

Abstract
The emergence of bacterial resistance due to the indiscriminate use of antibiotics warrants the need for developing new bioactive agents. In this context, antimicrobial peptides are highly useful for managing resistant microbial strains. In this study, we report the isolation and characterization of peptides obtained from the venom of the toadfish Thalassophryne nattereri. These peptides were active against Gram-positive and Gram-negative bacteria and fungi. The primary amino acid sequences showed similarity to Cocaine and Amphetamine Regulated Transcript peptides, and two peptide analogs—Tn CRT2 and Tn CRT3—were designed using the AMPA algorithm based on these sequences. The analogs were subjected to physicochemical analysis and antimicrobial screening and were biologically active at concentrations ranging from 2.1 to 13 µM. Zeta potential analysis showed that the peptide analogs increased the positive charge on the cell surface of Gram-positive and Gram-negative bacteria. The toxicity of Tn CRT2 and Tn CRT3 were analyzed in vitro using a hemolytic assay and tetrazolium salt reduction in fibroblasts and was found to be significant only at high concentrations (up to 40 µM). These results suggest that this methodological approach is appropriate to design novel antimicrobial peptides to fight bacterial infections and represents a new and promising discovery in fish venom.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-2151-4) contains supplementary material, which is available to authorized users.
Keywords: Thalassophryne nattereri, Venom, Antimicrobial peptide, Cytotoxicity
Introduction
The study of bioactive compounds has significantly increased knowledge in biochemistry and pharmacology for academic and biotechnological applications. Traditionally, the clinical and pharmaceutical industries use natural products as starting points to search for novel compounds to search for new products, and advances in biochemical synthesis have fostered the development of new products (Mathur and Hoskins 2017). In recent years, laboratories and industries realized the potential of natural products as the basis for further research. This approach is promising because of technological advances in these areas through increased capacity for information processing and sharing (Reid et al. 1993).
Toxins from aquatic animals are a valuable source of natural products for academic and applied research (Church and Hodgson 2002; Paul et al. 2003; Xie et al. 2017). The production of toxins by aquatic animals is crucial for their survival in highly competitive ecosystems. These organisms produce molecules with pharmacological properties different from those found in the venom of terrestrial animals (Kyte and Doolittle 1982; Borges et al. 2018). In this respect, fish venom constitutes a rich source of highly active compounds, increasing the relevance of this field of research.
The toadfish Thalassophryne nattereri (family Batrachoididae) is a venomous fish found in the Western Atlantic Ocean and is commonly known as Niquim. The species of this genus developed a unique venom inoculation apparatus (Collette 2006; Bücherl et al. 1968). The biochemical properties of the venom and the envenoming process have been studied since 1996. It is known that the composition of the venom is unique, and envenomation symptoms include edema, excruciating pain, ischemia, necrosis, and slow healing (Lopes-Ferreira et al. 1998, 2014; Spencer et al. 2005). The distinctiveness of the venom apparatus of T. nattereri is attractive to prospect for bioactive molecules. It is possible that the evolutionary context that resulted in the development of this apparatus promoted the emergence of unique molecules with high biological activity (Lopes-Ferreira et al. 2014). Nonetheless, few studies have identified and characterized multifunctional molecules in this fish species.
Natural products, such as animal venoms, are common and rich sources of bioactive peptides, which can be screened from chemical and recombinant libraries and are potential candidates for developing new medicines (Bailey and Wilce 2001). However, despite the large number of drug discovery projects, few cases have been successful (Zambelli et al. 2016). Furthermore, bioprospecting for pharmaceuticals has increased, because fewer drugs have been approved in recent years.
Bioactive peptides play a crucial role in many physiological processes. One of the primary functions of peptides is cell signaling for the translation and transport of molecules through multiple mechanisms. Given the different modes of action and physiological and pathological roles of peptides, the relationship between structure and function has been studied extensively (Ziegman and Alewood 2015; Robinson et al. 2017). Peptides are highly useful in human health diagnosis and therapy because of their multiple functions, reduced size, low immunogenicity, structural stability, and the development of effective strategies for chemical synthesis (Usmani et al. 2017).
Among the classes of bioactive peptides isolated from natural sources, multifunctional defense peptides—antimicrobial peptides (AMPs)—constitute a class of biomolecules little explored by the pharmaceutical industry. The high incidence of pathogenic microbial infections in humans, combined with resistance to antibiotic therapy, limits antimicrobial treatment and motivates research in this field. Few AMPs from fish venom have been isolated and characterized (Conceição et al. 2012; Houyvet et al. 2018). The advantage of AMPs over traditional antibiotics is the unique effect on the bacterial membrane (Hancock and Rozek 2002; Joo et al. 2016), reducing the likelihood of developing antimicrobial resistance.
Remarkable sources of venom peptides include ants, bees, centipedes, cone snails, scorpions, snakes, spiders, and wasps, and many of these peptides are active against bacteria, protozoans, fungi, and viruses (Primon-Barros and José Macedo 2017). However, few studies have examined venom AMPs from aquatic animals, especially from fish. In addition, given that the amount of native antimicrobial compounds obtained in vivo is limited, bioinformatics is crucial for developing antimicrobial drugs (Afacan et al. 2012).
In this study, we isolated and characterized a family of AMPs obtained from the venom of T. nattereri and active against Gram-positive and Gram-negative bacteria and fungi. The primary amino acid sequences showed similarity to Cocaine and Amphetamine Regulated Transcript (CART) peptides identified by Magalhães et al. (2006). The length of these peptides (56–51 amino acid residues) makes their pharmaceutical development and production unfeasible because of the high production costs when compared with synthetic antibiotics (Wimley and Hristova 2011).
Bioinformatics has helped overcome these limitations by serving as a tool to identify and design small bioactive molecules and investigate mechanisms of action (Chang et al. 2015). Here, the Antimicrobial Sequence Scanning System (AMPA) algorithm (Di Tommaso et al. 2011) was used to design and synthesize peptides homologous to CART peptides. This method is unconventional, because it uses high-throughput substitution data of bactenecin 12-mer against Pseudomonas aeruginosa to identify the active stretches of AMPs (Torrent et al. 2009). We designed two analogs and evaluated their biological activity using this methodology.
To our knowledge, this study is the first to identify active AMPs obtained from T. nattereri toxins homologous to CART peptides. Moreover, we designed analogs and evaluated their secondary structure, net charge, amphipathicity, antimicrobial activity, and cytotoxicity. These analogs are promising for developing new classes of multifunctional peptides because of their potent antimicrobial activity and low cellular toxicity.
