
Pharmacokinetic Profile and the Protein Corona of Gd-Cy5.5-PhMV-mPEG NPs.
Pharmacokinetic (PK) analysis of the Gd-Cy5.5-PhMV-mPEG NPs was carried out following the intravenous injection of BALB/c mice (n = 6) with 200 μg Gd-Cy5.5-PhMV-mPEG NPs, as described above for in vivo imaging. Blood was collected prior to injection, and up to 72 h postadministration, sera were prepared from the blood, and the ID% was determined by comparing the fluorescence to a standard curve based on Gd-Cy5.5-PhMV-mPEG samples of known concentration (Figure 8A). The data were fitted for a two-phase decay using GraphPad Prism to determine the circulation half-life, revealing a t1/2 fast of 133 min and a t1/2 slow of 2616 min (p < 0.001). This biphasic decay is consistent with RES clearance via liver and spleen.52 The PK analysis of our NPs revealed a longer circulation half-time in mice compared with that of other plant virus NPs or VLPs, such as potato virus X (12.5 min),53 CPMV (20.8 min),37 TMV-PEG (~10 min), and serum-albumin-modified TMV (~100 min),54 and synthetic NPs, such as dextran-coated Fe3O4 (37.4–150 min).55
Figure 8.

(A) Pharmacokinetic analysis of Gd-Cy5.5-PhMV-mPEG NPs in BALB/c mice. The NPs were administered intravenously at 200 μg/mouse (n = 6 mice per group). Blood was collected before and after injection (at 10 min, 30 min, 60 min, 2 h, 4 h, 6 h, 8 h, 12 h, 30 h, 48 h, 72 h). (B) SDS-PAGE analysis of the protein corona of Gd-Cy5.5-PhMV-mPEG NPs after incubation for 1 h in ~100% human plasma.
To gain further insight into the in vivo fate of the PhMV NPs, we studied their hard protein corona in human plasma (Figure 8B). Nanomaterials in biological fluids are known to interact with proteins, sugars, and lipids to form a corona, the outer layer of which is dynamic, whereas the inner layer (or hard corona) remains associated with the NP surface and confers a new biological identity upon the particle of high scientific relevance.56 Although the composition of the protein corona is strongly dependent on the material, size, and surface properties of the NP, dissecting the composition of the corona in a given biological fluid can help to determine the fate of NPs by predicting their interactions with different cell types and surface receptors, and also their half-life in vivo. We incubated Gd-Cy5.5-PhMV-mPEG particles with ~100% human plasma for 1 h followed by centrifugation to pellet the particles and extensive washing to remove all unbound or weakly bound proteins (soft corona), allowing us to focus on the strongly bound hard corona. The hard corona proteins were fractionated by SDS-PAGE as previously described and analyzed by liquid chromatography mass spectrometry.57
Among the ~3700 known human plasma proteins,58 some of which have previously been isolated from the hard corona of NPs,59,60 we detected and quantified 64 different proteins, as summarized in Table S2. The most abundant components of the hard corona were complement factors, accounting for ~34.5% of the total corona protein mass, followed by immunoglobulins (22.0%), serum albumin (5.6%), coagulation factors (2.3%), acute-phase proteins (2.2%), and lipoproteins (1.8%). The binding of opsonins such as complement factors and immunoglobulins is expected to promote phagocytosis and the eventual removal of NPs from the circulation via the RES, leading to NP accumulation in the liver and spleen,42,61 but also in the tumor.2 The complement system is a critical component of the innate immune system in the blood (5% of globulins) comprising more than 30 proteins that have been consistently identified in NP coronas by proteomic analysis.62,63 Complement factors have been found in the coronas of various NP formulations, including carbon nanotubes, micelles, liposomes, polymeric nanospheres, iron oxide, silica, gold nanoparticles, and TMV nanorods.57,64,65 The complexity of the plasma corona is driven by the dynamic exchange of proteins in the soft corona, as well as thousands of proteins competing for limited