

Abstract
The rabies and Ebola viruses recruit the highly conserved host protein LC8 for their own reproductive success. In vivo knockouts of the LC8 recognition motif within the rabies virus phosphoprotein (RavP) result in completely non-lethal viral infections. In this work, we examine the molecular role LC8 plays in viral lethality. We show that RavP and LC8 co-localize in rabies infected cells, and that LC8 interactions are essential for efficient viral polymerase functionality. NMR, SAXS, and molecular modeling demonstrate that LC8 binding to a disordered linker adjacent to an endogenous dimerization domain results in restrictions in RavP domain orientations. The resulting ensemble structure of RavP-LC8 tetrameric complex is similar to that of a related virus phosphoprotein that does not bind LC8, suggesting that with RavP, LC8 binding acts as a switch to induce a more active conformation. The high conservation of the LC8 motif in Lyssavirus phosphoproteins and its presence in other analogous proteins such as the Ebola virus VP35 evinces a broader purpose for LC8 in regulating downstream phosphoprotein functions vital for viral replication.
Keywords: Virus, NMR, SAXS, Intrinsically disordered proteins, protein interactions
Graphical Abstract
Introduction
The replication and transmission of a virus is dependent on a multitude of interactions between host and viral proteins. In general, proteins that are frequently co-opted by viruses are key to viral growth or host defense. One such protein is the highly conserved hub LC8, which binds to multiple viral partners. The rabies virus phosphoprotein (1), the Ebola virus VP35 (2), and the human immunodeficiency virus integrase (3), represent a small percentage of the more than 100 proteins known or predicted to bind LC8 in a wide array of cellular systems (4–6). LC8 binds its partners at a well-characterized and conserved recognition motif (4, 7, 8). Intriguingly, a null mutation in the LC8 binding motif of the rabies virus phosphoprotein (RavP) results in completely non-lethal viral infections in mice, compared to 100% lethality for rabies infections involving wild type RavP (9).
The rabies virus (RAV) is a nonsegmented, negative-sense single-stranded RNA virus of the Rhabdoviridae family and Mononegavirales order, which has afflicted humans since before its first mention in the Codex of Eshnunna in 1930 B.C. (10). Although the disease is preventable with vaccination and treatable with a post-exposure prophylaxis, the virus has a nearly 100% lethality rate once symptoms arise. Impressively, this rate is achieved with only 5 virally encoded genes, including an RNA-dependent RNA polymerase (RavL), a nucleoprotein that encapsulates and protects the viral RNA (RavN), and a non-catalytic phosphoprotein (RavP). RavP contains an LC8 binding motif, and is essential for viral transcription/replication via interactions with RavL and RavN as well as for host immune suppression by inhibiting host Signal Transducer and Activator of Transcription 1 (STAT1) (11–14). These essential roles make RavP an ideal target for elucidating the importance of LC8 interactions with viral proteins.
LC8 is an 89 amino acid dimeric protein (Fig. 1A) that is conserved in eukaryotes as evolutionarily distant as Homo sapiens and Giardia lamblia. Its numerous binding partners include the dynein intermediate chain (15), the transcription factor ASCIZ (16), and the tumor suppressor p53 binding protein (17). LC8 recognizes a conserved 8–10 amino acid motif in intrinsically disordered proteins (IDPs) and facilitates partner dimerization by forming a tetrameric complex with two IDP chains (4). While LC8 can accommodate a wide variety of residues, binding sequences often contain a KxTQT motif, where x is a small, hydrophobic amino acid (6, 8, 18, 19). Amino acids within the LC8 binding motif adopt a β-strand conformation, and are integrated into LC8’s central β-sheet (Fig. 1A) (20, 21), while segments neighboring the LC8 site remain largely disordered. Interestingly, whereas previously described LC8 partners are primarily monomeric until bound by LC8, RavP is a dimer in the absence of LC8 with residues 90–133 corresponding to the dimerization domain (22). The atypical oligomerization state of RavP suggests that LC8 is required for some other function than to facilitate dimerization.
Fig. 1. LC8 and RavP colocalize in cells and form a tight complex in vitro.

(A) Representative crystal structure of an LC8 dimer (protomers shown in shades of red). Partners bind along the noted groove and form a β-strand in each groove. (B) Domain organization for RavP showing the dimerization domain (DD, PDB 3L32) and the C-terminal domain (CTD, PDB 1VYI) in orange and green, respectively, as well as the predicted N-terminal helix (NTH, grey). Interacting partners are listed below the domain architecture diagram. The recognition motif for LC8 is underscored in red, and expanded above. Black lines denote predicted disorder (Linkers 1 and 2). (C) Confocal analysis of representative HeLa-LC8-GFP cells with RavP staining. (Top) Cells were infected with RAV-CVS11 at an MOI of 10 for 48 hours. Co-transfection of pTIT-RavN and pTIT-RavL with pTIT-RavP-WT (Middle) or pTIT-RavP-AAA (Bottom) proceeded for 48 hours. P was stained with a rabbit polyclonal antibody followed by incubation with Alexa-568 goat anti-rabbit IgG. White arrows denote the smaller Negri body-like structures seen during transfections, which are magnified in the inset. DAPI was used to stain the nuclei (Blue). (D) ITC thermogram for LC8 interactions with RavP, collected at 25 °C in 50 mM NaCl, 50 mM sodium phosphate, 1 mM NaN3, pH 7.5, and fit to a single-site binding model. This is a tight interaction with an affinity of 82 nM (See also Table S1). (E) Molecular mass of RavP and LC8-RavP complex measured by SEC-MALLS. The lines show the elution profile monitored by refractive index (left axis) for RavP (black) and the RavP-LC8 complex (red). The crosses show the molecular mass (right axis) derived from MALLS and refractometry measurements. The molecular mass calculated for dimeric RavP is 70.7 kDa (black line and crosses) and the heterotetrameric LC8-RavP is 94.8 kDa (red line and crosses).
