

Abstract
Background:
The link between modifiable breast cancer risk factors and tumor genomic alterations remains largely unexplored. We evaluated the association of pre-diagnostic body mass index (BMI), cigarette smoking, and alcohol consumption with somatic copy number variation (SCNV), total somatic mutation burden (TSMB), seven single base substitution (SBS) signatures (SBS1, SBS2, SBS3, SBS5, SBS13, SBS29, and SBS30), and nine driver mutations (CDH1, GATA3, KMT2C, MAP2K4, MAP3K1, NCOR1, PIK3CA, RUNX1, and TP53), in a subset of The Cancer Genome Atlas (TCGA).
Method:
Clinical and genomic data were retrieved from the TCGA database. Risk factor information was collected from four TCGA sites (n=219 women), including BMI (one year before diagnosis), cigarette smoking (smokers/non-smokers), and alcohol consumption (current drinkers/non-drinkers). Multivariable regression analyses were conducted in all tumors and stratified according to estrogen receptor (ER) status.
Results:
Increasing BMI was associated with increasing SCNV in all women (p=0.039) and among women with ER− tumors (p=0.031). Smokers had higher SCNV and TSMB versus non-smokers (p<0.05 all women). Alcohol drinkers had higher SCNV versus non-drinkers (p<0.05 all women and among women with ER+ tumors). SBS3 (defective homologous recombination-based repair) was exclusively found in alcohol drinkers with ER− disease. GATA3 mutation was more likely to occur in women with higher BMI. No association was significant after multiple testing correction.
Conclusion:
This study provides preliminary evidence that BMI, cigarette smoking, and alcohol consumption can influence breast tumor biology, in particular, DNA alterations.
Impact:
This study demonstrates a link between modifiable breast cancer risk factors and tumor genomic alterations.
Keywords: epidemiology, TCGA, total somatic mutational burden, somatic copy number variation, mutation
Introduction
Breast cancer is the most common cancer diagnosed in women in the United States (1). Understanding the molecular effects of epidemiological risk factors can provide insights into breast cancer etiology and progression, and may lead to better prevention efforts and treatment guidelines. Breast cancer risk factors can be classified as non-modifiable (e.g., family history (2) and previous diagnosis of benign breast disease (3)) or modifiable (e.g., being overweight (4), low physical activity (5), high alcohol consumption (6), and cigarette smoking (7,8)).
Using gene expression data, we previously evaluated the molecular influence of adiposity and alcohol consumption on breast cancer biology (9,10). Among postmenopausal women, we found a positive association between body mass index (BMI) and cellular proliferation pathways in estrogen receptor positive (ER+) breast tumors, whilst there were positive associations with inflammation pathways in women with ER− disease (9). Breast tumors from women who consumed more than 10 grams of alcohol per day displayed increased cellular proliferation compared to non-drinkers (10). Our work may partly explain why women with ER+ disease and high BMI have poorer prognosis (11,12), and provide new insights into alcohol-related breast tumorigenesis. Some studies have reported the association of breast cancer risk factors with breast tumor genomic alterations. Germline BRCA1/2 mutation, a hereditary breast cancer risk factor, is associated with increased genomic instability (13). TP53 somatic mutations are more frequent in breast tumors of early parous women and current smokers (14,15). However, the link between modifiable breast cancer risk factors and tumor genomic alterations remains largely unexplored.
The Cancer Genome Atlas (TCGA) is a large scale effort by the National Cancer Institute and National Human Genome Research Institute to understand the molecular basis of cancers (16). To date, over 1200 breast tumors have been characterized in TCGA using genomic, transcriptomic and proteomic profiling technologies (17). We obtained breast cancer risk factor information from a subset of 219 TCGA women. In this study, we evaluated the association of modifiable breast cancer risk factors with (1) somatic copy number variation (SCNV), (2) total somatic mutation burden (TSMB), (3) single base substitution (SBS) signatures, and (4) breast cancer driver mutations.
Materials and Methods
Study population
The Committee on the Use of Human Subjects in Research at Brigham and Women’s Hospital, Boston, MA reviewed and approved this study (Protocol Number: 2010P001641). Breast cancer risk factor data was sought from participants contributed by collaborators at four of the largest TCGA sites (the University of Pittsburgh (UP), Roswell Park Comprehensive Cancer Center (RPCCC), the Mayo Clinic (MC), and Memorial Sloan Kettering Cancer Center (MSKCC)) as previously detailed (9,10). Data were ultimately collected from 229 participants. Ten patients were subsequently excluded (1 male, 8 females redacted by TCGA, and 1 female with stage IV disease), resulting in 219 female breast cancer cases for this study.
Clinical and Epidemiological Variables
Age at diagnosis, year of diagnosis, race, disease stage, tumor grade, menopausal status, and immunohistochemistry results for ER, progesterone receptor (PR) and HER2 were retrieved from the TCGA clinical database. RPCCC and UP (53.4%) collected breast cancer risk data using a de novo self-administered questionnaire. Data from MC were previously collected as part of a case-control study (26.0%). Data from MSKCC were abstracted from patient medical records (20.5%). Three modifiable breast cancer risk factors (main exposures) were investigated in this study: BMI (kg/m2; one year before diagnosis), pre-diagnostic cigarette smoking use (smokers/non-smokers), and pre-diagnostic alcohol consumption (current drinkers/non-drinkers; Table 1).
