

Abstract
Catalytic hydrogenation or transfer hydrogenation of quinolines was thought to be a direct strategy to access dihydroquinolines. However, the challenge is to control the chemoselectivity and regioselectivity. Here we report an efficient partial transfer hydrogenation system operated by a cobalt-amido cooperative catalyst, which converts quinolines to 1,2-dihydroquinolines by the reaction with H3N·BH3 at room temperature. This methodology enables the large scale synthesis of many 1,2-dihydroquinolines with a broad range of functional groups. Mechanistic studies demonstrate that the reduction of quinoline is controlled precisely by cobalt-amido cooperation to operate dihydrogen transfer from H3N·BH3 to the N=C bond of the substrates.
Subject terms: Catalytic mechanisms, Homogeneous catalysis, Synthetic chemistry methodology
Controlling the partial reduction of quinolines is challenging, given the competing overreduction to tetrahydroquinolines. Here, the authors report a cobalt-amido cooperative catalyst for the selective, partial transfer hydrogenation of quinolines to 1,2-dihydroquinolines with H3N·BH3 as reductant.
Introduction
Dihydroquinolines (DHQs) are a potential surrogate of H2 for sustainable transformations, reminiscent of the structure of reduced nicotinamide adenine dinucleotide phosphate (NADPH)1–4, More importantly, 1,2-DHQs are versatile synthons leading to bioactive molecules5, pharmaceuticals6,7, natural products8–10, and organic electroluminescent materials11. They can be conveniently transformed by C–H functionalization12 to complex organic structures, and can achieve asymmetric olefin difunctionalization13,14, and N-functionalization15 (Fig. 1a). Catalytic regioselective hydrogenation and transfer hydrogenation of N-heteroarenes have attracted intense interest16–25, and could provide the most straightforward protocol with which to harvest the desired DHQs. However, catalytic transformation of quinolines to DHQs is extremely challenging.
Fig. 1. Conversion of quinolines to THQs and 1,2-DHQs.

a Diverse functionalization of 1,2-DHQ. b Hydrogenation and transfer hydrogenation of quinolines to THQs. c The present work on transfer hydrogenation of quinolines with H3N·BH3 to 1,2-DHQs by a cobalt-amido cooperative catalyst.
Significant progress has been made in hydrogenation of quinolines to tetrahydroquinolines (THQs)26–29, and transfer hydrogenation reactions with formates30, alcohols31,32 or Hantzsch esters33–35 (Fig. 1b), but the related catalysis to access DHQs has not been revealed to date. The difficulty associated with such catalysis to access 1,2-DHQs is control of chemoselectivity and regioselectivity since these reactions always suffer from over-reduction of the more reactive DHQs to THQs16,36. The breakthrough in this field was catalytic hydrosilylation and hydroboration of quinolines to the N-silylated or N-borylated 1,2-DHQs37–44 respectively, using transition metal catalysts or metal-free organocatalysts45,46. Deprotection of the silyl or boryl groups of the N-protected DHQs can provide an alternative route to 1,2-DHQs, however, such an N-protection/hydrolysis strategy encounter issues related to functional group compatibility and the complicated purification procedures39, which limits the expansion of 1,2-DHQ chemistry.
Synergism of metal-ligand reactivity is a practical strategy with which to design new catalysts to perform precise transformations47–54. For example, cooperative Ru–S reactivity allows for activation of the Si–H bond of hydrosilanes and catalytic intermolecular electrophilic C−H silylation of N-protected indoles to afford the corresponding C3-silylated indoles as single regioisomers55–57. In particular, such metal-ligand cooperation strategy greatly promotes the utility of the abundant metals for hydrogenation and transfer hydrogenation of unsaturated hydrocarbons58–63. However, the related catalysis has not been reported for the direct synthesis of DHQs.
In this work, we report an efficient cobalt-amido cooperative catalyst for the controlled, partial transfer hydrogenation of quinolines to 1,2-DHQs with H3N∙BH3 (Fig. 1c). Compared with the previous methods of catalytic hydrosilylation and hydroboration, the functional group tolerance in the present catalysis is exceedingly broad, in particular, with respect to ester, amide, and alkenyl moieties. Notably, tetrahydrogenation of the quinoline substrate can be also realized by using two equiv. of reducing agent under the identical reaction conditions for the catalytic dihydrogenation. Additionally, we have demonstrated that 1,2-DHQs are valuable starting material to synthesize chiral THQs, and they can serve as reducing agent for dihydrogen transfer.
Results
Synthesis and reactivity of 1
The phosphinoamido cobalt(II) half-sandwich complex (1) was easily prepared by treatment of [Cp*CoCl]2 with lithium (2-(diphenylphosphanyl)-4-methyl-phenyl)amide in THF64. Single crystal X-ray diffraction confirmed its structure as a neutral cobalt(II) compound with a 17-electron configuration. This compound was initially found to react gently with H3N∙BH3, gradually releasing H2 at room temperature. In particular, 1 is capable of catalyzing this dehydrogenation reaction. With a catalyst loading of 0.5 mol%, a solution in THF of H3N∙BH3 in 10 h released a nearly equimolar quantity of H2 based on GC quantification (see Supplementary Fig. 1).
Partial transfer hydrogenation of 4-methylquinoline
Based on the reactivity of 1 toward H3N∙BH3, the transfer hydrogenation of 4-methylquinoline (2a) with H3N∙BH3 was examined using 1 as a catalyst (see Supplementary Table 1). With 5 mol% of 1 in d8-toluene, the reaction of the quinoline (2a) with 1.1 equiv of H3N∙BH3 was conducted in a Young NMR tube. According to the 1H NMR spectroscopic analysis, the reaction conducted at 25 oC provided 4-methyl-1,2-dihydroquinoline (3a) in 36% yield in 4 h. Control experiments indicated that the cobalt complex is responsible for the catalysis, no reaction being observed in the absence of 1. Remarkably, in THF the quinoline substrate was converted nearly quantitatively to 3a within 3 h. This system is extremely effective; with even 0.5 mol% of 1, the yield of 3a reaches 99% within 20 h. Scaling up of the transfer hydrogenation failed to decrease the yield and pure 3a was isolated in 95% yield (see Fig. 2).
Fig. 2. Substrate scope.

Reaction conditions: substrate (0.5 mmol), H3N∙BH3 (0.55 mmol) and 1 (0.5 mol%) were stirred in THF (2 mL) at 25 oC for 20 h. Isolated yields are given. aTwo equiv. of H3N·BH3 (1 mmol) was used for reduction of 3-acetylquinoline. bRatios in parentheses refer to product ratios of 1,2-DHQ and THQ determined by 1H NMR spectroscopy.