The LC8 recognition motif is located within a disordered linker connecting the RavP dimerization domain (DD) with the C-terminal domain (CTD), and contains the canonical “TQT” binding motif (Fig. 1B). Viral transcription and replication require that RavP imposes the correct spatiotemporal arrangement on RavN and RavL at its N-terminus, and RavNRNA at its C-terminus (23). Therefore, elucidating the structure of RavP and how it is modulated by LC8 is key to understanding the viral replication complex.
In vivo, inhibiting LC8 binding to RavP does not stop viral infection and replication, but rather limits it to levels that do not kill the host organism (9). Here we show, via a minigenome luciferase assay, that a loss of LC8 interaction leads to a 70% decrease in viral polymerase activity. While it is clear that LC8 interactions with RavP are essential for rabies virus virulence, the molecular basis for why this interaction leads to increased lethality is unknown. To help address this question, we report the ensemble structure of the 90 kDa RavP-LC8 complex using nuclear magnetic resonance (NMR), small angle X-ray scattering (SAXS), and molecular dynamics simulations (MDS). We show that RavP is a dynamic duplex, with two chains held together by a central dimerization domain that is linked to a folded C-terminal domain by a long disordered linker. LC8 binds tightly to this linker, effectively lengthening the dimerization domain and shifting the population of the ensemble towards structures with more restricted mobility of both ordered and disordered domains distant from the LC8 recognition site. We propose that this change in the orientation and domain mobility rather than a dramatic change in the 3D structure could explain the functional consequences of viral incorporation of LC8, and that this conformational selection mechanism is shared among viral phosphoproteins that bind LC8, such as the Ebola virus VP35.
Results
LC8 and RavP colocalize in cells and form a tight complex in vitro
The LC8 binding site on RavP was identified by deletion experiments (9) and homology modeling (24) to correspond to amino acid positions 140–149, a region that contains a TQT motif; however, no experimental characterizations of this interaction in cells or in vitro was previously reported. Here, we demonstrate the co-localization of LC8 and RavP in both RAV infected and RavP transfected cells. In infected cells, RAV forms large ribonucleoparticle inclusions known as Negri bodies (NBs), which contain RavL, RavN, and RavP, as well as a variety of host proteins (25). NBs are sites for viral transcription and replication, and similar inclusions can be formed from minimal transfections with RavN and RavP (25).
Using confocal microscopy, we show that LC8 is localized inside NBs in infected cells (Fig. 1C, Top). Additionally, transfection using only RavP, RavN, and RavL led to puncta typical of smaller NB-like structures that exhibited colocalization between RavP and LC8 (Fig. 1C, Middle). To verify that the colocalization represented a direct interaction, we made a triple mutant of RavP where the TQT motif was replaced with three alanines (RavP-AAA). Although transfections with RavP-AAA still led to NB-like inclusion formation, LC8 was no longer localized to these regions, confirming that the TQT motif is the only site of interaction with LC8 (Fig. 1C, Bottom). Additionally, this suggests that although RavP localizes LC8 to NBs, LC8 is not essential for NB formation.
ITC and multi-angle laser light scattering (MALLS) show that LC8 binds RavP with an apparent 1:1 ratio (Fig. 1D), and forms a complex with a MW of approximately 95 kDa (Fig. 1E), indicating that the dimeric LC8 (~24 kDa) binds tightly to a RavP dimer (~66 kDa). Interestingly, the interaction between RavP and LC8 is among the tightest yet described for a naturally occurring LC8 partner, with a Kd of 82 ± 8 nM (Table S1)(6, 8). Unlike most LC8 binding partners, RavP forms a stable dimer in solution, and therefore the entropic cost of bridging two chains is already paid. The tight binding of LC8 to RavP is likely due to the lower entropic cost of binding compared to other, primarily monomeric partners.
