

Abstract
Recent evidence indicates that elevated placental adenosine signaling contributes to preeclampsia (PE). However, the molecular basis for the chronically enhanced placental adenosine signaling in PE remains unclear. Here we report that hypoxia inducible factor-1α (HIF-1α) is crucial for the enhancement of placental adenosine signaling. Utilizing a pharmacologic approach to reduce placental adenosine levels, we found that enhanced adenosine underlies increased placental HIF-1α in an angiotensin receptor type 1 receptor agonistic autoantibody (AT1-AA)-induced mouse model of PE. Knockdown of placental HIF-1α in vivo suppressed the accumulation of adenosine and increased CD73 and adenosine A2B receptor (ADORA2B) in the placentas of PE mouse models induced by AT1-AA or LIGHT, a TNF superfamily cytokine (TNFSF14). Human in vitro studies using placental villous explants demonstrated that increased HIF-1α resulting from ADORA2B activation facilitates the induction of CD73, ADORA2B, and FLT-1 expression. Overall, we demonstrated that i) elevated placental HIF-1α by AT1-AA or LIGHT upregulates CD73 and ADORA2B expression and ii) enhanced adenosine signaling through upregulated ADORA2B induces placental HIF-1α expression, which creates a positive feedback loop that promotes FLT-1 expression leading to disease development. Our results suggest that adenosine-based therapy targeting the malicious cycle of placental adenosine signaling may elicit therapeutic effects on PE.
Keywords: preeclampsia, adenosine signaling, hypoxia inducible factor-1α, angiotensin II type I receptor agonistic autoantibody (AT1-AA), tumor necrosis factor superfamily member 14 (TNFSF14)
Introduction
Preeclampsia (PE) is a systemic obstetric complication, characterized by new onset hypertension during pregnancy and primarily associated with serious perinatal outcomes (1, 2). Although abnormal placentation and associated placental dysfunction are known to be important factors in the pathogenesis of PE, the molecular mechanisms involved remain largely unknown, and currently there is no effective treatment for PE. Therefore, the elucidation of the underlying molecular mechanisms of PE leading to the development of novel treatment strategies is desperately needed to achieve the better perinatal outcomes.
Adenosine, an endogenous nucleoside, is a signaling molecule that serves an important function in physiological responses in the body and in the pathogenesis of various diseases (3, 4). Hypoxia and other stress conditions result in the release of ATP that is converted to adenosine by the sequential action of CD39 and CD73, two ecto-nucleotidases anchored on the cell surface (5). The function of extracellular adenosine is exerted via activation of adenosine receptors, which are of four types. Adenosine has a short half-life, and rapidly elevated adenosine signaling generally plays a protective role in the body, such as protecting against ischemic stress-induced tissue injury in the brain and heart. However, chronically elevated adenosine signaling contributes to the onset of chronic disease by triggering tissue injury and dysfunction, including chronic kidney disease, chronic pulmonary fibrotic diseases, priapism, and sickle cell disease (5–7). Intriguingly, levels of adenosine were reportedly increased in the circulation of both mother and fetus in PE and were correlated with disease severity (8, 9). Using a genetically-engineered mouse model with placenta-specific elevated adenosine and a pathogenic autoantibody-induced PE mouse model, we recently showed that placental adenosine levels and the expression of adenosine A2B receptor (ADORA2B) are elevated and that a local increase of adenosine in the placenta is sufficient to trigger key PE features through the activation of ADORA2B (10). Enhanced placental adenosine signaling via activation of ADORA2B impairs placenta development and induces the PE phenotype. Furthermore, as reported in our study involving a murine model and the placentas of PE patients (10), placental upregulation of CD73 is a key molecular mechanism that results in excessive placental accumulation of adenosine leading to PE development. Thus, our prior report provided solid evidence that enhanced adenosine signaling in the placenta is an important pathogenic factor for PE. However, the mechanisms by which the placental expression of CD73 and ADORA2B are regulated and elevated in the placentas of PE patients remains unknown. In other words, the molecular mechanisms that underlie how placental adenosine signaling is amplified and persistently enhanced in the pathogenesis of PE are not yet determined.
Hypoxia-inducible factor-1α (HIF-1α) is a key transcription factor that orchestrates the cellular response to hypoxia under physiologic and pathologic conditions (11). HIF-1α is elevated in the placentas of PE patients and promotes enhanced transcription of genes encoding soluble fms-like tyrosine kinase-1 (sFlt-1), soluble endoglin (sEng) and endothelin-1 (ET-1), all of which play key roles in the pathogenesis of PE (12–14). A growing body of evidence indicates that hypoxia-independent factors, including inflammatory cytokines and growth factors, also stimulate HIF-1α activity (13, 15, 16). Recently, we reported that hypoxia-independent upregulation of placental HIF-1α is induced by two independent pathogenic factors for PE, an angiotensin II type 1 receptor agonistic autoantibody (AT1-AA) (17, 18) and the inflammatory cytokine tumor necrosis factor superfamily member 14 (LIGHT) (19, 20). The resulting elevation in HIF-1α contributes to the development of PE through the production of sFlt-1 (21). However, the pathologic roles of sustained elevated placental HIF-1α in PE are not fully understood. Intriguingly, adenosine signaling is reportedly controlled by HIF-1α in various chronic pathologic conditions (22). HIF-1α directly regulates the transcription of both CD73 and ADORA2B, and several reports have indicated that transcriptionally upregulated CD73 and ADORA2B by HIF-1α plays important roles in the pathophysiology of chronic kidney disease, inflammatory bowel disease, intestinal ischemia, and acute lung injury (22–26). Thus, we hypothesized that placental upregulated HIF-1α underlies the elevation of CD73 and ADORA2B in PE placentas and contributes to sustain persistently enhanced adenosine signaling, which leads to disease progression. Here we sought to assess this hypothesis by conducting both mouse and human studies.
Methods
Animals
Wild type (WT) eight to ten-week-old timed pregnant C57BL/6 mice (mated with syngeneic males) were obtained from Harlan Laboratories (Indianapolis, IN, USA). The mice were housed in the animal care facility of the University of Texas McGovern Medical School and had access to food and water ad libitum. All protocols were reviewed and approved by the Institutional Animal Welfare Committee.