Materials and methods
Venom collection and purification
Venom samples were collected in the state of Alagoas, at Lagoa Mundaú, with a license from the Brazilian government for collection and maintenance of fish (Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis Protocol No. 2435609). Samples were obtained from 20 fish of the species Thalassophryne nattereri through compression of venom glands, as previously described (Lopes-Ferreira et al. 2004), and were lyophilized and stored at − 20 °C until use. The lyophilized venom was resuspended in ultrapure water (Millipore, USA), homogenized, and centrifuged at 1000 rpm for 1 min at room temperature (25 °C). A 3 mg aliquot was diluted in 3 mL of 0.1% trifluoroacetic acid (TFA), loaded onto a solid-phase extraction (SPE) Sep-Pak C18 cartridge (Waters Corporation, MA, USA), eluted with 80% of acetonitrile (ACN), and further concentrated in a vacuum centrifuge. The SPE fraction was separated by reverse-phase high-performance liquid chromatography (RP-HPLC) (Dionex Ultimate 3000 Thermo, CA, USA) on an Acclaim® C18 column (4.6 mm × 150 mm, μm, Dionex Corporation, Sunnyvale, CA, USA) and eluted using a two-solvent system composed of solvent A (TFA/H2O, 1:1000) and solvent B (TFA/ACN/H2O, 1:900:100) at a flow rate of 1.0 mL/min in a linear gradient from 10 to 80% of solvent B over 40 min. UV absorbance was monitored at 214 nm. The eluted fractions were collected, lyophilized, and solubilized in 100 µL of ultrapure water.
Mass spectrometry
HPLC-eluted fractions were analyzed by MALDI-TOF/MS using an Ettan MALDI/TOF/Pro system (Amersham Biosciences, Sweden) mass spectrometer. Each fraction was mixed with cyano-4-hydroxycinnamic acid matrix (10 mg/mL) (1:1 v/v), and 1 µL of this mixture was applied to the mass spectrometer plate and dried at room temperature. The mass spectra were identified as a function of the ionization intensity (%) of the samples, and the peaks with intensity between 30 and 70% at m/z of 200–8000 were analyzed.
N-terminal sequence analysis and structure prediction
To determine the primary amino acid sequence, the purified samples of native proteins were subjected to Edman degradation using a Shimadzu PPSQ-21 automated protein sequencer, following the manufacturer’s instructions. The sequences were aligned using Clustal Omega software (Sievers and Higgins 2014) and searched against the SWISS-MODEL Template Library (SMTL version 2019-02-20) using Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1997). The search identified four peptide templates. The quality of each template was predicted by analyzing some features of the target–template alignment. The PDB template ID 1hy9.1A, which was homologous to a CART protein (Uniprot Q16568), was used for constructing 3D models (Guex and Peitsch 1997), and Chimera version 1.13 (https://www.rbvi.ucsf.edu/chimera) was used for visualizing and analyzing the models (Pettersen et al. 2004).
Peptide design and synthesis
After peptide isolation and characterization, new peptides were designed using the AMPA algorithm based on the primary amino acid sequence of Niq CART3. The sequences were analyzed by the software in FASTA format, and antimicrobial domains were detected for the design and synthesis of AMPs (Torrent et al. 2011). Subsequently, amidated peptides were synthesized in a solid-phase multiple synthesizer (Peptide Synthesizer PSSM 8-Shimadzu) using the N-9-fluorophenylmethoxy-carbonyl (Fmoc) procedure (Atherton and Sheppard 1989) and the Nova PEG rink amide resin. The peptides were purified by RP-HPLC, and purity and identity were confirmed by mass spectrometry. The peptides were dissolved in ultrapure water (Millipore, USA) and stored at − 20 °C until use.
Physicochemical and structural analysis of peptides
The physicochemical parameters of peptide sequences were calculated using the ProtParam tool available on the bioinformatics resource portal ExPASy of the Swiss Institute of Bioinformatics website (https://web.expasy.org/protparam, accessed on 25 September 2018). The amino acid sequences of pepR and pepM were sent to the I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER) for predicting 3D structures (Roy et al. 2010).
Circular dichroism spectra of 50 µM Tn CRT2 or Tn CRT3 in 10 mM HEPES buffer, 50 mM NaF, pH 7.4, in the absence or presence of 2 mM of POPC and 2 mM POPC/POPG (1:1) large unilamellar vesicles (LUVs), were acquired at 25 °C in the 195–300 nm wavelength range using 0.1 cm quartz cells in a JASCO model J-815 spectropolarimeter (Tokyo, Japan). Each final spectrum corresponded to an average of ten scans, which were subsequently corrected for buffer or LUV baseline. LUVs were used as a membrane model system. These vesicles have a diameter of approximately 100 nm and were prepared by extrusion, as described elsewhere (The et al. 2002).
Measurement of bacterial ζ potential
Zeta potential was measured to evaluate changes in the surface charge of E. coli (ATCC 25922) and S. aureus (ATCC 6538) cells in the presence of Tn CRT2 and Tn CRT3. Assays were performed in a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) equipped with a 633 nm HeNe laser and disposable ζ cells with gold electrodes. Bacterial suspensions were prepared in Mueller–Hinton broth (MHB) to a final concentration of 1 × 108 CFU/mL. The pellet was centrifuged and resuspended in 10 mM HEPES buffer, 150 mM NaCl, pH 7.4, to a final concentration of 1 × 107 CFU/mL. Each peptide solution, at the respective MIC100 concentration, was added to the bacterial cells. Peptide-treated bacterial suspensions were dispensed into ζ cells and allowed to equilibrate for 15 min at 25 °C. The suspensions (2 × 107 cells per mL) were mixed with peptides at MIC100 concentrations for 30 min at 37 °C. The electrophoretic mobility of each sample was calculated, and the ζ potential was measured using the Smoluchowski equation, as previously described (Domingues et al. 2008). All experiments were repeated independently three times using freshly grown bacteria.
Antimicrobial assay
The antimicrobial activity and minimum inhibitory concentration (MIC) were determined using a modified NCCLS broth microdilution method (Pfaller et al. 2002). Antimicrobial activity was monitored using a liquid growth inhibition assay against four bacterial strains (E. coli [ATCC 25922], P. aeruginosa [ATCC 15442], S. aureus [ATCC 6538], and E. faecalis [ATCC 4083]) and one fungal strain (C. albicans [ATCC 10231]). The pre-inoculum of the strains was prepared in MHB (for bacteria) and brain heart infusion (for fungi) for approximately 12 h at 37 °C. The inoculum was standardized for 103 cells/mL at an absorbance of 595 nm. The lyophilized peptide fractions (40, 20, 10, 5 and 2.5 µM) were resuspended in ultrapure water and tested against different inocula in sterile 96-well plates at a final volume of 200 µL in each well (10 µL of diluted peptide or fraction and 190 µL of inoculum in culture medium). The samples were incubated for 18 h at 37 °C, and the inhibition of bacterial growth was determined by measuring absorbance at 595 nm. Chloramphenicol and amphotericin (Sigma-Aldrich, United States) were used as a positive control for bacteria and fungi, respectively. The inoculum of each microorganism was used as a negative control. The results were obtained from three independent experiments.