Using 0.5 mol% of 1 in THF at 25 oC, under the optimized reaction conditions, we investigated the substrate scope for the catalysis (Fig. 2). After changing the methyl group to a phenyl or bromo group, the corresponding 4-substituted-1,2-DHQs (3b, 3c) were obtained with good yields. Notably, quinolines with a variety of functional groups such as Cl (2d), Br (2e), CH2OH (2f), COOC2H5 (2g), NHCOCH3 (2j), Bpin (2h), C=CPh (2l) and C≡CPh (2k) at the 3-position all underwent smooth transfer hydrogenation to furnish the dearomatized products (3d-3l). The synthesis of 1,2-DHQs with ester substituents (3g and 3h) or an amide group (3j) is challenging since they are prone to hydrolysis during the deprotection of the N-silylated or N-borylated dihydroquinoline precursors39. Unlike the ester groups, the acyl group was hydrogenated in the course of partial transfer hydrogenation of the N-heterocycle. The reaction of 3-acetylquinoline (2m) with two equiv of H3N∙BH3 produced the corresponding hydroxyl compound (3m) in 89% yield. The reduction of 2-methylquinoline failed, probably due to the steric effect on the 1,2-hydrogenation (3n)41. Transfer hydrogenation of quinoline produced 1,2-DHQ (3o), which was obtained as solid.
In general, the regioselective 1,2-reduction of quinolines is not impeded by the electronic or steric nature of functional groups on the aryl ring. A variety of functionalized 1,2-DHQs (3p-3ad) were obtained in good to excellent yield with this mild catalytic protocol. In particular, halogen substituted quinolines tolerate the catalysis, as exemplified by 3c (86%), 3d (83%), 3e (80%), 3q (71%), 3r (82%), 3s (88%), 3t (81%), 3z (76%), 3aa (73%), 3ab (78%), 3ac (81%) and 3ad (89%). Although cobalt-based transfer hydrogenations of alkynes and alkenes with H3N∙BH3 have been reported65,66, the dihydrogenation reaction occurs selectively at the C=N bond of the N-heterocyclic ring (3k, 3l, and 3v). This cobalt-based catalytic reaction is not limited only to quinolines, and it can also efficiently process the transfer hydrogenation of benzo[d]oxazole, benzothiazole, phenanthridine and acridine, providing the desired products (3ae–3ah) with excellent yields. This protocol is extremely efficient and most of 1,2-DHQs were conveniently synthesized on a large scale and purified by crystallization. Crystal structures of 3h, 3u and 3ag were unequivocally established by single-crystal X-ray diffraction (Fig. 2). In the dihydrogenation of quinolines with substituents on the phenyl ring, THQs were also observed as by-products (<10% yield) which arise from further reduction of the resultant 1,2-DHQs (see below).
Transfer hydrogenation to THQs
DHQs are presumed to be key intermediates in the hydrogenation of quinolines to THQs16. Indeed, 1,2-DHQs can be smoothly further reduced to THQs by H3N∙BH3 in the presence of the cobalt catalyst. For example, the 6-methyldihydroquinoline (3u) was completely converted to the tetrahydrogenated product (4u) under the catalytic conditions. Use of two equiv of H3N∙BH3 under the identical conditions of partial reduction allowed the transfer hydrogenation of 2u to 4u with an excellent yield. To compare the differences in bond distances and angles between dihydrogenated and tetrahydrogenated quinolines, the structures of 3u and 4u were superimposed as shown in Fig. 3. Comparing 3u with 4u reveals significant differences in the heterocyclic ring of the compounds. As a result of the second reduction the C2-C3 distance increases from 1.330(3) Å to 1.527(3) Å, and the N-heterocyclic ring becomes puckered.
Fig. 3. Superposition of 3u (gray) with 4u (magenta).
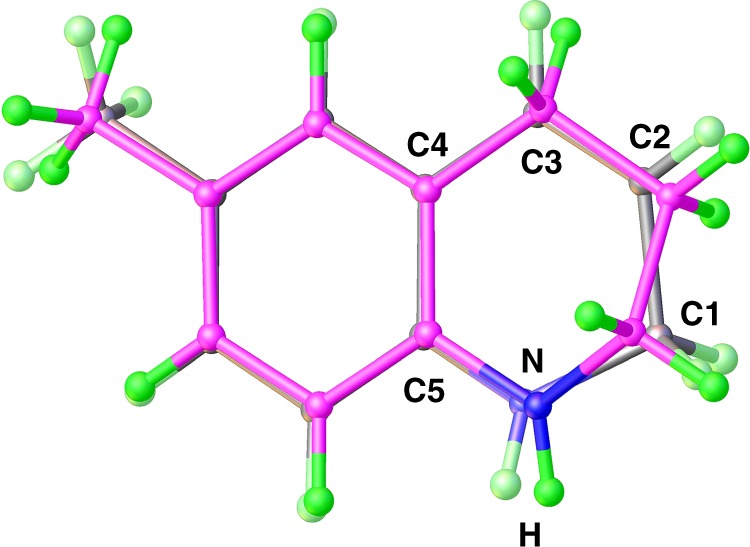
Selected bond distances (Å) for 4u: N1-C1, 1.452(3); C1-C2, 1.514(3); C2-C3, 1.527(3); C5-N1, 1.398 (3).
Synthetic applications
As a partially saturated heteroaromatic compound, the 1,2-dihydroquinoline system could act as both hydride acceptor and hydride donor, resembling the oxidized and reduced nicotinamide adenine dinucleotide respectively. We found 1,2-DHQs can be employed as dihydrogen sources for sustainable transformations. For example, phenanthridine (2ag) is reduced to 5,6-dihydrophenanthridine (3ag) quantitatively by 3u in the presence of 3 mol% of CF3COOH (TFA), and acridine (2ah) is hydrogenated to 9,10-dihydroacridine (3ah) by 3u under the identical reaction conditions (Fig. 4a). Disproportionation of 3u was not observed during the acid-catalyzed H-transfer reaction. The reaction profile monitored by 1H NMR spectroscopy clearly showed that the increase in the concentration of 3ah over time is consistent with the consumption of 3u (Fig. 4b). Interestingly, 3u is stable toward 2, 6-dimethylpyridine-3, 5-dicarboxylate. These results demonstrate that 3u is a mild organo-hydride reagent, and its hydride-donating ability (ΔHH-) is weaker than that of the Hantszch ester (ΔHH− = 69.3 kcal/mol)67.
Fig. 4. Synthetic Applications.