RavP construct design and domain structure
The high percentage of disorder in RavP makes it not amenable to study via crystallography, leaving NMR as the only viable method for residue specific structural information. NMR spectra of the 90 kDa LC8/RavP complex, however, show considerably broadened peaks that are difficult to assign using standard NMR techniques. Additionally, the large number of residues (297 amino acids per monomer) leads to significant peak overlap. Therefore, to simplify the resonance assignment process, we created constructs containing either the N-terminal half of the protein (RavP-N, residues 1–152), or the C-terminal half (RavP-C, residues 140–297; Fig. 1B, 2A). Both constructs include the LC8 binding site (residues 140–149). RavP-N contains the DD and the RavL and RavN interaction sites (12), and RavP-C contains the STAT1 and RavNRNA interaction sites (26, 27) (Fig. 1B,2A).
Fig. 2. Structural analysis and assignments of RavP constructs.

(A) Domain organization for RavP-N (1–152) and RavP-C(140–297) constructs. 15N,1H-HSQC spectra for RavP-N (B) and RavP-C (C) showing assignments. The spectra were acquired at 25 °C on a 700 MHz Bruker NMR spectrometer. (D) Secondary chemical shift values determined from Cα and Cβ shifts, showing two long disordered linkers and a well-ordered CTD, consisting of α-helices and β-sheets. The structural organization shown above was obtained from crystal structures of the CTD (PDB 1VYI) and dimerization domain (PDB 3L32), where cylinders represent α-helices; arrows represent β-strands, and dashed lines represent disordered regions.
Sequence analysis indicates that amino acids 135–155 are part of the disordered Linker 2, and previous work has demonstrated that the DD and CTD are both independently folding domains (28), justifying the design of smaller constructs that begin or end in the Linker 2 region. The CD spectra of both constructs corroborate the predicted structure via the characteristic negative ellipticity around 205 and 222 nm, indicative of both disordered and helical regions. The summation of the CD spectra for the individual RavP-N and RavP-C constructs yields a spectrum that matches one recorded for full-length RavP, indicating that these constructs collectively contain the same amount of secondary structure as full-length RavP (Fig. S1). RavP-N and RavP-C have similar number of residues (155 versus 158), but size exclusion chromatograms show that RavP-N migrates closer to RavP-FL than to RavP-C. This demonstrates that RavP-N, which contains the DD and the long intrinsically disordered N-terminal region, forms a dimer with highly flexible linkers in solution, whereas RavP-C is a monomer with a shorter flexible tail (Fig. S1).
As with RavP-FL, we used ITC to determine binding affinities for LC8 with RavP-N (Fig. S1), and RavP-C (Fig. S1). RavP-FL and RavP-N, which are dimers, have a much higher affinity for LC8 than the monomeric RavP-C (82 nM, 30 nM, and 4190 nM respectively; Table 1). The higher affinity for the dimeric constructs demonstrates the contribution of bivalency to affinity, as described previously for LC8 binding partners (29, 30), and shown here by the significant contribution of entropy to the difference in affinities (Table S1).
Resonance assignments of the 66 kDa RavP dimer
Backbone assignments of all observable peaks of RavP-N reveal that peaks corresponding to the helical dimerization domain and the eight amino acids at the N-terminus are not visible (Fig. 2B). Extreme peak broadening leading to complete loss of intensity has also been observed for the dimerization domain of a closely related vesicular stomatitis virus phosphoprotein (VSVP) (23). RavP-C spectra, on the other hand, show wide proton chemical shift dispersion, with a large number of peaks in the 8–8.5 ppm region (Fig. 2C). Secondary chemical shifts for RavP (Fig. 2D; Fig. S2) confirm the structure of the folded CTD, and identify boundaries for two long disordered linkers (Linker 1, residues 9–86; Linker 2, residues 138–190). Interestingly, there is no evidence for the N-terminal helix, predicted for positions 8–31 by PSIPRED (31), in contrast to what is seen in VSVP, in which residues 2–16/25–31 show a helical propensity (32). The difference between RavP and VSVP is accurately recognized by the program Agadir (33), which predicts significantly lower helical propensities for the N-terminus of RavP than VSVP (data not shown). By overlaying the spectra for RavP-FL with those of the constructs, we assigned 191 of 282 non-Proline residues (Fig. 3A,B). The missing assignments are for residues that have considerably broadened peaks: those within the dimerization domain (residues 87–137), near the C-terminus (residues 287–293) and at the N-terminus (residues 1–8). Assignments were verified by HNCO spectra of the full-length protein. It is of note that peaks corresponding to a helix between residues 247 and 253 either shift or disappear in RavP-FL compared to RavP-C, suggesting some structural rearrangement of these residues in RavP-FL compared to the monomeric CTD (Fig. 3A–C). RavP-FL peaks that do not overlay with the RavP-N peaks were not observed in 3D spectra, and were therefore not assigned. These peaks presumably represent residues in the DD or the first eight amino acids.
Fig. 3. RavP NMR relaxation experiments.