space on the NP surface. The immunological protein corona that forms around NPs is kinetically unstable due to dynamic exchange in vivo, as shown by the injection of Fe3O4 nanoworms precoated with human plasma into mice and the subsequent rapid loss of complement component C3 and other adsorbed proteins.64 Therefore, we postulate that our NPs should undergo a similar dynamic process in vivo. This could help to explain why our NPs have a prolonged circulation time and sustained tumor-homing capability in vivo even though more than 50% of the in vitro protein corona consists of complement proteins and immunoglobulins: some NPs lose their initial corona and absorb dysopsonins to escape clearance by macrophages, e.g., serum albumin and coagulation protein have been shown to extend NP circulation times in the blood by antagonizing the activity of NP-bound opsonins.65 Lipoproteins can also govern the in vitro and in vivo fate of NPs, e.g., ApoE carries NPs through the blood–brain barrier and ApoA4/ApoC3 may act as dysopsonins.66 Furthermore, nonspecific cellular uptake can be inhibited if polymer-modified nanocarriers are precoated with clusterin.66 All of these studies demonstrated the ability of protein coronas to increase the circulation half-life of NPs and facilitate their uptake by specific cells in vivo. Therefore, our PK and protein corona data confirm that long-circulating PhMV NPs are systemically taken up by phagocytes and their efficient accumulation in tumors reflects a combination of passive homing (EPR and cell transportation) and the active targeting of integrins.
CONCLUSIONS
We have developed an integrated NIRF and MRI bimodal contrast agents based on PhMV NPs. These NPs persist in the circulation, can target specific cells, and achieve efficient tumor delivery in vivo due to their special structure and surface modifications, the composition of the protein corona, and the delivery by cells of the innate immune system. PhMV NPs therefore offer an excellent platform for the future long-term monitoring of lesions and the development of innovative drug delivery vehicles.
METHODS
Preparation of PhMV VLPs.
The PhMV VLPs were prepared by expressing the coat protein subunits in E. coli as previously described, with modifications.7
Bioconjugation Reactions.
Internal cysteine residues in the PhMV particles were conjugated to maleimide-functionalized Cy5.5 and DOTA with further Gd3+ coordination. Purified PhMV particles were mixed with a 0.2 molar equiv per coat protein (Eq/CP) of Cy5.5-maleimide at a concentration of 1 mg/mL in borate buffer (0.1 M, pH 6.5) and 15% dimethyl sulfoxide overnight at room temperature and then 5 Eq/CP of DOTA-maleimide overnight. The resulting product (DOTA-Cy5.5-PhMV) was purified by sucrose cushion ultracentrifugation (0.1 mL of 30% sucrose cushion, 51 000 rpm, 1 h). The pellet was dissolved in borate buffer (0.05 M, pH 6.0), and a molar excess 10 Eq/CP of Gd3+ solution was added for coordination with DOTA at room temperature overnight. The products were purified by ultracentrifugation as above and dialysis in 0.01 M phosphate buffer (pH 7.0) overnight. The resulting Gd-Cy5.5-PhMV particles were dispersed in phosphate buffer at 1 mg/mL for external modification. For the control particles, the reaction was carried out with 10 Eq/CP poly(ethylene glycol) succinimidyl ester (mPEG-NHS, 2000 Da; Nanocs Inc., New York, NY) to generate the product Dy-Cy5.5-PhMV-mPEG. For the targeted particles, the reaction was carried out with 10 Eq/CP azide poly(ethylene glycol) maleimide (NHS-PEG-Mal, MW 2000 Da; Nanocs Inc.) to generate an intermediate that was centrifuged as above and redispersed in 10 mM phosphate buffer (pH 6.5) before reacting with 0.5 Eq/CP DGEA-cys for 3 h at room temperature. An excess of ethanethiol was added and incubated for a further 2 h to block the PEG-maleimide group. The final Gd-Cy5.5-PhMV-mPEG and Gd-Cy5.5-PhMV-DGEA particles were dialyzed against PBS (pH 7.4) for 4 h, concentrated, and stored in PBS.
