

Abstract
Background and Purpose
The worldwide prevalence of herpes simplex virus (HSV) and shortage of efficient therapeutic strategies to counteract it are global concerns. In terms of treatment, the widely utilized anti‐HSV drugs such as acyclovir have serious limitations, for example, drug resistance and side effects. Here, we have identified the guanidine‐modified (E,E)‐4,6‐bis(styryl)‐pyrimidine (BS‐pyrimidine) derivative compound 5d as an inhibitor of HSV and further elucidated the anti‐HSV mechanisms of compound 5d both in vitro and in vivo.
Experimental Approach
Cytopathic effect inhibition assay, plaque assay, and immunofluorescence assay were used to evaluate the anti‐HSV effects of compound 5d in vitro. Membrane fusion assays, immunofluorescence assays, Western blotting assays, and pull‐down assays were used to explore the anti‐HSV mechanisms of compound 5d. HSV‐1‐infected mice, combined with haematoxylin–eosin staining and quantitative RT‐PCR, were used to study the anti‐HSV effects of compound 5d in vivo.
Key Results
The guanidine‐modified compound 5d rather than the un‐modified compound 3a effectively inhibited both HSV‐1 and HSV‐2 multiplication in different cell lines, more effectively than acyclovir. Compound 5d may block virus binding and post‐binding processes such as membrane fusion, by targeting virus gB protein. In addition, compound 5d may also down‐regulate the cellular PI3K/Akt signalling pathway to interfere with HSV replication. Treatment with compound 5d also markedly improved survival and decreased viral titres in HSV‐infected mice.
Conclusions and Implications
Thus, the guanidine‐modified BS‐pyrimidine derivatives have the potential to be developed into novel anti‐HSV agents targeting both virus gB protein and cellular PI3K/Akt signalling pathways.
Abbreviations
- BCIP
5‐bromo‐4‐chloro‐3‐indolyl phosphate toluidine
- BS‐pyrimidine
(E,E)‐4,6‐bis(styryl)‐pyrimidine
- CPE
cytopathic effect
- DARTS
drug affinity responsive target stability assay
- gB
glycoprotein B
- gD
glycoprotein D
- HSV
herpes simplex virus
- MOI
multiplicity of infection
- NBT
p‐nitro blue tetrazolium chloride
- RFP
red fluorescent protein
What is already known
Guanidinium compounds possess antiviral activities and display high selectivity for cell surface heparan sulfate proteoglycan.
HSV infection requires initial contacts between virus gB protein and cell surface heparan sulfate.
What this study adds
Guanidine modifications enhance the anti‐HSV activity of BS‐pyrimidine derivatives in vitro and in vivo.
Guanidine‐modified compound 5d can block HSV binding and membrane fusion by targeting virus gB protein.
What is the clinical significance
Guanidine‐modified compound 5d may be a promising antiviral candidate for HSV treatment with novel targets.
1. INTRODUCTION
The herpes simplex virus (HSV) is a common, double‐stranded DNA virus belonging to the Herpesviridae family encompassing more than 100 viruses, eight of which affect humans (Fatahzadeh & Schwartz, 2007). HSV‐1 infections cause skin lesions that are generally localized at the oral, nasal, and ocular sites, whereas HSV‐2 infections most commonly occur in genital skin and mucosa (Smith & Robinson, 2002; Szczubiałka, Pyrć, & Nowakowska, 2016). Although HSV‐induced lesions are generally benign, in some instances, HSV infection is also associated with potentially fatal viral encephalitis and stromal keratitis, which is a leading cause of cornea‐derived blindness in developed countries (Farooq & Shukla, 2012; Suzich & Cliffe, 2018). The antiviral agents approved for herpesviruses are mainly nucleoside analogues, for example, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4829 and its derivatives, which interfere with viral DNA synthesis to reduce HSV multiplication. Despite these successes, drug resistance and side effects remain unresolved issues in the fight against HSV infection (Blot et al., 2000; Morfin & Thouvenot, 2003). Hence, the development of novel anti‐HSV agents with different mechanisms of action is of high importance.
Chemically synthesized https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4783 derivatives have been reported to possess many physiological properties, including anti‐inflammatory and antiviral activities (Berlinck & Romminger, 2016). Especially zanamivir (compound 1 in Figure 1a), a guanidine‐derived inhibitor of influenza A virus, selectively binds to the negatively charged amino acids at the neuraminidase activity site of virus via its guanidine group, thus inhibiting neuraminidase activity and release of the virus (Laver, Bischofberger, & Webster, 1999). Guanidino‐glycosides (compound 2 in Figure 1a), a family of cellular transporters, display high selectivity for cell surface https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4214 sulfate proteoglycans (HSPGs) and promote their clustering (Inoue, Wexselblatt, Esko, & Tor, 2014). Moreover, our previous studies have shown that a series of (E,E)‐4,6‐bis(styryl)‐pyrimidine (BS‐pyrimidine) analogues (compound 3 in Figure 1a) possessed good solubility and chemical stability and excellent fluorescence properties (Qiu et al., 2013). HSV infection requires initial contacts between virus glycoprotein B (gB) protein and heparan sulfate proteoglycans, followed by more specific interaction between virus glycoprotein D (gD) protein and cellular receptors such as https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1887, nectins‐1, and 3‐O‐sulfated heparan sulfate (Weed, Dollery, Komala, & Nicola, 2018). Thus, we hypothesize that the introduction of guanidine groups to BS‐pyrimidine derivatives may be able to enhance the anti‐HSV actions of these derivatives by enhancing the interaction with virus surface proteins or cell surface sulfated glycans.
Figure 1.
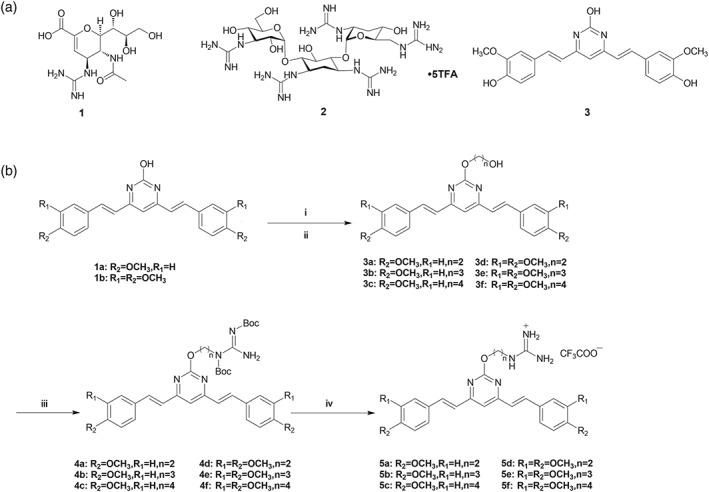
Chemical structures of zanamivir, guanidinoglycoside, and BS‐pyrimidine analogue and the synthetic strategy of BS‐pyrimidine derivatives. (a) Structures of zanamivir (1), guanidinoglycoside (2), and BS‐pyrimidine analogue (3). (b) Synthetic strategy of targeted compounds. Reagents and conditions: (i) K2CO3, DMF, different α‐bromo‐hydroxyl alkane protected by TBDMS, 12 hr, 80°C; (ii) TBAF, THF, 2 hr, room temperature (RT); (iii) 1,3‐di(t‐butyloxycarboryl)‐guanidine, DIAD, PPh3, anhydrous THF, 5 hr, 0°C, RT; and (iv) TFA/DCM (1/10), 15 hr, 0°C, RT
To test this hypothesis, we designed and successfully synthesized novel BS‐pyrimidine derivatives without (compounds 3a–f) and with guanidine modifications (compounds 5a–f), as shown in Figure 1b. We then explored the anti‐HSV activities and mechanisms of these pyrimidine derivatives both in vitro and in vivo. Guanidine modification was found to greatly improve the anti‐HSV activity of these pyrimidine derivatives, and the guanidine‐modified compound 5d effectively blocked virus binding and membrane fusion by targeting virus surface gB protein. In addition, treatment with compound 5d also markedly improved survival and decreased viral titres in HSV‐infected mice.
2. METHODS
2.1. Synthesis of guanidine‐modified (E,E)‐4,6‐bis(styryl)‐pyrimidine derivatives
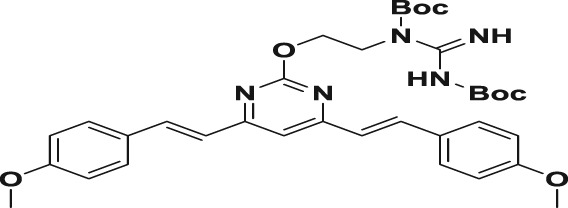
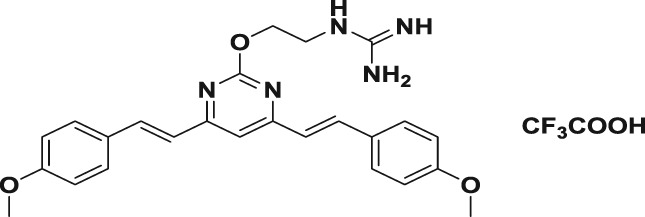
To synthesize the guanidine‐modified BS‐pyrimidine derivatives, the starting materials, compounds 1a,b, were prepared using different methoxyl‐substituted aromatic aldehydes and 4,6‐dimethyl‐2‐hydroxyl‐pyrimidine hydrochloride, as described previously (Gao et al., 2012). The compounds were then reacted with an excess of α‐bromo‐ω‐hydroxyl alkane protected by tert‐butyldimethylsilyl (TBDMS) in the presence of K2CO3 to yield the TBDMS‐protected ω‐hydroxyl alkyl derivatives, and then tetra‐n‐butylammonium fluoride (TBAF) was used to remove the protective TBDMS group to obtain compounds 3a–f. The hydroxyl group of compounds 3a–f reacted with 1,3‐di(t‐butyloxycarbonyl)guanidine using di‐isopropyl azodicarboxylate (DIAD) and triphenylphosphine (PPh3), and the Mitsunobu reaction was then carried out to form the intermediates 4a–f with yields of 80–85%. The final products, 5a–f, were obtained by removing the t‐butyloxycarbonyl (Boc) protection with trifluoroacetic acid giving yields of 30–60% (Figure 1b). All the structures of compounds were characterized by NMR spectroscopy and high‐resolution mass spectrometer analysis (see the Supporting Information).
2.1.1. General procedures for the preparation of compounds 3a–f
To a stirred solution of 1a–b (1.67 mmol) in https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7045 was added K2CO3 (3.33 mmol, 460 mg, 2.0 equiv) at room temperature (RT) for 1 hr. Different α‐bromo‐hydroxyl alkane protected by TBDMS (2.51 mmol, 1.5 equiv) was added to the resulting mixture and stirred at 85°C for 12 hr, then cooled to room temperature, diluted with EtOAc, washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel chromatography to furnish the intermediate compound. After that, to a stirred solution of the intermediate compound (1.18 mmol) in THF was added the TBAF (2.36 mmol, 2.0 equiv) dissolved in THF. The resulting solution was stirred at room temperature for 2 hr, then diluted with EtOAc, washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel chromatography to furnish the desired compounds 3a–f.
2.1.2. General procedures for the preparation of compounds 4a–f
A solution of 3a–f (0.72 mmol), 1,3‐di(t‐butyloxycarbonyl)guanidine (1.42 mmol, 372 mg, 2.0 equiv), and triphenylphosphine (1.08 mmol, 282 mg, 0.72 mmol) in anhydrous THF and DIAD (1.08 mmol, 0.2 ml, 1.5 equiv) was added at 0°C, and then the resulting solution was stirred at room temperature for 5 hr. The solvent was concentrated in vacuo, and the residue with EtOAc was washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel chromatography to furnish the desired compounds 4a–f.
2.1.3. General procedures for the preparation of compounds 5a–f
To a stirred solution of 4a–f (0.42 mmol) in DCM was added a 1:1 mixture (30 ml) of TFA and DCM dropwise at 0°C, and then the resulting solution was stirred at room temperature for 15 hr. The solvent was concentrated in vacuo, and the residue with EtOAc was washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel chromatography to furnish the desired compounds 5a–f.