Cellular viability
Cell viability was monitored using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay with mice embryonic fibroblasts (MEF). A total of 1 × 104 MEF per mL3 were sown in a 96-well microplate with Dulbecco’s Modified Eagle Medium (DMEM) culture medium. The cells were maintained for 24 h in DMEM in the incubator with 5% CO2 at 37 °C. After this period, the medium was removed, and the peptides were added in triplicate into microplate wells together with 240 µL of DMEM. Dimethyl sulfoxide (DMSO) was used as the negative control, and culture medium was used as the positive control. The plate was incubated for 24 h at 37 °C and 5% CO2. The culture medium was removed, and the cells were washed in phosphate-buffered saline (PBS). MTT (5 mg/mL, prepared in PBS) was added to each well, and the plates were incubated at 37 °C and 5% CO2 for 3 h. Then, MTT was removed, and DMSO was added to each well. Absorbance was measured in a plate reader (Synergy H1 Hybrid Reader, BIOTEK, EUA) at 540 nm.
Hemolysis test
The hemolytic activity of the synthetic peptides were evaluated on human erythrocytes of a healthy donor (type A). Blood was collected in 0.15 M citrate buffer, pH 7.4, and washed three times with 0.15 M PBS, pH 7.4, by centrifugation. To determine the hemolytic activity, a 3% erythrocyte suspension in 10 mL of each peptide fraction was transferred to wells of U-shaped bottom plates and incubated for 3 h at room temperature. Hemolysis was determined by absorbance measurement in a plate reader (Synergy H1 Hybrid Reader, BIOTEK, EUA) at 595 nm. An erythrocyte suspension in ultrapure water was used as a positive control (100% hemolysis).
Statistical analysis
Data analysis was performed using Prism software version 6 (GraphPad). Analysis of variance was used to analyze data, and p values < 0.05 were considered statistically significant.
Results
Purification of venom peptides and measurement of biological activity
Thalassophryne nattereri venom was fractionated by SPE, and aliquots were loaded onto the HPLC system. Seven fractions were identified (Fig. 1). The molecular mass of the HPLC fractions was determined using a MALDI-ToF mass spectrometer (Amersham Biosciences, Sweden). The mass to charge (m/z) ratio of each fraction was obtained by MALDI-ToF/MS analysis (Supplementary Table 1).
Fig. 1.

Chromatogram of reverse-phase high-performance liquid chromatography of the SPE 80% ACN sample of the Thalassophryne nattereri venom obtained from the solid phase extraction. UV absorbance was monitored at 214 nm
The eluted fractions were pooled and tested for antibacterial activity against Gram-positive and Gram-negative bacteria (E. coli ATCC 25922, P. aeruginosa ATCC 15442, S. aureus ATCC 6538, and E. faecalis ATCC 2912) and fungi (C. albicans ATCC 10231). Based on the antimicrobial assay results, fraction 6 was selected for further characterization (Supplementary Table 2).
Mass spectra of this fraction (Supplementary Fig. 1) presented peptides with a molecular mass close to 5000 Da with relatively high abundance and low complexity. Based on this finding and the antimicrobial assay results, this fraction was further characterized.
Peptide sequencing and structure prediction
The molecular mass and N-terminal amino acid sequence of the purified peptides were determined. The m/z was 5075.276, and this peptide was sequenced by Edman degradation. The 46-amino-acid NH2 terminal had 100% homology with sequences from the T. nattereri expressed sequence tag database described by Magalhães et al. (2006) and 99% homology with CART peptides. The comparison with the sequences of the other two CART peptides and the obtained molecular masses suggested that m/z 5593.132 and 5724.383 indicated an increase in mass of 506.55 and 637.75 Daltons, corresponding to fragments MNQMN and MNQMN, respectively. These peptides were designated Niq CART1 to 3. Sequence similarity search with BLAST and Clustal alignment indicated 68% homology with CART from Danio rerio and 66% with CART from Homo sapiens (Supplementary Fig. 2A). The alignments with CARTs from T. nattereri, goldfish, human, and rats described by Magalhães et al. (2006) are shown in Supplementary Fig. 2B. The degree of homology with CARTs from T. nattereri and the other species was 37–40%. The N-terminal region contained six conserved cysteine residues, which were linked by disulfide bridges in the recombinant CART peptide. The cysteine residues of the peptide selected in this study were not linked by disulfide bridges, because different motifs were selected.
Homology modeling depends on the alignment between target and template sequences whose 3D structures were determined experimentally. The 3D structure of Niq CART3 was modeled using SWISS-MODEL software and was comprised of an antiparallel β-sheet and an α-helix (Fig. 2).
Fig. 2.
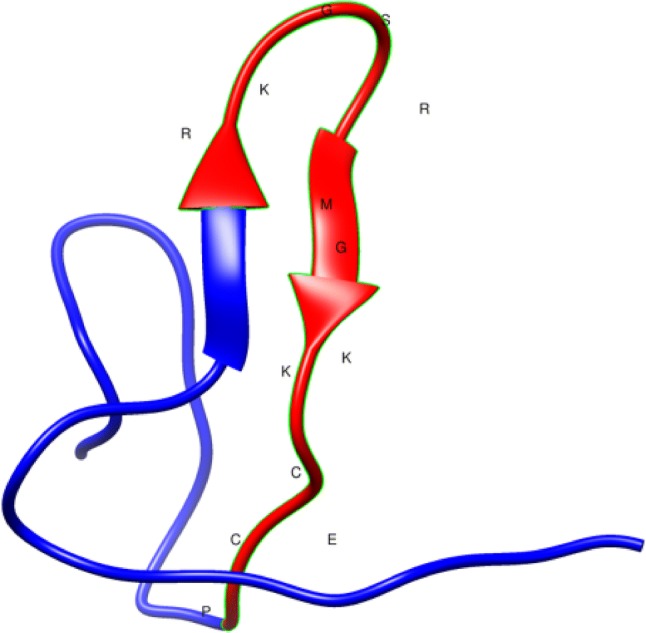
Prediction of the structure of Niq CART3 using a Cocaine and amphetamine regulated transcript (CART) protein (Uniprot Q16568, PDB 1hy9.1A) as template. Models were generated using SWISS-MODEL software. The domain shown in red with the respective amino acid sequence was used on AMPA for designing peptides Tn CRT2 and Tn CRT3. The structure was created using Chimera software (Pettersen et al. 2004)
Peptide design and evaluation of physicochemical properties
The antimicrobial activity of protein and peptides can be predicted using different databases and algorithms. The AMPA algorithm assigns an antimicrobial index to each amino acid and constructs a protein profile through a sliding window procedure (Torrent et al. 2009). The antimicrobial index was calculated based on 50% the maximum inhibitory concentration (IC50) and was obtained experimentally by single amino acid substitutions of bactenecin 2A (Hilpert et al. 2005). The sequences of the predicted structure were experimentally validated to determine the antimicrobial activity of the whole protein (Di Tommaso et al. 2011). One sequence—Tn CRT2 (RKGSRMGKKCECP)—was predicted using the AMPA algorithm. Considering that positively charged residues are present in AMPs, another analog was designed by exchanging arginine for glutamine and was named Tn CRT3 (RKGSQMGKKCECP). Both peptides were synthesized by the Fmoc procedure, and their C-termini were blocked with NH2 to improve stability and reduce sensitivity to proteases.