Particle Characterization.
The ζ potential of the VLPs (25 mg/mL) was determined using a 90Plus device (Brookhaven Instruments). Surface modification was confirmed by TEM, SDS-PAGE, and agarose gel electrophoresis (see the Supporting Information). UV–vis absorption and fluorescence emission were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). The concentration of Gd3+ ions was determined using a 730-ES inductively coupled plasma optical emission spectrometry (ICP-OES) instrument (Agilent Technologies, Santa Clara, CA).
Quantitative T1 Mapping by MRI.
The ionic relaxivity of the VLP-based contrast agents was determined using a Brukman Minispec mq60 relaxometer (60 MHz) and a BioSpec 70/30USR preclinical 7.0 T (300 MHz) (Bruker Inc., Billerica, MA). For MRI phantom studies, the same Biospec 7 T scanner equipped with a 3 cm birdcage 1H coil (Bruker, Erlangen, Germany) was used. The imaging parameters are detailed in the Supporting Information.
In Vivo NIRF Imaging and MRI.
Mice were anesthetized for all procedures (2.5% isoflurane, O2 flow 2.0 L/min), and their respiration, body temperature, and heart rate were monitored in real time. They were scanned before and 1, 4, 8, 32, 48, 72, 144, 192, and 240 h after the injection of 100 μL nanoparticles (2 mg/mL) in PBS through the tail vein (n = 6 for each time point, dose = 8 mg/kg). Both in vivo NIRF imaging and in vivo MR imaging were performed at each time point. NIRF imaging was scanned by using an IVIS 200 small-animal imaging system (Xenogen, Alameda, CA). Images were acquired and analyzed using Living Image v2.5 software (Xenogen). During MR imaging, mice were anesthetized with isoflurane, and the respiration rate and body temperature were monitored and maintained at 70–80/min and 35 ± 0.1 °C, respectively. The in vivo MRI parameters were the same as those used for the phantom scan (see the Supporting Information). However, NAV = 4 was used in spiral SRLL sequence for in vivo studies to improve the signal-to-noise ratio. Images were acquired before and after injection. T1-mapping images were statistically analyzed using Matlab software (MathWorks, Natick, MA). After bimodal imaging, the mice were euthanized immediately, the tumor and major organs (brain, lung, heart, liver, spleen, and kidneys) were removed, and ex vivo fluorescence images were obtained using the same settings described for in vivo fluorescence imaging. The tissues were weighed and digested in concentrated nitric acid overnight and heated to 90 °C for a further 6 h before the Gd content was determined by ICP-OES. Values are presented as means plus standard deviations for n = 6 mice per group.
In Vivo Intravital Imaging.
A Leica SP5 confocal microscope equipped with a ~20 Å water immersion lens (Leica HCX-APO-L, N.A. 1.0) and a tunable 16 W Ti/sapphire IR laser tuned to 800 nm (Chameleon Coherent, Inc.) was used for 2PLSM intravital imaging. Four-dimensional XYZT images with an XY dimension of 760 Å × 760 μm and a Z dimension of 100–130 μm (collected at 5 μm Z intervals) were obtained at 20 s intervals. In a parallel study, an independent cohort of nude mice (nu/nu, n = 3) was used to image the CNS vasculature. These mice were maintained on a Teklad 2018 s alfalfa free diet (Harlan Lab) starting 2 weeks before imaging to reduce autofluorescence. Open cranial windows were prepared, as previously described.51,67 Each mouse was administered 200 μg of A647-PhMV-mPEG (~2.56 × 1013 particles). Intravital imaging was carried out from preinjection to 2 h postinjection. The data sets were analyzed using Imaris (BitPlane, Inc.) to generate images and to determine clearance.
In Vivo PK Analysis.