a 3u serving as reducing agent for dihydrogen transfer. b Reaction profile of transfer hydrogenation of acridine (2ah, 0.32 M) to 9,10-dihydroacridine (3ah) by 3u (0.32 M) catalyzed by CF3COOH (3 mol%) in THF-d8 at 25 oC. c Asymmetric functionalization of 3aa through enantioselective borylation and amination.
1,2-DHQs are important synthetic intermediates that can lead to versatile N-heterocyclic compounds, such as chiral THQs and N-functionalized DHQs, which are common in pharmaceuticals and natural products68. For instance, through acylation and enantioselective borylation69, 6-trifluoromethyl-dihydroquinoline (3aa) was conveniently transformed to enantioenriched 3-boryl-tetrahydroquinolines R-6 (92% yield, 94% ee) and S-6 (87% yield, 95% ee), respectively (Fig. 4c). Such chiral N-heterocyclic organoboron compounds are amenable to diverse stereospecific functionalization at the stereogenic C-B bond70,71. For example, subsequent amination of the two enantiomers allows for the construction of a new C-N bond72, affording R-7 and S-7 compounds without loss of enantiomeric purity (94% ee). In particular, enantiomer S-7 is a structural analog of positive inotropic agent (S)-903, and the potential agent Sumanirole for the treatment of Parkinson’s disease73.
Discussion
To probe the mechanism of quioline reduction, the regio-specificity of hydrogen transfer with the selectively deuterated ammonia borane derivatives D3N∙BH3 and H3N∙BD3 was investigated for 2u (Fig. 5). Tracing the regioselectivity of the deuterium addition to 2u with D3N∙BH3, deuterium incorporation at the 1-position, the N atom in the desired product (d-3u) was found (see Supplementary Fig. 2). In the case of H3N∙BD3 as the dihydrogen source, 2H NMR spectroscopic analysis established that d-3u′ was D-labeled entirely at the 2-position. Consecutive regioselective transfer hydrogenation of 2u with two equiv of D3N∙BH3 afforded the bis-deuterated quinoline (d2-4u) deuterated at the 1- and 3-positions (Supplementary Fig. 4). In comparison with d2-4u, the reaction of 2u with 2 equiv of H3N∙BD3 resulted in formation of d2-4u′ which has deuterium exclusively at the 2- and 4-positions.
Fig. 5. Deuterium labeling experiments.

a Reactions of 2u with D3N·BH3. b Reactions of 2u with H3N·BD3.
To understand the relationship between the dihydrogenation and tetrahydrogenation catalysis performed by 1, we followed the kinetics of the reactions of 6-methylquinoline (2u) with one or two equiv of H3N∙BH3. The changes in concentrations of reaction components over time are shown in Fig. 6. Using one equiv of H3N∙BH3 as the hydrogen source, the reaction profile indicates that the dihydroproduct (3u) alone was produced in the initial period of transfer hydrogenation (Fig. 6a). Although 7% of tetrahydrogenated product (4u) was observed, the major product of the reaction was the dihydrogenated compound (3u). In contrast, when starting with two equiv of H3N∙BH3, both 3u and 4u were produced in the initial stage of the catalysis but the reaction profile unambiguously reveals the intermediacy of 3u in the tetrahydrogenation reaction (Fig. 6b).
Fig. 6. Reaction profiles.

a Catalytic transfer hydrogenations of 6-methylquinoline (2u) with one equiv of H3N·BH3. b 2u with two equiv of H3N·BH3. Conditions: [2u] = 0.32 M, [1] = 0.0016 M, [H3N·BH3] = 0.35 M for (a) and 0.70 M for (b) in THF-d8 at 25 oC.
The kinetic order in each reaction component was established for the transformation of 2u to 3u at 25 oC. For reactions carried out at constant [2u], i.e. [H3N∙BH3] = 0.32 M, the initial rate (vi) for the product formation against the concentration of catalyst varied between 0.8 and 4.8 mM and showed first-order kinetics in [1], leading to a rate constant, kobs = 4 M min−1 (Supplementary Fig. 5). At constant [H3N∙BH3] = 0.32 M and [1] = 1.6 mM, performing the dihydrogenation reaction with concentrations of 2u ranging from 0.16 to 0.64 M led to a linear increase in vi (Supplementary Fig. 6), which leads to a first-order reaction rate constant of 3.7 M min−1. This value is consistent with kobs deduced from the plot of vi ~ [1]. No inhibition was observed for vi at high initial concentrations of the substrate. Since 2u can be easily reduced to 4u at high concentrations of H3N∙BH3, the kinetic experiments were carried out at constant [2u] = 0.32 M and [1] = 1.6 mM by varying [H3N∙BH3] in the range of 0.08–0.32 M. A linear plot of vi ~ [H3N∙BH3] established a first-order dependence on the dihydrogen source at this stage (Supplementary Fig. 7). These results indicate that all the three components are involved in the turnover-limiting formation of 3u. The overall rate law of this dihydrogenated reaction is thus expressed by rate = kobs[1][2u][H3N∙BH3].
Deuterium kinetic isotope effects (DKIE) were further probed for the dihydrogenation reaction (Supplementary Fig. 8). According to the rate of formation of 3u measured for H3N∙BH3 versus H3N∙BD3, a KIE of kNH·BH/kNH·BD = 1.55 was calculated. A larger KIE with kNH·BH/kND·BH = 3.63 was found for 2u with D3N∙BH3. For the double DKIE reaction of D3N∙BD3 with 2u, the KIE value was determined to be 5.73 (kNH·BH/kND·BD). These results indicate that hydrogen transfers clearly participate in the turnover-determining step.
Density functional calculations74 were performed to explore the detailed mechanism and regioselectivity of the reaction. As a 17-electron neutral compound, the catalyst (1) favors the low spin doublet, which is 7.4 kcal/mol lower in energy than the quartet state (Fig. 7). These calculations suggest that the catalysis is initiated by the reaction of 1 with H3N∙BH3 through an isoenergetic complex intermediate (Int1), in which a hydrogen bond NH---H-NH2 is formed by the interaction of the amide group with the NH3 moiety. A concerted proton transfer/hydride transfer75–77 from H3N∙BH3 to the amido-cobalt unit in Int1 yields a cobalt(II)-hydride species (Int2) by release of H2N-BH2. This step was calculated to be endergonic by 4.8 kcal/mol in the doublet state. Binding of the substrate to Int2 forming Int3, was predicted to proceed via H-bonding between the N atom of the quinoline and the H atom of amino group of the ligand.
Fig. 7. Gibbs energy diagram.

For computational details, see Supplementary Information.