Introduction of human autoantibody (AT1-AA) or LIGHT into pregnant mice
PE mouse models induced by AT1-AA or LIGHT were conducted. Briefly, purified IgGs were isolated from preeclamptic (PE) or normotensive (NT) pregnant patient sera (PE-IgG, NT-IgG respectively) as previously described (10, 17, 21). Eight to ten-week-old timed-pregnant wild type dams mated with wild type males were treated with NT-IgG or PE-IgG (0.8mg) on E13.5 and E14.5 by retro-orbital sinus injection. Some wild type dams also received polyethylene glycol-linked adenosine deaminase (PEG-ADA) (5 units) on E13.5 together with IgG injection by retro-orbital sinus injection. For LIGHT injection experiments, recombinant mouse LIGHT (2 ng; R&D Systems, Minneapolis, MN, USA) or the same volume of saline was introduced into pregnant mice by retro-orbital sinus injection on E13.5 and E14.5 as previously reported (19, 21). For neutralization experiments, anti-mouse lymphotoxin β receptor (LTβR) monoclonal antibody and anti-mouse herpes virus entry mediator (HVEM) monoclonal antibody were utilized. Briefly, hybridoma cells that specifically secrete rat anti-mouse HVEM monoclonal antibody or rat anti-mouse LTβR monoclonal antibody were cultured in DMEM/F12 supplemented with 10% low-IgG FBS (Life Technologies Inc., Carlsbad, CA, USA) and 25 mM HEPES as previously described (19). Supernatant was harvested and monoclonal antibodies were purified using G-Sepharose column chromatography (GE Healthcare, Chicago, IL, USA). Either LTβR monoclonal antibody (100 μg) or HVEM monoclonal antibody (100 μg) was simultaneously coinjected with LIGHT. All mice were sacrificed on E18.5 prior to delivery, and their blood and organs were collected.
In vivo siRNA-induced knockdown of Hif-1α in pregnant mice
To knockdown Hif-1α mRNA levels in PE-IgG or LIGHT-injected pregnant mice, a siRNA knockdown method (Altogen Biosystems, Las Vegas, NV, USA) using nanoparticles to surround siRNA constructs was used as previously reported (21, 27). Briefly, scrambled siRNA control or Hif-1α siRNA (Sigma, St. Louis, MO, USA) encapsulated in nanoparticles was prepared according to instructions from the company (Altogen Biosystems) and was administered on E13.5 and E14.5 by retro-orbital sinus injection together with PE-IgG or LIGHT.
The measurement of blood pressure and proteinuria
The systolic blood pressure of all mice was measured at the same time daily by tail cuff plethysmography using a carotid catheter-calibrated system (CODA, Kent Scientific, Torrington, CT, USA) as previously described(17, 21). The mice were kept warm using a warming pad (AD Instruments Co, Dunedin, New Zealand). For the measurement of proteinuria, urine was collected for analysis using metabolic cages. Total microalbumin and creatinine in the urine were determined by ELISA (Exocell, Philadelphia, PA, USA) and the ratio of urinary albumin to creatinine was calculated as an index of proteinuria as previously described (17, 21).
Measurement of adenosine levels
Samples were rapidly removed and collected in liquid nitrogen. Adenosine was extracted from frozen placenta tissues or plasma using 0.6 N perchloric acid, separated and quantified using reversed phase high-performance liquid chromatography (HPLC) as previously described (6, 10, 24).
Real-time RT-PCR analysis
RNA isolation and real-time RT-PCR were conducted as previously described (21). Syber green was used for the analysis of all transcripts measured using the primers listed in Table. 1.
Table 1.
PCR primers used in this study.
| Mouse Hif-1α | forward | 5’-GAAATGGCCCAGTGAGAAAA-3’ |
| reverse | 5’-CTTCCAVGTTGCTGACTTGA-3’ | |
| Mouse Cd73 | forward | 5’-CAGATCCGCAAGGAAGAACC-3’ |
| reverse | 5’-ATGGTGCCCTGGTACTGGTC-3’ | |
| Mouse Adora2b | forward | 5’-GCGAGAGGGATCATTGCTG-3’ |
| reverse | 5’- CAGGAACGGAGTCAATCCAA-3’ | |
| Mouse Gapdh | forward | 5’-TGACCTCAACTACATGGTCTACA-3’ |
| reverse | 5’- CTTCCCATTCTCGGCCTTG-3’ | |
| Human HIF-1α | forward | 5’-TGCTCATCAGTTGCCACTTC-3’ |
| reverse | 5’-TCCTCACACGCAAATAGCTG-3’ | |
| Human CD73 | forward | 5’-ACCACGTATCCATGTGCATTT-3’ |
| reverse | 5’-AAAGGGCAATACAGCAGCCAG-3’ | |
| Human ADORA2B | forward | 5’-TGCACTGACTTCTACGGCTG-3’ |
| reverse | 5’-GGTCCCCGTGACCAAACTT-3’ | |
| Human FLT-1 | forward | 5’-TTTGCCTGAAATGGTGAGTAAGG-3’ |
| reverse | 5’-TGGTTTGCTTGAGCTGTGTTC-3’ | |
| Human GAPDH | forward | 5’-TGCACCACCAACTGCTTAGC-3’ |
| reverse | 5’-ACAGTCTTCTGGGTCGCAGTG-3’ | |
Immunohistochemistry
Formalin fixed tissue blocks were cut into 4-μm thick sections and subjected to immunohistochemistry as previously described (21). Briefly, endogenous peroxidase activity was quenched by 10 min incubation in a 3% hydrogen peroxide/methanol buffer. Antigen retrieval was conducted by incubating slides in sodium citrate buffer (pH 6.0) at 89°C for 15 min. After blocking with the normal goat serum, the slides were then incubated with antibody against mouse HIF-1α (1:100, LS-B2823, Lifespan Biosciences, Seattle, WA, USA) in a humidified chamber at 4°C overnight. After the primary antibody incubation, ABC staining system kit (VEACTASTAIN, VECTOR LAB, Burlingame, CA, USA) was used according to the manufacturer’s suggested protocol. Antigen-antibody reactions were visualized with a dual alkaline phosphatase/fluorescence development system (VECTOR Red Substrate Kit, VECTORLAB). The slides were counterstained with Mayer’s hematoxylin.