Particles (4 mg/mL stock in 10 mM PBS pH 7.4) at a dose of 200 μg/mouse were administered by tail vein injection to BALB/c mice (Charles River). Blood (100 μL) of each mouse was collected before and after injection via retro-orbital bleed. Blood collection was at the following time points: t = 0 min, 10 min, 30 min, 60 min, 2 h, 4 h, 6 h, 8 h, 12 h, 30 h, 48 h, and 72 h, with n = 6 for each time point. A total of 24 mice were divided into four groups; each group was bled at only three time points, and the interval between each time point was at least 4 h in accordance with our IACUC protocol. Blood was collected into heparin-coated capillaries and plasma separated by centrifugation (2000g for 10 min). The fluorescence (λex = 573 and λem = 637 nm) of the plasma samples was determined using a Tecan Infinite M200 plate reader; a standard curve was established to determine the concentration of NPs and then normalized to the %ID.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by a grant from the National Institutes of Health (R01-CA202814) to N.F.S. The authors thank Drs. Jay T. Myers and Alex Y. Huang (Case Western Reserve University) for their help with the intravital imaging. H.M. acknowledges USIEF (United States–India Education Foundation, New Delhi), Department of Biotechnology (DBT, New Delhi), Prof. H.S. Savithri, and Prof. M.R.N. Murthy (IISc, Bengaluru) for the support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.9b03956.
Detailed schematic of chemical modification of PhMV nanoparticles, tumor xenografts, and MRI parameters, dynamic light scattering (DLS), comparison of r1-relaxivity of Gd3+-based nanoparticles, molecular structure of the macrocyclic Gd complexes, cytotoxicity assay, histological analysis, identified proteins in the corona surrounding Gd-Cy5.5-PhMV-mPEG (PDF) Intravital imaging (MPG)
The authors declare no competing financial interest.
REFERENCES
- (1).Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater 2016, 1, No. 16014. [Google Scholar]
- (3).Wang S; Huang P; Chen X Hierarchical Targeting Strategy for Enhanced Tumor Tissue Accumulation/Retention and Cellular Internalization. Adv. Mater 2016, 28, 7340–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wen AM; Steinmetz NF Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem. Soc. Rev 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rohovie MJ; Nagasawa M; Swartz JR Virus-Like Particles: Next-Generation Nanoparticles for Targeted Therapeutic Delivery. Bioeng. Transl. Med 2017, 2, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ranjith-Kumar CT; Gopinath K; Jacob ANK; Srividhya V; Elango P; Savithri HS Genomic Sequence of Physalis Mottle Virus and Its Evolutionary Relationship with Other Tymoviruses. Arch. Virol 1998, 143, 1489–1500. [DOI] [PubMed] [Google Scholar]
- (7).Sastri M; Kekuda R; Gopinath K; Ranjith Kumar CT; Jagath R; Savithri HS Assembly of Physalis Mottle Virus Capsid Protein in Escherichia coli and the Role of Amino and Carboxy Termini in the Formation of the Icosahedral Particles. J. Mol. Biol 1997, 272, 541–552. [DOI] [PubMed] [Google Scholar]
- (8).Krishna SS; Hiremath CN; Munshi SK; Prahadeeswaran D; Sastri M; Savithri HS; Murthy MRN Three-Dimensional Structure of Physalis Mottle Virus: Implications for the Viral Assembly. J. Mol. Biol 1999, 289, 919–934. [DOI] [PubMed] [Google Scholar]