From Int3, four different mechanistic pathways have been located, and the two with lower barriers are presented here. The first of these involves proton transfer from the amino group to the quinoline nitrogen (TS1) coupled with an electron transfer from the CoII center to the quinoline moiety. TS1 is a doublet whose barrier was calculated to be 12.7 kcal/mol relative to Int3. The resultant CoIII-H species with an N-hydrogenated quinoline radical (Int4) is +3.5 kcal/mol higher in energy than Int3. Due to the directing effect of the hydrogen bond, C8H7N---H-NH between the substrate and the amino group of the ligand, the hydrogen atom transfer from the metal center to the ortho-position of the substrate via TS2C1 (1,2-hydrogenation pathway, 19.8 kcal/mol) was found to be preferred by 2.3 kcal/mol over the H-transfer to the para-position (TS2C3, 1,4-hydrogenation pathway, 22.1 kcal/mol, Supplementary Fig. 99). TS1 was calculated to be the turnover-determining transition state (TDTS) with a total barrier of 21.6 kcal/mol relative to Int1 (the turnover determining intermediate or TDI)77. The calculated barrier is also in reasonable agreement with the experimental rate constant of 4 min−1, which can be converted by classical transition state theory to an energy barrier of 19.0 kcal/mol. Importantly, TS2 is the regioselectivity determining step and the regioselectivity is kinetically controlled. As a consequence, 1,2-DHQ is the major product, with a calculated product ratio 1,2-/1,4-DHQ of 48.6:1.
Such a stepwise H+-e−/H∙ pathway can only take place in the doublet state, while in the quartet state the hydrogen transfers proceed via a concerted H+/H– pathway (TS1’). The concerted transfer of the hydridic Co(III)-H and the protic NH-H to the C=N bond in the doublet state is energetically slightly favorable (+0.6 kcal/mol) over the quartet state. Related to the stepwise pathway, the total barrier of the concerted pathway is 2.0 kcal/mol higher. For both pathways, the ammonia proton is transferred to the quinoline nitrogen and the hydride of the BH3 unit is delivered to the 2-position of quinoline. This agrees well with the results obtained from deuterium isotope labeling experiments (Fig. 5). Other mechanisms through different H3N∙BH3 activation modes have also been considered (Supplementary Figs. 100–108), but are associated with much higher barriers.
DFT predictions imply a stepwise H+-e−/H∙ pathway for the partial transfer hydrogenation. To gain more evidence, we calculated the kinetic isotope effects of deuterated ammonia boranes on the transition states of TS1, TS2C1, and TS1’ (Table 1). The calculated DKIE effects for TS1 using H3N∙BD3 or D3N∙BH3 are 3.57 and 1.67, respectively. Since TS1 involves a proton-coupled electron transfer process, it is reasonable that a larger KIE should come from deuterated D3N∙BH3 with respect to H3N∙BD3. The DKIE effect observed for H3N∙BD3 is dominated by an equilibrium isotope effect, resulting from the formation of a Co-D bond in Int2 when compared with the B-D bond in Int1. For the double DKIE reaction of D3N∙BD3, the total KIE was predicted to be 6.01. Compared with the transition state of TS1, TS2C1 was found to exhibit a much larger DKIE of 3.21 using H3N∙BD3 and only an equilibrium isotope effect (1.36) for D3N∙BH3. These DKIEs arise from transferring a deuterium atom from Co-D to C1, and the formation of N-D versus N-H bonds in TS2C1 and Int1. In contrast, the DKIE increases to 6.09 when D3N∙BH3 is used in the concerted pathway involving TS1’ while an inverse KIE of 0.50 was calculated for H3N∙BD3. The calculated DKIE values for TS2C1 and TS1’ are inconsistent with the experimental results.
Table 1.
Computational and experimental kinetic isotope effects.
| KIE | H3N·BD3 | D3N·BH3 | D3N·BD3 |
|---|---|---|---|
| Cal.(TS1) | 1.67 | 3.57 | 6.01 |
| Cal.(TS2C1) | 3.21 | 1.36 | 4.35 |
| Cal.(TS1’) | 0.50 | 6.09 | 3.06 |
| Exp. | 1.55 | 3.63 | 5.73 |
The calculated DKIE values for TS1 are consistent with the experimental values (1.55 for H3N∙BD3, 3.63 for D3N∙BH3, and 5.73 for D3N∙BD3), and this validates the suggested reaction pathway via TS1 as the TDTS. Accordingly, a proposed mechanism of the catalytic partial transfer hydrogenation of quinoline is shown in Fig. 8. Complemented by the basic site, the cobalt-amido complex (Co-NH) is able to activate H3N∙BH3, generating a hydride-proton species (HCo-NH(H), Int2) for hydrogen transfers75–77. Combining the experimental data with theoretical studies, a stepwise H+-e−/H∙ mechanism is proposed for the subsequent reactions. Transfer of the proton from the amino group of the ligand to the N atom of quinoline, induced by the H-bonding C8H7N---H-NH (Int3) and accompanied by an electron transfer from the metal center, generates Int4. Through the H-bonding interaction with the N atom of the substrate, the amido site not only assists the proton transfer from the NH3 moiety to the N atom but also directs the hydrogen atom transfer from Co(III)-H to the 2-position to furnish 1,2-regioselective reduction.
Fig. 8. Proposed Mechanism.
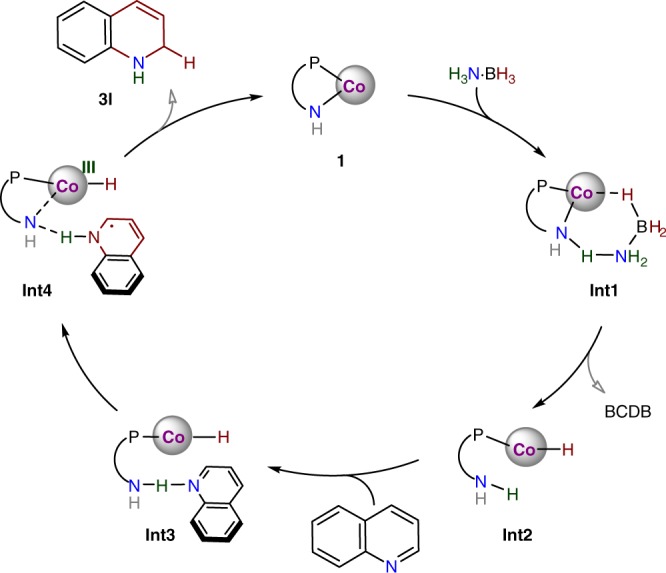
Catalytic cycle for partial transfer hydrogenation of quinoline.