Table 1.
Demographic and lifestyle characteristics of 219 The Cancer Genome Atlas (TCGA) women in this study.
| Clinical Characteristics | ||
| TCGA site, n (%) | Mayo Clinic | 57 (26.0) |
| Memorial Sloan Kettering Cancer Center | 45 (20.5) | |
| University of Pittsburgh | 44 (20.1) | |
| Roswell Park Comprehensive Cancer Center | 73 (33.3) | |
| Age at initial diagnosis, n (%) | <45 years | 45 (20.5) |
| ≥45 to <55 years | 69 (31.5) | |
| ≥55 to <65 years | 53 (24.2) | |
| ≥65 years | 52 (23.7) | |
| Disease stage, n (%) | I | 50 (22.8) |
| II | 130 (59.4) | |
| III | 39 (17.8) | |
| Tumor grade, n (%) | 1 | 37 (16.9) |
| 2 | 63 (28.8) | |
| 3 | 47 (21.5) | |
| Missing | 72 (32.9) | |
| Race, n (%) | White | 192 (87.7) |
| Non White | 20 (9.1) | |
| Missing | 7 (3.2) | |
| Estrogen receptor status, n (%) | Positive | 179 (81.7) |
| Negative | 40 (18.3) | |
| Progesterone receptor status, n (%) | Positive | 158 (72.1) |
| Negative | 61 (27.9) | |
| HER2 status, n (%) | Positive | 28 (12.8) |
| Negative | 186 (84.9) | |
| Missing | 5 (2.3) | |
| Modifiable Risk Factors | ||
| BMI, n (%) | <25 | 71 (32.4) |
| ≥25 to <30 | 53 (24.2) | |
| ≥30 | 77 (35.2) | |
| Missing | 18 (8.2) | |
| Pre-diagnostic cigarette smoking, n (%) | Smoker | 81 (37.0) |
| Non-smoker | 104 (47.5) | |
| Missing | 34 (15.5) | |
| Pre-diagnostic alcohol consumption, n (%) | Current drinker | 129 (58.9) |
| Non-drinker | 46 (21.0) | |
| Missing | 44 (20.1) |
BMI data were available for 43/44 (97.7%) UP participants, 62/73 (84.9%) RPCCC participants, 42/46 (91.3%) MSKCC participants, and 55/57 (96.5%) MC participants. For RPCCC and UP participants, BMI was calculated using self-reported weight at one year prior to breast cancer diagnosis and present height (without shoes) from the questionaire. For MSKCC participants, BMI was calculated using the weight (up to one year prior to breast cancer diagnosis) and height extracted from medical records. For MC participants, BMI was extracted from their previous case-control study database.
Smoking data were available for 44/44 (100%) UP participants, 63/73 (86.3%) RPCCC participants, 46/46 (100%) MSKCC participants, and 33/57 (57.9%) MC participants. At RPCCC and UP, participants self-reported their smoking status as never smoked, quit before diagnosis, or smoking at diagnosis on the questionaire. MSKCC abstracted smoking information from medical records as never smoked, quit before diagnosis, or smoking at diagnosis. For MC, we abstracted smoking information from their case-control study database: smoking up to one year prior to diagnosis or smoking currently at diagnosis. In total, 81 women reported smoking, 69 (85.2%) were past smokers, 11 (13.6%) were current smokers, and 1 (1.2%) reported smoking but current status was unknown. Cigarette smoking exposure was re-categorized into smokers (n=81) and non-smokers (n=104).
Alcohol data were available for 44/44 (100%) UP participants, 62/73 (84.9%) RPCCC participants, 36/46 (78.3%) MSKCC participants, and 33/57 (57.9%) MC participants. For RPCCC and UP, patients selected one out of seven categories on the questionaire that reflected the average number of alcohol drinks they consumed at ages 18, 30, 45, and 60: none, <1 drink per week, 1–6 drinks per week, 1 drink per day, 2 to 3 drinks per day, >3 drinks per day, and not applicable. MSKCC abstracted alcohol consumption up to the year before breast cancer diagnosis from medical records. Alcohol data from MC were the total number of alcohol drinks consumed in the year before breast cancer diagnosis and recorded as a continuous number ranging from 0 to 14. Data from MC were recoded to match the categories in our questionnaire: 0 remains coded as none (n=5), 0.5 drinks was recoded as <1 drink per week (n=5), 1 to 5 drinks was recoded as 1–6 drinks per week (n=18), 7 drinks were recoded as 1 drink per day (n=2), 8 to 14 drinks were recoded as 2 to 3 drinks per day (n=3), and 24 patients were coded as missing. In this manuscript, alcohol exposure wasdefined as the alcohol intake closest to but before diagnosis of breast cancer. The total numbers collected in the seven categories were: none (n=46; 21.0%), <1 drink per week (n=54; 24.7%), 1–6 drinks per week (n=57; 26.0%), 1 drink per day (n=10; 4.6%), 2 to 3 drinks per day (n=6; 2.7%), >3 drinks per day (n=2; 0.9%), and unknown (n=44; 20.1%). Alcohol exposure was also re-categorized into current drinkers (n=129) and non-drinkers (n=46).