(A) 1H-15N TROSY-HSQC spectral overlay of RavP-FL (black) with RavP-C (green) and RavP-N (orange) demonstrating the strong agreement in chemical shifts between the constructs and the full-length protein. Peaks that shift between RavP-FL and RavP-C spectra are labelled in black, and peaks that disappear or decrease in intensity are labeled in grey. Normalized intensities for all three proteins are shown with the domain architecture in (B). (C) Crystal structure for the RavP CTD (PDB 1VYI) highlighting residues with peaks that shift in HSQCs for RavP-C and RavP-FL (green). (D) BEST 1H-15N TROSY-HSQC for free (black) and LC8 bound (red) RavP-FL collected at 800 MHz. Example peaks showing large attenuations in peak intensity are labelled. (E, Top) Ratios of Bound:Free peak intensities, generated using the spectra shown in (D). Error bars were calculated using the intensities of the baseline noise. Peaks that disappear in the bound sample in (D) are marked by an *, and are localized to the LC8 site. Relaxation times (T1, and T2) for free (black) and bound (red) are shown overlaid on the same axis, as are J0 values from the spectral density analysis. Residues with fit errors greater than 25% of their value are not shown. HSQC-based 1H-15N NOEs (Isat/Iunsat) demonstrate the difference between linker and CTD residues. The CLEANEX intensities (E, Bottom) for free and bound RavP show regional differences, and were compared as a CLEANEX ratio (bound:free) in the graph above. Note that LC8 interactions lead to decreased CLEANEX intensities from residues 55–70, and across the majority of Linker 2. The segments most changed by LC8-binding are highlighted in grey.
Dynamics measurements and structural heterogeneity in free and LC8-bound RavP
Dynamics measurements for RavP-FL were determined using T1, T2, steady-state het-NOE, peak intensity, and amide proton H/H exchange CLEANEX experiments (Fig. 3E, black). Due to peak overlap, heteronuclear NOE and CLEANEX experiments were collected using an HNCO-based pulse sequence (Fig. S3) in addition to the more sensitive HSQC-based. T1 values do not show any variation within the linkers or CTD (average of 0.7 s for the linkers, and approximately 1.0 s for the CTD). The longer relaxation time for the structured region is expected for a large protein at 800 MHz field strength (34). T2 on the other hand, shows heterogeneity in the linkers, suggesting multiple conformations (Fig. 3E; Fig. S4). As expected, T2 values decrease for residues closer to the structured regions, and become too low for reliable fitting for residues in the ordered CTD. Het-NOE experiments corroborate the disorder in the linker regions, with values near 0.2–0.3. Despite relatively large error bars, HSQC-based het-NOEs for the ordered CTD have values distinctly higher than those of the linkers, closer to 1, as expected for folded proteins (Fig. 3E). RavP-FL normalized intensities (Fig. 3B) for CTD peaks show greatly diminished intensities compared to the linkers. T1, T2, and Het-NOE data are further analyzed using spectral density mapping (35). Variabilities in J0, support our conclusion that the linkers in RavP are likely in exchange between multiple conformations on an intermediate timescale (Fig. 3E; Fig. S4). For the CLEANEX experiment, which measures fast amide proton exchange with water, peaks are observed only for amide protons that are readily exchangeable (36); therefore, higher peak intensities identify residues that are most solvent exposed. Linker 1 residues are all visible in the spectrum, but residues 55–70 show lower intensity peaks. In the CTD, peaks for residue 221 and the C-terminal residue 297 are both visible. Residue 221 is part of the longest turn in the structure, and residue 297 is disordered. Linker 2 peaks vary in intensity; residues 160–180 are either absent or much less intense, indicating some order. In summary, for free RavP, variations in CLEANEX intensities, peak intensities, T2 relaxation times, and J0 values (Fig. 3) indicate that the linkers are largely disordered, but have significant structural heterogeneity on the microsecond to millisecond timescale. As a final measure of structural propensities within the linkers, we collected 1H-15N RDCs, which can provide long-ranged information on relative orientations of backbone bond vectors within the protein. While the CTD peaks are not visible due to the substantially decreased peak intensities, 1DNH values for the linkers cluster around 0, confirming that the disordered linkers are sampling many conformations with no significant secondary structure (Fig. S4).
A comparison of HSQC peak intensities between free and LC8-bound RavP-FL reveals peak attenuation for all residues within the LC8 binding motif (residues 140–149, Fig. 3), similar to other LC8 binding partners (7, 37). Indeed, the only peaks that disappear in the HSQC are those within the LC8 binding region, and there are no peaks that shift (Fig. 3D,E). Carbonyl chemical shifts reiterate these findings, and additionally show some peak disappearance between residues 160–170. Further, RDC experiments indicate that 1DNH values for the linkers are unchanged by LC8 interactions (Fig. S4). To test the effect of LC8 on RavP-FL dynamics, we collected the same experiments as above on the LC8-RavP complex (Fig. 3). Spectral density analyses suggest that the N-terminus of Linker 1 is somewhat more restricted and undergoing slower exchange in the complex, evidenced by the small increase in J0 in this region (Fig. 3E). Additionally, CLEANEX intensity ratios for residues 55–70, 140–180, and 221 in the CTD are significantly less than 1. These long-range effects suggest a decrease in flexibility, without a change in secondary structure, as shown by similar carbonyl chemical shifts as well as similar CD spectra (Fig. S3). Taken together, these data demonstrate that LC8 binding does not change the secondary structure of RavP (aside from the binding site which is too small to contribute to a change in the CD signal), but instead may impact domain orientations and flexibility within the conformational ensemble.