Assessment of histopathologic changes in kidneys by periodic acid-Schiff (PAS) staining
Kidneys were dissected from the mice on E18.5, fixed in 4% formaldehyde and embedded in paraffin. Tissue blocks were cut into 4μm sections and stained with periodic acid-Schiff (PAS) by standard procedures as previously described (10). The extent of renal damage was assessed by quantifying the glomeruli that showed characteristic features of damage in PE such as decreased Bowman’s space and occlusion of capillary loop spaces. To examine those features, the glomeruli were counted in 5 fields of randomized and blinded slides (10x magnification), with each field having at least 10 glomeruli. A highest score of 5 was accorded to glomeruli with a normal amount of capillary space within Bowman’s capsule. A score of 1 was assigned to the glomeruli that showed complete loss of capillary space and an intermediate score of 3 was assigned to the glomeruli that displayed reduced, but not completely obliterated, capillary space.
Immunoblotting
Immunoblotting was performed as previously described (21). Briefly, samples were lysed with RIPA lysis buffer (Santa Cruz, Dallas, TX, USA) in the presence of proteinase inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Lysates were resolved on SDS–PAGE and electroblotted onto polyvinylidene difluoride membranes. After blocking with Odyssey Blocking Buffer (LI-COR, Lincoln, NE, USA), the membranes were probed with antibody against human/mouse HIF-1α (1:1000, LS-B2823, Lifespan Biosciences), and then probed with secondary antibodies labeled with IRDye fluorophores (LI-COR). The antibody/antigen complexes were scanned and detected using the ODYSSEY infrared imaging system and software (LI-COR).
Measurement of circulating sFlt-1 levels
sFlt-1 levels in mouse plasma were quantitatively determined by using ELISA kits which are commercially available (Qantikine ELISA) (R&D Systems) as previously described (21).
Patients
Patients who were admitted to Memorial Hermann Hospital were identified by the obstetrical faculty of the University of Texas McGovern Medical School at Houston. Preeclampsia patients were diagnosed with severe disease based on the definition set by the National High Blood Pressure Education Program Working Group Report (28). Preeclampsia patients with the presence of new-onset severe hypertension after 20 weeks of gestation (systolic blood pressure >160 and/or diastolic blood pressure >110 mmHg), and/or organ dysfunction such as neurological complication, acute kidney injury, elevated liver enzymes, and low platelets were included in this study. Control pregnant women were selected on the basis of having an uncomplicated, normotensive (NT) pregnancy, with an expected normal-term delivery. The research protocol was approved by the Institutional Committee for the Protection of Human Subjects.
Human Placental Villous Explant Culture
Human placentas were obtained from normal pregnant women without any complications who delivered vaginally between 38 weeks and 40 weeks of gestation at Memorial Hermann Hospital in Houston. The explant culture system was conducted as described previously (21) On delivery, the placentas were immediately placed on ice and submerged in phenol red–free DMEM containing 10% BSA and antibiotics. Villous explant fragments weighing 50mg were dissected from the placenta and transferred to 24-well plates at 37°C under 5% CO2. The explants were incubated for 24 hours and then treated with NT- or PE-IgG (100μg /ml) or recombinant human LIGHT (100 pg/mL) (R&D Systems). The explants were incubated for 24 hours and then pretreated with or without HIF-1α inhibitors (10μM CAY10585 or 1μM Chrysin) (Santa Cruz) for 15 min and then treated with ADORA2B agonist (1μM BAY60–6583) (Tocris Biosciences, Bristol, UK).
Statistical analysis
All data are expressed as the mean ± SEM. Mann-Whitney’s U test was applied in two-group analysis. Comparison of the data obtained at different time points from multiple groups as repeated measurements were analyzed by two-way repeated measures analysis of variance, followed by the Bonferroni post hoc test. Differences among the means of multiple groups in every analysis except for ones described above were compared by the one-way analysis of variance, followed by a Tukey’s post hoc test. Statistical significance was set as P<0.05. Statistical programs were run by GraphPad Prism 5 statistical software (GraphPad, San Diego, CA, USA).
Results
Enhanced adenosine signaling underlies the upregulation of placental HIF-1α in AT1-AA-induced mouse model of PE.
To examine the possibility that HIF-1α might be controlled by chronically elevated placental adenosine signaling in PE, we took advantage of an established experimental mouse PE model induced by adoptive transfer of patient-derived-IgG (PE-IgG) known to contain the pathogenic autoantibodies, AT1-AA. Additionally, we took a pharmacological approach by using polyethylene glycol-modified adenosine deaminase (PEG-ADA), a safe drug used to treat ADA-deficient patients to lower adenosine levels resulting from their enzyme deficiency. First, we found that PEG-ADA treatment significantly suppressed the PE-IgG-induced increase in placental adenosine (Figure 1A). Importantly, PE-IgG-induced key PE features, hypertension and proteinuria, were significantly attenuated by treatment with PEG-ADA (Figure 1, B and C). Histologic analysis revealed that pathologic changes seen in the kidneys of PE-IgG-infused mice (i.e., smaller glomeruli with swelling and narrowing of capillary spaces) were attenuated in the kidneys of PEG-ADA-treated mice (Figure 1, D and E). Placental distress is often associated with PE and is observed in PE-IgG-injected pregnant mice (29). We found that placentas of PE-IgG-injected mice treated with PEG-ADA displayed significantly less prominent placental calcifications, a hallmark of placental distress observed in placentas of PE patients, as compared with those of PE-IgG-injected mice without PEG-ADA treatment (Figure 1F and G). We found that levels of circulating sFlt-1, an antiangiogenic factor that plays central roles in the development of systemic endothelial dysfunction leading to the manifestation of PE features (30), induced by PE-IgG were significantly reduced by treatment with PEG-ADA (Figure 1H). Thus, these results revealed not only the pathophysiological significance of enhanced adenosine signaling in PE, but also the therapeutic possibilities against PE by using PEG-ADA to suppress adenosine signaling.
Figure 1. Suppression of placental adenosine levels by the treatment of PEG-ADA ameliorates the preeclamptic features in pregnant mice induced by AT1-AA.

Pregnant mice were injected with purified IgG from normotensive pregnant women (NT-IgG) or PE patients (PE-IgG) on E13.5 and E14.5. PEG-ADA (5 units per mouse) was co-injected with PE-IgG on E13.5. Samples were collected on E18.5.