Tumor genomic data
Tumor genomic data were downloaded from (http://cancergenome.nih.gov/): normalized SCNV (version 2024; mapped using GRCh38; Affymetrix Genome-Wide Human single nucleotide polymorphism Array 6.0) and somatic mutations (version gdc-1.0.0; derived from Illumina Genome Analyzer II whole exome sequencing and MuTect somatic mutation calls (18)). Of 219 participants, 218 had SCNV data and 192 had somatic mutation data. For SCNV, chromosomal segments that were quantitated by <10 probes were excluded and a cutoff of 0.2 was used to indicate a gain or loss segment (19); total SCNV was obtained by summing all the segments that passed the cutoff (i.e, ≥|0.2|). TSMB was calculated by summing all significant synonymous and nonsynonymous mutations for each case. SCNV and TSMB data were winsorized to 96% where data below the 2nd percentile were set to the value at the 2nd percentile and data above the 98th percentile were set to the value at the 98th percentile.
Twenty five distinct SBS signatures derived using TCGA whole exome sequencing data were available for 186 breast cancer cases (20–22). SBS signatures were derived by applying a previously established methodology (23) to the MC3 release of TCGA somatic mutations (24). Briefly, the activity of each of the consensus COSMICv3 SBS signatures (25) was quantified in each of the examined TCGA breast cancer samples. For each SBS signature, every participant is represented by a score which reflects the numbers of mutations per megabase attributed to an SBS signature. The scores for each signature were winsorized to 96%. To reduce spurious findings, only seven SBS signatures with at least 15 cases with non-zero scores were analyzed. The seven SBS signatures investigated were: SBS1 (cell-division/mitotic clock), SBS2 (hyperactivity of AID/APOBEC enzymes), SBS3 (defective homologous recombination-based DNA repair), SBS5 (ERCC2 mutations/cigarette smoking), SBS13 (similar to SBS2), SBS29 (putative etiology of tobacco chewing attributed in oral cancer), and SBS30 (deficiency in base excision repair due to inactivating mutations in NTHL1).
Forty three somatic mutations implicated in breast cancer (i.e., driver genes) were first identified using Mutation Significance version 2 (26) and Genomic Identification of Significant Targets in Cancer (27): AFF2, AKT1, ARID1A, BRCA1, BRCA2, CASP8, CBFB, CDH1, CDKN1B, CTCF, CUL4B, ERBB2, FOXA1, GATA3, GPRIN2, GPS2, HIST1H3B, KMT2A, KMT2C, KRAS, MAP2K4, MAP3K1, MED23, MYB, NBL1, NCOR1, NF1, PIK3CA, PIK3R1, PTEN, PTPN22, PTPRD, RAB40A, RB1, RUNX1, SF3B1, SPEN, STAG2, TBL1XR1, TBX3, TMEM151B, TP53, and ZFP36L1 (28,29). To reduce spurious findings, analyses were only performed in nine mutations that occurred in at least 10 cases for each exposure: CDH1, GATA3, KMT2C, MAP2K4, MAP3K1, NCOR1, PIK3CA, RUNX1, and TP53 (Supplementary Table S1).
Statistical Analyses
SCNV and TSMB were log10 transformed. SBS scores were cube root transformed. The Mann-Whitney test, Kruskal-Wallis test, Fisher’s test, and Spearman’s rho were used to evaluate the relationships between clinical characteristics (i.e., age and year of diagnosis, race, ER/PR/HER2, grade, and stage) and SCNV, TSMB, or somatic mutations. To evaluate the relationship between the exposures (BMI, cigarette smoking, and alcohol intake) and SCNV, TSMB, or a SBS signature, multivariable linear regression models were run, adjusted for the following clinical characteristics: age and year of diagnosis (model 1); age and year of diagnosis, ER/PR/HER2, grade, stage, and TCGA site (model 2). Secondary analyses stratified the women by ER status. BMI (per 5 kg/m2 increase) was analyzed as a continuous variable; cigarette smoking and alcohol consumption were analyzed as categorical variables.
To evaluate the relationship between each exposure and a driver mutation, we performed multivariable logistic regression, adjusting for two sets of covariates. Model 1 adjusted for age and year of diagnosis as above; model 2 adjusted for age and year of diagnosis, ER/PR, stage, TSMB, and TCGA site. HER2 was not included due to small numbers of HER2+ cases. Secondary analyses were restricted to women with ER+ tumors only. Sensitivity analyses were conducted by restricting to the White population to determine whether the results were influenced by race.
Data are presented as the mean counts ± standard deviation or β estimate ± standard error in the text, or unless otherwise stated. Analyses were conducted using R, version 3.4.2. Statistical significance tests were two-sided. Statistical significance was defined as p<0.05. None of the reported p-values were formally adjusted for multiple testing.
Results
These 219 TCGA participants were diagnosed with invasive breast cancer between 2001 and 2011 (median year=2008). The majority were white post-menopausal women, between 45 to 55 years old with predominantly ER+/PR+/HER2− early stage breast cancer (Table 1). They were also predominantly never smokers but were current drinkers and 35.2% were overweight as defined by BMI ≥30. When stratified by TCGA site, patient and tumor characteristics were similar (Supplementary Table S2). Tumor grade was not available (88.9%) for the majority of MSKCC participants. The number of UP, MC, and MSKCC participants were similar across the three BMI categories. Most RPCCC participants were either underweight (BMI <25; 32.9%) or overweight (BMI ≥30; 41.1%). Most of the participants from MSKCC and UP were non-smokers; there were more smokers among RPCCC participnts; and the number of smokers and non-smokers were similar among MC participants. Most of the participants from each site were current drinkers.