Based on the deuterium labeling studies, we propose that the 1,2-DHQ product is the intermediate in the transfer hydrogenation of quinolines to THQs16,36,38. The conversion of 1,2-DHQ to 1,4-DHQ was not observed under the catalytic conditions, and we speculate that the partially saturated product is further reduced by the “hydride-proton” species through Int2, which is able to transfer the hydride of Co(II)-H to the 4-position, and the proton of the amino ligand to the C-3 position20,22. This hypothesis is further supported by the transfer hydrogenation of 3u with the selectively deuterated ammonia boranes (Fig. 9, Supplementary Fig. 3). For the reaction of 3u with H3N∙BD3, the 2H NMR spectrum exhibits only a characteristic 2H signal at δ 2.73 for d-4u, indicating deuterium labeling at the 4-position. In the case of D3N∙BH3, deuterium incorporation was found only at the 3-position in the desired product, d-4u′. These results also suggest that reversible dehydrogenation of 3u or 4u does not occur in the transfer hydrogenations18,20.
Fig. 9. Deuterium labeling.
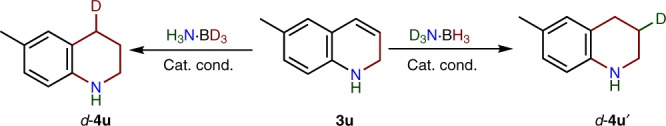
Reductions of 3u with deuterated ammonia borane derivatives.
We have developed a highly efficient transfer hydrogenation system to convert quinolines to 1,2-DHQs. Through cobalt-amido cooperation, the N-heteroarene system smoothly undergoes 1,2-reduction by H3N∙BH3 which serves as a proton/hydride source. The reaction is conveniently controlled by the use of equimolar amounts of reducing agent at room temperature. This catalysis exhibits broad functional group compatibility and enables the large-scale synthesis of 1,2-DHQs. Experimental and theoretical studies reveal that the catalysis invokes a key intermediate, HCo-NH(H) for the hydrogen transfers. The reactive nature of such a “hydride-proton” intermediate is reflected by the dehydrogenation of H3N∙BH3, catalyzed by the cobalt-amino complex to release H2 at room temperature. Through H-bonding with the substrate, the amido site is essential in assisting proton transfer and thus directs the 1,2-hydrogenation.
Methods
Complex 1
n-BuLi (2.5 mol/L) (0.29 mL, 0.72 mmol) was added to a solution of 1,2-Ph2P(p-CH3C6H4)NH2 (210 mg, 0.72 mmol) in THF (20 mL) at 0 °C. After stirring for 3 h, the solution was allowed to warm to room temperature, and then [Cp*CoCl]2 (165 mg, 0.36 mmol) was added. The color of the mixture immediately turned from yellow to rose-Bengal. The solvent was removed under vacuum, and the residue was extracted with hexane (3 × 10 mL). After recrystallization from hexane at −30 °C, compound 1 was obtained as red microcrystals. Yield: 282 mg (81%). HRMS (m/z): [M]+ calcd for C29H32CoNP, 484.1604; found, 484.1610; analysis (calcd., found for C29H32CoNP): C(71.89, 71.68), H (6.66, 6.52), N (2.89, 2.97).
Partial transfer hydrogenation
In a N2-filled glovebox, a scintillation vial (with a magnetic stir bar) was charged with quinoline (0.5 mmol), and H3N·BH3 (0.55 mmol, 17 mg). Then, catalyst 1 (1.2 mg, 2.5 µmol) and THF (2 ml) were added. The mixture was stirred at 25 oC. After the indicated time, the product was isolated by chromatography on silica gel eluting with EtOAc/petroleum ether.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank the Natural Science Foundation of China (21871166 and 21873031) and Natural Science Foundation of Shandong Province (ZR2019ZD45) for their financial support.
Author contributions
W.W., M.P., and C.-H.T. conceived and designed the project. M.P. S.Z., and W.W. conducted the experiments and analyzed the data. J.-Y.C. and R.-Z.L. carried out the computational analyses. W.W. and R.-Z.L. wrote the manuscript. All authors provided comments on the experiments and the manuscript during its preparation.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 1935438-1935441, 1935443, 1970225 and 1970226. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the article and Supplementary Information.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Sukbok Chang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rong-Zhen Liao, Email: rongzhen@hust.edu.cn.
Wenguang Wang, Email: wwg@sdu.edu.cn.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-15118-x.
References
- 1.Stout DM, Meyers AI. Recent advances in the chemistry of dihydropyridines. Chem. Rev. 1982;82:223–243. doi: 10.1021/cr00048a004. [DOI] [Google Scholar]
- 2.McSkimming A, Colbran SB. The coordination chemistry of organo-hydride donors: new prospects for efficient multi-electron reduction. Chem. Soc. Rev. 2013;42:5439–5488. doi: 10.1039/c3cs35466k. [DOI] [PubMed] [Google Scholar]
- 3.Bull JA, Mousseau JJ, Pelletier G, Charette AB. Synthesis of pyridine and dihydropyridine derivatives by regio- and stereoselective addition to N-activated pyridines. Chem. Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]
- 4.Zheng C, You SL. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012;41:2498–2518. doi: 10.1039/c1cs15268h. [DOI] [PubMed] [Google Scholar]
- 5.Zeng X, Frey GD, Kinjo R, Donnadieu B, Bertrand G. Synthesis of a simplified version of stable bulky and rigid cyclic (alkyl)(amino) carbenes, and catalytic activity of the ensuing gold (I) complex in the three-component preparation of 1, 2-dihydroquinoline derivatives. J. Am. Chem. Soc. 2009;131:8690–8696. doi: 10.1021/ja902051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson WS, Buell BG. 1, 2-Dihydroquinoline. J. Am. Chem. Soc. 1952;74:4517–4520. doi: 10.1021/ja01138a015. [DOI] [Google Scholar]
- 7.Dillard RD, Pavey DE, Benslay DN. Synthesis and antiinflammatory activity of some 2, 2-dimethyl-1, 2-dihydroquinolines. J. Med. Chem. 1973;16:251–253. doi: 10.1021/jm00261a019. [DOI] [PubMed] [Google Scholar]
- 8.Mizoguchi H, Oikawa H, Oguri H. Biogenetically inspired synthesis and skeletal diversification of indole alkaloids. Nat. Chem. 2014;6:57–64. doi: 10.1038/nchem.1798. [DOI] [PubMed] [Google Scholar]
- 9.Engler TA, LaTessa KO, Iyengar R, Chai W, Agrios K. Stereoselective syntheses of substituted pterocarpans with anti-HIV activity, and 5-aza-/5-thia-pterocarpan and 2-aryl-2, 3-dihydrobenzofuran analogues. Bioorg. Med. Chem. 1996;4:1755–1769. doi: 10.1016/0968-0896(96)00192-7. [DOI] [PubMed] [Google Scholar]
- 10.Roche SP, Porco JA., Jr Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed. 2011;50:4068–4093. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim, B. Y., Ahn, J. B., Hyun, S. H. & Kang, J. S. Organic electroluminescent material as blue-emitting dopant for organic light emitting device. Repub. Korean Kongkae Taeho Kongbo, KR2013096647A (2013).