(A) Levels of adenosine in mouse placentas were measured by HPLC. (NT-IgG: n=4, PE-IgG: n=8, PE-IgG+PEG-ADA: n=5, **P<0.01 vs NT-IgG, ##P<0.01 vs PE-IgG)
(B) Blood pressure was measured by tail-cuff plethysmography on a daily base. (n=5–8 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG at the same time point)
(C) Proteinuria was determined as urine albumin to creatinine ratio by ELISA. (n=5–8 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG)
(D) Renal histology assessed by PAS staining. Pathologic changes in kidneys of PE-IgG-treated WT mice (swollen glomeruli with narrowed capillary and Bowman’s spaces) were suppressed by PEG-ADA. Scale bar, 100μm.
(E) Histological changes were quantified as the highest score of 5 accorded to a normal glomerulus to the lowest score of 1 assigned to the glomerulus that showed complete loss of capillary space. (n=4–6 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG)
(F) Placental histology assessed by H&E staining. Placental damage in the labyrinth zone of PE-IgG-treated WT mice (calcification; arrows, the disorganization of tissue resulting in abnormal blood pooling; inset box) was reduced by treatment with PEG-ADA. Scale bar, 100 μm.
(G) The number of calcifications obtained per field under X10 magnification is quantified. (n=4–6 per group, **P<0.01 vs NT-IgG, ##P<0.01 vs PE-IgG)
(H) The level of circulating sFlt-1 in mouse plasma was determined by ELISA. (n=4–6 mice per group, **P<0.01 vs NT-IgG, ##P<0.01 vs PE-IgG)
Next, we examined the effect of adenosine suppression by PEG-ADA on the placental expression of HIF-1α. Immunohistochemical analysis revealed that the PE-IgG induced placental HIF-1α protein, largely concentrated in nuclei, was suppressed by PEG-ADA treatment (Figure 2A). We also found that PEG-ADA treatment significantly suppressed the increased placental HIF-1α mRNA expression induced by PE-IgG (Figure 2B). These results provide significant in vivo evidence that placental upregulation of HIF-1α is closely associated with chronically elevated adenosine signaling in the AT1-AA-induced PE mouse model.
Figure 2. Elevated placental adenosine underlies the increased placental expression of HIF-1α in AT1-AA-induced PE mouse model.
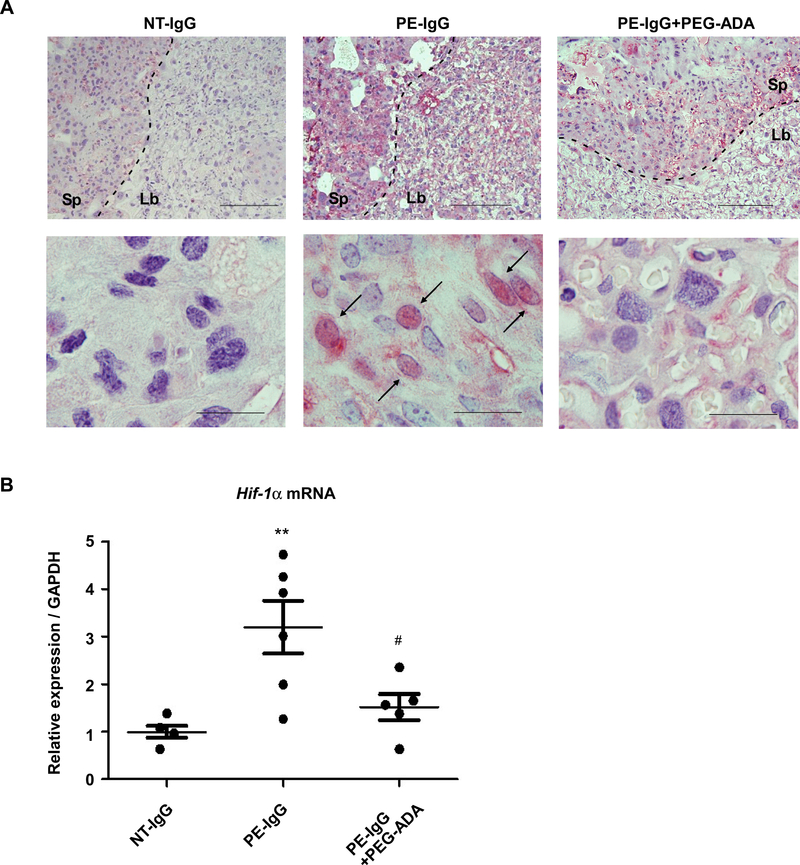
Pregnant mice were injected with purified IgG from normotensive pregnant women (NT-IgG) or PE patients (PE-IgG) on E13.5 and E14.5. PEG-ADA (5 units per mouse) was co-injected with PE-IgG on E13.5. Samples were collected on E18.5.
(A) HIF-1α protein in mouse placentas was detected by immunohistochemistry. Antigen-antibody reactions were visualized with an alkaline phosphatase (pink). Arrows indicate the cells with positive staining in the nucleus. Scale bar: upper, 500μm; lower, 50μm. Sp: spongiotrophoblast zone, Lb: labyrinth zone.
(B) Placental Hif-1α mRNA levels were quantified by real-time RT-PCR. (n=4–6 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG).
Increased placental HIF-1α induced by AT1-AA contributes to excess accumulation of adenosine, upregulation of Cd73 and Adora2b in the placenta.
To examine a potential involvement of HIF-1α in enhanced placental adenosine signaling and PE, we conducted siRNA-induced in vivo knockdown of Hif-1α. As shown in Figure 3A, Hif-1α siRNA injection successfully suppressed placental HIF-1α protein levels in PE-IgG-treated mice compared with those mice injected with control scrambled siRNA. Real time RT-PCR of mouse placentas revealed that HIF-1α knockdown repressed the elevation of Cd73 and Adora2b gene expression induced by PE-IgG (Figure 3, B and C). As shown in Figure 3D, we also found that the excess accumulation of adenosine caused by injection of PE-IgG was suppressed significantly in placentas of Hif-1α siRNA-injected pregnant mice. These results suggest that the induction of HIF-1α in the placenta is responsible for the elevation of placental adenosine, increased CD73 (which could contribute to adenosine accumulation), and enhanced ADORA2B signaling.