Ensemble structure from SAXS and molecular dynamics simulations
To assess the effect of LC8 on RavP domain orientations, we performed SAXS experiments on RavP and the RavP-LC8 complex (Fig. 4A). The SAXS-determined molecular weights of 69 kDa and 91 kDa for free and bound RavP are consistent with the theoretical MW for a dimer (68 kDa with 6xHis tag) and a heterotetramer (92 kDa) respectively (Table S2). The radius of gyration (Rg) calculated using the Guinier approximation for RavP-free was 4.9 ± 0.1 nm, and 5.0 ± 0.1 nm for the RavP-LC8 complex (Fig. 4B). Interestingly, although the Rg values are approximately the same for bound and free, the Dmax is significantly smaller for the complex (210 Å for RavP and 173.5 Å for the complex). This indicates that although the structures occupy a similar volume on average, RavP-free can inhabit more extended conformations (Fig. 4C). The Kratky plots for both free and bound RavP are typical of a multi-domain protein with flexible linkers (Fig. 4D), presenting a maximum at low q values, followed by a gradual decrease as q increases.
Fig. 4. SAXS data and selected ensembles analysis.

(A) Fitted SAXS profiles for RavP (bottom) and LC8-RavP (top). The experimental curve is shown as a black line and the EOM selected ensemble as a red line. (B) Guinier plot for RavP (in black) and LC8-RavP complex (in red). (C) Pair distribution function for RavP (in black) and LC8-RavP complex (in red). (D) Kratky plot for RavP (in black) and LC8-RavP complex (in red). (E) Radius of gyration distributions for the initial pool (grey) and EOM-selected RavP ensembles (black histogram). RavP models from the EOM are shown as cartoon with their central dimerization domains colored in orange and their C-terminal domains in green. The intrinsically disordered regions are shown in grey. The relative contribution of each model to the EOM calculated SAXS profile is indicated as a percentage. The RavP selected ensemble is overlaid and aligned on the DD in (F). (G) Radius of gyration distributions for the initial pool (grey) and EOM-selected LC8-RavP ensembles (red histogram). Models are shown with LC8 in red, and overlaid in (H). (I) PRE intensity ratios of the paramagnetic (oxidized) to diamagnetic (reduced) of MTSL labelled Rav-C261S free (black) or RavP-C261-LC8 complex (red). Values are only shown for residues 1–196, as the CTD peaks were not visible in this experiment. (J, Top) Ensemblator analysis of models generated via SBM and MDS for RavP (black) and LC8-RavP (red). Models for each sample were compared in one group, with automatic clustering turned off. Residues with a higher LODR in the complex represent regions that are less restricted in motion when LC8 is bound to RavP. Lower LODR regions, on the other hand, are those that are more rigidified within the complex ensemble. A comparison of the differences is shown below. Note the large decrease in variability for residues in the LC8 binding site (residues 140–149). Differences in the CTD, particularly in the loop containing residue 221, are distal from the LC8 site and therefore unexpected. (K) VSVP ensemble overlay of 5 structures, adapted from Leyrat et al., 2012 (23), with conformers aligned using the DD.
A mixture of structure-based models (SBM) and molecular dynamics simulations (MDS) was used to create an ensemble to fit the SAXS profile. In classical MDS, the LC8-RavP interaction was stable over the course of the simulation (data not shown). With the GAJOE program, the number of conformers was decreased until a minimized weighted ensemble of five conformers was obtained to fit the SAXS data (Fig. 4E–H). The ensemble predicted SAXS scattering profile fit well to the data, with χEOM values of 0.64 for the RavP-free ensemble, and 0.49 for the complex. The Rg range for RavP-free and bound is very similar in the selected ensemble, and both can access an extended (Rg range 5–6.8 nm) and a compact (Rg range 3.5–4.2 nm) conformation. Conformers with an intermediate Rg are underrepresented in the selected ensembles, indicating that there is a thermodynamic bias for the extended and compact conformations. Interestingly, the gap between these two broadly defined conformations is much more dramatic for the complex, signifying that some intermediate conformations are prohibited in the presence of LC8. It is of note that the selected ensemble for both samples has a strong enrichment for the compact structure when compared to the initial pool, demonstrating the importance of the compact structure within the ensemble.
Weights for selected structures are shown by a percentage next to each structure. A comparison of these weighted structures reveals that the CTDs of unbound RavP are free to rotate around the DD, while the CTDs within the complex are kept in closer proximity to one another (Fig. 4E–H). Additionally, CTDs in the complex have a relatively fixed orientation with respect to the DD, and the distance between the CTDs is the main distinction between the compact (Rg 3.8–4.1 nm) and extended (5.1–6.8 nm) conformations in the complex. The Rg gap between 4.1 and 5.1 nm may therefore represent the relative benefits of a slight attraction between the CTDs versus an increased freedom of motion. Another interesting point is that Linker 1 is often excluded from the CTD region in the bound complex, most likely because LC8 creates a situation where there is no space for this linker.