- 12.Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Aryl−aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 13.Wedek V, Van Lommel R, Daniliuc CG, De Proft F, Hennecke U. Organocatalytic, enantioselective dichlorination of unfunctionalized alkenes. Angew. Chem. Int. Ed. 2019;58:9239–9243. doi: 10.1002/anie.201901777. [DOI] [PubMed] [Google Scholar]
- 14.Yuan YA, Lu DF, Chen YR, Xu H. Iron‐catalyzed direct diazidation for a broad range of olefins. Angew. Chem. Int. Ed. 2016;55:534–538. doi: 10.1002/anie.201507550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duttwyler S, et al. Regio- and stereoselective 1, 2–dihydropyridine alkylation/addition sequence for the synthesis of piperidines with quaternary centers. Angew. Chem. Int. Ed. 2014;53:3877–3880. doi: 10.1002/anie.201310517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobereiner GE, et al. Iridium-catalyzed hydrogenation of N-heterocyclic compounds under mild conditions by an outer-sphere pathway. J. Am. Chem. Soc. 2011;133:7547–7562. doi: 10.1021/ja2014983. [DOI] [PubMed] [Google Scholar]
- 17.Adam R, et al. A general and highly selective cobalt–catalyzed hydrogenation of N-heteroarenes under mild reaction conditions. Angew. Chem. Int. Ed. 2017;56:3216–3220. doi: 10.1002/anie.201612290. [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty S, Brennessel WW, Jones WD. A molecular iron catalyst for the acceptorless dehydrogenation and hydrogenation of N-heterocycles. J. Am. Chem. Soc. 2014;136:8564–8567. doi: 10.1021/ja504523b. [DOI] [PubMed] [Google Scholar]
- 19.Wang T, et al. Highly enantioselective hydrogenation of quinolines using phosphine-free chiral cationic ruthenium catalysts: scope, mechanism, and origin of enantioselectivity. J. Am. Chem. Soc. 2011;133:9878–9891. doi: 10.1021/ja2023042. [DOI] [PubMed] [Google Scholar]
- 20.Baralt E, Smith SJ, Hurwitz J, Horvath IT, Fish RH. Homogeneous catalytic hydrogenation. 6. synthetic and mechanistic aspects of the regioselective reductions of model coal nitrogen, sulfur, and oxygen heteroaromatic compounds using the (η5-pentamethylcyclopentadienyl)rhodium tris(acetonitrile) dication complex as the catalyst precursor. J. Am. Chem. Soc. 1992;114:5187–5196. doi: 10.1021/ja00039a033. [DOI] [Google Scholar]
- 21.Li X, et al. Spiro bicyclic bisborane catalysts for metal-free chemoselective and enantioselective hydrogenation of quinolines. Angew. Chem. Int. Ed. 2019;131:4712–4716. doi: 10.1002/ange.201900907. [DOI] [PubMed] [Google Scholar]
- 22.Mahdi T, Stephan DW. Facile protocol for catalytic frustrated Lewis pair hydrogenation and reductive deoxygenation of ketones and aldehydes. Angew. Chem. Int. Ed. 2015;54:8511–8514. doi: 10.1002/anie.201503087. [DOI] [PubMed] [Google Scholar]
- 23.Lam J, Szkop KM, Mosaferi E, Stephan DW. FLP catalysis: main group hydrogenations of organic unsaturated substrates. Chem. Soc. Rev. 2019;48:3592–3612. doi: 10.1039/C8CS00277K. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Loose F, Bezdek MJ, Wang X, Chirik PJ. Hydrogenation of N-heteroarenes using rhodium precatalysts: Reductive elimination leads to formation of multimetallic clusters. J. Am. Chem. Soc. 2019;141:17900–17908. doi: 10.1021/jacs.9b09540. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, et al. Unmasking the ligand effect in manganese-catalyzed hydrogenation: Mechanistic insight and catalytic application. J. Am. Chem. Soc. 2019;141:17337–17349. doi: 10.1021/jacs.9b09038. [DOI] [PubMed] [Google Scholar]
- 26.Wang DS, Chen QA, Lu SM, Zhou YG. Asymmetric hydrogenation of heteroarenes and arenes. Chem. Rev. 2011;112:2557–2590. doi: 10.1021/cr200328h. [DOI] [PubMed] [Google Scholar]
- 27.Yang ZP, Wu QF, You SL. Direct asymmetric dearomatization of pyridines and pyrazines by iridium–catalyzed allylic amination reactions. Angew. Chem. Int. Ed. 2014;53:6986–6989. doi: 10.1002/anie.201404286. [DOI] [PubMed] [Google Scholar]
- 28.Meng W, Feng X, Du H. Frustrated Lewis pairs catalyzed asymmetric metal-free hydrogenations and hydrosilylations. Acc. Chem. Res. 2017;51:191–201. doi: 10.1021/acs.accounts.7b00530. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Chen F, He YM, Fan QH. Asymmetric ruthenium-catalyzed hydrogenation of 2, 6-disubstituted 1, 5-naphthyridines: Access to chiral 1, 5-diaza-cis-cecalins. Angew. Chem. Int. Ed. 2015;54:4622–4625. doi: 10.1002/anie.201411105. [DOI] [PubMed] [Google Scholar]
- 30.Wang C, Li C, Wu X, Pettman A, Xiao J. pH–Regulated asymmetric transfer hydrogenation of quinolines in water. Angew. Chem. Int. Ed. 2009;48:6524–6528. doi: 10.1002/anie.200902570. [DOI] [PubMed] [Google Scholar]
- 31.Mai VH, Nikonov GI. Transfer hydrogenation of nitriles, olefins, and N-heterocycles catalyzed by an N-heterocyclic carbene-supported half-sandwich complex of ruthenium. Organometallics. 2016;35:943–949. doi: 10.1021/acs.organomet.5b00967. [DOI] [Google Scholar]
- 32.Wang Y, et al. Transfer hydrogenation of alkenes using ethanol catalyzed by a NCP pincer iridium complex: scope and mechanism. J. Am. Chem. Soc. 2018;140:4417–4429. doi: 10.1021/jacs.8b01038. [DOI] [PubMed] [Google Scholar]
- 33.Tu XF, Gong LZ. Highly enantioselective transfer hydrogenation of quinolines catalyzed by gold phosphates: achiral ligand tuning and chiral–Anion control of stereoselectivity. Angew. Chem. Int. Ed. 2012;51:11346–11349. doi: 10.1002/anie.201204179. [DOI] [PubMed] [Google Scholar]