- (9).Frietze KM; Peabody DS; Chackerian B Engineering Virus-Like Particles as Vaccine Platforms. Curr. Opin. Virol 2016, 18, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shirbaghaee Z; Bolhassani A Different Applications of Virus-Like Particles in Biology and Medicine: Vaccination and Delivery Systems. Biopolymers 2016, 105, 113–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lua LHL; Connors NK; Sainsbury F; Chuan YP; Wibowo N; Middelberg APJ Bioengineering Virus-Like Particles as Vaccines. Biotechnol. Bioeng 2014, 111, 425–440. [DOI] [PubMed] [Google Scholar]
- (12).Hu H; Zhang Y; Shukla S; Gu Y; Yu X; Steinmetz NF Dysprosium-Modified Tobacco Mosaic Virus Nanoparticles for Ultra-High-Field Magnetic Resonance and Near-Infrared Fluorescence Imaging of Prostate Cancer. ACS Nano 2017, 11, 9249–9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Masarapu H; Patel BK; Chariou PL; Hu H; Gulati NM; Carpenter BL; Ghiladi RA; Shukla S; Steinmetz NF Physalis Mottle Virus-Like Particles as Nanocarriers for Imaging Reagents and Drugs. Biomacromolecules 2017, 18, 4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Datta A; Hooker JM; Botta M; Francis MB; Aime S; Raymond KN High Relaxivity Gadolinium Hydroxypyridonate–Viral Capsid Conjugates: Nanosized MRI Contrast Agents1. J. Am. Chem. Soc 2008, 130, 2546–2552. [DOI] [PubMed] [Google Scholar]
- (15).Garimella PD; Datta A; Romanini DW; Raymond KN; Francis MB Multivalent, High-Relaxivity MRI Contrast Agents Using Rigid Cysteine-Reactive Gadolinium Complexes. J. Am. Chem. Soc 2011, 133, 14704–14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Prasuhn DE Jr.; Yeh RM; Obenaus A; Manchester M; Finn MG Viral MRI contrast agents: coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cyclo-addition. Chem. Commun 2007, 1269–1271. [DOI] [PubMed] [Google Scholar]
- (17).Dharmarwardana M; Martins AF; Chen Z; Palacios PM; Nowak CM; Welch RP; Li S; Luzuriaga MA; Bleris L; Pierce BS; Sherry AD; Gassensmith JJ Nitroxyl Modified Tobacco Mosaic Virus as a Metal-Free High-Relaxivity MRI and EPR Active Superoxide Sensor. Mol. Pharmaceutics 2018, 15, 2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li X; Kim J; Yoon J; Chen X Cancer-Associated, Stimuli-Driven, Turn on Theranostics for Multimodality Imaging and Therapy. Adv. Mater 2017, 29, No. 1606857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hu H; Zhou H; Du J; Wang Z; An L; Yang H; Li F; Wu H; Yang S Biocompatiable Hollow Silica Microspheres as Novel Ultrasound Contrast Agents for in Vivo Imaging. J. Mater. Chem 2011, 21, 6576–6583. [Google Scholar]
- (20).Odedina FT; Ogunbiyi JO; Ukoli FAM Roots of Prostate Cancer in African-American Men. J. Natl. Med. Assoc 2006, 98, 539–543. [PMC free article] [PubMed] [Google Scholar]
- (21).Grzesiak JJ; Bouvet M The α2β1 Integrin Mediates the Malignant Phenotype on Type I Collagen in Pancreatic Cancer Cell Lines. Br. J. Cancer 2006, 94, 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hu H; Li D; Liu S; Wang M; Moats R; Conti PS; Li Z Integrin α2β1 Targeted GdVO4:Eu Ultrathin Nanosheet for Multimodal PET/MR Imaging. Biomaterials 2014, 35, 8649–8658. [DOI] [PubMed] [Google Scholar]
- (23).Sri Krishna S; Sastri M; Savithri HS; Murthy MRN Structural Studies on the Empty Capsids of Physalis Mottle Virus. J. Mol. Biol 2001, 307, 1035–1047. [DOI] [PubMed] [Google Scholar]