2.2. Cell culture and virus infection
Vero E6 cells (ATCC CRL‐1586; RRID:CVCL_0574) were routinely maintained in DMEM (Life Technologies, USA) supplemented with 10% FBS (ExCell Bio, China), penicillin (100 U·ml−1), and streptomycin (100 μg·ml−1) at 37°C in an atmosphere containing 5% CO2. Hep‐2 (RRID:CVCL_1906) and HeLa (RRID:CVCL_0030) cells were grown in DMEM supplemented with 10% FBS, 100 U·ml−1 of penicillin, and 100 μg·ml−1 of streptomycin.
HSV‐1 strain F was purchased from ATCC (ATCC VR‐734). HSV‐2 strain 333 was obtained from Wuhan Institute of Virology, Chinese Academy of Sciences. Virus stocks were generated by infecting Vero (RRID:CVCL_0059) cell monolayers for 48 hr and then lysing the cells with two freeze–thaw cycles. Collected lysates were aliquoted and stored at −80°C. Virus stocks were quantified by titration on Vero E6 cells (48‐hr infection, 37°C), according to Reed and Muench method (Reed & Muench, 1938). For virus infection, virus propagation solution was diluted in PBS containing 0.2% BSA and was added to cells at the indicated multiplicity of infection (MOI). Virus was allowed to adsorb for 1 hr at 4°C. After removing the virus inoculum, cells were maintained in infecting media (DMEM, 2% FBS) at 37°C in 5% CO2.
2.3. Cytotoxicity assay
The cytotoxicity of compounds was measured by the MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; Sigma‐Aldrich, USA) assay. Briefly, confluent Vero, HeLa, or Hep‐2 cell cultures in 96‐well plates were exposed to different concentrations of compounds in triplicate and then incubated at 37°C for 48 hr. Next, 10 μl of PBS containing MTT (final concentration: 0.5 mg·ml−1) was added to each well. After 4‐hr incubation at 37°C, the supernatant was removed, and 200 μl of DMSO was added to each well to solubilize the formazan crystals. After vigorous shaking, absorbance values were measured in a microplate reader (Bio‐Rad, USA) at 570 nm. Cell viability was expressed as a percentage of non‐treated control.
2.4. Cytopathic effect inhibition assay
The antiviral activity was evaluated by the cytopathic effect (CPE) inhibition assay (Wang, Yin, et al., 2011). Briefly, Vero cells in 96‐well plates were infected with HSV‐1 or HSV‐2 (MOI = 0.1) and then treated with compounds at indicated concentrations in triplicate after removal of the virus inoculum. After 24‐hr incubation, the cells were fixed with 4% formaldehyde for 20 min at room temperature. After removal of the formaldehyde, the cells were stained with 0.1% (w/v) crystal violet for 30 min at 37°C. The plates were washed and dried, and the intensity of crystal violet staining for each well was measured at 570 nm. The concentration required for a test compound to reduce the CPE of HSV by 50% (IC50) was determined.
2.5. Plaque assay
Confluent Vero cell monolayers in six‐well plates were incubated with 10‐fold serial dilutions of HSV‐1 or HSV‐2 at 4°C for 1 hr. The inoculum was removed; cells were washed with PBS and overlaid with maintenance DMEM containing 1.5% agarose, 0.02% DEAE‐dextran, 1 mM of l‐glutamine, 0.1 mM of non‐essential amino acids, 100 U·ml−1 of penicillin, and 100 μg·ml−1 of streptomycin. After incubation for 3 days at 37°C in a humidified atmosphere of 5% CO2, cells were fixed with 0.05% glutaraldehyde, followed by staining with 1% crystal violet in 20% ethanol for plaque counting.
For plaque reduction assay, HSV‐1 or HSV‐2 (100–200 PFU per well) was firstly pre‐incubated with or without compound 5d for 60 min at 37°C before infection. Then, the virus– compound 5d mixtures were transferred to confluent Vero cell monolayers in six‐well plates, incubated at 37°C for 1 hr, and then subjected to plaque assay for counting the number of plaques.
2.6. Time‐of‐addition assay
Vero cells were infected with HSV‐1 or HSV‐2 (MOI = 1.0) under four different treatment conditions: (a) pretreatment of virus: HSV was pretreated with compound 5d (20 μM) at 37°C for 1 hr before infection; (b) pretreatment of cells: Vero cells were pretreated with 20 μM of compound 5d before infection; (c) adsorption: Vero cells were infected in media containing compound 5d (20 μM) at 4°C for 1 hr; and (d) post‐adsorption: after removal of unabsorbed virus, compound 5d (20 μM) was added to the cells. At 24 hr post‐infection (p.i.), virus yields were determined by plaque assay.
Moreover, another time‐dependent study was also performed to explore which viral stage after adsorption is inhibited by compound 5d as described previously (Li et al., 2017). Briefly, HSV‐1 (MOI = 1.0)‐infected Vero cells were treated with 30 μM of compound 5d for different time intervals (0–4 hr p.i., 4–8 hr p.i., 8–12 hr p.i., 0–8 hr p.i., and 0–12 hr p.i.), after which (at 24 hr p.i.) the virus yields were determined via plaque assay.
2.7. Confocal analysis
For imaging purposes, Vero cells were seeded overnight on glass coverslips in complete cell culture media before treatment with the compounds of interest (3a, 5a, or 5d) for 1 hr at 37°C. For co‐localization analysis of compounds 3a or 5d with virus gB protein, HSV‐2 (MOI = 1.0) was pretreated with compound 3a or 5d at 37°C for 1 hr before infection. Then, after removal of the virus inoculum, media containing compound 3a or 5d (20 μM) were added to HeLa cells. At 1 hr p.i., the localization of virus gB protein was evaluated via immunofluorescence assay using anti‐gB primary antibody (Abcam Cat#ab6506, RRID:AB_305528) and DyLight 649‐conjugated secondary antibody (Abbkine, CA, USA). The fluorescence from the compounds and secondary antibodies were measured using excitation wavelengths of 488 and 550 nm, respectively, using a confocal laser scanning microscope (Zeiss LSM 710, Germany).
For co‐localization analysis of compound 5d with endolysosome compartments markers such as Rab5 (early endosome marker), Rab7 (late endosome marker), and LAMP1 (late endosome/lysosome marker), HeLa cells were firstly transfected with plasmids expressing red fluorescent protein (RFP)‐coupled Rab5, Rab7, and LAMP1, respectively. After 24‐hr incubation, compound 5d (20 μM) was added to HeLa cells and incubated at 37°C for another 1 hr. The fluorescence from compound 5d and RFP were measured using excitation wavelengths of 488 and 550 nm, respectively, using a confocal laser scanning microscope (Zeiss LSM 710, Germany).
2.8. Syncytium formation inhibition assay
Syncytium assays were performed using the method described by Du et al., (2017), with some modifications. Briefly, monolayers of Vero cells grown in 12‐well plates were infected with HSV‐1 or HSV‐2 (MOI = 3.0) for 2 hr at 4°C. After removal of the virus inoculum, media containing compound 3a, 5a, or 5d at indicated concentrations were added to cells. After 16‐hr incubation at 37°C, cells were fixed and then stained with haematoxylin–eosin solution (Beyotime, Nantong, China). The inhibition of syncytium formation was observed using an optical microscope. To quantify the degree of syncytium inhibition, the relative syncytium areas of different fields were measured using ImageJ (NIH) v.1.33u (RRID:SCR_003070). The percentage syncytium formation was determined relative to the non‐treated virus control group.
2.9. Drug affinity responsive target stability assay
Drug affinity responsive target stability assay (DARTS) experiments for identifying the targets of compound 5d were performed as previously reported (Lomenick et al., 2009; Lomenick, Jung, Wohlschlegel, & Huang, 2011). Briefly, HSV‐2 (MOI = 1.0)‐infected Vero cells were lysed with NP‐40 cell lysis buffer (Beyotime, Nantong, China) and treated with or without compound 5d (60 μM) for 1 hr at room temperature followed by digestion with different concentrations of pronase (1, 2.5, and 5 μg·ml−1) in reaction buffer (50 mM of Tris–HCl (pH 8.0), 50 mM of NaCl, and 10 mM of CaCl2) for 30 min at room temperature. The digestion was stopped by directly adding 5× SDS‐PAGE loading buffer and inactivation by boiling. Protein samples were separated with 10% SDS‐PAGE and analysed by Western blot with antibodies against HSV gB protein or cellular GAPDH protein as control.
2.10. Indirect immunofluorescence assay
The indirect immunofluorescence assay was performed as previously described (Wang, Wu, et al., 2017). HSV‐1‐infected or HSV‐2‐infected (MOI = 1.0) cells were treated with compound 5d (20 μM) under different conditions. At 0, 1, or 7 hr p.i., cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min. Then cells were permeabilized using 0.5% (v/v) Triton X‐100 in PBS for 5 min before incubated with 2% BSA/PBS for 1 hr at 37°C. Cells were washed and incubated with anti‐HSV gB (Cat#ab6506, RRID:AB_305528), ICP27 (Cat#ab53480, RRID:AB_881581), or ICP5 (Cat#ab6508, RRID:AB_305530) antibodies (Abcam, USA) overnight at 4°C. After washing, the cells were incubated with DyLight 649‐conjugated secondary antibody (Abbkine, USA) for 50 min at 37°C. Nuclear DNA was labelled with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5498 (Sigma‐Aldrich, USA). Finally, cells were washed and observed using confocal laser scanning microscope (Zeiss LSM 710, Germany).
2.11. Pull‐down assay
The heparin‐coupled magnetic beads (BeaverBeads™ Magrose Heparin, Beaverbio, Suzhou, China) and cell lysates containing HSV‐2 surface gB protein were used to perform gB‐mediated binding assays. In brief, the cell lysate of HSV‐2‐infected Vero cells was first incubated with heparin‐coupled magnetic beads and a cocktail of protease inhibitors (Roche, Basel, Switzerland) at room temperature for 1 hr to form gB‐linked magnetic beads (gB‐beads). After that, compound 3a or 5d was added to the gB‐beads and incubated at room temperature for another 1 hr. After washing three times with PBS, a confocal laser scanning microscope (Zeiss LSM 510, Jena, Germany) was used to detect the fluorescence from the magnetic beads to indicate the presence of compounds bound to the gB‐beads. The binding of the HSV‐2 gB protein to the magnetic beads was also verified using Western blot analysis.
2.12. Real‐time RT‐PCR
Total RNA was extracted from HSV‐infected Vero cells using an RNAiso™ Plus Kit (Takara, Japan) and analysed by using the One Step SYBR PrimeScript RT‐PCR Kit (Takara, Japan). The real‐time RT‐PCR was performed using the following primers: gD mRNA, 5′‐AGCATCCCGATCACTGTGTACTA‐3′ and 5′‐GCGATGGTCAGGTTGTACGT‐3′, and monkey β‐actin mRNA, 5′‐CTCCATCCTGGCCTCGCTGT‐3′ and 5′‐GCTGTCACCTTCACCGTTCC‐3′. The real‐time RT‐PCR was performed at 42°C for 5 min, 95°C for 10 s, 40 cycles of 95°C for 5 s, and 60°C for 34 s, followed by melting curve analysis, according to the instrument documentation (ABI PRISM 7500, Applied Biosystems, USA). The relative amounts of HSV RNA molecules were determined using the comparative (2−ΔΔCT) method, as previously described (Livak & Schmittgen, 2001).
2.13. Western blot assay
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. After drug treatment, the cell lysates were separated by SDS‐PAGE and transferred to nitrocellulose membrane. After being blocked in Tris‐buffered saline containing 0.1% Tween 20 (v/v) and 5% non‐fat milk (w/v) at 4°C overnight, the membranes were rinsed and incubated at room temperature for 2 hr with antibodies against cellular phosphorylated https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=781#2503 (Cat#4228, RRID:AB_659940), https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 (Cat#9271, RRID:AB_329825) and NF‐κB (Cat#3031, RRID:AB_330559) proteins, or β‐actin (Cat#3700, RRID:AB_2242334) and GAPDH (Cat#2118, RRID:AB_561053) proteins (Cell Signaling Technology, Danvers, USA) as control. The membranes were washed and incubated with AP‐labelled secondary antibody (1:2,000 dilutions) at room temperature for 2 hr. The protein bands were then visualized by incubating with the developing solution (p‐nitro blue tetrazolium chloride [NBT] and 5‐bromo‐4‐chloro‐3‐indolyl phosphate toluidine [BCIP]) at RT for 30 min. The relative densities of proteins were all determined by using ImageJ (NIH) v.1.33u (USA).