First, the association of clinical characteristics (i.e., age and year of diagnosis, race, ER/PR/HER2 status, grade, stage, and TCGA site) with SCNV counts or TSMB was evaluated. Year of diagnosis, ER/PR/HER2 status, and grade were significantly associated with SCNV and/or TSMB (p<0.05; Supplementary Table S3). Based on these results and following the approach of Zhu et al. (19), we adjusted for age and year of diagnosis, ER/PR/HER2 status, grade, stage, and TCGA site in the multivariable regression analyses comparing exposures with SCNV or TSMB.
In all women, increasing BMI was significantly associated with increased SCNV after adjusting for age and year of diagnosis, ER/PR/HER2 status, tumor grade, and stage (model 2 p=0.039; Table 2). Increasing BMI was also significantly associated with increased SCNV among women with ER− tumors after adjusting for age and year of diagnosis (model 1 p=0.031; Table 2). When further stratified by menopausal status, the association between BMI and SCNV did not change appreciably among all post-menopausal women or among post-menopausal women with ER− tumors. There was no relationship between BMI and SCNV in pre-menopausal women (Supplementary Table S4).
Table 2.
Multivariable analyses with somatic copy number variation (SCNV) and total somatic mutation burden (TSMB).
| Log10 SCNV | Log10 TSMB | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | n | Estimate | Standard Error | p-value | n | Estimate | Standard Error | p-value | |
| BMI per 5 kg/m2 increase | |||||||||
| All tumors | 1 | 200 | 0.060 | 0.032 | 0.062 | 178 | 0.006 | 0.019 | 0.761 |
| 2 | 130 | 0.077 | 0.037 | 0.039 | 117 | 0.013 | 0.020 | 0.522 | |
| ER positive tumors | 1 | 163 | 0.039 | 0.033 | 0.245 | 144 | −0.007 | 0.019 | 0.706 |
| 2 | 106 | 0.068 | 0.039 | 0.082 | 94 | 0.004 | 0.022 | 0.841 | |
| ER negative tumors | 1 | 37 | 0.212 | 0.094 | 0.031 | 34 | 0.091 | 0.054 | 0.103 |
| 2 | 24 | 0.170 | 0.131 | 0.218 | 23 | 0.117 | 0.062 | 0.085 | |
| Pre-diagnostic cigarette smoking | |||||||||
| All tumors | 1 | 184 | 0.152 | 0.087 | 0.082 | 159 | 0.135 | 0.052 | 0.010 |
| 2 | 120 | 0.252 | 0.105 | 0.018 | 107 | 0.145 | 0.061 | 0.020 | |
| ER positive tumors | 1 | 148 | 0.125 | 0.095 | 0.192 | 127 | 0.114 | 0.058 | 0.053 |
| 2 | 97 | 0.227 | 0.115 | 0.051 | 85 | 0.150 | 0.070 | 0.036 | |
| ER negative tumors | 1 | 36 | 0.262 | 0.208 | 0.218 | 32 | 0.150 | 0.114 | 0.199 |
| 2 | 23 | 0.257 | 0.354 | 0.483 | 22 | 0.132 | 0.172 | 0.462 | |
| Pre-diagnostic alcohol consumption | |||||||||
| All tumors | 1 | 174 | 0.261 | 0.100 | 0.010 | 155 | 0.027 | 0.065 | 0.671 |
| 2 | 119 | 0.241 | 0.129 | 0.064 | 107 | 0.032 | 0.077 | 0.680 | |
| ER positive tumors | 1 | 141 | 0.230 | 0.108 | 0.035 | 124 | 0.026 | 0.071 | 0.719 |
| 2 | 96 | 0.169 | 0.137 | 0.219 | 85 | 0.079 | 0.084 | 0.354 | |
| ER negative tumors | 1 | 33 | 0.364 | 0.253 | 0.161 | 31 | 0.091 | 0.140 | 0.519 |
| 2 | 23 | 0.513 | 0.497 | 0.324 | 22 | −0.204 | 0.241 | 0.418 | |
Model 1 adjusted for age and year of diagnosis. Model 2 adjusted for age and year of diagnosis, estrogen receptor (ER), progesterone receptor, HER2 status, tumor grade, stage, and study site.
Smokers had higher SCNV compared to non-smokers (mean counts 160.0±150.7 vs 147.3±162.5; all women model 2 p=0.018; Table 2); the magnitude of this association is similar among women with ER+ tumors. After further adjusting for alcohol consumption, the association between cigarette smoking and SCNV for all women did not change appreciably (β= 0.101±0.092, p=0.274 for model 1; β = 0.208±0.109, p=0.059 for model 2). Smokers also had higher TSMB compared to non-smokers (mean counts 109.7±116.9 vs 73.9±67.3; all women model 1 p=0.010 and model 2 p=0.020; Table 2). Likewise, the magnitude of this association is similar among women with ER+ tumors (model 1 p=0.053 and model 2 p=0.036). The association between cigarette smoking and TSMB remained significant after further adjusting for alcohol consumption (β = 0.136±0.055, p=0.014 for model 1; β = 0.145±0.063, p=0.022for model 2).