- (24).Tsoi KM; MacParland SA; Ma X-Z; Spetzler VN; Echeverri J; Ouyang B; Fadel SM; Sykes EA; Goldaracena N; Kaths JM; Conneely JB; Alman BA; Selzner M; Ostrowski MA; Adeyi OA; Zilman A; McGilvray ID; Chan WCW Mechanism of Hard-Nanomaterial Clearance by the Liver. Nat. Mater 2016, 15, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Trevaskis NL; Kaminskas LM; Porter CJH From Sewer to Saviour — Targeting the Lymphatic System to Promote Drug Exposure and Activity. Nat. Rev. Drug Discovery 2015, 14, 781. [DOI] [PubMed] [Google Scholar]
- (26).Hema M; Nagendrakumar SB; Yamini R; Chandran D; Rajendra L; Thiagarajan D; Parida S; Paton DJ; Srinivasan VA Chimeric Tymovirus-Like Particles Displaying Foot-and-Mouth Disease Virus Non-Structural Protein Epitopes and Its Use for Detection of FMDV-NSP Antibodies. Vaccine 2007, 25, 4784–4794. [DOI] [PubMed] [Google Scholar]
- (27).Hauck TS; Ghazani AA; Chan WCW Assessing the Effect of Surface Chemistry on Gold Nanorod Uptake, Toxicity, and Gene Expression in Mammalian Cells. Small 2008, 4, 153–159. [DOI] [PubMed] [Google Scholar]
- (28).Helm L Optimization of Gadolinium-Based MRI Contrast Agents for high Magnetic-Field Applications. Future Med. Chem 2010, 2, 385–396. [DOI] [PubMed] [Google Scholar]
- (29).Hu H; Arena F; Gianolio E; Boffa C; Di Gregorio E; Stefania R; Orio L; Baroni S; Aime S Mesoporous Silica Nanoparticles Functionalized with Fluorescent and MRI Reporters for the Visualization of Murine Tumors Overexpressing avp3 Receptors. Nanoscale 2016, 8, 7094–7104. [DOI] [PubMed] [Google Scholar]
- (30).An L; Hu H; Du J; Wei J; Wang L; Yang H; Wu D; Shi H; Li F; Yang S Paramagnetic Hollow Silica Nanospheres for in Vivo Targeted Ultrasound and Magnetic Resonance Imaging. Biomaterials 2014, 35, 5381–5392. [DOI] [PubMed] [Google Scholar]
- (31).Le DHT; Hu H; Commandeur U; Steinmetz NF Chemical Addressability of Potato Virus X for Its Applications in Bio/Nanotechnology. J. Struct. Biol 2017, 200, 360–368. [DOI] [PubMed] [Google Scholar]
- (32).Caravan P; Farrar CT; Frullano L; Uppal R Influence of Molecular Parameters and Increasing Magnetic Field Strength on Relaxivity of Gadolinium- and Manganese-Based T1 Contrast Agents. Contrast Media Mol. Imaging 2009, 4, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hefferon K Repurposing Plant Virus Nanoparticles. Vaccines 2018, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Huang CW; Li Z; Conti PS In Vivo Near-Infrared Fluorescence Imaging of Integrin α2β1 in Prostate Cancer with Cell-Penetrating-Peptide–Conjugated DGEA Probe. J. Nucl. Med 2011, 52, 1979–1986. [DOI] [PubMed] [Google Scholar]
- (35).Salaam AD; Hwang P; McIntosh R; Green HN; Jun H-W; Dean D Nanodiamond-DGEA Peptide Conjugates for Enhanced Delivery of Doxorubicin to Prostate Cancer. Beilstein J. Nanotechnol 2014, 5, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hartshorn CM; Bradbury MS; Lanza GM; Nel AE; Rao J; Wang AZ; Wiesner UB; Yang L; Grodzinski P Nanotechnology Strategies To Advance Outcomes in Clinical Cancer Care. ACS Nano 2018, 12, 24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shukla S; Ablack AL; Wen AM; Lee KL; Lewis JD; Steinmetz NF Increased Tumor Homing and Tissue Penetration of the filamentous Plant Viral Nanoparticle Potato Virus X. Mol. Pharmaceutics 2013, 10, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Niu M; Naguib YW; Aldayel AM; Shi Y.-c.; Hursting SD; Hersh MA; Cui Z Biodistribution and in Vivo Activities of Tumor-Associated Macrophage-Targeting Nanoparticles Incorporated with Doxorubicin. Mol. Pharmaceutics 2014, 11, 4425–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dahlberg AM; Kaminskas LM; Smith A; Nicolazzo JA; Porter CJH; Bulitta JB; McIntosh MP The Lymphatic System Plays a Major Role in the Intravenous and Subcutaneous Pharmacokinetics of Trastuzumab in Rats. Mol. Pharmaceutics 2014, 11, 496–504. [DOI] [PubMed] [Google Scholar]