2.14. Molecular docking analysis
Molecular docking was conducted in MOE v2014.09011. The 2D structures of compounds 3a and 5d were drawn in ChemBioDraw 2014 and converted to 3D structure in MOE through energy minimization. The crystal structures of HSV‐1 gB protein (PDB code: 5FZ2) and HSV‐2 gD protein (PDB code: 4MYW) were obtained from the protein data bank (http://www.rcsb.org). Prior to docking, the force field of AMBER10: EHT and the implicit solvation model of reaction field (R‐field) were selected. MOE‐Dock was used for molecular docking simulations of molecules with proteins. The docking workflow followed the “induced fit” protocol, in which the side chains of the receptor pocket were allowed to move according to ligand conformations, with a constraint on their positions. The weight used for tethering side chain atoms to their original positions was 10. For each ligand, all docked poses of which were ranked by London dG scoring first, and then a force field refinement was carried out on the top 20 poses followed by a rescoring of GBVI/WSA dG. The conformations with the lowest free energies of binding were selected as the best (probable) binding modes. Molecular graphics were generated by PyMOL (RRID:SCR_000305).
2.15. Animal experiments
All animal care and experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Ocean University of China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010 and with the recommendations made by the British Journal of Pharmacology. The current experiment is based on the rule of the replacement, refinement, or reduction (the 3Rs). Three‐week‐old female BALB/c mice (BALB/cAnNCrl, average weight, 11.0 ± 2.0 g; RRID:IMSR_CRL:028) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and raised in a pathogen‐free environment (23 ± 2°C and 55 ± 5% humidity). The experimental groups were designed as follows: 96 mice were divided randomly into six groups (n = 16 each group): (a) normal control (without HSV infection), (b) virus control (placebo), (c) acyclovir (10 mg·kg−1), (d) compound 3a (5 mg·kg−1), (e) compound 5d (5 mg·kg−1), and (f) compound 5d (10 mg·kg−1). Mice were anaesthetized and infected via the intranasal route with 106 PFU of HSV‐1 (F strain) in 50 μl of PBS. Twenty‐four hours after inoculation, mice received treatment (i.p.) with acyclovir (10 mg·kg−1), compound 3a (5 mg·kg−1), compound 5d (5 mg·kg−1), compound 5d (10 mg·kg−1), or placebo, and the treatments were repeated once daily for 5 days.
Each day, mice were weighed and monitored for signs of clinical disease, including inactivity, ruffled fur, hunched back, laboured respiration, and huddling behaviour for 14 days. Animals were killed when a >20% weight loss or a combination of two other obvious sickness signs was observed. To determine virus titres in organs, mice (n = 5 each group) were killed, and the lungs and brains were removed, homogenized, and clarified by centrifugation on Day 6 after inoculation. Samples were assayed for virus titres by quantitative RT‐PCR assay and plaque assay. The relative amounts of HSV gD mRNA molecules were determined using the comparative (2−ΔΔCT) method. Histopathological analysis was also performed using haematoxylin–eosin staining on lung and brain samples collected on Day 6.
2.16. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. Randomization was used to assign samples to the experimental groups and treatment conditions for all in vivo studies. Data collection and acquisition of all in vitro and in vivo experimental paradigms were performed in a blinded manner. Unless indicated otherwise, experiments were carried out at n ≥ 5, where n = number of independent experiments. Each independent experiment consisted of six technical replicates (i.e., six wells of a 96‐well plate for CPE inhibition assay), unless stated otherwise. Data are presented as mean ± SD. Some results were normalized to control for unwanted sources of variation. Statistical significance was calculated by GraphPad Prism 5.0 (San Diego, CA, USA; RRID:SCR_002798) using one‐way ANOVA analysis followed by post hoc Tukey's tests if F achieved statistical significance (P < .05) and there was no significant variance in homogeneity. Differences in mouse survival rates were compared using a log‐rank (Mantel–Cox) test. Statistical significance was considered to be P < .05.
2.17. Materials
Acyclovir was purchased from Sigma (St. Louis, USA). Anti‐phosphorylated PI3K, Akt, and NF‐κB antibodies and anti‐β‐actin and GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, USA). Anti‐HSV gB, ICP27, and ICP5 antibodies were obtained from Abcam (Cambridge, MA, USA). DyLight 649‐conjugated and AP‐labelled secondary antibodies were obtained from Abbkine (CA, USA). The plasmids expressing RFP‐coupled Rab5 (mRFP–Rab5), Rab7 (mRFP–Rab7), and LAMP1 (LAMP1–RFP) were purchased from Addgene (Cambridge, MA, USA). All other reagents were from Sigma (USA) unless stated otherwise.
Additional figures and tables, NMR spectra, and other data related to characterization of compounds presented in the current manuscript are included in the Supporting Information.
2.18. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski, et al., 2019; Alexander, Fabbro et al., 2019).
3. RESULTS
3.1. Guanidine modification enhanced the inhibitory effects of BS‐pyrimidine derivatives against both HSV‐1 and HSV‐2
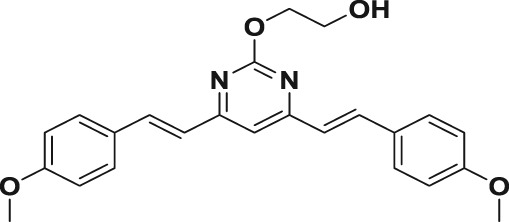
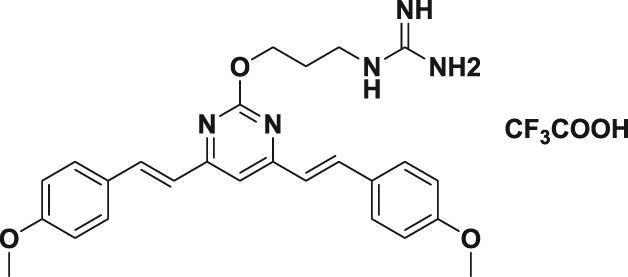
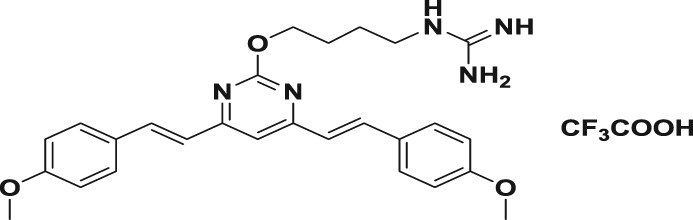
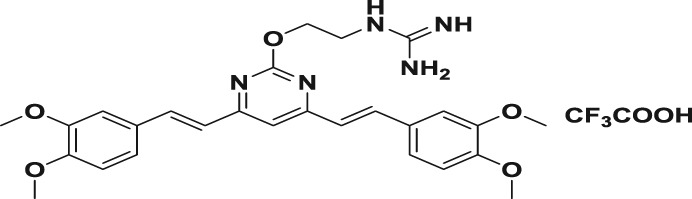
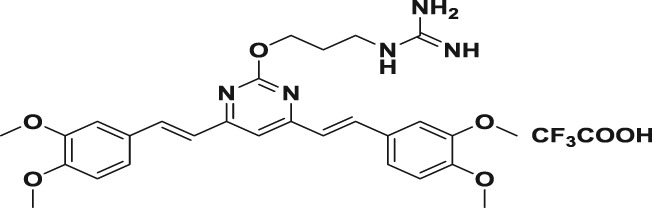
In this study, we first designed and successfully synthesized a serial of BS‐pyrimidine derivatives without or with guanidine modifications (5a–f; Figure 1b). Then the anti‐HSV activities of these BS‐pyrimidine derivatives were evaluated by CPE inhibition assay in both HSV‐1‐ and HSV‐2‐infected Vero cells. The results showed that compounds 5a, 5d, 5e, and 5f (with guanidine modification) possessed marked anti‐HSV activity, giving IC50 values < 11 μM (Table 1). In contrast, the similar compound 3a without guanidine modification or 4a with Boc protected guanidine, all showed very weak anti‐HSV activity (IC50 > 360 μM; Table 1). This strongly suggests that guanidine modification greatly enhances the anti‐HSV activity of BS‐pyrimidine derivatives. Moreover, if one more methoxyl group is introduced into 5a to produce 5d, the anti‐HSV activity increases slightly, and the toxicity decreases (Figure 2 and Table 1). In addition, by comparing two series of compounds (5a–c and 5d–f), we found that compounds with two methylene spacer groups (i.e., −(CH2)2−) between the guanidine and BS‐pyrimidine moieties (such as 5a and 5d) were most active in their inhibition of HSV, suggesting that the elongation of the methylene spacer in the guanidine‐modified pyrimidine derivatives may weaken their anti‐HSV effects.
Table 1.
The cytotoxicity and anti‐HSV effects of compounds in vitro (CC50, IC50, and SI)
| Compound | Structure | CC50 (μM)a | IC50 (μM)b | SIc | ||
|---|---|---|---|---|---|---|
| HSV‐1 | HSV‐2 | HSV‐1 | HSV‐2 | |||
| 3a |
|
80.7 ± 1.7 | 365.9 ± 38.3 | 621.7 ± 18.2 | 0.2 | 0.1 |
| 4a |
|
118.2 ± 3.6 | 378.1 ± 19.3 | 528.7 ± 28.1 | 0.3 | 0.2 |
| 5a |
|
95.6 ± 2.6 | 2.2 ± 0.6 | 3.4 ± 0.3 | 43.5 | 28.1 |
| 5b |
|
69.6 ± 2.1 | 30.5 ± 1.2 | 37.4 ± 1.0 | 2.3 | 1.9 |
| 5c |
|
109.6 ± 3.8 | 62.1 ± 0.4 | 59.7 ± 2.1 | 1.8 | 1.8 |
| 5d |
|
119.9 ± 2.7 | 1.8 ± 0.1 | 2.2 ± 0.4 | 66.6 | 54.5 |
| 5e |
|
119.2 ± 1.4 | 5.4 ± 0.8 | 8.5 ± 0.9 | 22.1 | 14.0 |
| 5f |
|
111.0 ± 2.6 | 9.5 ± 1.8 | 10.8 ± 1.4 | 11.7 | 10.3 |
| Acyclovir | 459.5 ± 17.3 | 9.6 ± 0.8 | 12.5 ± 2.5 | 47.9 | 36.8 | |
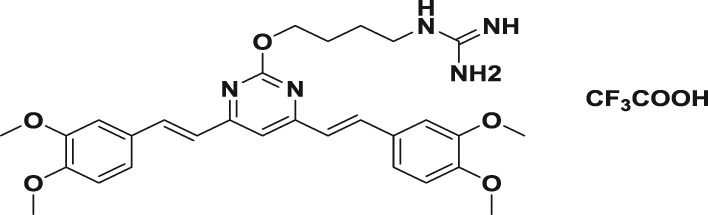
Note. The inhibition effects on HSV‐1 (F strain) and HSV‐2 (333 strain; multiplicity of infection = 0.1) multiplication in Vero cells were evaluated by cytopathic effect inhibition assay. Results shown are means ± SD from five independent experiments.
Abbreviations: HSV, herpes simplex virus; SI, selectivity index.
Concentration required to reduce cell viability by 50% in Vero cells.
Concentration required to reduce the cytopathic effect of the virus by 50% at 24 hr p.i.
The ratio of CC50 to IC50 (SI = CC50/IC50).
Figure 2.