Analyses were repeated by comparing never-smokers (n=104) and past smokers (n=69). The results were similar to smokers vs non-smokers. Past smokers had higher SCNV compared to never smokers (mean counts 159.3±148.0 vs 147.3±162.5; all women model 1 p=0.054 and model 2 p=0.013; women with ER+ tumors model 2 p=0.030; Supplementary Table S5). Past smokers had higher TSMB compared to never smokers (mean counts 111.7±119.0 vs 73.9±67.3; all women model 1 p=0.007 and model 2 p=0.005; women with ER+ tumors model 2 p=0.005; Supplementary Table S5).
Alcohol drinkers had higher SCNV compared to non-drinkers (mean counts 158.0±156.3 vs 118.3±163.6; all women model 1 p=0.010; Table 2). Higher SCNV was also observed among drinkers with ER+ disease (mean counts 137.1±141.3 vs 104.0±161.5; model 1 p=0.035). The association between alcohol and SCNV did not change appreciably after accounting for cigarette smoking in the multivariable models (all women model 1 β=0.230±0.104, p=0.029 and model 2 β=0.177±0.131, p=0.182; ER+ tumors model 1 β=0.204±0.113, p=0.072 and model 2 β=0.105±0.141, p=0.457).
Neither BMI nor cigarette smoking were associated with any SBS signature (Supplementary Tables S6 and S7). Alcohol consumption was associated with SBS3 in women with ER− tumors (model 1 β=2.13±0.66, p=0.004; model 2 β=3.45±0.84, p=0.004; Supplementary Table S8). SBS3 was exclusively detected in alcohol drinkers with ER− tumors – 11/16 (68.8%) drinkers versus 0/8 non-drinkers (Figure 1). BRCA1/2 was not detected in the ER− tumors of these 16 drinkers.
Figure 1.
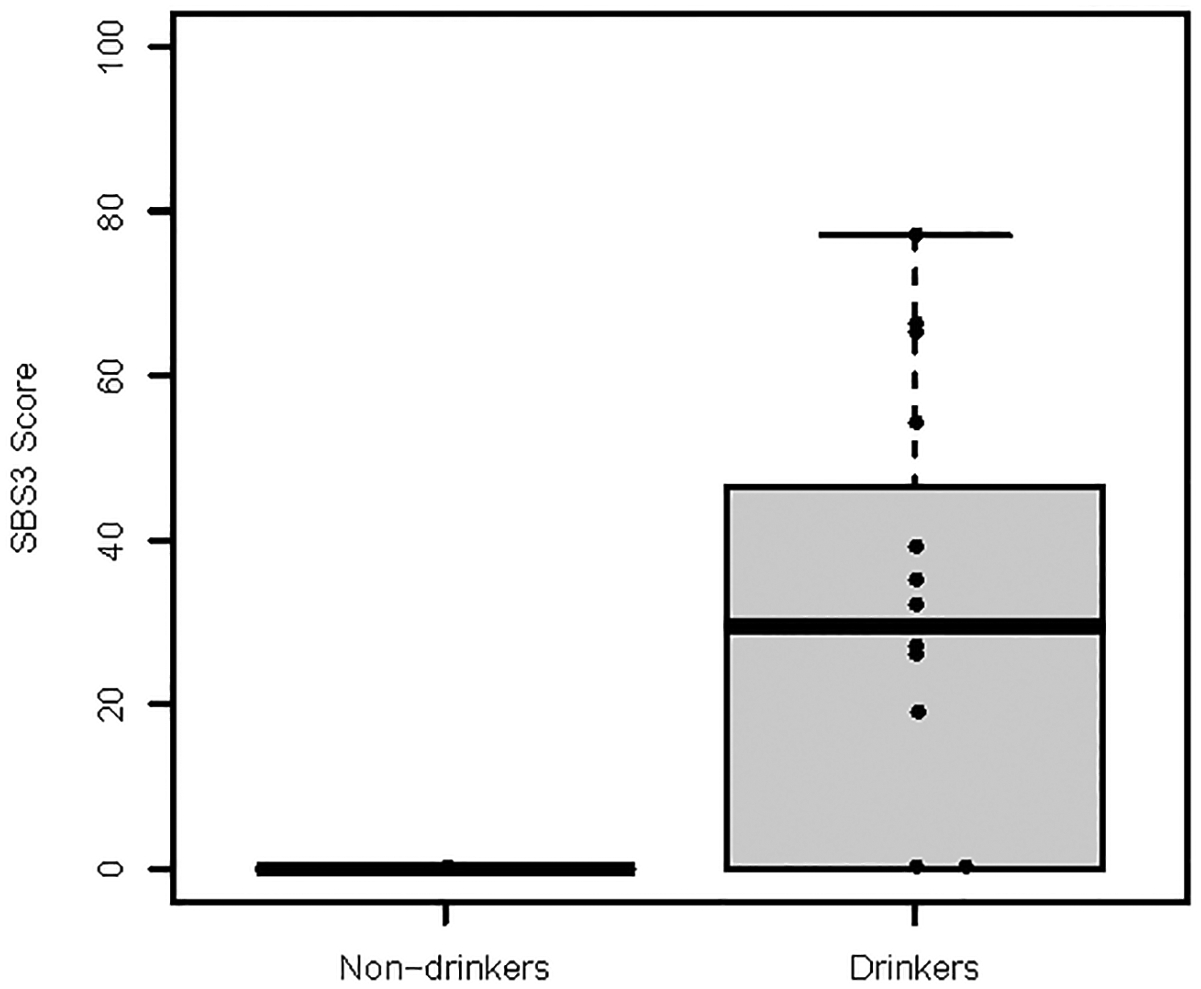
Alcohol consumption was associated with single base substitution signature 3 (SBS3) in women with ER− tumors (model 1 β=2.13±0.66, p=0.004; model 2 β=3.45±0.84, p=0.004). SBS3 was exclusively detected in alcohol drinkers: 11/16 (68.8%) drinkers versus 0/8 non-drinkers.