Figure 3. Increased placental HIF-1α induced by AT1-AA is responsible for the excess accumulation of adenosine and increased expression of Cd73 and Adora2b in the placenta.

Pregnant mice were injected with purified IgG from normotensive pregnant women (NT-IgG) or PE patients (PE-IgG) on E13.5 and E14.5, and in vivo siRNA-mediated Hif-1α mRNA knockdown was performed. Samples were collected on E18.5.
(A) HIF-1α protein in the placentas of PE-IgG treated pregnant mice co-injected with control siRNA (Con-siRNA) or Hif-1a siRNA was detected by immunoblotting.
(B) Placental Cd73 mRNA levels were quantified by real-time RT-PCR. (n=5 or 6 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG+con-si)
(C) Placental Adora2b mRNA levels were quantified by real-time RT-PCR. (n=5 or 6 mice per group, **P<0.01 vs NT-IgG, #P<0.05 vs PE-IgG+con-si)
(D) Adenosine levels in mouse placentas were quantified by HPLC. (n=4 or 5 mice per group, **P<0.01 vs NT-IgG, ##P<0.01 vs PE-IgG+con-si)
Increased placental HIF-1α mediates LIGHT-induced excess accumulation of adenosine and upregulation of Cd73 and Adora2b in the placenta.
TNF superfamily member 14, LIGHT, is elevated in the circulation and placentas of PE patients, and the injection of LIGHT into pregnant mice induces features of PE including the overproduction of sFlt-1 (19). Additionally, we recently reported that elevated placental HIF-1α contributes to the development of LIGHT-induced PE features by using a LIGHT infusion mouse model of PE and siRNA-induced in vivo knockdown of Hif-1α (21). Thus, to determine whether elevated HIF-1α and excessive production of placental adenosine is a general pathogenic mechanism for PE, we took advantage of using a PE mouse model based on infusion of LIGHT. First, we found that LIGHT injection into pregnant mice resulted in increased levels of adenosine in placentas (Figure 4A). Next, we found that LIGHT-induced placental adenosine production was significantly reduced by neutralizing antibodies specific for LIGHT receptors: lymphotoxin β receptor (LTβR) and herpes virus entry mediator (HVEM) (Figure 4A). We have already reported that LIGHT injection into pregnant mice resulted in increased expression of HIF-1α (21). Similarly, injection of LIGHT into pregnant mice induced Cd73 and Adora2b gene expression in mouse placentas and their induction was inhibited by neutralizing antibodies to LIGHT receptors (Figure 4, B and C). These results demonstrated that LIGHT signaling via its receptors also contributes to excess placental adenosine production and elevated expression of Cd73 and Adora2b.
Figure 4. LIGHT signaling via its receptors contributes to excess placental adenosine production and elevated expression of Cd73 and Adora2b in the placenta.

Saline or LIGHT with or without neutralizing antibodies against lymphotoxin β receptor (anti-LTβR) or herpes virus entry mediator (anti-HVEM) was injected into pregnant mice on E13.5 and E14.5. In vivo siRNA-mediated Hif-1α mRNA knock down was conducted. Samples were collected on E18.5.
(A) Adenosine levels in mouse placentas were determined by HPLC. (n=4–7 per group, **P<0.01 vs Saline, #P<0.05 vs LIGHT, ✝P<0.05 vs LIGHT+con-si)
(B) Placental Cd73 mRNA levels were quantified by real-time RT-PCR. (n=3–6 per group, **P<0.05 vs Saline, #P<0.05 vs LIGHT, ✝P<0.05 vs LIGHT+con-si)
(C) Placental Adora2b mRNA levels were quantified by real-time RT-PCR. (n=3–6 per group, **P<0.01 vs Saline, #P<0.05 vs LIGHT, ✝✝P<0.01 vs LIGHT+con-si)
To assess whether HIF-1α contributes to placental excess adenosine in the LIGHT-injection model of PE as it does in PE-IgG-injected mice, we conducted in vivo knock-down of HIF-1α by injecting siRNA-embedded nanoparticles, together with LIGHT, into pregnant mice. Hif-1α siRNA injection successfully reduced the levels of placental Hif-1α mRNA (data not shown) and suppressed the LIGHT-induced elevation of Cd73 and Adora2b mRNA levels, as well as excess accumulation of adenosine in mouse placentas (Figure 4, A–C). Altogether, these findings indicate that enhanced placental adenosine signaling, which contributes to the development of PE features, is mediated by HIF-1α in experimental models of PE using either PE-IgG or LIGHT infusion.
HIF-1α induced by the activation of ADORA2B facilitates the activation of CD73, ADORA2B, and FLT-1 gene expression in human villous explants.
To extend our mouse findings to humans, we cultured human villous explants isolated from normal pregnant women and then treated the placental explants with PE-IgG or LIGHT under ambient oxygen levels. First, we found that PE-IgG induced the expression of CD73 mRNA in cultured human villous explants independent of hypoxia (Figure 5A). PE-IgG-induced expression of CD73 was significantly suppressed by the inhibition of AT1 receptors by losartan or the AT1-AA epitope peptide, AFHYESQ. We also found that PE-IgG induced the expression of ADORA2B mRNA and that the PE-IgG-induced expression of ADORA2B was significantly inhibited by losartan or the AT1-AA epitope peptide in cultured human villous explants (Figure 5A). In addition, we found that LIGHT induced the expression of CD73 and ADORA2B in cultured human villous explants (Figure 5B). These results indicate that enhanced placental adenosine signaling through the upregulation of CD73 and activation of ADORA2B is closely related to PE pathophysiology induced by AT1-AA or LIGHT in women with PE.
Figure 5. Upregulated HIF-1α via ADORA2B activation is responsible for the induction of CD73, ADORA2B, and FLT-1 gene expression in human villous explants.