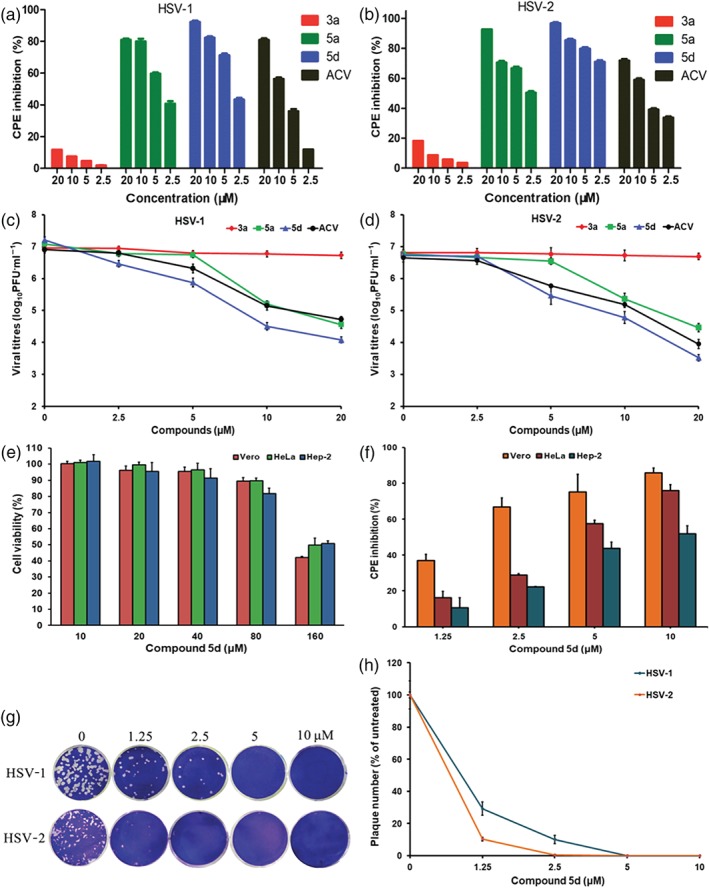
Effect of guanidine modification on the inhibitory effect of BS‐pyrimidine derivatives on herpes simplex virus (HSV)‐1 and HSV‐2. (a) HSV‐1‐infected or (b) HSV‐2‐infected (multiplicity of infection [MOI] = 0.1) Vero cells were treated with compounds 3a, 5a, 5d, or acyclovir (ACV; 2.5, 5, 10, and 20 μM) for 24 hr at 37°C. After that, the anti‐HSV effects were evaluated by cytopathic effect (CPE) inhibition assay. Results shown are means ± SD from six independent experiments. (c) HSV‐1 or (d) HSV‐2 (MOI = 1.0) was pretreated with compounds 3a, 5a, 5d, or ACV (2.5, 5, 10, and 20 μM) for 1 hr at 37°C before infection. Then, after adsorption, the media containing each compound were added to cells, respectively. At 24 hr p.i., the virus yields were determined by plaque assay. Values are means ± SD (n = 5). (e) After 48‐hr exposure to compound 5d at the indicated concentrations, the cell viability of Vero, HeLa, and Hep‐2 cells was measured by MTT assay, respectively. Values are means ± SD (n = 6). (f) HSV‐1 (MOI = 0.1)‐infected Vero, HeLa, and Hep‐2 cells were treated with compound 5d (1.25, 2.5, 5, and 10 μM) for 24 hr at 37°C. Then the antiviral effects were determined by CPE inhibition assay. Values are means ± SD (n = 5). (g) Approximately 100–200 PFU per well of HSV‐1 or HSV‐2 virus were pre‐incubated with compound 5d (0, 1.25, 2.5, 5, and 10 μM) for 1 hr at 37°C before infection, respectively. Then the virus–compound 5d mixture was transferred to Vero cells, incubated at 4°C for 1 hr and subjected to plaque reduction assay. (h) Plots of the plaque numbers from the plaque reduction assays performed on Vero cells infected with HSV and treated with the indicated concentrations of compound 5d (1.25, 2.5, 5, and 10 μM). Results shown are means ± SD from five independent experiments
To further evaluate the inhibition by compounds 3a, 5a, and 5d on HSV infection, the virus yield reduction assay was performed in HSV (MOI = 1.0)‐infected Vero cells. As shown in Figure 2c (HSV‐1) and Figure 2d (HSV‐2), the virus yields decreased with increasing compound 5a and 5d concentrations in a dose‐dependent manner, and the IC50 values of compound 5d were lower than those of acyclovir, and compounds 5a and 3a (Table S1). The cytotoxicity assays in Vero, HeLa, and Hep‐2 cells showed that compound 5d had no obvious cytotoxicity below 160 μM (Figure 2e). The SI values of compound 5d for inhibition of HSV‐1 and HSV‐2 were about 66.6 and 54.5, respectively, greater than those of acyclovir and compound 5a (Table 1). Moreover, viral multiplication was also inhibited in Hep‐2 and HeLa cells by compound 5d or acyclovir in a dose‐dependent manner (Figures 2f and S1 and Table S2).
To explore whether compound 5d had direct inhibitory effects on viral particles, plaque reduction assays were performed as described previously (Wang et al., 2017). As shown in Figure 2g,h, pre‐incubation of the HSV‐1 or HSV‐2 with compound 5d at concentrations of 1.25–10 μM markedly reduced the number of plaques, suggesting that compound 5d may be able to interact with viral particles directly.
3.2. Influence of different treatment conditions of compound 5d on HSV infection
Time‐of‐addition assay was further performed to determine the stage(s) at which compound 5d exerts its inhibitory effect in vitro. As shown in Figure 3a, pretreating the HSV‐1 and HSV‐2 with 20 μM of compound 5d for 1 hr before infection significantly reduced the virus titres of the HSV compared with the non‐treated virus control group. Addition of compound 5d during adsorption or post‐adsorption also significantly reduced virus titres of both HSV‐1 and HSV‐2. However, pretreatment of the cells scarcely decreased the virus titres in vitro at all (Figure 3a). Furthermore, the results of indirect immunofluorescence assays also showed that pretreatment of HSV with compound 5d before infection markedly reduced the fluorescence of ICP27 and gB proteins in the HSV‐infected cells (Figure S2). Taken together, compound 5d may be able to inactivate virus particles directly or block some steps in the HSV life cycle after adsorption.
Figure 3.
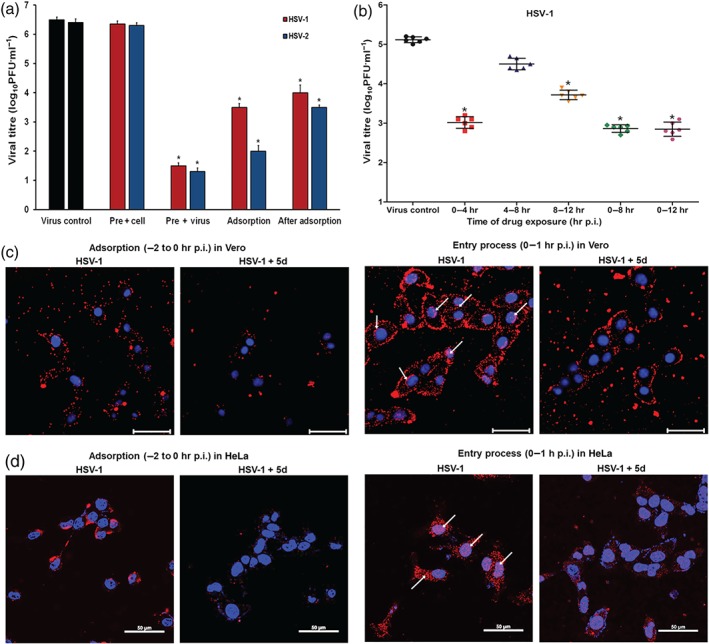
Influence of different treatment conditions of compound 5d on herpes simplex virus (HSV) infection. (a) Vero cells were infected with HSV‐1 or HSV‐2 (multiplicity of infection [MOI] = 1.0) using four different treatment conditions: (i) Pre + virus: HSV was pretreated with compound 5d (20 μM) at 37°C for 1 hr before infection; (ii) Pre + cell: Vero cells were pretreated with 20 μM of compound 5d before infection; (iii) Adsorption: Vero cells were infected in media containing compound 5d (20 μM) at 4°C for 1 hr; and (iv) Post‐adsorption: After removal of unabsorbed virus, compound 5d (20 μM) was added to the cells. At 24 hr p.i., virus yields were determined by plaque assay. The results were presented as mean ± SD from five independent experiments. *P < .05, significantly different from virus control group. (b) HSV‐1 (MOI = 1.0)‐infected Vero cells were treated with 30 μM of compound 5d for the specified time period, and then the media were removed and cells were overlaid with compound‐free media. Then at 24 hr p.i., the cell supernatants were collected and the virus yields were determined by plaque assay. Values are means ± SD (n = 6). *P < .05, significantly different from virus control group. (c, d) HSV‐1 (MOI = 1.0)‐infected (c) Vero cells or (d) HeLa cells were treated with or without compound 5d (20 μM) during the adsorption process or entry process. After washing with PBS, immunofluorescence assays were performed using anti‐ICP5 antibodies to highlight the number of virions binding to or entering into the cells. The scale bar represents 50 μm. The results shown are representative of five independent experiments
Another time‐dependent study was also performed to explore which viral stage after adsorption is inhibited by compound 5d, as described by Li et al., (2017). As shown in Figure 3b, treatment with compound 5d during the first 4 hr (0–4 hr p.i.) after adsorption resulted in a significant reduction in the virus titre (about 125‐fold). However, much less inhibition was noted (about fourfold) when compound 5d was added 4 hr after infection (4–8 hr p.i; Figure 3b). In addition, compound 5d treatment for the first 8 hr (0–8 hr p.i.) or 12 hr (0–12 hr p.i.) also resulted in a significant reduction in the virus titre (about 180‐fold; Figure 3b). Thus, compound 5d may also inhibit the early steps (0–4 hr p.i.) of the HSV life cycle after adsorption.
Moreover, we further explored which of the early steps in HSV life cycle are the main ones in which compound 5d exerts its inhibition effect by using immunofluorescence assay. As shown in Figure 3c, obvious signs of red fluorescence can be seen from the cell membranes of HSV‐infected Vero cells without compound 5d treatment. However, compound 5d treatment (20 μM) significantly reduced the fluorescence on the cell surfaces, suggesting that compound 5d may interfere with the virus adsorption process. During entry stage (0–1 hr p.i.), the fluorescence from HSV‐1 ICP5 proteins can be clearly observed in both the cell cytoplasm and nuclei. However, with compound 5d treatment, the ICP5 fluorescence could only be found on the cell surface, and virtually no fluorescence could be found coming from the cytoplasm and nuclei (Figure 3c). This suggests that compound 5d may be able to block HSV entry into Vero cells. Similarly, the results in HeLa cells indicated that compound 5d also markedly blocked both adsorption and entry process of HSV‐1 in HeLa cells (Figure 3d).
3.3. Guanidine modification may enhance the interaction between BS‐pyrimidine derivatives and HSV glycoproteins
Since compound 5d may interact with virus particles and block HSV entry process, it is relevant to ask whether compound 5d is able to inhibit the HSV‐induced membrane fusion process. As can be seen from Figure 4a–d, obvious signs of syncytia with multinuclear cells could be observed in the non‐treated virus control group. However, treatment with compound 5d (5–20 μM) after adsorption led to a marked inhibition of syncytium formation in both HSV‐1‐ and HSV‐2‐infected Vero cells, compared with the virus control group (Figure 4a–d). Moreover, as shown in Figure 4e, compound 3a was a very weak inhibitor of HSV‐2 induced syncytium formation in the Vero cells, while treatment with compounds 5d or 5a (with guanidine) after adsorption all markedly blocked syncytium formation only with a limited number of small syncytia (red arrow indicated), suggesting that the guanidine‐modified compound 5d can truly inhibit HSV‐induced membrane fusion.
Figure 4.