All clinical characteristics, except race and study site, was significantly associated with at least one driver mutation (Supplementary Table S9). Age and year of diagnosis, ER/PR status, stage, TSMB, and TCGA site, were accounted for in the multivariable binary logistic model analyses between each exposure and somatic mutation. Increasing BMI was associated with GATA3 mutation (all women model 2 odds ratio (OR)=1.43, 95% confidence interval (CI)=1.02–2.01; women with ER+ tumors model 2: OR=1.43, 95% CI=1.02–2.01; Supplementary Table S10). The ORs for all women and within women with ER+ tumors were identical since GATA3 was only detected in ER+ tumors (also refer to Supplementary Table S1 for frequency numbers). No driver mutation was associated with cigarette smoking or alcohol consumption.
Sensitivity analyses were conducted for the main findings by restricting the dataset to White participants (Supplementary Table S11). The association between BMI and GATA3, and alcohol with SCNV and SBS3 remained significant. The relationship between BMI or smoking with SCNV and TSMB did not change appreciably.
Discussion
Little is known about the molecular influence of breast cancer risk factors on tumor genomic profiles. Understanding the molecular impact of breast cancer risk factors, particularly modifiable risk factors, can enhance our knowledge of breast cancer etiology and point to new avenues for prevention strategies and treatment. In this pilot subset of 219 TCGA participants, we evaluated the association of three modifiable breast cancer risk factors and tumor genomic alterations. Higher BMI was positively associated with increased SCNV in all women and among women with ER− tumors. Higher BMI was also associated with GATA3 mutation among women with ER+ tumors. Cigarette smoking was positively associated with increased SCNV and TSMB in all women. Current alcohol consumption was positively associated with increased SCNV for all women and among women with ER+ tumors. Current alcohol drinkers with ER− disease exclusively expressed mutations associated with defective homologous recombination-based repair (SBS signature 3). The collective association of these exposures with elevated SCNV suggests that they enhance DNA genomic instability in breast tumors. Our study is the first to provide a preliminary direct molecular link between modifiable risk factors (BMI, cigarette smoking, and alcohol consumption) and breast tumor biology at the DNA level.
The relationship between BMI and breast cancer risk varies according to menopausal and ER status. High BMI is consistently associated with decreased risk of pre-menopausal breast cancer (both ER+ and ER-) but increased risk of ER+ post-menopausal breast cancers (30–32). One possible mechanism linking high BMI and breast cancer risk is the exposure of breast tissues to high levels of estrogen produced by adipose tissues in overweight/obese women. High estrogen levels can increase cellular proliferation and initiate breast tumorigenesis (33). Indeed, we reported that tumors from post-menopausal women with higher BMI (versus lower BMI) were enriched for cellular proliferation (ER+ and ER− tumors), and interferon alpha and gamma pathways (ER− tumors) (9). Gene networks involved in cellular proliferation were overexpressed in triple negative breast cancers from pre-menopausal obese patients (34). GATA3 mutation, frequently observed in ER+ breast tumors, is associated with enhanced tumor growth (35,36). Our current work suggests that the positive correlations between BMI and tumor proliferation may be in part driven by the presence of GATA3 mutation, and contributes additional evidence that BMI increases DNA genomic instability.
Cigarette smoking is associated with an increased (10–20%) breast cancer risk, especially among women who started smoking at a young age (37) or smoked at least >10 years before their first birth (7,8,38). Current evidence suggests the association of cigarette smoking and breast cancer risk is limited to ER+ disease, not confounded by alcohol (8,38), and the risk is proportional to smoking intensity (38). In our study, the observation of increased SCNV and TSMB in breast tumors of smokers (versus non-smokers), unmodified by alcohol, is in line with tobacco carcinogenesis – inducing DNA damage, leading to misreplication, and subsequently increasing TSMB (20). We were unable to determine the association between pre-pregnancy smoking and TSMB as we did not collect adolescence and pre-pregnancy smoking information. However, in our sub analysis, our preliminary finding suggests that past smoking versus never smoking was also associated with increased SCNV and TSMB, especially in ER+ tumors. Among our 81 smokers, 37 (45.7%) smoked <1 cigarette pack/day, 25 (30.9%) smoked ≥1 pack/day, and 19 (23.4%) responses were missing. Thus, we were unable to evaluate the link between SCNV or TSMB and smoking intensity in our study due to small numbers. In addition, our small sample size may have reduced our ability to observe an association between cigarette smoking and TP53 mutations as previously reported by Conway et al (15).