- 34.Guo QS, Du DM, Xu J. The development of double axially chiral phosphoric acids and their catalytic transfer hydrogenation of quinolines. Angew. Chem. Int. Ed. 2008;47:759–762. doi: 10.1002/anie.200703925. [DOI] [PubMed] [Google Scholar]
- 35.Rueping M, Antonchick AP. Organocatalytic enantioselective reduction of pyridines. Angew. Chem. Int. Ed. 2007;46:4562–4565. doi: 10.1002/anie.200701158. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H, et al. Hydrogenation of quinolines using a recyclable phosphine-free chiral cationic ruthenium catalyst: enhancement of catalyst stability and selectivity in an ionic liquid. Angew. Chem. Int. Ed. 2008;47:8464–8467. doi: 10.1002/anie.200802237. [DOI] [PubMed] [Google Scholar]
- 37.Park S, Chang S. Catalytic Dearomatization of N-Heteroarenes with Silicon and Boron Compounds. Angew. Chem. Int. Ed. 2017;56:7720–7738. doi: 10.1002/anie.201612140. [DOI] [PubMed] [Google Scholar]
- 38.Dudnik AS, Weidner VL, Motta A, Delferro M, Marks TJ. Atom-efficient regioselective 1, 2-dearomatization of functionalized pyridines by an earth-abundant organolanthanide catalyst. Nat. Chem. 2014;6:1100–1107. doi: 10.1038/nchem.2087. [DOI] [PubMed] [Google Scholar]
- 39.Arrowsmith M, Hill MS, Hadlington T, Kociok-Köhn G, Weetman C. Magnesium-catalyzed hydroboration of pyridines. Organometallics. 2011;30:5556–5559. doi: 10.1021/om2008138. [DOI] [Google Scholar]
- 40.Tamang SR, Singh A, Unruh DK, Findlater M. Nickel catalyzed regioselective 1,4-hydroboration of N-heteroarenes. ACS Catal. 2018;8:6186–6191. doi: 10.1021/acscatal.8b01166. [DOI] [Google Scholar]
- 41.Zhang F, Song H, Zhuang X, Tung CH, Wang W. Iron-catalyzed 1, 2-selective hydroboration of N-heteroarenes. J. Am. Chem. Soc. 2017;139:17775–17778. doi: 10.1021/jacs.7b11416. [DOI] [PubMed] [Google Scholar]
- 42.Lortie JL, Dudding T, Gabidullin BM, Nikonov GI. Zinc-catalyzed hydrosilylation and hydroboration of N-heterocycles. ACS Catal. 2017;7:8454–8459. doi: 10.1021/acscatal.7b02811. [DOI] [Google Scholar]
- 43.Liu H, Khononov M, Eisen MS. Catalytic 1, 2-regioselective dearomatization of N-heteroaromatics via a hydroboration. ACS Catal. 2018;8:3673–3677. doi: 10.1021/acscatal.8b00074. [DOI] [Google Scholar]
- 44.Liu J, et al. Ni-O cooperation versus nickel(II) hydride in catalytic hydroboration of N-heteroarenes. ACS Catal. 2019;9:3849–3857. doi: 10.1021/acscatal.8b05136. [DOI] [Google Scholar]
- 45.Gandhamsetty N, Joung S, Park SW, Park S, Chang S. Boron-catalyzed silylative reduction of quinolines: selective sp3 C–Si bond formation. J. Am. Chem. Soc. 2014;136:16780–16783. doi: 10.1021/ja510674u. [DOI] [PubMed] [Google Scholar]
- 46.Rao B, Chong CC, Kinjo R. Metal-free regio-and chemoselective hydroboration of pyridines catalyzed by 1, 3, 2-diazaphosphenium triflate. J. Am. Chem. Soc. 2018;140:652–656. doi: 10.1021/jacs.7b09754. [DOI] [PubMed] [Google Scholar]
- 47.Ikariya T, Murata K, Noyori R. Bifunctional transition metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem. 2006;4:393–406. doi: 10.1039/B513564H. [DOI] [PubMed] [Google Scholar]
- 48.Peters, R. Cooperative Catalysis: Designing Efficient Catalysts for Synthesis. (Wiley-VCH Verlag, 2015).
- 49.Wang D, Astruc D. The golden age of transfer hydrogenation. Chem. Rev. 2015;115:6621–6686. doi: 10.1021/acs.chemrev.5b00203. [DOI] [PubMed] [Google Scholar]
- 50.Higashi T, Kusumoto S, Nozaki K. Cleavage of Si–H, B–H, and C–H bonds by metal–ligand cooperation: focus review. Chem. Rev. 2019;119:10393–10402. doi: 10.1021/acs.chemrev.9b00262. [DOI] [PubMed] [Google Scholar]
- 51.Drover MW, Love JA, Schafer LL. 1, 3-N, O-Complexes of late transition metals. Ligands with flexible bonding modes and reaction profiles. Chem. Soc. Rev. 2017;46:2913–2940. doi: 10.1039/C6CS00715E. [DOI] [PubMed] [Google Scholar]
- 52.Drover MW, Schafer LL, Love JA. Capturing HBCy2: Using N., O‐chelated complexes of rhodium (I) and iridium (I) for chemoselective hydroboration. Angew. Chem. Int. Ed. 2016;55:3181–3186. doi: 10.1002/anie.201511448. [DOI] [PubMed] [Google Scholar]
- 53.Zell T, Milstein D. Hydrogenation and dehydrogenation iron pincer catalysts capable of metal–ligand cooperation by aromatization/dearomatization. Acc. Chem. Res. 2015;48:1979–1994. doi: 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
- 54.Drummond MJ, Ford CL, Gray DL, Popescu CV, Fout AR. Radical rebound hydroxylation versus H-atom transfer in non-heme iron (III)-hydroxo complexes: reactivity and structural differentiation. J. Am. Chem. Soc. 2019;141:6639–6650. doi: 10.1021/jacs.9b01516. [DOI] [PubMed] [Google Scholar]
- 55.Stahl T, Müther K, Ohki Y, Tatsumi K, Oestreich M. Catalytic generation of borenium ions by cooperative B–H bond activation: the elusive direct electrophilic borylation of nitrogen heterocycles with pinacolborane. J. Am. Chem. Soc. 2013;135:10978–10981. doi: 10.1021/ja405925w. [DOI] [PubMed] [Google Scholar]
- 56.Omann L, Königs CDF, Klare HF, Oestreich M. Cooperative catalysis at metal–sulfur bonds. Acc. Chem. Res. 2017;50:1258–1269. doi: 10.1021/acs.accounts.7b00089. [DOI] [PubMed] [Google Scholar]