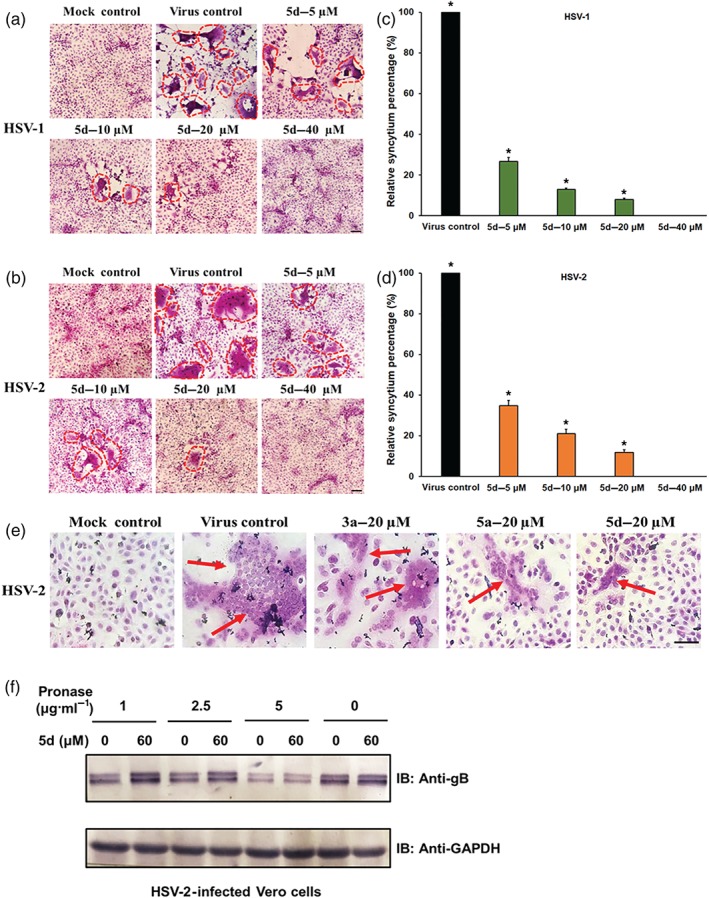
Compound 5d inhibits syncytium formation mediated by herpes simplex virus (HSV) surface glycoproteins. (a, b) HSV‐1‐infected or HSV‐2‐infected (multiplicity of infection = 3.0) Vero cells were treated with or without compound 5d (5, 10, 20, and 40 μM) after virus adsorption. At 16 hr p.i., the cells were fixed and then stained with haematoxylin–eosin solution. The inhibition of syncytium formation of (a) HSV‐1 and (b) HSV‐2 was observed using light microscopy. Bar represents 50 μm. (c, d) Plots quantifying syncytium formation in (c) HSV‐1‐ and (d) HSV‐2‐infected cells with different treatments. The relative areas of the syncytium in the different fields were measured using ImageJ (NIH) v.1.33u (USA), and the relative syncytium percentages were determined relative to the non‐treated virus control group. The data shown represent the results of five independent experiments. (e) HSV‐2 (multiplicity of infection = 3.0)‐infected Vero cells were added with compound 3a, 5a, or 5d (20 μM) after adsorption and incubated at 37°C for 16 hr. Then the cells were fixed and stained with haematoxylin–eosin solution. Bar represents 50 μm. (f) Drug affinity responsive target stability assay analysis for 5d. HSV‐2 infected Vero cell lysates were incubated with or without compound 5d (60 μM) for 1 hr and then digested with pronase (1, 2.5, and 5 μg·ml−1) for 30 min. Protein samples were separated by SDS‐PAGE and immunoblotted with the indicated antibodies. The results shown are representative of five independent experiments. *P < 0.05, significantly different from virus control group
As compound 5d directly inhibited HSV adsorption and membrane fusion, we then questioned whether compound 5d directly interacts with virus core fusogen gB protein. We employed a DARTS (Lomenick et al., 2009; Lomenick et al., 2011), which relies on the reduction of protease susceptibility of the target protein upon drug binding, to detect the potential interaction between compound 5d and gB protein. As shown in Figure 4f, virus gB protein was protected by protease digestion in the extracts of compound 5d (60 μM)‐treated cells especially under pronase treatment at 1 or 2.5 μg·ml−1, suggesting that compound 5d may interact with virus gB protein in HSV‐infected Vero cells.
3.4. Subcellular localization of BS‐pyrimidine derivatives with or without guanidine modification
The BS‐pyrimidine derivatives have been reported to have intrinsic fluorescent properties (Tong et al., 2016). Thus, the intracellular localization of compounds 3a, 5a, and 5d can be investigated using confocal microscopy. As shown in Figure 5a, green fluorescence from compound 5d could be observed coming from the cell cytoplasm after 1‐hr incubation, and most fluorescence could be detected from locations close to the nuclear membrane. However, compound 3a (without guanidine modification) produced less fluorescence from the cytoplasm, while compound 5a largely localized to the cytoplasm, similar to compound 5d (Figure 5a). Thus, the guanidine modification may change the intracellular localization of the BS‐pyrimidine derivatives.
Figure 5.
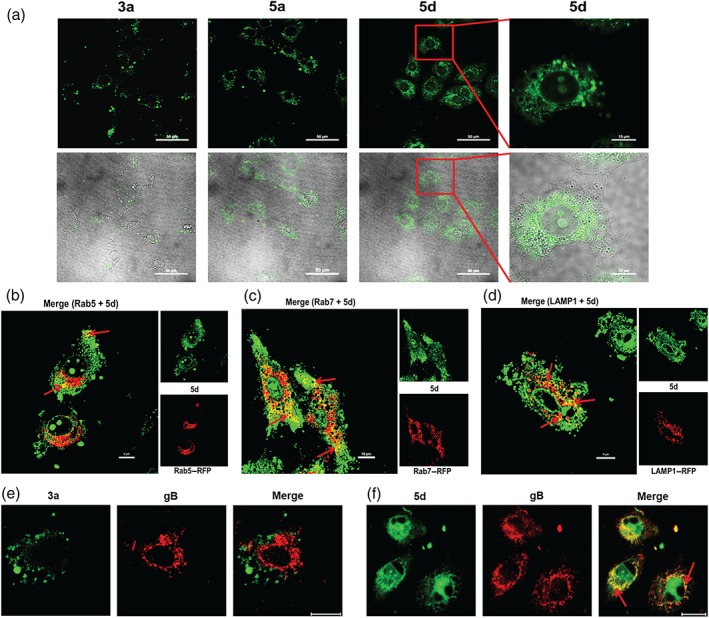
Subcellular localization of BS‐pyrimidine derivatives in Vero and HeLa cells. (a) Vero cells were incubated with compounds 3a, 5a, or 5d (30 μM) for 1 hr at 37°C. The fluorescence was then detected by confocal laser scanning microscopy. The scale bar represents 50 μm. An enlarged view of part of one field (highlighted by the red rectangle) is shown to indicate the localization of compound 5d within the Vero cells. The scale bar represents 10 μm in this case. (b‐d) HeLa cells were transfected with expression plasmids for red fluorescent protein (RFP)‐coupled (b) Rab5, (c) Rab7, or (d) LAMP‐1. At 24 hr post‐transfection, cells were added with compound 5d (20 μM) and incubated for 1 hr at 37°C. After that, the fluorescence was detected via confocal microscopy. The scale bar represents 10 μm. (e, f) Herpes simplex virus (HSV)‐2 (multiplicity of infection = 1.0) was pretreated with (e) compound 3a or (f) 5d at 37°C for 1 hr before infection. Then, after removal of the virus inoculum, media containing compound 3a or 5d (20 μM) were added to cells. At 1 hr p.i., the localization of virus gB protein was evaluated via immunofluorescence assay. The fluorescence was detected via confocal microscopy. The scale bar represents 20 μm
As compound 5d can enter into cells and inhibit HSV entry process in HeLa cells, we further explored the association of compound 5d with viral endosomal transport systems by detecting the co‐localization of compound 5d with Rab5, Rab7, and LAMP1 in HeLa cells. The results showed that the co‐localization signal (yellow spot) of compound 5d with Rab5, Rab7, and LAMP1 could be clearly detected after 1‐hr incubation in HeLa cells (red arrow indicated; Figure 5b–d). Especially, there was obvious co‐localization between compound 5d and LAMP1 around the cell nucleus in a punctate pattern (the yellow dot; Figure 5d), suggesting that compound 5d may be able to localize to endolysosome compartments after entry into HeLa cells.
As compound 5d can block virus‐induced membrane fusion (Figure 4e) and co‐localize with endolysosome compartments in HeLa cells (Figure 5d), we further explored the localization of compounds 3a, 5d, and virus gB protein in HSV‐infected HeLa cells. The results shown in Figure 5e indicated that the red fluorescence produced by HSV‐2 gB protein (at 1 hr p.i.) was emitted from the cytoplasm around the nuclei. However, the green fluorescence from compound 3a was not co‐localized with the gB protein. In contrast, cells treated with compound 5d (20 μM) showed obvious co‐localization signal (many yellow spots) around the cell nuclei (Figure 5f), suggesting that compound 5d (rather than 3a) becomes co‐localized with virus gB protein after HSV endocytosis in HeLa cells. Thus, the co‐localization of compound 5d with virus gB protein and endolysosome compartments may be related to its ability to inhibit gB‐mediated membrane fusion before capsid egress from endolysosome in HeLa cells.
3.5. Compound 5d may inactivate HSV infectivity by interaction with virus gB protein
The initial binding of HSV to cellular heparan sulfate is mainly mediated by gB, followed by the interaction of gD with one of three receptors: herpesvirus entry mediator, nectin‐1, or 3‐O‐sulfated heparan sulfate (Shukla et al., 1999; Spear & Longnecker, 2003). Therefore, it is interesting to see if compound 5d can directly interact with HSV surface glycoprotein gB or gD by performing pull‐down assays using heparin‐coupled magnetic beads (Beaverbio, China). As shown in Figure 6a,b, virus gB protein rather than gD protein in HSV‐2‐infected cell lysates could specifically bind to heparin‐coupled magnetic beads, suggesting that the interaction of gB protein to heparin is much stronger than that of gD protein. Thus, the formed gB‐linked magnetic bead can be used for further pull‐down assay with compounds 5d and 3a.
Figure 6.