In order to test this model, where LC8 interactions lead to increased proximity of the CTDs to each other, and decreased proximity of the CTDs to Linker 1, we performed Paramagnetic Relaxation Enhancement (PRE) NMR experiments by attaching an MTSL spin label to C297. RavP contains two endogenous Cys, C261 on the surface of the CTD, and the C-terminal residue C297 adjacent to the CTD. C261 was mutated to a Serine (C261S) to simplify the PRE analysis. Effects of the mutagenesis were monitored by NMR, which indicated no change in structure based on chemical shifts (data not shown). In paramagnetic samples, CTD peaks were not visible due to the innate broadness of the peaks, as well as their proximity to the MTSL attached to this domain. Instead, we monitored the impact of labelling on the easily observed peaks in linker regions (Fig. 4I, Fig. S4). Supporting the MDS-generated models, linker intensities are significantly lower in the free RavP sample, particularly at the N-terminus of Linker 1, indicating that LC8-binding results in the exclusion of Linker 1 from the binding region (Fig. 4I). It is not possible to verify from our PRE data whether the CTDs are held in closer proximity in the complex, as those peaks were not visible in either spectrum.
To compare structures within the ensembles, we used the Ensemblator program (38) which has a built-in Locally Overlaid Dipole Residual (LODR) tool to evaluate residue-level structural heterogeneity by comparing individually overlaid dipeptides (Fig. 4J). In general, peptides with perfect alignment will have an RMSD of 0, and peptides that show an approximately 180° rotation in the ensemble will have an RMSD of ~5 Å. For RavP, the Ensemblator recognizes the linkers as highly variable, and the domains as mostly static structures (Fig. 4J). Although these models are independent from the NMR data, with the exception that the linkers were modeled as disordered segments due to the near-zero ΔCα-ΔCβ and 1DNH values (Fig. 2D, Fig. S4), they recapitulate multiple findings from NMR. As expected, the difference curve between RavP-free and the complex (Fig. 4J) reveals a common structure for the LC8 binding site (Residues 140–149), consistent with the formation of a β-strand. Additionally, the sole residue visible in the CTD in the ClEANEX spectrum (Fig. 3E, G221) is pinpointed in the models as the most variable portion of the CTD (Fig. 4J).
Conservation of modular structure and LC8 binding
Phosphoprotein (P) expression and function is conserved across viruses in the Rhabdoviridae family (28). Previous work comparing Ps in the Mononegavirales order, which includes the Rhabdoviridae family, has demonstrated that, although there is little sequence conservation across families, Ps are consistently organized in a modular manner, with disordered linkers connecting independently folding domains (28). In order to test if LC8 binding C-terminal of the DD is also conserved, we scanned phosphoproteins from different genera in the Rhabdoviridae family, as well as different Lyssaviruses (the rabies genus), for potential binding motifs. The LC8 motif in Lyssavirus phosphoproteins is stringently conserved (Fig. 5A,B); however, no phosphoproteins from other Rhabdoviridae genera contain the motif.
Fig. 5. Phylogenetic and functional analysis of the Rhabdoviridae family and Lyssavirus genus Phosphoprotein.

(A) ConSurf analysis of P proteins within the Lyssavirus genus, with conserved residues shown in blue, and variable residues shown in red, mapped onto a EOM-generated RavP protomer. The LC8 binding sequence is highlighted, with example sequences from viruses within the Lyssavirus genus shown in (B). Residues in bold are amino acids frequently present in LC8 binding partners. *Denotes sequences verified to bind LC8 (1). Blue ovals denote approximate binding sites of known partners (see Discussion). (C) Luciferase reporter assays for IFNα-treated Hek-293T cells (2,000 U/mL, 6 hours). Cells were transfected with luciferase assay components, as well as a plasmid encoding RavP-WT, RavP-AAA, or an empty vector control. (D) Luciferase expression from the RAV minigenome system. BSR-T7 cells were transfected with either RavP constructs shown, as well as RavN, RavL or RavL-AAA, and luciferase reporter assay components. All luciferase values are normalized to the RavP-WT transfection, with error bars shown as the standard deviation from the mean for two independent experiments, run in triplicate. A negative control is shown ((−) P), in which the RavP plasmid was not transfected. (E) Predicted domain architectures for RavP, VSVP, Ebola virus VP35, and Marburg virus VP35. LC8 sites are marked by red ovals.
Mapping the conserved regions of the RavP sequence within the Lyssavirus genus on one chain of the RavP dimer (Fig. 5A) shows that the DD and CTD are both relatively conserved (blue), while the linkers are generally more variable (red and blue). Interestingly, within Linker 2, only the LC8 binding sequence is conserved, indicating that there is evolutionary pressure selecting for this binding sequence. Additionally, the highly conserved N-terminal region of Linker 1 contains binding sites for L and N, which are both essential for viral proliferation (12).