Mutational signatures created using SBS, small insertion and deletion, or double base substitution have been characterized across a wide spectrum of human cancers to better understand the diversity of mutational processes underlying cancer development (20–23,39,40). Specifically, signature SBS4 represents mutations occurring in epithelial cells directly exposed to cigarette smoke while SBS5 represents smoking-associated mutations occurring in cells not directly exposed to cigarette smoke. SBS4 was not detected in any of our samples. SBS5 was not associated with cigarette smoking in breast cancer. These findings are consistent with previous work where smoking-associated mutational signatures were not enriched in breast cancer (20). It could also be speculated that cigarette smoking influences breast carcinogenesis in a different manner, or breast cancer may harbor an undiscovered smoking-specific mutational signature.
Alcohol consumption is a well-established breast cancer risk factor (6,41,42). Proposed mechanisms for alcohol-induced breast carcinogenesis include elevated oxidative stress (43–45), DNA damage (46,47), and estrogen metabolism (48,49). To the best of our knowledge, this is the first study to demonstrate a relationship between alcohol consumption and genome-wide SCNV in breast tumors. TP53 protects breast tumor cells from alcohol-induced DNA damage in vitro (47). Since we observed mutations related to DNA damage and repair (i.e., SBS3) exclusively in ER− tumors of alcohol drinkers, we would expect ER− tumors to be enriched for TP53 mutations. Indeed, 14/15 drinkers and 3/7 non-drinkers with ER− disease had TP53 mutations. More studies are warranted to investigate the link between alcohol consumption, TP53 mutation, dysfunctional DNA damage and repair, and ER− breast carcinogenesis to better understand the complex interplay of mechanisms attributed to alcohol consumption in breast tumors.
Limitations of our study include small sample size, especially for ER− and HER2+ disease, thus limiting analyses for dose-response assessments of exposure. We analyzed three exposures and performed subtype analyses by ER and menopausal status. The observed associations may reflect chance findings as no analysis survived multiple hypothesis testing after Bonferroni correction. We could not assess other mutational signatures derived using small insertion and deletion or double base substitution because only 23 of our participants had whole genome sequencing data. Finally, other modifiable risk factors such as physical activity were not collected.
In conclusion, our study provides preliminary evidence that BMI, cigarette smoking, and alcohol consumption, can influence breast tumor biology, in particular, DNA alterations. A deeper understanding of the molecular impact of breast cancer risk factors will enhance our knowledge of breast cancer etiology, and create opportunities for prevention strategies and treatment. Future larger epidemiological studies are required to confirming these findings.
Supplementary Material
ACKNOWLEDGEMENTS
The data used in this study were in whole or in part based on the data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We thank the TCGA participants and staff at the University of Pittsburgh, Roswell Park Comprehensive Cancer Center, the Mayo Clinic, and Memorial Sloan Kettering Cancer Center.
Financial Information
Funding for this project was provided by the Klarman Family Foundation (YJH), University of Pittsburgh School of Medicine Dean’s Faculty Advancement Award (FM), Susan G Komen SAC110014 (SEH), and the NIH Support Grant P30 CA016056 to Roswell Park Comprehensive Cancer Center.
Abbreviations list:
- BMI
body mass index
- ER
estrogen receptor
- MC
Mayo Clinic
- MSKCC
Memorial Sloan Kettering Cancer Center
- PR
progesterone receptor
- RPCCC
Roswell Park Comprehensive Cancer Center
- SBS
single base substitution
- SCNV
somatic copy number variation
- TCGA
The Cancer Genome Atlas
- TSMB
total somatic mutation burden
- UP
University of Pittsburgh
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest
REFERENCES
- 1.American Cancer Society. Cancer Facts & Figures 2016. Atlanta: American Cancer Society; 2016. [Google Scholar]
- 2.Collaborative group on hormonal factors in breast cancer. Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358:1389–99. [DOI] [PubMed] [Google Scholar]
- 3.Tamimi RM, Rosner B, Colditz GA. Evaluation of a breast cancer risk prediction model expanded to include category of prior benign breast disease lesion. Cancer. 2010. [cited 2016 Nov 9];116:4944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. [DOI] [PubMed] [Google Scholar]
- 5.Thune I, Brenn T, Lund E, Gaard M. Physical activity and the risk of breast cancer. N Engl J Med. 1997;336:1269–75. [DOI] [PubMed] [Google Scholar]
- 6.Chen WY, Rosner B, Hankinson SE, Colditz GA, Willett WC. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA. 2011;306:1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaudet MM, Gapstur SM, Sun J, Ryan Diver W, Hannan LM, Thun MJ. Active smoking and breast cancer risk: Original cohort data and meta-analysis. J Natl Cancer Inst. 2013;105:515–25. [DOI] [PubMed] [Google Scholar]
- 8.Gaudet MM, Carter BD, Brinton LA, Falk RT, Gram IT, Luo J, et al. Pooled analysis of active cigarette smoking and invasive breast cancer risk in 14 cohort studies. Int J Epidemiol. 2017;46:881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heng YJ, Wang J, Ahearn TU, Boyer S, Zhang X, Ambrosone CB, et al. Molecular mechanisms linking high body mass index to breast cancer etiology in post-menopausal tumor and tumor-adjacent tissues. Breast Cancer Res Treat. 2019;173:667–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Heng YJ, Eliassen AH, Tamimi RM, Hazra A, Carey VJ, et al. Alcohol consumption and breast tumor gene expression. Breast Cancer Res. 2017;19:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewertz M, Jensen M-BB, Gunnarsdóttir KÁ, Højris I, Jakobsen EH, Nielsen D, et al. Effect of obesity on prognosis after early-stage breast cancer. J Clin Oncol. 2011;29:25–31. [DOI] [PubMed] [Google Scholar]