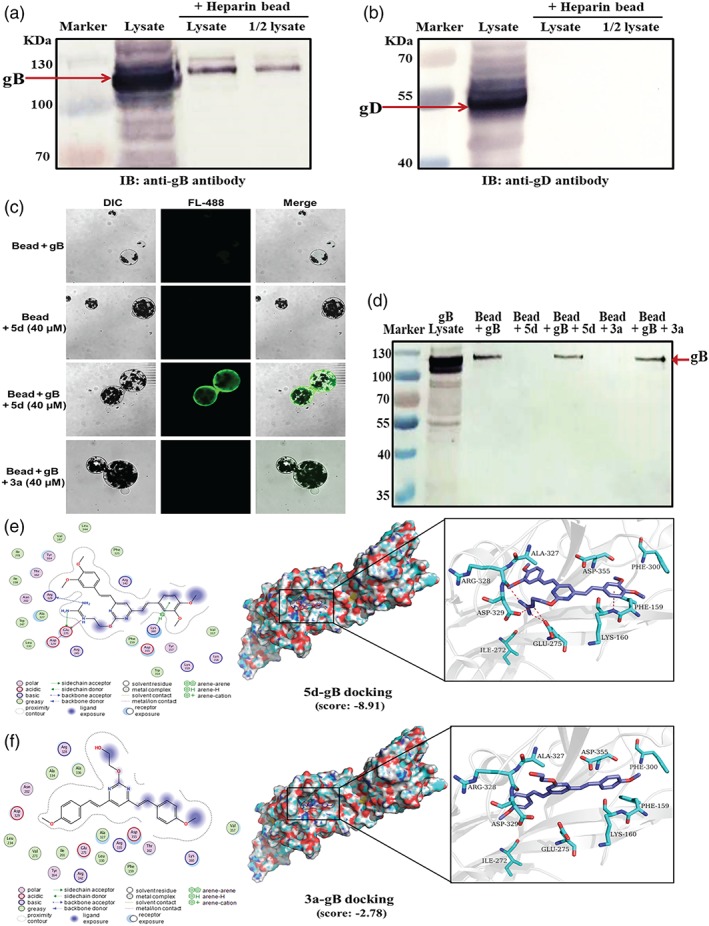
Compound 5d may inactivate herpes simplex virus (HSV) infectivity by interaction with virus gB protein. (a, b) The cell lysate of HSV‐2‐infected Vero cells was incubated with heparin‐coupled magnetic beads (Beaverbio, China) at room temperature for 1 hr. Then the binding of virus gB or gD protein to heparin beads and the input cell lysate were detected by Western blotting with (a) anti‐gB or (b) gD antibody, respectively. (c) The cell lysate of HSV‐2‐infected Vero cells containing gB protein was incubated with the heparin‐coupled magnetic beads at room temperature for 1 hr to form gB‐linked magnetic beads (gB‐beads). Then, compound 3a or 5d (40 μM) was added to the gB‐beads and incubated at room temperature for another 1 hr. After washing, the fluorescence from the magnetic beads was detected via confocal microscopy to indicate the presence of compound 5d bound to gB‐beads. (d) The binding of HSV‐2 gB protein to the magnetic beads in the different groups was verified using Western blot. (e, f) Predicted binding modes of compounds (e) 5d or (f) 3a on HSV‐1 gB protein (PDB code: 5FZ2). Compound 5d or 3a is coloured in purple, and the surrounding residues in the binding pockets are coloured in cyan. The backbone of the receptor is depicted as light blue cartoon. The main binding amino acids of gB protein to compound 5d (Glu275, Arg328, Asp329, and Lys160) were also shown in the 3D interaction map, which are defined based on the docking score in MOE
Then, compound 3a or 5d was incubated with the HSV‐2 gB‐linked magnetic beads at RT for 1 hr, respectively. The amount of compound binding to the magnetic beads was then detected using fluorescence microscopy. As shown in Figure 6c, the magnetic beads treated with 40 μM of compound 5d (“bead + gB + 5d”) produced copious amounts of green fluorescence. In contrast, virtually, no fluorescence could be detected from non‐treated beads (“bead + gB”) or compound 5d‐treated beads without gB lysate (“bead + 5d”). In addition, there was virtually no specific fluorescence from compound 3a‐treated gB‐beads (“bead + gB + 3a”), suggesting that guanidine modification may enhance the interaction between gB and BS‐pyrimidine derivatives. Moreover, the results of Western blotting in Figure 6d clearly showed that HSV‐2 gB protein definitely became bound to the heparin‐coupled magnetic beads to form gB‐beads. It is also clear that treatment with compound 5d also reduced the amount of gB binding to heparin beads (bead + gB + 5d) compared with non‐treated beads (bead + gB). Besides that, compound 5d rather than compound 3a could also specifically bind to HSV‐1 gB‐linked magnetic beads (Figure S3).
To further investigate the binding mode of compound 5d with gB, docking simulation studies were carried out using the crystal structure of HSV‐1 gB protein (PDB code: 5dZ2). The results indicated that the two nitrogen atoms of guanidine group of compound 5d can form two hydrogen bonds with the oxygen atom of carboxyl group of Glu275 in gB. Another nitrogen atom of guanidine group of compound 5d can form a hydrogen bond with the oxygen atom of backbone of Arg328 in gB (Figure 6e). The interaction energy between compound 5d and the residues in gB was shown in Table S3, and the binding ability between compound 5d and the residues is Glu275 > Arg328 > Asp329 > Lys160. However, there was no specific interaction between compound 3a and gB with very low docking score (−2.78 kcal·mol−1; Figure 6f). In addition, both compound 3a and compound 5d can interact with gD protein with low docking score (Figure S4), suggesting that gD binding may not be the main reason for anti‐HSV actions of guanidine‐modified pyrimidine derivatives.
3.6. Compound 5d may also inhibit PI3K/Akt signal pathway to reduce HSV replication
As compound 5d can markedly block the adsorption and entry process of HSV in both Vero and HeLa cells, we further explored if this compound could influence the cellular signalling pathways required for HSV infection. The https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=673/https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 signalling pathway was reported to be required for efficient HSV infection, and the inhibitors of PI3K signalling could reduce both entry and fusion of HSV (Tiwari & Shukla, 2010). Herein, the cellular PI3K protein was significantly activated in the HSV‐2‐infected control group to higher level than that in the normal control group at 2 hr p.i. (Figure 7a,b). However, the level of phosphorylated PI3K was decreased, concentration‐dependently, after treatment with compound 5d for 2 hr (Figure 7a,b). Moreover, treatment with compound 5d (2.5, 5, 10, and 20 μM) for 2 hr also significantly decreased the level of phosphorylated Akt (Figure 7c,d). The activation of PI3K and Akt proteins was also inhibited by compound 5d treatment in HSV‐1‐infected Vero cells (Figure S5). However, compound 5d treatment did not significantly influence the activation of the PI3K/Akt pathway in the non‐infected Vero cells (Figure S6), suggesting that the inhibition of PI3K/Akt pathway by compound 5d may be related to its inhibition of HSV infection. In addition, the level of phosphorylated NF‐κB protein was only significantly reduced by the highest concentration of compound 5d (20 μM; Figure 7e,f). Furthermore, treatment with compound 5d also significantly reduced the levels of virus gD mRNA in both HSV‐1‐ and HSV‐2‐infected cells (Figure 7g,h).
Figure 7.
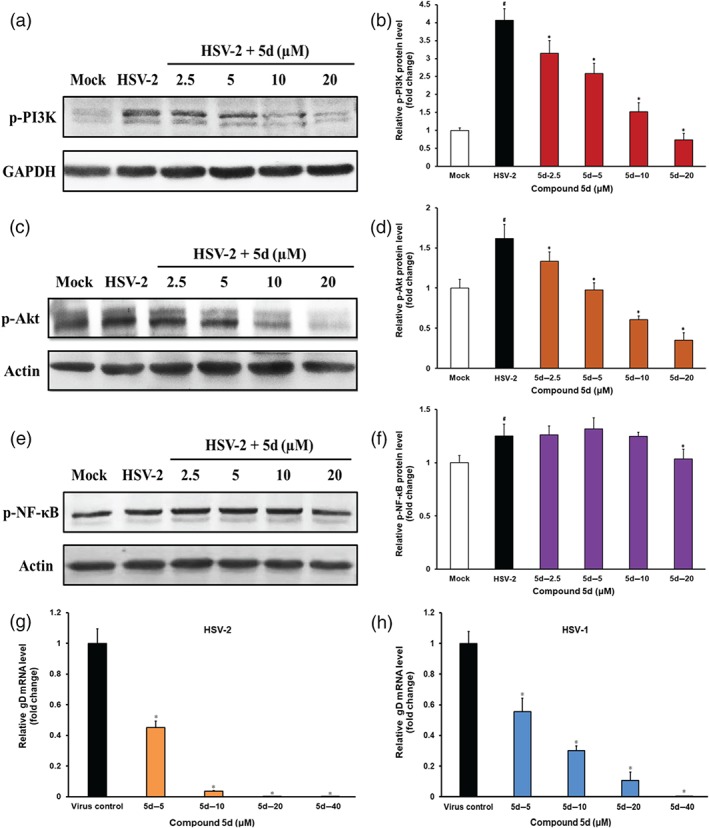
Involvement of PI3K/Akt signalling pathway in the anti‐herpes simplex virus (HSV) actions of compound 5d. (a, c, e) HSV‐2 (multiplicity of infection = 1.0)‐infected cells were treated with or without compound 5d (2.5, 5, 10, 20 μM) for 2 hr after adsorption, and then the phosphorylation of (a) PI3K, (c) Akt, and (e) NF‐κB proteins was evaluated via Western blotting. Blots were also probed for GAPDH and β‐actin proteins as loading controls. The results shown are representative of five independent experiments. (b, d, f) Plots quantifying the immunoblots (as ratios to GAPDH or β‐actin) for: (b) p‐PI3K, (d) p‐Akt, and (f) p‐NF‐κB proteins. The ratios for non‐infected cells (Mock) were assigned values of 1.0, and the data were presented as mean ± SD (n = 5). # P < .05, significantly different from normal control group (Mock); *P < .05, significantly different from virus control group (HSV‐2). (g, h) HSV (multiplicity of infection = 1.0)‐infected Vero cells were treated with or without compound 5d (5, 10, 20, and 40 μM) for 8 hr at 37°C. After that, the total RNA was extracted for real‐time RT‐PCR assay of (g) HSV‐2 and (h) HSV‐1 gD mRNAs and cellular β‐actin mRNA. The relative amounts of virus gD mRNAs were determined using the comparative (2−ΔΔCT) method. The RNA levels for non‐drug‐treated cells (virus control) were assigned values of 1.0. Results shown are means ± SD from five independent experiments. *P < .05, significantly different from virus control group
3.7. Compound 5d exhibited greater in vivo antiviral activity than compound 3a, in HSV‐1‐infected mice
The antiviral activities of BS‐pyrimidine derivatives compounds 3a and 5d were further tested in a murine intranasal model of HSV pneumonia and encephalitis (De Clercq & Luczak, 1976). Briefly, mice infected with HSV‐1 (F strain) received intraperitoneal treatment with acyclovir (10 mg·kg−1), compound 3a (5 mg·kg−1), compound 5d (5 or 10 mg·kg−1), or placebo (PBS) once daily for 5 days. The survival curves showed that on Day 14 p.i., only 20% of the mice in the placebo group survived, whereas 100% of animals in the compound 5d (10 mg·kg−1)‐treated group survived, a higher survival that that in acyclovir (10 mg·kg−1)‐treated or compound 5d (5 mg·kg−1)‐treated group (90%). However, in the group treated with compound 3a (5 mg·kg−1)‐, only 30% of mice survived (Figure 8a).
Figure 8.
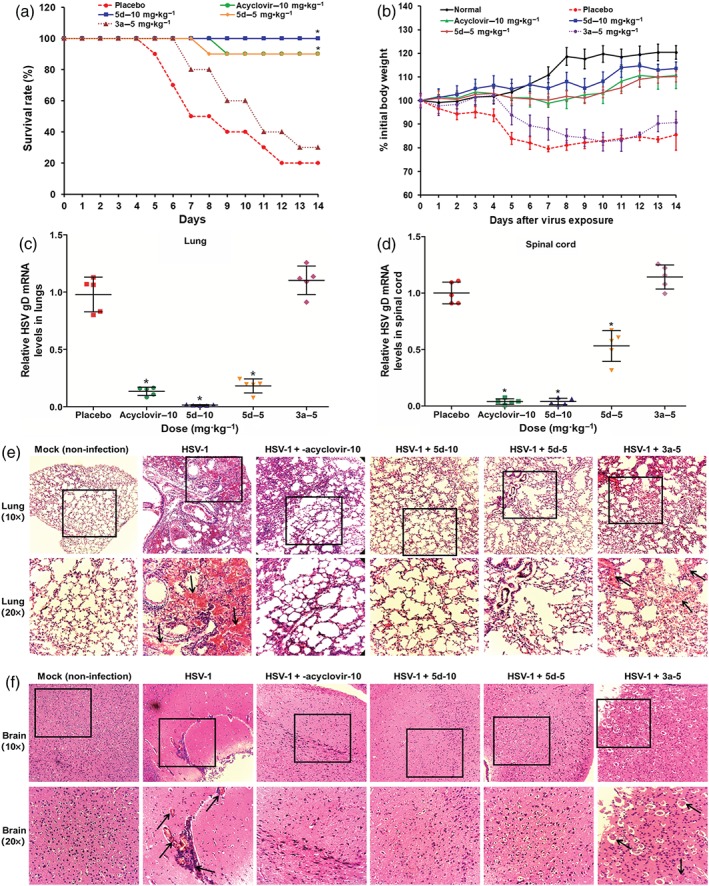
The anti‐herpes simplex virus (HSV) actions of compound 5d in vivo. (a) Survival rate. HSV‐1‐infected mice were received intraperitoneally treatment of acyclovir (10 mg·kg−1), compound 3a (5 mg·kg−1), compound 5d (5 or 10 mg·kg−1), or placebo (PBS) once daily for 5 days. Results are expressed as percentage of survival, evaluated daily for 14 days. *P < .05, significantly different from virus control group (placebo; n = 10 per group). (b) HSV‐infected mice received intraperitoneally treatment with indicated compounds for 5 days. The body weights of 10 mice in each group were monitored daily for 14 days and are expressed as a percentage of the initial value. Values are means ± SD (n = 10 per group). (c, d) Viral titres in lungs and spinal cord. After intraperitoneal therapy with indicated compounds for 5 days, the viral titres in (c) lungs and (d) spinal cords were evaluated by quantitative RT‐PCR assay of HSV gD mRNA on Day 6 p.i. Data are expressed as means ± SD for five mice per group. *P < .05, significantly different from virus control group. (e, f) Histopathological analyses of lung and brain tissues on Day 6 p.i. by haematoxylin–eosin staining (×10 and ×20). The representative micrographs from each group were shown. HSV‐1 + compound 3a‐5, HSV‐1 infected tissues with compound 3a (5 mg·kg−1) treatment; HSV‐1 + 5d‐10, HSV‐1 infected tissues with compound 5d (10 mg·kg−1) treatment; HSV‐1 + 5d‐5, HSV‐1 infected tissues with compound 5d (5 mg·kg−1) treatment; HSV‐1 + acyclovir‐10, HSV‐1 infected tissues with acyclovir (10 mg·kg−1) treatment; HSV‐1, HSV‐1 infected tissues without drugs; Mock, non‐infected mice tissues. The black arrows indicate the presence of hyperaemia and infiltration of inflammatory cells in the lumen
Moreover, the weight of mice was monitored as a measure of disease progression (Figure 8b). In the virus control group (placebo), the weights of the mice began to decrease at 4 days p.i., losing up to 18% of initial weight, before gradually recovering. Infected mice treated with compound 5d (5 or 10 mg·kg−1) gradually increased their body weights, without weight loss. In contrast, mice receiving compound 3a (5 mg·kg−1) showed a steady loss of weight from 5 days p.i. until the end of the experiment 14 days p.i. In addition, the group treated with acyclovir (10 mg·kg−1) gradually increased their body weights only with a limited weight loss of less than 5% during the infection (Figure 8b).