It is surprising that Lyssaviruses are the only genus within the Rhabdoviridae family to appropriate LC8, especially when the majority of genera display a similar domain architecture (Fig. S5), consisting of two disordered linkers (humps in prediction values) between ordered domains. This suggests that LC8 interactions are not required for stabilization of the RavP dimeric structure, but have instead evolved as an additional switch to regulate downstream functions for RavP. Future work on the pathway(s) affected by LC8 binding can illuminate not only the importance of LC8 for the whole Lyssavirus genus, but also a key difference between viruses within the Rhabdoviridae family.
LC8 is not involved in RAV suppression of the STAT1-mediated immune response
STAT1 is a RavP binding partner that plays an essential role in the innate immune response by regulating the transcriptional response to interferons (IFNs). During a typical innate immune response, activated STAT1 translocates to the nucleus and binds to IFN-activated sequences within target gene promoters, stimulating transcription of these genes. During a RAV infection, on the other hand, RavP binds to cytosolic STAT1, inhibiting STAT1 translocation and activity. Here we use an IFN-meditated luciferase assay to probe the effect of LC8-binding to RavP on the suppression of the host immune system. The exposure of HEK-293T cells to IFNα in control, RavP-WT, or RavP-AAA transfected cells reveals that LC8 interactions do not affect the ability of RavP to suppress STAT1 activity (Fig. 5C). While control cells demonstrated a potent response to IFNα, transfections with either RavP-WT or RavP-AAA containing plasmids significantly suppressed this response, indicating that removal of the LC8 binding motif has no impact on RavP suppression of the STAT1-mediated innate immune response pathway.
LC8 is required for efficient rabies polymerase activity via interactions with RavP, but not RavL
The presence of LC8 within the ribonucleoparticle inclusions (Fig. 1C) suggests it has a critical role in viral polymerase activity. To test this hypothesis, we used a minireplicon system, wherein the viral polymerase components (RavP, RavN, and RavL) are transfected in eukaryotic cells, alongside a luciferase reporter gene framed by the RAV 3’ leader and 5’ trailer sequences (39). Therefore the production of luciferase, measured via a luminescence assay, is directly related to viral polymerase activity. A comparison of transfections using WT RavP or RavP-AAA shows that mutations of the LC8 motif lead to a 70% decrease in luciferase intensity (Fig. 5D), demonstrating that LC8 plays a role in viral transcription/replication.
Previous work has suggested that RavL also interacts with LC8, and that this interaction is required for efficient polymerase activity (40); however, this assertion was based largely on focus forming unit assays, which do not directly measure polymerase activity. To directly test the importance of the LC8 motif in RavP, we mutated the TQT motif in RavL to an AAA (RavL-AAA), and performed a minigenome luciferase assay with RavP-WT, RavN, and RavL-AAA (Fig 5D). These data indicate that the LC8 binding sequence in RavL is not important for polymerase activity, as there is no statistically significant difference between these samples (Fig. 5D). Consistently, mutation of the LC8 site in RavP eliminates LC8 localization to the NB-like inclusions (Fig. 1C), suggesting that RavL is insufficient for LC8 recruitment to the sites of viral transcription.
Discussion
RavP is an essential component of the rabies virus, as it interacts with RavL (12), RavN (11), RavNRNA (11, 14), and a multitude of host proteins (41). These interactions implicate RavP in many diverse roles, including viral transcription, controlled replication, immune evasion (13, 42), and nucleocytoplasmic transport (43), as well as host apoptosis/autophagy regulation (44) and mitochondrial dysfunction (45). Prior research demonstrates that interactions between RavP and LC8 are imperative for viral lethality (9). Cell-based experiments suggest roles for LC8 in primary transcription (9) and intracellular localization (43); however molecular level descriptions of the role of LC8 in viral lethality have been hampered by the structural complexity of the multi-domain, partially disordered RavP.
Here we show by CD, SAXS, MALLS, and NMR, that RavP is an IDP duplex composed of two structured domains and two long disordered linkers. Linker 1 (residues 1–86) and Linker 2 (residues 138–191, connecting the central DD to the CTD) are highly dynamic on the nanosecond to picosecond timescale (homogeneous T1 and het-NOEs), but have considerable structural heterogeneity on the millisecond to microsecond time scale, demonstrated by variations in the amide proton solvent exchange, transverse relaxation rates, and J0 values (Fig. 4E). While the structure of the CTD in full length RavP matches crystallographic studies on the individual domain, the SAXS selected ensemble of the full length RavP emphasizes the motional freedom available to the CTDs: either proximal (CTDs are essentially adjacent), or distal (CTDs are on opposite sides of the dimerization domain). These multiple orientations contrast with the homologous VSVP, for which the CTD conformations are primarily proximal (23), while the N-terminal Linker 1 samples a wide range of conformations. Regulation of these CTD orientations by LC8 could explain the importance of LC8 in rabies virulence as discussed below.