The physicochemical characteristics of the Tn CRTs were predicted using the ProtParam tool available on the bioinformatics resource portal ExPASy. The net charge, grand average of hydropathicity (GRAVY), and theoretical isoelectric point (pI), among other properties, were evaluated (Table 1). These peptides were cationic (net charge of 3.9 and 2.9, respectively) because of the presence of five positively charged residues, including lysine and arginine. Furthermore, the peptides had similar pI (approximately 9), indicating that the net charge was equal to 0 at this pH value. In addition, the GRAVY score, a metric of the overall hydrophobicity/hydrophilicity of polypeptides, was negative for Tn CRT2 and Tn CRT3 (− 1.577 and − 1.500, respectively), suggesting that these AMPs were hydrophilic. The value of the Boman index, which is a measure of peptide affinity to proteins and its ability to establish biological interactions, was 3.83 and 3.11 for Tn CRT2 and Tn CRT3, respectively.
Table 1.
Physicochemical parameters of peptides
| Tn CRT2 | Tn CRT3 | |
|---|---|---|
| Sequence | RKGSRMGKKCECP | RKGSQMGKKCECP |
| Molecular weight | 1478.83 g/mol | 1450.77 g/mol |
| Net charge at pH7 | + 3.9 | + 2.9 |
| Theoretical isoelectric point (pI) | 9.85 | 9.39 |
| GRAVY (grand average of hydropathicity)a | − 1.577 | − 1.500 |
| Instability indexb | 59.60 | 59.60 |
| Boman Index (kcal/mol) | 3.83 | 3.11 |
| Hydrophobicity | + 25.74 kcal mol−1 | + 24.70 kcal mol−1 |
Physicochemical parameters were obtained using the ProtParam tool in ExPASy (Gasteiger et al. 2003) (https://web.expasy.org/protparam/; accessed on 18 February 2019)
aThe negative GRAVY value suggests that this peptide is hydrophilic
bThe instability index value indicates that this peptide is unstable
Secondary structure analysis
The I-Tasser server was used to predict secondary structure (Fig. 3a, b). The model suggested that both Tn CRT peptides tended to form a small helix between serine, glycine, and lysine and that the rest of the structure was an α-helix, suggesting the absence of a regular secondary structure in the overall peptide sequence. Tn CRT3 presented a β-sheet between residues glutamine, serine, and glycine. CD spectra of Tn CRT peptides in aqueous buffer exhibited a negative minimum at ≈ 198 nm, suggesting the formation of a random coil (Fig. 3c, d). In the presence of POPC and POPC:POPG LUVs, the spectral profile indicated that the peptide was unstructured and not toxic to mammalian cells. The incubation of both peptides with POPC model membranes resulted in a less-intense CD signal, which might be the result of vesicle aggregation and precipitation, increasing light scattering, and decreasing peptide concentration in solution, as observed for AMP Sub3 (Torcato et al. 2013).
Fig. 3.

Secondary structures of peptides. a The secondary structure of Tn CRT2 and b Tn CRT3 was predicted using the I-TASSER algorithm (https://zhanglab.ccmb.med.umich.edu/I-TASSER/), and the best model (highest C-Score) was represented using Chimera software (Pettersen et al. 2004). Circular dichroism spectra of c Tn CRT2 (50 µM) and d Tn CRT3 (50 µM) in aqueous buffer (black line) and in the presence of POPC (gray line) and 2 mM POPC/POPG (1:1) large unilamellar vesicles (dotted line)
MIC and cytotoxicity of peptides
The antimicrobial activity of Tn CRT2 and Tn CRT3 was assessed against two species of Gram-negative bacteria, three species of Gram-positive bacteria, and one species of fungi (Table 2). All Gram-negative bacteria were sensitive to Tn CRT at a concentration of 2.1–13.0 μM. This peptide was active against P. aeruginosa and E. coli at the same concentration range. For Gram-positive bacteria, Tn CRT2 was active against S. hominis and E faecalis, with a MIC of 8.3 μM, and Tn CRT3 was active against S. aureus and C. albicans at 3.2 μM (Table 2).
Table 2.
Antimicrobial activity spectrum of Tn CRT2 and Tn CRT3-MIC, minimum inhibitory concentration
| MIC100 (μM) | ||||||
|---|---|---|---|---|---|---|
| Microorganism | ||||||
| Gram-negative bacteria | Gram-positive bacteria | Yeast | ||||
| E. coli (ATCC 25922) | P. aeruginosa (ATCC 15442) | S. aureus (ATCC 6538) | S. hominis (ATCC 27844) | E. faecalis (ATCC 2912) | C. albicans (ATCC 10231) | |
| Tn CRT2 | 2.1 ± 0.2 | 2.1 ± 0.3 | > 40 | 8.3 ± 0.0 | 8.3 ± 0.0 | 13.5 ± 0.0 |
| Tn CRT3 | 13 ± 0.4 | 13 ± 0.8 | 3.2 ± 1.0 | > 40 | > 40 | 3.2 ± 0.6 |
The hemolytic activity of Tn CRT2 and Tn CRT3 was measured using human erythrocytes. Erythrocytes were incubated with the peptides for 3 h at 37 °C, and the highest concentration (40 μM) of Tn CRT2 and Tn CRT3 caused hemolysis of 50.38% and 34.49%, respectively (Fig. 4).
Fig. 4.
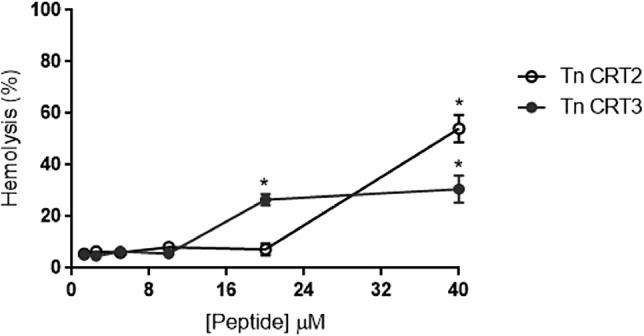
Hemolytic activity of Tn CRT peptides in human erythrocytes. The mean standard deviation of three independent experiments is presented. *p < 0.005
Cytotoxicity against MEF was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The assay results showed that the peptides at a concentration ranging from 5 to 40 μM were not cytotoxic to these cells (Fig. 5). Nonetheless, at these concentrations, the toxicity of Tn CRT2 was non-significantly higher than that of Tn CRT3 (Fig. 5).
Fig. 5.