- (40).Volpes R; van den Oord JJ; Desmet VJ Distribution of the VLA Family of Integrins in Normal and Pathological Human Liver Tissue. Gastroenterology 1991, 101, 200–206. [DOI] [PubMed] [Google Scholar]
- (41).Maeda H The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: the Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzyme Regul 2001, 41, 189–207. [DOI] [PubMed] [Google Scholar]
- (42).Dobrovolskaia MA; McNeil SE Immunological Properties of Engineered Nanomaterials. Nat. Nanotechnol 2007, 2, 469. [DOI] [PubMed] [Google Scholar]
- (43).Viswanathan S; Kovacs Z; Green KN; Ratnakar SJ; Sherry AD Alternatives to Gadolinium-Based Metal Chelates for Magnetic Resonance Imaging. Chem. Rev 2010, 110, 2960–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mi P; Kokuryo D; Cabral H; Wu H; Terada Y; Saga T; Aoki I; Nishiyama N; Kataoka K A pH-Activatable Nanoparticle with Signal-Amplification Capabilities for Non-Invasive Imaging of Tumour Malignancy. Nat. Nanotechnol 2016, 11, 724. [DOI] [PubMed] [Google Scholar]
- (45).Bruckman MA; Jiang K; Simpson EJ; Randolph LN; Luyt LG; Yu X; Steinmetz NF Dual-Modal Magnetic Resonance and Fluorescence Imaging of Atherosclerotic Plaques in Vivo Using VCAM-1 Targeted Tobacco Mosaic Virus. Nano Lett. 2014, 14, 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Drugs.com. Feridex Dosage. https://www.drugs.com/dosage/feridex.html (accessed Oct 25, 2016).
- (47).Huang CW; Li Z; Cai H; Shahinian T; Conti PS Novel α2β1 Integrin-Targeted Peptide Probes for Prostate Cancer Imaging. Mol. Imaging 2011, 10, 284–294. [DOI] [PubMed] [Google Scholar]
- (48).Li S-D; Huang L Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharmaceutics 2008, 5, 496–504. [DOI] [PubMed] [Google Scholar]
- (49).Shukla S; Eber FJ; Nagarajan AS; DiFranco NA; Schmidt N; Wen AM; Eiben S; Twyman RM; Wege C; Steinmetz NF The Impact of Aspect Ratio on the Biodistribution and Tumor Homing of Rigid Soft-Matter Nanorods. Adv. Healthcare Mater 2015, 4, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Leong HS; Steinmetz NF; Ablack A; Destito G; Zijlstra A; Stuhlmann H; Manchester M; Lewis JD Intravital Imaging of Embryonic and Tumor Neovasculature Using Viral Nanoparticles. Nat. Protoc 2010, 5, 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Shukla S; Dorand RD; Myers JT; Woods SE; Gulati NM; Stewart PL; Commandeur U; Huang AY; Steinmetz NF Multiple Administrations of Viral Nanoparticles Alter in Vivo Behavior—Insights from Intravital Microscopy. ACS Biomater. Sci. Eng 2016, 2, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Li S-D; Huang L Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharmaceutics 2008, 5, 496–504. [DOI] [PubMed] [Google Scholar]
- (53).Lee KL; Shukla S; Wu M; Ayat NR; El Sanadi CE; Wen AM; Edelbrock JF; Pokorski JK; Commandeur U; Dubyak GR; Steinmetz NF Stealth Filaments: Polymer Chain Length and Conformation Affect the in Vivo Fate of PEGylated Potato Virus X. Acta Biomater. 2015, 19, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pitek AS; Jameson SA; Veliz FA; Shukla S; Steinmetz NF Serum Albumin ‘Camouflage’ of Plant Virus Based Nanoparticles Prevents Their Antibody Recognition and Enhances Pharmacokinetics. Biomaterials 2016, 89, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Abdollah MRA; Carter TJ; Jones C; Kalber TL; Rajkumar V; Tolner B; Gruettner C; Zaw-Thin M; Baguña Torres J; Ellis M; Robson M; Pedley RB; Mulholland P; de Rosales RTM; Chester KA Fucoidan Prolongs the Circulation Time of Dextran-Coated Iron Oxide Nanoparticles. ACS Nano 2018, 12, 1156–1169. [DOI] [PubMed] [Google Scholar]