Furthermore, 6 days p.i., six mice of each treatment group were killed to determine viral titres in the lung and spinal cord. After treatment with compound 5d for 5 days, the relative virus gD mRNA levels in both lungs and spinal cord was lower than those in samples from the virus control group, suggesting that intraperitoneal therapy with compound 5d could inhibit HSV multiplication in mice (Figure 8c,d). Acyclovir (10 mg·kg−1) treatment also showed significant reduction of virus titres in mouse lungs and spinal cord (Figure 8c,d). However, compound 3a (5 mg·kg−1) treatment did not significantly reduce HSV multiplication in samples of lungs and spinal cord (Figure 8c,d). In addition, the results of plaque assays also indicated that compound 5d, rather than compound 3a, reduced the pulmonary virus titres, compared with the virus control group (Figure S7).
To further evaluate the effects of compound 5d on viral pneumonia and encephalitis in mice, tissue samples were analysed by histopathological methods. As shown in Figure 8e, lung tissues in the virus control group showed marked infiltration of inflammatory cells in the alveolar walls and the presence of massive hyperaemia in the lumen. However, mice treated with compound 5d (5 or 10 mg·kg−1) following infection had intact columnar epithelium in the bronchiole even in the presence of tiny serocellular exudates in the lumen (Figure 8e). However, lung tissues in infected mice treated with compound 3a (5 mg·kg−1) showed marked infiltration of inflammatory cells in the alveolar walls (Figure 8e). In samples of brain tissues from the virus‐infected mice, treatment with compound 5d (5 or 10 mg·kg−1) showed very little hyperaemia and infiltration of inflammatory cells, compared with brain tissues from the HSV‐1‐infected control group. This was comparable to the effects of acyclovir (10 mg·kg−1; Figure 8f). Thus, compound 5d, rather than compound 3a, may be able to attenuate symptoms of pneumonia and encephalitis in HSV‐infected mice.
4. DISCUSSION
Entry of HSV into target cells is the first step for virus infection, which depends upon multiple cell surface receptors and virus surface glycoproteins such as gB, gD, gH, and gL (Majmudar et al., 2019; Spear, 2004; Spear & Longnecker, 2003). Thus, targeting virus entry process seems to be a promising approach to limit HSV infection. In the present study, we have demonstrated that guanidine modifications greatly improve the anti‐HSV activities of BS‐pyrimidine derivatives. The guanidine‐modified derivative compound 5d can effectively block virus binding and membrane fusion by targeting virus gB protein, which is quite different from the antiviral mechanism by which acyclovir acts. Most importantly, compound 5d treatment also markedly improved survival and decreased viral titres in HSV‐infected mice.
Time‐of‐addition assays showed that pretreatment of HSV with compound 5d before infection significantly reduced the virus titres of HSV, suggesting that compound 5d may be able to directly interact with HSV particles. Moreover, compound 5d also inhibited HSV‐mediated membrane fusion process and markedly blocked virus adsorption and entry processes in both HeLa and Vero cells (Figure 3), suggesting that compound 5d may interact with some virus surface glycoproteins such as gB and gD. The results of the DARTS assay indicated that compound 5d may have interactions with virus gB protein in HSV‐infected Vero cells (Figure 4). Furthermore, compound 5d treatment also inhibited activation of the PI3K/Akt signalling pathway and reduced the mRNA levels of virus proteins in HSV‐infected cells (Figure 7). As the PI3K/Akt pathway is required for efficient HSV replication (Liu & Cohen, 2015; Vanhaesebroeck, Stephens, & Hawkins, 2012), it is possible that compound 5d may be able to interact with HSV particles to interfere with the activation of PI3K/Akt pathway, so as to further inhibit subsequent replication of HSV (Figure 9).
Figure 9.
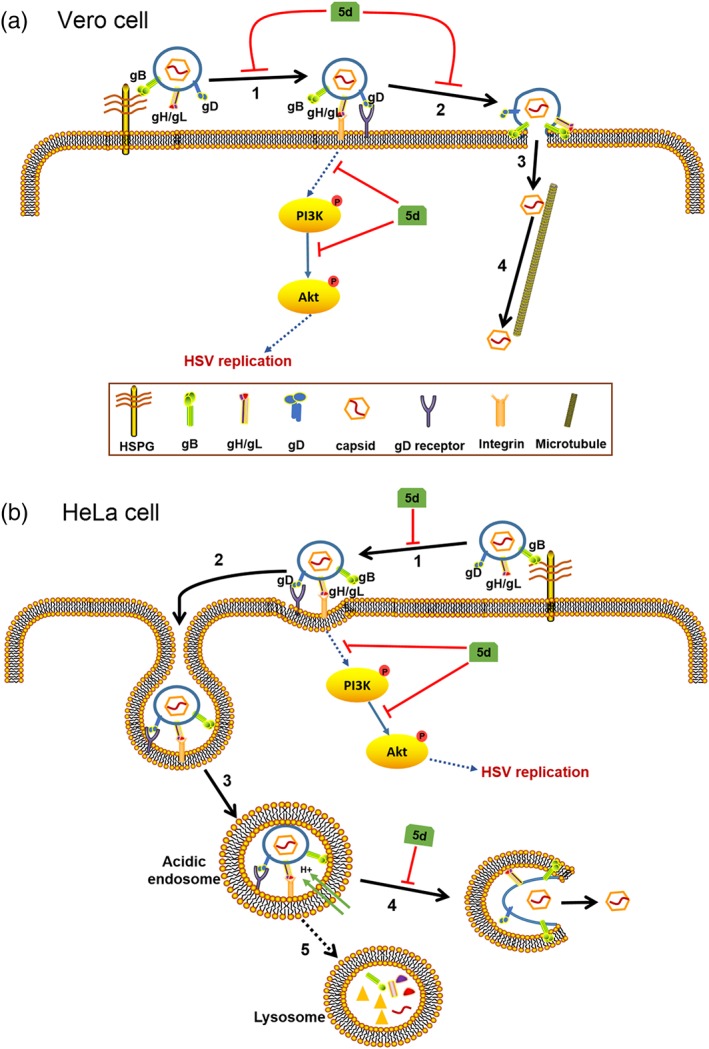
Mechanism of action of compound 5d in herpes simplex virus (HSV)‐infected Vero and HeLa cells. (a) In Vero cells, compound 5d may block the initial binding of HSV gB protein to cellular heparan sulfate glycosaminoglycan (HSPG; 1) and interfere with the fusion process at plasma membrane (2), so as to inhibit the adsorption and entry process of HSV. compound 5d also inhibits the activation of the PI3K/Akt pathway in early stage of HSV life cycle to reduce virus replication. (b) In HeLa cells, compound 5d may block the initial attachment of HSV particle to cellular HSPG (1) before gD protein binding to its receptor. After virus endocytosis (2), and endosomal acidification (3), compound 5d may block gB‐mediated fusion at endosomal membrane (4), so as to inhibit the capsid egress from endolysosome compartments. Compound 5d may also block the activation of the PI3K/Akt pathway to inhibit subsequent virus replication
BS‐pyrimidine derivatives have been shown to produce intrinsic fluorescence, which can be used to probe their subcellular localization (Tong et al., 2016). Here, the guanidine‐modified pyrimidine derivative compound 5d, rather than compound 3a, accessed the cell cytoplasm and co‐localized with both endolysosome compartments and virus gB protein in HeLa cells (Figure 5), suggesting that compound 5d may interact with gB protein to interfere with virus entry process. The results of pull‐down assays confirmed that compound 5d (rather than the non‐modified compound 3a could directly interact with virus gB protein (Figures 6 and S3). Docking simulation studies indicated that compound 5d may interact with Glu275, Arg328, and Asp329 of gB via hydrogen bond and slat bridge, while compound 3a had almost no interaction with gB protein. These potential binding sites (E275, R328, and D329) are highly conserved in different HSV strains, which may be the reason why compound 5d can bind to the gB protein of both HSV‐1 and HSV‐2. In addition, compounds 3a and 5d may weakly interact with gD protein with comparable low docking scores (Figure S4), suggesting that gD binding may not be the main reason for anti‐HSV actions of guanidine‐modified pyrimidine derivatives. Thus, guanidine modification may enhance the interaction between HSV surface gB protein and BS‐pyrimidine derivatives to efficiently block HSV adsorption and entry processes in both Vero and HeLa cells (Figure 9).
The mouse model of HSV intranasal infection has been used for studying HSV‐induced pneumonia and encephalitis (Drummond, Eglin, & Esiri, 1994; Gamba, Cavalieri, Courreges, Massouh, & Benencia, 2004). The in vitro antiviral effects of compound 5d were mirrored in a murine intranasal model of HSV‐1 pneumonia and encephalitis. Intraperitoneal therapy of HSV‐1‐infected mice with compound 5d markedly improved their survival and inhibited HSV multiplication in both lungs and spinal cord (Figure 8). Moreover, the histopathological analysis indicated that compound 5d treatment also attenuated the pneumonia and encephalitis symptoms in HSV‐infected mice, comparable with the effects of acyclovir. However, treatment with compound 3a (without guanidine modification) had almost no protective effects against HSV‐1 infection‐induced weight loss and mortality (Figure 8). Thus, guanidine modification can also improve the anti‐HSV activities of BS‐pyrimidine derivatives in vivo.
In conclusion, our studies not only revealed that guanidine modification can enhance the anti‐HSV effect of BS‐pyrimidine derivatives both in vitro and in vivo but also identified that guanidine‐modified compound 5d may be a promising anti‐HSV candidate targeting both virus gB protein and cellular PI3K/Akt signal pathway. Further studies of the anti‐HSV effects of compound 5d against clinical strains especially the acyclovir‐resistant strains will be required to advance it for drug development. Nevertheless, the guanidine‐modified compound 5d may be a promising antiviral candidate for therapy of herpes simplex encephalitis and pneumonia in the future.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
W.W. and T.J. conceived and designed the experiments. C.X., J.Z., J.W., R.Y., D.W., R.Y., and W.L. performed the experiments. W.W., C.X., J.Z., and T.J. interpreted the data and wrote the original manuscript. All authors had the final approval of the submitted and published versions.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14206 and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Anti‐HSV effects of BS‐pyrimidine derivatives and Acyclovir in vitro.