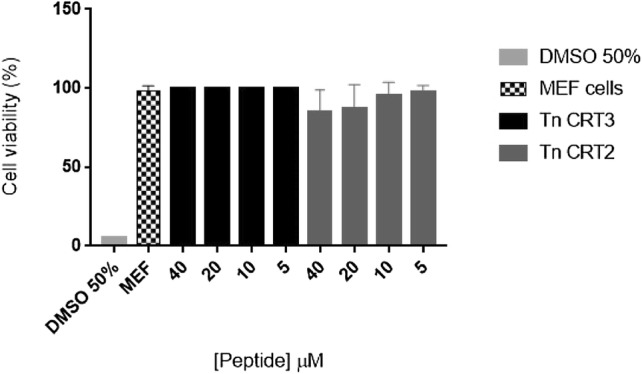
Cell viability of mice embryonic fibroblasts (MEFs) treated with Tn CRT peptides. MEFs were incubated with different concentrations of peptides for 24 h at 37 °C. The effects on cell viability were determined using an MTT assay. Untreated MEFs were used as a negative control and MEFs treated with 50% DMSO were used as the positive control. The results correspond to the mean ± standard deviation of three independent experiments
Potential zeta measurements (ζ)
To further determine whether Tn CRT peptides could target the bacterial outer membrane and whether electrostatic attractions were involved in the antimicrobial activity of Tn CRT peptides, the membrane surface potential of E. coli and S. aureus cells was monitored by ζ potential analysis. The zeta potential provides information about the surface charge of the cells and is calculated from the mobility of cells in the presence of an electrical field under specific pH and salt concentrations. Microorganisms were incubated with the peptides at MIC100 values. The controls had a negative surface charge: − 24.55 mV for E. coli and − 25.93 mV for S. aureus, as expected (Table 3). The peptides significantly changed the surface charge of E. coli and S. aureus (Table 3). All zeta potential values became less negative in these bacterial species after contact with the peptides, and changes were more pronounced in S. aureus (− 4.11) using Tn CRT2 and E. coli (− 8.27) using Tn CRT3.
Table 3.
Zeta potential (mV) of E. coli and S. aureus after exposure to Tn CRT2 and Tn CRT3
| Zeta potential (mV) at MIC100 | |||
|---|---|---|---|
| Microorganism | Control | Tn CRT2 | Tn CRT3 |
| E. coli | − 24.55 ± 1.23 | − 17.35 ± 0.78 | − 10.69 ± 0.80 |
| S. aureus | − 25.93 ± 2.43 | − 4.11 ± 1.21 | − 8.27 ± 1.25 |
Discussion
AMPs are a major topic of research, because they act mainly by directly disrupting the membrane structure. Given the biological importance of cell membranes, the development of AMP-resistant microorganisms is slow (Campagna et al. 2007; Taheri et al. 2018). In the present study, we report the isolation and characterization of a family of peptides obtained from the venom of T. nattereri and active against Gram-positive and Gram-negative bacteria and fungi. The amino acid sequences were compared with those from the Uniprot database (Bateman et al. 2017), and data analysis indicated homology with CART peptides identified by Magalhães et al. (2006).
These authors found a large cluster homologous to a CART peptide mainly involved in regulating food intake. Whether the CART peptide in the T. nattereri venom is involved in feeding is unknown. T. nattereri uses venom primarily for defense, and its release is involuntary, in contrast to terrestrial animals. The presence of CART suggests that the venom gland has functions other than the production of toxins (Magalhães et al. 2006).
The obtained T. nattereri sequence, found in the present study, was aligned with CART peptide sequences from Homo sapiens and Danio rerio (Supplementary Fig. 1). It is worth noting that the sequences were highly homologous, and the cysteine residues were conserved and, therefore, might play a fundamental role in the function and structure of these peptides (Thim et al. 1998). MS analysis revealed the presence of three major peptides with m/z of 5075.276, 5593.132, and 5724.383. The m/z of 5593.132 and 5724.383 indicated an increase in molecular mass of 506.55 and 637.75 Daltons, corresponding to fragments NQMN and MNQMN, respectively. The results suggest that the analyzed peptide has a mass of 5724.383, which agrees with the findings by Magalhães et al. (2006). The two fragments may have been generated by the action of venom proteases (Lopes-Ferreira et al. 2004). Furthermore, these proteases may be critical in the immune response of T. nattereri fish by functioning as a substrate for the generation of AMPs.
CART peptides were initially described in 1995 and were shown to have only neuromodulatory activity (Douglass and Daoud 1996) but not other pharmacological activities. T. nattereri Niq CART peptides have relatively low toxicity to eukaryotic cells and limited spectrum of action but can be used as templates for the rational design of analogs with enhanced properties for these endpoints (Gaspar et al. 2013). These data encourage searching for peptides that may serve as models for developing new drugs.
The AMPA algorithm is useful for searching and design of AMPs. This algorithm uses a propensity score to generate an antimicrobial profile. The score is calculated on the basis of antimicrobial indices defined for individual residues (Dziuba and Dziuba 2014). The AMPA server found one peptide (Tn CRT2) with biological activity. Amidated peptides have a higher positive charge—and higher activity—than their non-amidated counterparts (Mura et al. 2016). For this reason, Tn CRT peptides were synthesized with an amidated C-terminus. Furthermore, it has been proposed that amidation increases biological activity by improving structural stability at the membrane interface (Dennison and Phoenix 2011).
Some physical and chemical characteristics of Tn CRT peptides were predicted by sequence analysis. Many AMPs are cationic, including fish peptides (Pan et al. 2015; Houyvet et al. 2018). This feature facilitates their interaction with anionic membranes, leading to multiple effects on membranes, including permeabilization, depolarization, leakage, and lysis, culminating in cell death. However, other evidence indicates that some anionic AMPs interact with targets other than cell membranes (Harris et al. 2009). Therefore, the net positive charge of Tn CRT2 and Tn CRT23 and high pI demonstrate that these peptides are cationic. Cationic peptides are known to be an essential part of the innate immunity of vertebrates, invertebrates, and plants (Stocklin et al. 2004).
The aliphatic index is strongly correlated with thermostability and is defined as the relative volume of a protein occupied by amino acids with aliphatic side chains (alanine, valine, isoleucine, and leucine). The aliphatic index of the analyzed Tn CRT peptides was zero, indicating that these amino acids were absent (Dziuba and Dziuba 2014). The value of the GRAVY index, a measure of peptide solubility, was negative for both peptides, indicating their hydrophilic nature (Kyte and Doolittle 1982). The value of the Boman index, which is a measure of peptide affinity to proteins and its ability to establish biological interactions (Boman 2003), ranged from 3.83 to 3.11 in these peptides. Boman index higher than 2.48 indicates that an AMP is multifunctional or plays different roles in the cell because of its ability to interact with several proteins. Similar results were observed for peptides predicted from the transcriptome of Paracoccidioides brasiliensis (Silva et al. 2012).
Structural analysis using I-Tasser, which uses a de novo approach to predict 3D peptide structures from amino acid sequence information (Shen et al. 2014), indicated that Tn CRT2 and Tn CRT3 adopted a helical conformation, including a short α-helix in Tn CRT2 and a short β-sheet in Tn CRT3 (Fig. 3a, b). The comparison of CD spectra indicated that the two peptides adopted random coil structures, under the conditions tested. However, little is known about the CD bands of very short helices, and their properties are expected to depend strongly on helix length for very short helices (Manning and Woody 1991). Short helices, as predicted for Tn CRT 2, are usually formed in peptides with low stability, and form partly frayed helices, still is difficult to study this problem (Chin et al. 2002). For Tn CRT 3, the prediction of β-sheet content by CD often fails. β-sheets can differ in the parallel–antiparallel orientation, length and number of strands, and twists. This structural variability is manifested in the CD spectral diversity, which is believed to be an inherent limitation of secondary structure estimation (Khrapunov 2009; Micsonai et al. 2018).