(A) Human villous explants were treated with 100μg/ml NT-IgG or PE-IgG for 24 hours in the presence or absence of 5uM Losartan (Los) or 1uM autoantibody-neutralizing 7 amino acid epitope peptide (AFHYESQ, termed 7a.a). CD73 or ADORA2B mRNA levels were quantified using real-time RT-PCR. (n=3 independent experiments, *P<0.05, **P<0.01 vs NT-IgG, #P<0.05, ##P<0.01 vs PE-IgG)
(B) CD73 or ADORA2B mRNA levels in cultured human villous explants treated with 100pg/ml LIGHT or PBS for 24 hours were quantified using real-time RT-PCR. (n=3 independent experiments, *P<0.05, **P<0.01)
(C) HIF-1α mRNA levels in cultured human villous explants treated with ADORA2B agonist (1μM BAY60–6583) for 24 hours were quantified using real-time RT-PCR. (n=3 independent experiments, **P<0.01)
(D) HIF-1α protein determined by immunoblotting. Human villous explants were treated with 1μM BAY60–6583 for indicated hours or 100μM CoCl2 for 30 min as a positive control of HIF-1α production.
(E) Human villous explants were incubated for 24 hours and then pretreated with or without HIF-1α inhibitors (10μM CAY10585 or 1μM Chrysin) for 15 min and then treated with ADORA2B agonist (1μM BAY60–6583) for 24 hours. CD73, ADORA2B, and FLT-1 mRNA levels were determined by real-time RT-PCR. (n=3 independent experiments, **P<0.01, #P<0.05, ##P<0.01)
(F) Working model: The reinforcing reciprocal regulation of HIF-1α and adenosine signaling is the key driving force for their persistent elevation in the placenta. The malicious cycle of enhanced adenosine signaling in which HIF-1α plays a central role is a common causative factor underlying the pathophysiology of PE induced by AT1-AA or LIGHT.
Next, to determine whether ADORA2B-mediated HIF-1α induction reciprocally upregulates CD73 and ADORA2B gene expression, we treated human villous explants with an ADORA2B agonist in the presence or absence of two specific HIF-1α inhibitors. First, we found that BAY60–6583, a specific ADORA2B agonist, significantly induced HIF-1α expression at both mRNA and protein levels in villous explants (Figure 5, C and D). And, BAY60–6583 induced both CD73 and ADORA2B gene expression in cultured human villous explants (Figure 5E). In contrast, treatment with HIF-1α inhibitors, CAY10585 or Chrysin, significantly reduced ADORA2B agonist-induced CD73 and ADORA2B gene expression (Figure 5E). Thus, these results provide direct evidence that ADORA2B activation positively controls the expression of CD73 and ADORA2B in the human placenta through HIF-1α.
Next, we assessed whether ADORA2B activation was involved in increased FLT-1 gene expression in cultured human villous explants. We found that BAY60–6583 directly induced FLT-1 mRNA gene expression (Figure 5E). Because HIF-1α is known to induce FLT-1 gene expression (31), to test the possibility that HIF-1α functions downstream of ADORA2B activation is responsible for increased FLT-1 gene expression, we treated human villous explants with ADORA2B agonist, BAY60–6583, in the presence or absence of HIF-1α inhibitors, CAY10585 or Chrysin. We found that ADORA2B activation-mediated increased FLT-1 gene expression was significantly attenuated by HIF-1α inhibitors (Figure 5E), indicating that ADORA2B activation induces FLT-1 gene expression in a HIF-1α dependent manner in cultured human villous explants.
Overall, our human studies support our mouse findings indicating that i) PE-IgG or LIGHT, as hypoxia-independent factors, directly induce HIF-1α expression, ii) elevated HIF-1α upregulates CD73 and ADORA2B gene expression and iii) enhanced adenosine signaling through upregulated ADORA2B induces placental HIF-1α expression, which in turn creates a positive feedback loop that promotes FLT-1 gene expression leading to disease development. (Figure 5F).
Discussion
Using two mouse models of PE and human in vitro studies, the current study revealed that placental HIF-1α controls the expression of CD73 and ADORA2B as well as adenosine levels in the placenta. Also, placental HIF-1α is reciprocally induced by chronically elevated adenosine signaling through the production of adenosine by CD73, and enhanced activation of ADORA2B. Our results indicate that HIF-1α plays a central role in triggering and persistently enhancing placental adenosine signaling, which contributes to the progression of PE.
Our previous report revealed that the pathogenic effects of elevated adenosine signaling in PE were mediated by the upregulation of placental CD73 leading to the excess accumulation of adenosine and the activation of the adenosine receptor, ADORA2B, which was also significantly elevated in placentas from women with PE and from the mouse models of PE (10). In the current study, our hypothesis that placental HIF-1α accounted for increased ADORA2B and CD73 gene expression was supported by findings from two PE mouse models (induced by AT1-AA or LIGHT) and human in vitro studies using villous explant cultures. Placental expression of CD73 and ADORA2B, along with adenosine accumulation induced by AT1-AA or LIGHT, was suppressed by the knockdown of HIF-1α. Results from human villous explants demonstrated that the activation of ADORA2B induces upregulation of HIF-1α, CD73, and ADORA2B, and that the increased expression of CD73 and ADORA2B was controlled in a HIF-1α-dependent manner. Thus, our findings indicate that elevated HIF-1α results in the transcriptional activation of CD73 leading to increased adenosine production in PE placentas. HIF-1α also drives increased production of ADORA2B which is activated by the excessive accumulation of adenosine. Although ADORA2B has the lowest affinity for adenosine among the four adenosine receptors (3), in the setting of PE, elevated CD73 produces excessive placental adenosine, resulting in enhanced activation of ADORA2B. Furthermore, the abundance of ADORA2B is also significantly increased in PE placentas, providing enhanced opportunity for ADORA2B signaling leading to increased HIF-1α production, sFlt-1 gene activation and disease progression.
Additionally, results from AT1-AA-induced PE mouse models in which pharmacologic approaches (i.e. PEG-ADA) were used to reduce placental adenosine revealed that enhanced placental adenosine accumulation leads to the upregulation of Hif-1α in the placenta. We also found that the activation of ADORA2B provokes the induction of HIF-1α, which leads to the transcriptional upregulation of CD73 and ADORA2B in human villous explants. These results indicate that ADORA2B signaling provides a positive feedback loop that promotes continued production of HIF-1α. The result is a perpetual malicious cycle of CD73-mediated adenosine production and ADORA2B signaling that contributes to persistent elevation of HIF-1α and chronic activation of the sFlt-1 gene, which causes the development of pre-eclamptic features. The current study provides valuable evidence regarding the role of HIF-1α in contributing to persistently enhanced placental adenosine signaling leading to the manifestation of PE features.