Table S2. Inhibition effects of 5d and Acyclovir on HSV multiplication in different cells.
Table S3. The interaction energy between 5d and the residues in gB protein
Figure S1. The cytotoxicity and anti‐HSV effects of Acyclovir in different cells.
Figure S2. Influence of different treatment conditions of compound 5d on HSV replication.
Figure S3. Compound 5d rather than 3a can specifically bind to HSV‐1 gB linked magnetic beads.
Figure S4. Predicted binding modes of the compound 3a or 5d on HSV‐2 gD protein.
Figure S5. Inhibition of compound 5d on PI3K/Akt signal pathway in HSV‐1‐infected cells.
Figure S6. Influence of compound 5d on PI3K/Akt signal pathway in non‐infected Vero cells.
Figure S7. The pulmonary viral titres in HSV‐1 infected mice lungs.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (81874320 and 81741146), the National Natural Science Foundation of China‐Shandong Joint Fund for Marine Science Research Centers (U1706210 and U1606403), the National Science and Technology Major Project of China (2018ZX09735‐004), the Marine S&T Fund of Shandong Province (2018SDKJ0403), and the Natural Science Foundation of Shandong Province (ZR2017MH013).
Wang W, Xu C, Zhang J, et al. Guanidine modifications enhance the anti‐herpes simplex virus activity of (E,E)‐4,6‐bis(styryl)‐pyrimidine derivatives in vitro and in vivo. Br J Pharmacol. 2020;177:1568–1588. 10.1111/bph.14918
Wei Wang, Cuijing Xu, and Jianqiang Zhang contributed equally to this work.
Contributor Information
Wei Wang, Email: wwwakin@ouc.edu.cn.
Tao Jiang, Email: jiangtao@ouc.edu.cn.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). The Concise Guide to Pharmacology 2019/20: Nuclear hormone receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). The Concise Guide to Pharmacology 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlinck, R. G. , & Romminger, S. (2016). The chemistry and biology of guanidine natural products. Natural Product Reports, 33, 456–490. 10.1039/c5np00108k [DOI] [PubMed] [Google Scholar]
- Blot, N. , Schneider, P. , Young, P. , Janvresse, C. , Dehesdin, D. , & Tron, P. (2000). Treatment of an acyclovir and foscarnet‐resistant herpes simplex virus infection with cidofovir in a child after an unrelated bone marrow transplant. Bone Marrow Transplantation, 26, 903–905. 10.1038/sj.bmt.1702591 [DOI] [PubMed] [Google Scholar]
- De Clercq, E. , & Luczak, M. (1976). Intranasal challenge of mice with herpes simplex virus: An experimental model for evaluation of the efficacy of antiviral drugs. The Journal of Infectious Diseases, 133(Supplement 2), A226–A236. 10.1093/infdis/133.Supplement_2.A226 [DOI] [PubMed] [Google Scholar]
- Drummond, C. W. , Eglin, R. P. , & Esiri, M. M. (1994). Herpes simplex virus encephalitis in a mouse model: PCR evidence for CNS latency following acute infection. Journal of the Neurological Sciences, 127, 159–163. 10.1016/0022-510x(94)90068-x [DOI] [PubMed] [Google Scholar]
- Du, R. K. , Wang, L. L. , Xu, H. , Wang, Z. Y. , Zhang, T. , Wang, M. L. , … Li, Y . (2017). A novel glycoprotein D‐specific monoclonal antibody neutralizes herpes simplex virus. Antiviral Research, 147, 131–141. 10.1016/j.antiviral.2017.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq, A. V. , & Shukla, D. (2012). Herpes simplex epithelial and stromal keratitis: An epidemiologic update. Survey of Ophthalmology, 57, 448–462. 10.1016/j.survophthal.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatahzadeh, M. , & Schwartz, R. A. (2007). Human herpes simplex virus infections: Epidemiology, pathogenesis, symptomatology, diagnosis, and management. Journal of the American Academy of Dermatology, 57, 737–763. 10.1016/j.jaad.2007.06.027 [DOI] [PubMed] [Google Scholar]
- Gamba, G. , Cavalieri, H. , Courreges, M. C. , Massouh, E. J. , & Benencia, F. (2004). Early inhibition of nitric oxide production increases HSV‐1 intranasal infection. Journal of Medical Virology, 73, 313–322. 10.1002/jmv.20093 [DOI] [PubMed] [Google Scholar]
- Gao, L. , Liu, Q. , Ren, S. , Wan, S. , Jiang, T. , Wong, I. L. K. , … Wang, S. (2012). Synthesis of a novel series of (E,E)‐4,6‐bis(styryl)‐2‐O‐glucopyranosyl‐pyrimidines and their potent multidrug resistance (MDR) reversal activity against cancer cells. Journal of Carbohydrate Chemistry, 31, 620–633. 10.1080/07328303.2012.689041 [DOI] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, M. , Wexselblatt, E. , Esko, J. D. , & Tor, Y. (2014). Macromolecular uptake of alkyl‐chain‐modified guanidinoglycoside molecular transporters. Chembiochem, 15(5), 676–680. 10.1002/cbic.201300606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver, W. G. , Bischofberger, N. , & Webster, R. G. (1999). Disarming flu viruses. Scientific American, 280, 78–87. 10.1038/scientificamerican0199-78 [DOI] [PubMed] [Google Scholar]
- Li, T. , Liu, L. B. , Wu, H. L. , Chen, S. D. , Zhu, Q. C. , Gao, H. , … Peng, T. (2017). Anti‐herpes simplex virus type 1 activity of Houttuynoid A, a flavonoid from Houttuynia cordata Thunb. Antiviral Research, 144, 273–280. 10.1016/j.antiviral.2017.06.010 [DOI] [PubMed] [Google Scholar]
- Liu, X. Q. , & Cohen, J. I. (2015). The role of PI3K/Akt in human herpesvirus infection: From the bench to the bedside. Virology, 479, 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lomenick, B. , Hao, R. , Jonai, N. , Chin, R. M. , Aghajan, M. , Warburton, S. , … Huang, J. (2009). Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences of the United States of America, 106, 21984–21989. 10.1073/pnas.0910040106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick, B. , Jung, G. , Wohlschlegel, J. A. , & Huang, J. (2011). Target identification using drug affinity responsive target stability (DARTS). Curr Protoc Chem Biol, 3(4), 163–180. 10.1002/9780470559277.ch110180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmudar, H. , Hao, M. , Sankaranarayanan, N. V. , Zanotti, B. , Volin, M. V. , Desai, U. R. , & Tiwari, V. (2019). A synthetic glycosaminoglycan mimetic blocks HSV‐1 infection in human iris stromal cells. Antiviral Research, 161, 154–162. 10.1016/j.antiviral.2018.11.007 [DOI] [PubMed] [Google Scholar]
- Morfin, F. , & Thouvenot, D. (2003). Herpes simplex virus resistance to antiviral drugs. Journal of Clinical Virology, 26, 29–37. 10.1016/s1386-6532(02)00263-9 [DOI] [PubMed] [Google Scholar]
- Qiu, P. , Xu, L. , Gao, L. , Zhang, M. , Wang, S. , Tong, S. , … Jiang, T. (2013). Exploring pyrimidine‐substituted curcumin analogues: Design, synthesis and effects on EGFR signaling. Bioorganic & Medicinal Chemistry, 21, 5012–5020. 10.1016/j.bmc.2013.06.053 [DOI] [PubMed] [Google Scholar]
- Reed, L. , & Muench, H. A. (1938). Simple method for estimating fifty percent endpoints. American Journal of Hygiene, 27, 943–947. [Google Scholar]
- Shukla, D. , Liu, J. , Blaiklock, P. , Shworak, N. W. , Bai, X. , Esko, J. D. , … Spear, P. G. (1999). A novel role for 3‐O‐sulfated heparan sulfate in herpes simplex virus 1 entry. Cell, 99, 13–22. 10.1016/s0092-8674(00)80058-6 [DOI] [PubMed] [Google Scholar]
- Smith, J. S. , & Robinson, N. J. (2002). Age‐specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. The Journal of Infectious Diseases, 186, S3–S28. 10.1086/343739 [DOI] [PubMed] [Google Scholar]
- Spear, P. G. (2004). Herpes simplex virus: Receptors and ligands for cell entry. Cellular Microbiology, 6, 401–410. 10.1111/j.1462-5822.2004.00389.x [DOI] [PubMed] [Google Scholar]
- Spear, P. G. , & Longnecker, R. (2003). Herpesvirus entry: An update. Journal of Virology, 77, 10179–10185. 10.1128/jvi.77.19.10179-10185.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzich, J. B. , & Cliffe, A. R. (2018). Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology, 522, 81–91. 10.1016/j.virol.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczubiałka, K. , Pyrć, K. , & Nowakowska, M. (2016). In search for effective and definitive treatment of herpes simplex virus type 1 (HSV‐1) infections. RSC Advances, 6, 1058–1075. 10.1039/C5RA22896D [DOI] [Google Scholar]
- Tiwari, V. , & Shukla, D. (2010). Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type‐1 entry. The Journal of General Virology, 91, 3002–3009. 10.1099/vir.0.024166-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, S. , Zhang, M. , Wang, S. X. , Yin, R. J. , Yu, R. L. , Wan, S. B. , & Jiang, T. (2016). Isothiouronium modification empowers pyrimidine‐substituted curcumin analogs potent cytotoxicity and Golgi localization. European Journal of Medicinal Chemistry, 123, 849–857. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B. , Stephens, L. , & Hawkins, P. (2012). PI3K signalling: The path to discovery and understanding. Nature Reviews. Molecular Cell Biology, 13, 195–203. 10.1038/nrm3290 [DOI] [PubMed] [Google Scholar]
- Wang, W. , Wu, J. D. , Zhang, X. S. , Hao, C. , Zhao, X. L. , Jiao, G. L. , … Yu, G. (2017). Inhibition of influenza A virus infection by fucoidan targeting viral neuraminidase and cellular EGFR pathway. Scientific Reports, 7, 40760 10.1038/srep40760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Yin, R. J. , Zhang, M. , Yu, R. L. , Hao, C. , Zhang, L. J. , & Jiang, T. (2017). Boronic acid modifications enhance the anti‐influenza A virus activities of novel quindoline derivatives. Journal of Medicinal Chemistry, 60, 2840–2852. 10.1021/acs.jmedchem.6b00326 [DOI] [PubMed] [Google Scholar]
- Wang, W. , Zhang, P. , Hao, C. , Zhang, X. E. , Cui, Z. Q. , & Guan, H. S. (2011). In vitro inhibitory effect of carrageenan oligosaccharide on influenza A H1N1 virus. Antiviral Research, 92, 237–246. 10.1016/j.antiviral.2011.08.010 [DOI] [PubMed] [Google Scholar]
- Weed, D. J. , Dollery, S. J. , Komala, S. T. , & Nicola, A. V. (2018). Acidic pH mediates changes in antigenic and oligomeric conformation of herpes simplex virus gB and is a determinant of cell‐specific entry. Journal of Virology, 92, e01034–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Anti‐HSV effects of BS‐pyrimidine derivatives and Acyclovir in vitro.
Table S2. Inhibition effects of 5d and Acyclovir on HSV multiplication in different cells.
Table S3. The interaction energy between 5d and the residues in gB protein
Figure S1. The cytotoxicity and anti‐HSV effects of Acyclovir in different cells.
Figure S2. Influence of different treatment conditions of compound 5d on HSV replication.
Figure S3. Compound 5d rather than 3a can specifically bind to HSV‐1 gB linked magnetic beads.
Figure S4. Predicted binding modes of the compound 3a or 5d on HSV‐2 gD protein.
Figure S5. Inhibition of compound 5d on PI3K/Akt signal pathway in HSV‐1‐infected cells.
Figure S6. Influence of compound 5d on PI3K/Akt signal pathway in non‐infected Vero cells.
Figure S7. The pulmonary viral titres in HSV‐1 infected mice lungs.