Native AMPs have random coil conformation and usually contain a high percentage of arginine, proline, tryptophan, and/or histidine residues (Takahashi et al. 2010; Nguyen et al. 2011). Indolicidin has this configuration (Powers and Hancock 2003).
The synthetic analogs of Tn CRT2 and Tn CRT3 had a relatively high antimicrobial activity against reference Gram-negative and Gram-positive bacteria (MIC of 2.1 µM and 8.3 µm, respectively). Moreover, Tn CRT3 was highly active against C. albicans at 3.2 µM. The discovery of bioactive peptides by Liu et al. (2018) using an integrated in silico–in vitro represents the success in conducting these methods for the design of new molecules more potent than native peptides. The MIC values found in our study were lower than those of peptides isolated from Pterois volitans and pardaxin analogs (Thennarasu and Nagaraj 2007; Houyvet et al. 2018). For C. albicans, the MIC values were lower than those of AMPs isolated from other venomous animals, including TistH from Tytius sigmuris (MIC of 128 µM/mL) and crotamine from the rattlesnake Crotalus durissus terrificus (MIC of 2.5–10.0 mM/mL) (Moreira et al. 2012; Torres-Rêgo et al. 2016). It is known that antifungal peptides can bind to chitin and disrupt the integrity of the fungal cell wall membrane, increasing its permeability or forming pores. Tn CRT3 was more active against C. albicans than Tn CRT2, suggesting that the former might present a higher affinity for chitin, as demonstrated for other marine peptides (Pushpanathan et al. 2012).
Considering the high cationicity of AMPs, electrostatic interactions are involved in binding to the cell membrane. Surface charge neutralization is important for the antimicrobial activity of AMPs that target the bacterial surface (Torcato et al. 2013). The positive charge on the surface of Gram-positive and Gram-negative bacteria increased in the presence of Tn CRT peptides (Table 3). For Tn CRT3, the surface charge neutralization in S. aureus was reached at a lower concentration when compared with E. coli. This result may be correlated with the higher density of anionic groups and the presence of O-antigen in LPS, preventing the AMPs to neutralize the negative charge in LPS lipid A in Gram-negative bacteria. As observed for rBPI21 (Domingues et al. 2008), these results suggest that higher AMP concentrations are needed to neutralize the negative charge of adjacent LPS molecules. However, the Tn CRT2 concentration that neutralized the surface charge in S. aureus and E. coli was approximately 40 μM and < 2.1 μM, respectively, considering the MIC results. In this case, a higher peptide concentration may be required for neutralizing surface charges and killing Gram-positive bacteria. Quantitative data on the affinity between AMPs and cell wall components will help determine whether these components act as electrostatic barriers to prevent the action of AMPs on the cell membrane.
A common strategy for improving the activity of AMPs is changing their amino acid sequence. The physiological characteristics of certain amino acids play important roles in the activity and target spectrum of AMPs. Changing the amino acid sequence may also affect cytotoxicity. Our results showed that replacing arginine with glutamine did not affect the activity of Tn CRT3. The hemolytic assay results showed that the highest concentration of Tn CRT2 and Tn CRT3 induced approximately 50% and 40% of hemolysis, respectively, suggesting a mechanism of action involving membrane disruption; however, this hypothesis needs to be further tested. These results are related to peptide structure, and the peptide with a higher propensity to form small helices and higher hydrophobicity, Tn CRT2, showed higher antimicrobial and hemolytic activity, whereas the peptide with a lower propensity to form small helices and lower hydrophobicity, Tn CRT3, presented lower antimicrobial and hemolytic activity. The incorporation of hydrocarbon staples significantly increased the hemolytic activity of AMPs, including polybia-MP1 (Luong et al. 2017). As some AMPs are toxic to different cell lines in vitro (Hoskin and Ramamoorthy 2008), synthetic Tn CRT2 and Tn CRT3 were incubated with MEF. The results indicated that Tn CRT3 slightly decreased the viability of MEF. We hypothesize that the slight change in peptide charge exerts different effects on the cell membrane of microorganisms, human erythrocytes, and mammalian cells. Nonetheless, further studies are necessary to elucidate the mechanism by which Tn CRT3 interacts with target microorganisms.
In summary, the isolation of new AMPs from the venom of the fish Thalassophryne nattereri was described. Synthetic cationicity-enhanced analogs were designed after native peptide purification by RP-HPLC, Edman degradation, MS analysis, sequencing, and structural characterization. The analysis of these two peptide analogs revealed that synthetic unmodified peptides had a potential spectrum of antimicrobial activity. In contrast, cationicity-enhanced analogs exhibited a broader spectrum of activity and higher antimicrobial activity. Furthermore, the unmodified analogs did not affect the proliferative capacity of MEF, demonstrating that native venom peptides are biologically active but can be chemically modified to increase biological activity. Both facilitations readily illustrate the potential of these peptide models for drug design and development.
Conclusion
In this study, we described the new AMP derived from a CART peptide from the venom of T. nattereri. The AMPA algorithm was used for the virtual screening of AMP candidates based on the amino acid sequence of CART peptides. The virtual screening of Tn CRT peptides, together with structural analysis and in vitro assays of the predicted AMPs, helped gain insight into the molecular basis of the interaction of peptides with prokaryotic and mammalian cell membranes. This understanding may allow enhancing the selectivity of AMPs, consequently decreasing cytotoxicity and increasing biological activity. The unique membrane disruption activity of AMPs can be synergistically improved by combining them with conventional antibiotics. Recent findings help design AMPs with enhanced activity to reverse multidrug resistance. However, further research is needed to overcome limitations and determine their scope.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This research was supported by FAPESP grant No. 2017/00032-0. In addition to funding, we acknowledge Ana Cruz for technical help on CD experiments and Carlos Eduardo Silva da Cruz for English revision.
Author contributions
KC participated in the study design, conducted analysis and manuscript preparation; VAS, VMA and GLC participated in the execution of antimicrobial, in silico experiments, data analysis and manuscript preparation; XAON performed the HPLC purification and mass analysis; SAC synthesized the peptides; MR performed Edman sequencing; SAD and MARBC participated in the execution of biophysical experiments; MLF collected venom and designed experiments.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Katia Conceição, Email: katia.conceicao@unifesp.br.
Gabrielle L. de Cena, Email: gabilupeticena@gmail.com
Verônica A. da Silva, Email: veronicasdiogo@gmail.com
Xisto Antonio de Oliveira Neto, Email: xistoneto9@gmail.com.
Vitor Martins de Andrade, Email: vitormartinsdeandrade@yahoo.com.br.
Dayane Batista Tada, Email: day.tada@gmail.com.
Michael Richardson, Email: mmikeana@yahoo.com.br.
Sonia A. de Andrade, Email: sonia.andrade@butantan.gov.br
Susana A. Dias, Email: susanadias@medicina.ulisboa.pt
Miguel A. R. B. Castanho, Email: macastanho@medicina.ulisboa.pt
Mônica Lopes-Ferreira, Email: monica.lopesferreira@butantan.gov.br.