- (56).Monopoli MP; Pitek AS; Lynch I; Dawson KA Formation and Characterization of the Nanoparticle–Protein Corona In Nanomaterial Interfaces in Biology: Methods and Protocols; Bergese P, Hamad-Schifferli K, Eds.; Humana Press: Totowa, NJ, 2013; pp 137–155. [DOI] [PubMed] [Google Scholar]
- (57).Pitek AS; Wen AM; Shukla S; Steinmetz NF The Protein Corona of Plant Virus Nanoparticles Influences their Dispersion Properties, Cellular Interactions, and In Vivo Fates. Small 2016, 12, 1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Anderson NL; Polanski M; Pieper R; Gatlin T; Tirumalai RS; Conrads TP; Veenstra TD; Adkins JN; Pounds JG; Fagan R; Lobley A The Human Plasma Proteome: A Non-redundant List Developed by Combination of Four Separate Sources. Mol. Cell. Proteomics 2004, 3, 311–326. [DOI] [PubMed] [Google Scholar]
- (59).Aggarwal P; Hall JB; McLeland CB; Dobrovolskaia MA; McNeil SE Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv. Drug Delivery Rev 2009, 61, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Dobrovolskaia MA; Patri AK; Zheng J; Clogston JD; Ayub N; Aggarwal P; Neun BW; Hall JB; McNeil SE Interaction of Colloidal Gold Nanoparticles with Human Blood: Effects on Particle Size and Analysis of Plasma Protein Binding Profiles. Nanomedicine 2009, 5, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dobrovolskaia MA; Germolec DR; Weaver JL Evaluation of Nanoparticle Immunotoxicity. Nat. Nanotechnol 2009, 4, 411. [DOI] [PubMed] [Google Scholar]
- (62).Lundqvist M; Stigler J; Elia G; Lynch I; Cedervall T; Dawson KA Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tenzer S; Docter D; Kuharev J; Musyanovych A; Fetz V; Hecht R; Schlenk F; Fischer D; Kiouptsi K; Reinhardt C; Landfester K; Schild H; Maskos M; Knauer SK; Stauber RH Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol 2013, 8, 772. [DOI] [PubMed] [Google Scholar]
- (64).Chen F; Wang G; Griffin JI; Brenneman B; Banda NK; Holers VM; Backos DS; Wu L; Moghimi SM; Simberg D Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat. Nanotechnol 2017, 12, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Tenzer S; Docter D; Rosfa S; Wlodarski A; Kuharev J; Rekik A; Knauer SK; Bantz C; Nawroth T; Bier C; Sirirattanapan J; Mann W; Treuel L; Zellner R; Maskos M; Schild H; Stauber RH Nanoparticle Size Is a Critical Physicochemical Determinant of the Human Blood Plasma Corona: A Comprehensive Quantitative Proteomic Analysis. ACS Nano 2011, 5, 7155–7167. [DOI] [PubMed] [Google Scholar]
- (66).Schöttler S; Becker G; Winzen S; Steinbach T; Mohr K; Landfester K; Mailänder V; Wurm FR Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly- (phosphoester)-coated nanocarriers. Nat. Nanotechnol 2016, 11, 372. [DOI] [PubMed] [Google Scholar]
- (67).Dorand RD; Barkauskas DS; Evans TA; Petrosiute A; Huang AY Comparison of Intravital Thinned Skull and Cranial Window Approaches to Study CNS Immunobiology in The Mouse Cortex. IntraVital 2014, 3, No. e29728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.