- 12.Sparano JA, Wang M, Zhao F, Stearns V, Martino S, Ligibel JA, et al. Obesity at diagnosis is associated with inferior outcomes in hormone receptor-positive operable breast cancer. Cancer. 2012;118:5937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen B, Venet D, Lambertini M, Desmedt C, Salgado R, Horlings HM, et al. Imprint of parity and age at first pregnancy on the genomic landscape of subsequent breast cancer. Breast Cancer Res. 2019;21:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conway K, Edmiston SN, Cui L, Drouin SS, Pang J, He M, et al. Prevalence and spectrum of p53 mutations associated with smoking in breast cancer. Cancer Res. 2002;62:1987–95. [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sougnez C, Gabriel S, Meyerson M, Lander ES, Cibulskis K, Lawrence MS, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu B, Mukherjee A, Machiela MJ, Song L, Hua X, Shi J, et al. An investigation of the association of genetic susceptibility risk with somatic mutation burden in breast cancer. Br J Cancer. 2016;115:752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petljak M, Alexandrov LB. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis. 2016;37:531–40. [DOI] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013;3:246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018;6:271–281.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv. 2019;322859. [Google Scholar]
- 26.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heng YJ, Lester SC, Tse GMK, Factor RE, Allison KH, Collins LC, et al. The molecular basis of breast cancer pathological phenotypes. J Pathol. 2017;241:375–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoemaker MJ, Nichols HB, Wright LB, Brook MN, Jones ME, O’Brien KM, et al. Association of Body Mass Index and Age With Subsequent Breast Cancer Risk in Premenopausal Women. JAMA Oncol. 2018;4:e181771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang XR, Chang-Claude J, Goode EL, Couch FJ, Nevanlinna H, Milne RL, et al. Associations of breast cancer risk factors with tumor subtypes: A pooled analysis from the breast cancer association consortium studies. J Natl Cancer Inst. 2011;103:250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78. [DOI] [PubMed] [Google Scholar]
- 33.Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013. page 161–70. [DOI] [PubMed] [Google Scholar]
- 34.Mamidi TKK, Wu J, Tchounwou PB, Miele L, Hicks C. Whole genome transcriptome analysis of the association between obesity and triple-negative breast cancer in caucasian women. Int J Environ Res Public Health. 2018;15:2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gustin JP, Miller J, Farag M, Marc Rosen D, Thomas M, Scharpf RB, et al. GATA3 frameshift mutation promotes tumor growth in human luminal breast cancer cells and induces transcriptional changes seen in primary GATA3 mutant breast cancers. Oncotarget. 2017;8:103415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takaku M, Grimm SA, Roberts JD, Chrysovergis K, Bennett BD, Myers P, et al. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat Commun. 2018;9:1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones ME, Schoemaker MJ, Wright LB, Ashworth A, Swerdlow AJ. Smoking and risk of breast cancer in the Generations Study cohort. Breast Cancer Res. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersen ZJ, Jørgensen JT, Grøn R, Brauner EV, Lynge E. Active smoking and risk of breast cancer in a Danish nurse cohort study. BMC Cancer. 2017;17:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singletary KW, Gapstur SM. Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms. JAMA. 2001;286:2143–51. [DOI] [PubMed] [Google Scholar]
- 42.Hirko KA, Chen WY, Willett WC, Rosner BA, Hankinson SE, Beck AH, et al. Alcohol consumption and risk of breast cancer by molecular subtype: Prospective analysis of the nurses’ health study after 26 years of follow-up. Int J Cancer. 2016;138:1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright RM, McManaman JL, Repine JE. Alcohol-induced breast cancer: A proposed mechanism. Free Radic Biol Med. 1999;26:348–54. [DOI] [PubMed] [Google Scholar]
- 44.Seitz HK, Pelucchi C, Bagnardi V, La Vecchia C. Epidemiology and pathophysiology of alcohol and breast cancer: Update 2012. Alcohol Alcohol. 2012;47:204–12. [DOI] [PubMed] [Google Scholar]
- 45.Shen J, Platek M, Mahasneh A, Ambrosone CB, Zhao H. Mitochondrial copy number and risk of breast cancer: A pilot study. Mitochondrion. 2010;10:62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks PJ. DNA Damage, DNA Repair, and Alcohol Toxicity-A Review. Alcohol Clin Exp Res. 2006;21:1073–82. [PubMed] [Google Scholar]
- 47.Zhao M, Howard EW, Guo Z, Parris AB, Yang X. P53 pathway determines the cellular response to alcohol-induced DNA damage in MCF-7 breast cancer cells. PLoS One. 2017;12:e0175121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorgan JF, Baer DJ, Albert PS, Judd JT, Brown ED, Corle DK, et al. Serum hormones and the alcohol-breast cancer association in postmenopausal women. J. Natl. Cancer Inst 2001. page 710–5. [DOI] [PubMed] [Google Scholar]
- 49.Reichman ME, Judd JT, Longcope C, Schatzkin A, Clevidence BA, Nair PP, et al. Effects of alcohol consumption on plasma and urinary hormone concentrations in premenopausal women. J Natl Cancer Inst. 1993;85:722–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.