References
- Afacan NJ, Yeung ATY, Pena OM, Hancock REW. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr Pharm Des. 2012;18:807–819. doi: 10.2174/138161212799277617. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, et al. Swiss-Prot Protein Knowledgebase, release 47.3 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton E, Sheppard RC. Solid phase peptide synthesis—a practical approach. Oxford: IRL Press; 1989. [Google Scholar]
- Bailey P, Wilce J. Venom as a source of useful biologically active molecules. Emerg Med. 2001;13:28–36. doi: 10.1046/j.1442-2026.2001.00174.x. [DOI] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, O’Donovan C, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman HG. Antibacterial peptides: basic facts and emerging concepts. J Intern Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]
- Borges MH, Andrich F, Lemos PH, et al. Combined proteomic and functional analysis reveals rich sources of protein diversity in skin mucus and venom from the Scorpaena plumieri fish. J Proteomics. 2018;187:200–211. doi: 10.1016/j.jprot.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Bücherl W, Buckley EE, Deulofeu V (1968) (eds) Academic Press, New York, pp xxiv + 707, illus
- Campagna S, Saint N, Molle G, Aumelas A. Structure and mechanism of action of the antimicrobial peptide piscidin. Biochemistry. 2007;46:1771–1778. doi: 10.1021/bi0620297. [DOI] [PubMed] [Google Scholar]
- Chang KY, Lin TP, Shih LY, Wang CK. Analysis and prediction of the critical regions of antimicrobial peptides based on conditional random fields. PLoS ONE. 2015;10:1–16. doi: 10.1371/journal.pone.0119490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin DH, Woody RW, Rohl CA, Baldwin RL. Circular dichroism spectra of short, fixed-nucleus alanine helices. Proc Natl Acad Sci USA. 2002;99(24):15416–15421. doi: 10.1073/pnas.232591399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church JE, Hodgson WC. The pharmacological activity of fish venoms. Toxicon. 2002;40:1083–1093. doi: 10.1016/S0041-0101(02)00126-5. [DOI] [PubMed] [Google Scholar]
- Collette BB. A review of the venomous toadfishes, subfamily Thalassophryninae. Copeia. 2006;1966:846. doi: 10.2307/1441412. [DOI] [Google Scholar]
- Conceição K, Monteiro-dos-Santos J, Seibert CS, et al. Potamotrygon cf. henlei stingray mucus: biochemical features of a novel antimicrobial protein. Toxicon. 2012;60:821–829. doi: 10.1016/j.toxicon.2012.05.025. [DOI] [PubMed] [Google Scholar]
- Dennison SR, Phoenix DA. Influence of C-terminal amidation on the efficacy of modelin-5. Biochemistry. 2011;50:1514–1523. doi: 10.1021/bi101687t. [DOI] [PubMed] [Google Scholar]
- Di Tommaso P, Torrent M, Boix E, et al. AMPA: an automated web server for prediction of protein antimicrobial regions. Bioinformatics. 2011;28:130–131. doi: 10.1093/bioinformatics/btr604. [DOI] [PubMed] [Google Scholar]
- Domingues MM, Santiago PS, Castanho MA, Santos NC. What can light scattering spectroscopy do for membrane-active peptide studies? J Pept Sci. 2008;14:394–400. doi: 10.1002/psc.1007. [DOI] [PubMed] [Google Scholar]
- Douglass J, Daoud S. Characterization of the human cDNA and genomic DNA encoding CART: a cocaine-and amphetamine-regulated transcript. Gene. 1996;169:241–245. doi: 10.1016/0378-1119(96)88651-3. [DOI] [PubMed] [Google Scholar]
- Dziuba B, Dziuba M. New milk protein-derived peptides with potential antimicrobial activity: an approach based on bioinformatic studies. Int J Mol Sci. 2014;15:14531–14545. doi: 10.3390/ijms150814531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar D, Salomé Veiga A, Castanho MARB. From antimicrobial to anticancer peptides. A review. Front Microbiol. 2013;4:1–16. doi: 10.3389/fmicb.2013.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Hancock REW, Rozek A. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett. 2002;206:143–149. doi: 10.1016/S0378-1097(01)00480-3. [DOI] [PubMed] [Google Scholar]
- Harris F, Dennison S, Phoenix D. Anionic antimicrobial peptides from eukaryotic organisms. Curr Protein Pept Sci. 2009;10:585–606. doi: 10.2174/138920309789630589. [DOI] [PubMed] [Google Scholar]
- Hilpert K, Volkmer-Engert R, Walter T, Hancock REW. High-throughput generation of small antibacterial peptides with improved activity. Nat Biotechnol. 2005;23:1008–1012. doi: 10.1038/nbt1113. [DOI] [PubMed] [Google Scholar]
- Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta Biomembr. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houyvet B, Bouchon-Navaro Y, Bouchon C, et al. Identification of a moronecidin-like antimicrobial peptide in the venomous fish Pterois volitans: functional and structural study of pteroicidin-α. Fish Shellfish Immunol. 2018;72:318–324. doi: 10.1016/j.fsi.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Joo HS, Fu CI, Otto M. Bacterial strategies of resistance to antimicrobial peptides. Philos Trans R Soc B Biol Sci. 2016 doi: 10.1098/rstb.2015.0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrapunov S. Circular dichroism spectroscopy has intrinsic limitations for protein secondary structure analysis. Anal Biochem. 2009;389:174–176. doi: 10.1016/j.ab.2009.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Liu S, Bao J, Lao X, Zheng H. Novel 3D structure based model for activity prediction and design of antimicrobial peptides. Sci Rep. 2018;25:11189. doi: 10.1038/s41598-018-29566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Ferreira M, Barbaro KC, Cardoso DF, et al. Thalassophryne nattereri fish venom: Biological and biochemical characterixation and serum neutralization of its toxic activities. Toxicon. 1998;36:405–410. doi: 10.1016/S0041-0101(97)00115-3. [DOI] [PubMed] [Google Scholar]
- Lopes-Ferreira M, Emim JADS, Oliveira V, et al. Kininogenase activity of Thalassophryne nattereri fish venom. Biochem Pharmacol. 2004;68:2151–2157. doi: 10.1016/j.bcp.2004.07.037. [DOI] [PubMed] [Google Scholar]
- Lopes-Ferreira M, Grund LZ, Lima C. Thalassophryne nattereri fish venom: from the envenoming to the understanding of the immune system. J Venom Anim Toxins Incl Trop Dis. 2014;20:1–12. doi: 10.1186/1678-9199-20-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong HX, Kim DH, Lee BJ, Kim YW. Antimicrobial activity and stability of stapled helices of polybia-MP1. Arch Pharm Res. 2017;40:1414–1419. doi: 10.1007/s12272-017-0963-5. [DOI] [PubMed] [Google Scholar]
- Magalhães GS, Junqueira-de-Azevedo IL, Lopes-Ferreira M, Lorenzini DM, Ho PL, Moura-da-Silva AM, et al. Transcriptome analysis of expressed sequence tags from the venom glands of the fish Thalassophryne nattereri. Biochimie. 2006;88:693–699. doi: 10.1016/j.biochi.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Manning MC, Woody RW. Theoretical CD studies of polypeptide helices: examination of important electronic and geometric factors. Biopolymers. 1991;31:569–586. doi: 10.1002/bip.360310511. [DOI] [PubMed] [Google Scholar]