Experimentally induced animal models of PE have played important roles in the identification and characterization of pathogenic factors contributing to the disease (2). However, because individual models have limitations, we utilized two independent mouse models in the present study, one based on injection of a pathogenic autoantibody (AT1-AA) and the other on the injection of an inflammatory cytokine (LIGHT). Previous studies have shown that both experimental models induce key features of PE, including hypertension, proteinuria and systemic endothelial dysfunction resulting from increased circulatory factors, such as sFlt-1 and endothelin-1 (17, 19). Moreover, these preeclamptic mice show glomerular endotheliosis, a characteristic renal pathology specifically seen in PE patients, which is caused by glomerular endothelial dysfunction triggered by factors such as sFlt-1 (17, 19). Because our two distinct experimental approaches led to the same results, we are confident that placental HIF-1α plays an essential role in the amplification of placental adenosine signaling in PE and that our result is valuable in elucidating mechanisms contributing to PE pathophysiology.
Roles of intracellular adenosine under the physiologic and pathologic conditions remain largely unknown. Recently Huang et al. reported that accumulated intracellular adenosine causes global placental DNA hypomethylation that is associated with PE (32). Aberrant gene expression in the placenta due to the epigenetic modulation caused by the changes in DNA methylation status is widely considered to be involved in the PE pathophysiology (33, 34). Huang et al. showed that placental DNA hypomethylation status related to PE can be induced independent of ADORA2B activation, through the uptake of adenosine into the cytoplasm by equilibrative nucleoside transporters (ENTs). Considering our current results, accumulated placental adenosine may contribute to PE by two distinct pathways of gene regulation: 1) Transcriptional regulation of genes related to PE by HIF-1α, through the elevated placental extracellular adenosine-mediated activation of ADORA2B, and 2) epigenetic changes of placental gene expression due to the accumulated intracellular adenosine-induced DNA hypomethylation. Our results indicate that these two pathways starting from elevated placental adenosine collaboratively work together to alter gene expression associated with PE development.
Our previous report revealed that enhanced placental adenosine signaling via adenosine ADORA2B activation induces the overproduction of sFlt-1, not only in the placentas but also in the maternal circulation (10). The current report adds new insight that ADORA2B activation is capable of inducing sFlt-1 in human placentas. Moreover, our previous report demonstrated that excess placental adenosine contributes to the failure in the formation of placental vasculature, which leads to placental dysfunction related to fetal growth restriction (10). Therefore, we expect that blocking the malicious cycle of adenosine signaling could elicit therapeutic benefit against PE not only through the recovery from the systemic endothelial dysfunction induced by the increased sFlt-1 released from the placenta to maternal circulation, but also through the amelioration of placental dysfunction. From this perspective interference with adenosine signaling, either by reducing adenosine levels or blocking ADORA2B activation, may reduce PE features and enable pregnancy to be extended.
It is well accepted that elevated HIF-1α plays an important role in regulating the expression of genes that maintain the proper trophoblast functions in the process of early placental development (35). Also, HIF-1α is considered critical for the cardiac development by regulating genes such as Hand1 (36). Therefore, considering the crucial roles of HIF-1α in the embryonic and placental development, it might be undesirable to block HIF-1α as a treatment option for PE patients. Instead, the current study demonstrated that suppression of placental adenosine levels by PEG-ADA treatment enabled the alleviation of key PE features, including hypertension and renal damage, in an experimental model of PE in pregnant mice. PEG-ADA is a drug which is approved by the FDA and has been used for the treatment of ADA-deficient individuals for more than three decades. Therapeutic strategies to suppress placental adenosine levels by PEG-ADA might elicit beneficial effects on PE not only by blocking the malicious cycle of enhanced placental extracellular adenosine signaling in which HIF-1α plays a central role, but also by hindering potentially detrimental epigenetic changes triggered by the increased intracellular adenosine. Based on the present study we hope that adenosine-based therapy can become a useful treatment option for PE.
Acknowledgements
This study was supported by National Institutes of Health Grant RC4HD067977, the Bob and Hazel Casey Endowed Chair in Biochemistry, and the Japan Society for the Promotion of Science grant 15H06171, 19K09774.
Abbreviation list:
- PE
preeclampsia
- HIF 1α
hypoxia inducible factor-1α
- AT1-AA
angiotensin receptor type 1 receptor agonistic autoantibody
- LIGHT
homologous to Lymphotoxin, exhibits Inducible expression and competes with HSV Glycoprotein D for binding to Herpesvirus entry mediator, a receptor expressed on T lymphocytes
- TNFSF14
tumor necrosis factor superfamily member 14
- ADORA2B
adenosine A2B receptor
- sFlt-1
soluble fms-like tyrosine kinase-1
- sEng
soluble endoglin
- ET-1
endothelin-1
- PEG-ADA
polyethylene glycol-modified adenosine deaminase
- LTβR
lymphotoxin β receptor
- HVEM
herpes virus entry mediator
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Steegers EA, von Dadelszen P, Duvekot JJ, and Pijnenborg R. (2010) Pre-eclampsia. Lancet 376, 631–644 [DOI] [PubMed] [Google Scholar]
- 2.Chaiworapongsa T, Chaemsaithong P, Yeo L, and Romero R. (2014) Pre-eclampsia part 1: current understanding of its pathophysiology. Nature reviews. Nephrology 10, 466–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen JF, Eltzschig HK, and Fredholm BB (2013) Adenosine receptors as drug targets--what are the challenges? Nature reviews. Drug discovery 12, 265–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karmouty-Quintana H, Xia Y, and Blackburn MR (2013) Adenosine signaling during acute and chronic disease states. Journal of molecular medicine 91, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, and Xia Y. (2012) Adenosine signaling in normal and sickle erythrocytes and beyond. Microbes and infection / Institut Pasteur 14, 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Zhang W, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, and Xia Y. (2011) Detrimental effects of adenosine signaling in sickle cell disease. Nature medicine 17, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mi T, Abbasi S, Zhang H, Uray K, Chunn JL, Xia LW, Molina JG, Weisbrodt NW, Kellems RE, Blackburn MR, and Xia Y. (2008) Excess adenosine in murine penile erectile tissues contributes to priapism via A2B adenosine receptor signaling. The Journal of clinical investigation 118, 1491–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espinoza J, Espinoza AF, and Power GG (2011) High fetal plasma adenosine concentration: a role for the fetus in preeclampsia? American journal of obstetrics and gynecology 205, 485 e424–487 [DOI] [PubMed] [Google Scholar]