- 57.Klare HF, et al. Cooperative catalytic activation of Si−H bonds by a polar Ru−S bond: regioselective low-temperature C−H silylation of indoles under neutral conditions by a Friedel−Crafts mechanism. J. Am. Chem. Soc. 2011;133:3312–3315. doi: 10.1021/ja111483r. [DOI] [PubMed] [Google Scholar]
- 58.Morris RH. Exploiting metal–ligand bifunctional reactions in the design of iron asymmetric hydrogenation catalysts. Acc. Chem. Res. 2015;48:1494–1502. doi: 10.1021/acs.accounts.5b00045. [DOI] [PubMed] [Google Scholar]
- 59.Chirik PJ. Iron- and cobalt-catalyzed alkene hydrogenation: catalysis with both redox-active and strong field ligands. Acc. Chem. Res. 2015;48:1687–1695. doi: 10.1021/acs.accounts.5b00134. [DOI] [PubMed] [Google Scholar]
- 60.MacNair AJ, Millet CR, Nichol GS, Ironmonger A, Thomas SP. Markovnikov-selective, activator-free iron-catalyzed vinylarene hydroboration. ACS Catal. 2016;6:7217–7221. doi: 10.1021/acscatal.6b02281. [DOI] [Google Scholar]
- 61.Liang Q, Osten KM, Song D. Iron-catalyzed gem-specific dimerization of terminal alkynes. Angew. Chem. Int. Ed. 2017;56:6317–6320. doi: 10.1002/anie.201700904. [DOI] [PubMed] [Google Scholar]
- 62.Papa V, et al. Efficient and selective hydrogenation of amides to alcohols and amines using a well-defined manganese-PNN pincer complex. Chem. Sci. 2017;8:3576–3585. doi: 10.1039/C7SC00138J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elangovan S, et al. Selective catalytic hydrogenations of nitriles, ketones, and aldehydes by well-defined manganese pincer complexes. J. Am. Chem. Soc. 2016;138:8809–8814. doi: 10.1021/jacs.6b03709. [DOI] [PubMed] [Google Scholar]
- 64.Pang M, et al. Addition of a B–H bond across an amido–cobalt bond: CoII–H-catalyzed hydroboration of olefins. Organometallics. 2018;37:1462–1467. doi: 10.1021/acs.organomet.8b00114. [DOI] [Google Scholar]
- 65.Fu S, et al. Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: stereodivergent synthesis of Z-and E-alkenes. J. Am. Chem. Soc. 2016;138:8588–8594. doi: 10.1021/jacs.6b04271. [DOI] [PubMed] [Google Scholar]
- 66.Han C, et al. Palladium/Graphitic carbon nitride (g‐C3N4) stabilized emulsion microreactor as a store for hydrogen from ammonia borane for use in alkene hydrogenation. Angew. Chem. Int. Ed. 2018;57:14857–14861. doi: 10.1002/anie.201809882. [DOI] [PubMed] [Google Scholar]
- 67.Zhu X-Q, Zhang M-T, Yu A, Wang C-H, Cheng J-P. Hydride, hydrogen atom, proton, and electron transfer driving forces of various five-membered heterocyclic organic hydrides and their reaction intermediates in acetonitrile. J. Am. Chem. Soc. 2008;130:2501–2516. doi: 10.1021/ja075523m. [DOI] [PubMed] [Google Scholar]
- 68.Sridharan V, Suryavanshi PA, Menendez JC. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011;111:7157–7259. doi: 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]
- 69.Kubota K, Watanabe Y, Hayama K, Ito H. Enantioselective synthesis of chiral piperidines via the stepwise dearomatization/borylation of pyridines. J. Am. Chem. Soc. 2016;138:4338–4341. doi: 10.1021/jacs.6b01375. [DOI] [PubMed] [Google Scholar]
- 70.Ding J, Hall DG. Concise synthesis and antimalarial activity of all four mefloquine stereoisomers using a highly enantioselective catalytic borylative alkene isomerization. Angew. Chem. Int. Ed. 2013;52:8069–8073. doi: 10.1002/anie.201303931. [DOI] [PubMed] [Google Scholar]
- 71.Ding J, Rybak T, Hall DG. Synthesis of chiral heterocycles by ligand-controlled regiodivergent and enantiospecific Suzuki Miyaura cross-coupling. Nat. Commun. 2014;5:5474–5482. doi: 10.1038/ncomms6474. [DOI] [PubMed] [Google Scholar]
- 72.Hupe E, Marek I, Knochel P. Diastereoselective reduction of alkenylboronic esters as a new method for controlling the stereochemistry of up to three adjacent centers in cyclic and acyclic molecules. Org. Lett. 2002;4:2861–2863. doi: 10.1021/ol0262486. [DOI] [PubMed] [Google Scholar]
- 73.Heier RF, et al. Synthesis and biological activities of (R)-5, 6-dihydro-N, N-dimethyl-4 H-imidazo [4, 5, 1-ij] quinolin-5-amine and its metabolites. J. Med. Chem. 1997;40:639–646. doi: 10.1021/jm960360q. [DOI] [PubMed] [Google Scholar]
- 74.Alonso DA, Brandt P, Nordin SJM, Andersson PG. Ru(arene)(amino alcohol)-catalyzed transfer hydrogenation of ketones: mechanism and origin of enantioselectivity. J. Am. Chem. Soc. 1999;121:9580–9588. doi: 10.1021/ja9906610. [DOI] [Google Scholar]
- 75.Yamakawa M, Yamada I, Noyori R. CH/π attraction: the origin of enantioselectivity in transfer hydrogenation of aromatic carbonyl compounds catalyzed by chiral η6-arene-ruthenium(II) complexes. Angew. Chem. Int. Ed. 2001;40:2818–2821. doi: 10.1002/1521-3773(20010803)40:15<2818::AID-ANIE2818>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 76.Yang X, Zhao L, Fox T, Wang Z-X, Berke H. Transfer hydrogenation of imines with ammonia–borane: a concerted double-hydrogen-transfer reaction. Angew. Chem. Int. Ed. 2010;49:2058–2062. doi: 10.1002/anie.200906302. [DOI] [PubMed] [Google Scholar]
- 77.Sebastian K, Shaik S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011;44:101–110. doi: 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 1935438-1935441, 1935443, 1970225 and 1970226. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the article and Supplementary Information.