- Mathur S, Hoskins C. Drug development: lessons from nature. Biomed Rep. 2017;6:612–614. doi: 10.3892/br.2017.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A, Wien F, Bulyáki É, Kun J, Moussong É, Lee YH, Goto Y, Réfrégiers M, Kardos J. BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018;46:W315–W322. doi: 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira JT, de Souza AO, Nunes GLC, et al. Unraveling the antifungal activity of a South American rattlesnake toxin crotamine. Biochimie. 2012;95:231–240. doi: 10.1016/j.biochi.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Mura M, Wang J, Zhou Y, Pinna M, Zvelindovsky AV, Dennison SR, Phoenix DA. The effect of amidation on the behaviour of antimicrobial peptides. Eur Biophys J. 2016;45:195–207. doi: 10.1007/s00249-015-1094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011;29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Pan C-Y, Yu CY, Chen J-Y, et al. The antimicrobial peptide pardaxin exerts potent anti-tumor activity against canine perianal gland adenoma. Oncotarget. 2015 doi: 10.18632/oncotarget.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul TJT, Asif MK, Vladimir B. Bioinformatics for venom and toxin sciences. Brief Bioinform. 2003;4:53–62. doi: 10.1093/bib/4.1.53. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Haturvedi V, Espinel-Ingroff A et al (2002) Reference method for broth dilution antifungal susceptibility testing of yeasts ; approved standard, 2nd edn. Serving the World ’ s Medical Science Community Through Voluntary Consensus
- Powers JPS, Hancock REW. The relationship between peptide structure and antibacterial activity. Peptides. 2003;24:1681–1691. doi: 10.1016/j.peptides.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Primon-Barros M, José Macedo MA. Animal venom peptides: potential for new antimicrobial agents. Curr Top Med Chem. 2017;17:1119–1156. doi: 10.2174/1568026616666160930151242. [DOI] [PubMed] [Google Scholar]
- Pushpanathan M, Jayachandran S, Jayashree S, et al. Identification of a novel antifungal peptide with chitin-binding property from marine metagenome. Protein Pept Lett. 2012;19:1289–1296. doi: 10.2174/092986612803521620. [DOI] [PubMed] [Google Scholar]
- Reid WV et al (1993) Using genetic resources for sustainable development
- Robinson SD, Undheim EAB, Ueberheide B, King GF. Venom peptides as therapeutics: advances, challenges and the future of venom-peptide discovery. Expert Rev Proteomics. 2017;14:931–939. doi: 10.1080/14789450.2017.1377613. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput. 2014;10:4745–4758. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- Sievers F, Higgins DG. Clustal Omega. Curr Protoc Bioinf. 2014 doi: 10.1002/0471250953.bi0313s48. [DOI] [PubMed] [Google Scholar]
- Silva ON, Franco OL, de Carvalho MJA, et al. Predicting antimicrobial peptides from eukaryotic genomes: in silico strategies to develop antibiotics. Peptides. 2012;37:301–308. doi: 10.1016/j.peptides.2012.07.021. [DOI] [PubMed] [Google Scholar]
- Spencer P, Juliano L, Junqueira de Azevedo I, et al. Natterins, a new class of proteins with kininogenase activity characterized from fish venom. Biochimie. 2005;87:687–699. doi: 10.1016/j.biochi.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Stocklin R, Menin L, Bulet P. Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. 2004;198:169–184. doi: 10.1111/j.0105-2896.2004.0124.x. [DOI] [PubMed] [Google Scholar]
- Taheri B, MohammadiI M, Nabipour I, et al. Identification of novel antimicrobial peptide from Asian sea bass (Lates calcarifer) by in silico and activity characterization. PLoS ONE. 2018;13:1–22. doi: 10.1371/journal.pone.0206578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D, Shukla SK, Prakash O, Zhang G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie. 2010;92:1236–1241. doi: 10.1016/j.biochi.2010.02.023. [DOI] [PubMed] [Google Scholar]
- The HAS, Bullet M, The HIT, Bullet M (2002) Nunes-Correia, Ramalho-Santos, Pedroso de Lima—1998—Sendai virus fusion activity as modulated by target membrane components. 25:1–10 [DOI] [PubMed]
- Thennarasu S, Nagaraj R. Specific antimicrobial and hemolytic activities of 18-residue peptides derived from the amino terminal region of the toxin pardaxin. Protein Eng Des Sel. 2007;9:1219–1224. doi: 10.1093/protein/9.12.1219. [DOI] [PubMed] [Google Scholar]
- Thim L, Nielsen PF, Judge ME, et al. Purification and characterisation of a new hypothalamic satiety peptide, cocaine and amphetamine regulated transcript (CART), produced in yeast. FEBS Lett. 1998;428:263–268. doi: 10.1016/S0014-5793(98)00543-2. [DOI] [PubMed] [Google Scholar]
- Torcato IM, Huang YH, Franquelim HG, et al. The antimicrobial activity of sub3 is dependent on membrane binding and cell-penetrating ability. ChemBioChem. 2013;14:2013–2022. doi: 10.1002/cbic.201300274. [DOI] [PubMed] [Google Scholar]
- Torrent M, Nogués VM, Boix E. A theoretical approach to spot active regions in antimicrobial proteins. BMC Bioinform. 2009;10:1–9. doi: 10.1186/1471-2105-10-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M, Pulido D, De La Torre BG, et al. Refining the eosinophil cationic protein antibacterial pharmacophore by rational structure minimization. J Med Chem. 2011;54:5237–5244. doi: 10.1021/jm200701g. [DOI] [PubMed] [Google Scholar]
- Torres-Rêgo M, Machado RJA, Carvalho E, et al. Characterization of TistH, a multifunctional peptide from the scorpion Tityus stigmurus: structure, cytotoxicity and antimicrobial activity. Toxicon. 2016;119:362–370. doi: 10.1016/j.toxicon.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Usmani SS, Bedi G, Samuel JS, et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS ONE. 2017;12:1–12. doi: 10.1371/journal.pone.0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC, Hristova K. Antimicrobial peptides: successes, challenges and unanswered questions. J Membr Biol. 2011;239:27–34. doi: 10.1007/s00232-011-9343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B, Huang Y, Baumann K, Fry B, Shi Q. From marine venoms to drugs: efficiently supported by a combination of transcriptomics and proteomics. Mar Drugs. 2017;15:103. doi: 10.3390/md15040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli VO, Pasqualoto KFM, Picolo G, et al. Harnessing the knowledge of animal toxins to generate drugs. Pharmacol Res. 2016;112:30–36. doi: 10.1016/j.phrs.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Ziegman R, Alewood P. Bioactive components in fish venoms. Toxins (Basel) 2015;7:1497–1531. doi: 10.3390/toxins7051497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.