- 9.Yoneyama Y, Suzuki S, Sawa R, Yoneyama K, Power GG, and Araki T. (2002) Increased plasma adenosine concentrations and the severity of preeclampsia. Obstetrics and gynecology 100, 1266–1270 [DOI] [PubMed] [Google Scholar]
- 10.Iriyama T, Sun K, Parchim NF, Li J, Zhao C, Song A, Hart LA, Blackwell SC, Sibai BM, Chan LN, Chan TS, Hicks MJ, Blackburn MR, Kellems RE, and Xia Y. (2015) Elevated placental adenosine signaling contributes to the pathogenesis of preeclampsia. Circulation 131, 730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajakumar A, Brandon HM, Daftary A, Ness R, and Conrad KP (2004) Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 25, 763–769 [DOI] [PubMed] [Google Scholar]
- 13.Pringle KG, Kind KL, Sferruzzi-Perri AN, Thompson JG, and Roberts CT (2010) Beyond oxygen: complex regulation and activity of hypoxia inducible factors in pregnancy. Human reproduction update 16, 415–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, and Caniggia I. (2006) Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. American journal of physiology. Regulatory, integrative and comparative physiology 291, R1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuschel A, Simon P, and Tug S. (2012) Functional regulation of HIF-1alpha under normoxia--is there more than post-translational regulation? Journal of cellular physiology 227, 514–524 [DOI] [PubMed] [Google Scholar]
- 16.Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, and Jelkmann W. (1999) Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 94, 1561–1567 [PubMed] [Google Scholar]
- 17.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, and Xia Y. (2008) Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nature medicine 14, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, and Luft FC (1999) Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. The Journal of clinical investigation 103, 945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, Parchim NF, Iriyama T, Luo R, Zhao C, Liu C, Irani RA, Zhang W, Ning C, Zhang Y, Blackwell SC, Chen L, Tao L, Hicks MJ, Kellems RE, and Xia Y. (2014) Excess LIGHT contributes to placental impairment, increased secretion of vasoactive factors, hypertension, and proteinuria in preeclampsia. Hypertension 63, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iriyama T, Wang G, Yoshikawa M, Mimura N, Matsui H, Sayama S, Kumasawa K, Nagamatsu T, Koga K, Kotani T, Niimi K, Yamamoto E, Kellems RE, Xia Y, Osuga Y, and Fujii T. (2019) Increased LIGHT leading to sFlt-1 elevation underlies the pathogenic link between hydatidiform mole and preeclampsia. Scientific reports 9, 10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iriyama T, Wang W, Parchim NF, Song A, Blackwell SC, Sibai BM, Kellems RE, and Xia Y. (2015) Hypoxia-independent upregulation of placental hypoxia inducible factor-1alpha gene expression contributes to the pathogenesis of preeclampsia. Hypertension 65, 1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poth JM, Brodsky K, Ehrentraut H, Grenz A, and Eltzschig HK (2013) Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. Journal of molecular medicine 91, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, and Eltzschig HK (2011) Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5’-nucleotidase (CD73) and the A2B adenosine receptor. Journal of immunology 186, 4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, Sun K, Glover L, Grenz A, Sun H, Tao L, Zhang W, Colgan SP, Blackburn MR, Eltzschig HK, Kellems RE, and Xia Y. (2013) Elevated ecto-5’-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circulation research 112, 1466–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckle T, Kewley EM, Brodsky KS, Tak E, Bonney S, Gobel M, Anderson D, Glover LE, Riegel AK, Colgan SP, and Eltzschig HK (2014) Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. Journal of immunology 192, 1249–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong T, Westerman KA, Faigle M, Eltzschig HK, and Colgan SP (2006) HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 20, 2242–2250 [DOI] [PubMed] [Google Scholar]
- 27.Liu C, Wang W, Parchim N, Irani RA, Blackwell SC, Sibai B, Jin J, Kellems RE, and Xia Y. (2014) Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination-preventing isopeptide modification. Hypertension 63, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts JM, Pearson G, Cutler J, Lindheimer M, and Pregnancy N. W. G. o. R. o. H. D. (2003) Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension 41, 437–445 [DOI] [PubMed] [Google Scholar]
- 29.Irani RA, Zhang Y, Blackwell SC, Zhou CC, Ramin SM, Kellems RE, and Xia Y. (2009) The detrimental role of angiotensin receptor agonistic autoantibodies in intrauterine growth restriction seen in preeclampsia. The Journal of experimental medicine 206, 2809–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maynard S, Epstein FH, and Karumanchi SA (2008) Preeclampsia and angiogenic imbalance. Annual review of medicine 59, 61–78 [DOI] [PubMed] [Google Scholar]
- 31.Gerber HP, Condorelli F, Park J, and Ferrara N. (1997) Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. The Journal of biological chemistry 272, 23659–23667 [DOI] [PubMed] [Google Scholar]
- 32.Huang A, Wu H, Iriyama T, Zhang Y, Sun K, Song A, Liu H, Peng Z, Tang L, Lee M, Huang Y, Ni X, Kellems RE, and Xia Y. (2017) Elevated Adenosine Induces Placental DNA Hypomethylation Independent of A2B Receptor Signaling in Preeclampsia. Hypertension 70, 209–218 [DOI] [PubMed] [Google Scholar]
- 33.Wilson SL, Leavey K, Cox BJ, and Robinson WP (2018) Mining DNA methylation alterations towards a classification of placental pathologies. Human molecular genetics 27, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Januar V, Desoye G, Novakovic B, Cvitic S, and Saffery R. (2015) Epigenetic regulation of human placental function and pregnancy outcome: considerations for causal inference. American journal of obstetrics and gynecology 213, S182–196 [DOI] [PubMed] [Google Scholar]
- 35.Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, and Post M. (2000) Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). The Journal of clinical investigation 105, 577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunwoodie SL (2009) The role of hypoxia in development of the Mammalian embryo. Developmental cell 17, 755–773 [DOI] [PubMed] [Google Scholar]