LC8 restricts dynamics of disordered linkers and the ordered CTD
Using a wide variety of dynamics experiments that probe for small differences between bound and free RavP, we demonstrate that LC8 binding to RavP does not change RavP secondary structure, but rather alters the dynamics of Linkers 1 and 2, and rigidifies portions of the CTD. The decrease in peak intensity within the LC8 binding site (residues 140–149) is typical for LC8 binding proteins, presumably due to formation of the interfacing β-strand; however, long-range effects on Linker 1, Linker 2 residues 150–180, and CTD residue 221, indicate that LC8 alters dynamics outside of the binding site. The linkers in RavP are the sites for interaction with a wide variety of proteins (Fig. 5A) including the ribosomal protein L9 (41), mitochondrial complex I (45), IRF-3 (42), STAT1 (13), RavNRNA (14), PML (46), and nuclear import and export factors (43). Additionally, a number of proteins bind RavP at unknown sites (e.g. BECN1 (44) and HSP90/Cdc37 (47)), and may also depend on the accessibility of these linkers. There is indeed evidence that LC8 binding causes changes in the nuclear/cytoplasmic localization of RavP (43), suggesting changes in accessibility of nuclear import and export sequences within these linkers. We have tested the impact of LC8 binding on two downstream interactions: the innate immune response protein STAT1, and the viral polymerase components. While LC8 does not affect RavP-STAT1 interactions, it enhances viral polymerase activity. Further studies will determine whether RavP interactions with LC8 affect other downstream partners.
LC8 elongates the RavP dimerization domain, facilitating CTD orientations conducive to transcription
Molecular dynamics simulations based on SAXS data, and supported by PRE experiments, reveal that the CTDs are more constricted upon LC8 binding, resulting in a more compact structural ensemble. A comparison of RavP to VSVP, another member of the Rhabdoviridae family that shares a common domain architecture (28), suggests an intriguing new role for LC8 in the restriction of the conformational ensemble, and potentially in the regulation of downstream interactions common to both. In the VSVP ensemble the CTD is more restricted with respect to the DD (23) (Fig. 4K) favoring orientations where both CTDs of the dimer are constrained on one side of the DD, similar to the LC8-bound RavP ensemble (Fig. 4G,H), and different from the free RavP ensemble (Fig. 4E,F). Contributing to this difference is the longer dimerization domain in VSVP, and the shorter Linker 2. Interestingly, LC8-binding in RavP would result in a dimerization domain that is lengthened to a similar extent as the VSV-DD, suggesting that the lengthening of the DD restricts CTD domain orientation. Another difference is in the topology of the peptide chains in the DD. In RavP, Linkers 1 and 2 extend from the same side of the DD, resulting in steric hindrance between the N-terminal and C-terminal region. In VSVP the linkers are on opposite faces of the DD. Our PRE data support a model where LC8 binding leads to increased distance between Linker 1 and the CTD of RavP, resulting in linker localizations to opposite faces of the central DD, similar to the native state of VSVP.
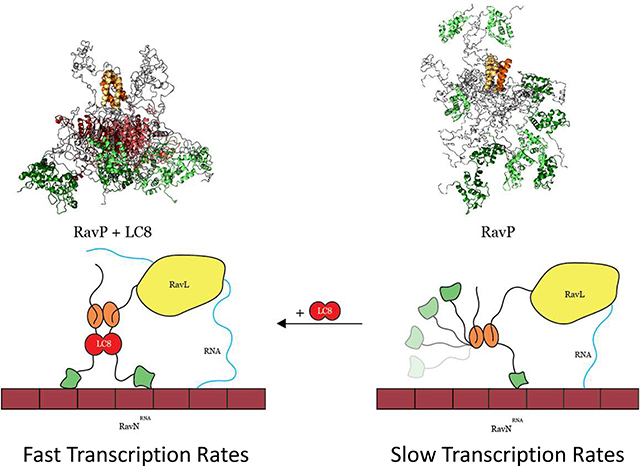
The striking similarity in tertiary orientations and CTD restriction between LC8/RavP and VSVP suggests that LC8 binding facilitates a phosphoprotein function common between VSVP and RavP. One common, essential P function is to connect the viral polymerase, L, to the nucleoprotein-RNA complex, NRNA, during viral transcription and replication. It is proposed that CTD binding NRNA could trigger some conformational change in NRNA, resulting in loosening the grip of N on the RNA to allow the polymerase access to the RNA (Albertini 2006). The polymerase requires the cooperation of P to synthesize long RNA molecules and/or to scan intergenic regions during viral transcription and replication. Two models have been proposed to explain how P assists the polymerase in its motion along the template RNA: (i) In the first model, P remains attached to L and walks along the N-RNA template with its CTD continuously and alternatively associating with and dissociating from N (48). (ii) In the second model, multiple P molecules are positioned through their CTD at regular intervals along the N-RNA template, and the polymerase jumps from one to another (49).
Our data support a model for the replication complex in which both CTDs of a phosphoprotein dimer must be oriented on the same side of the molecule for efficient activity of the replication complex (Fig. 6). In VSV, the architecture of P creates this situation in the free protein, so that VSVP is fully functional in its free form. In RAV, the higher flexibility of Linker 2 and the steric hindrance induced by the presence of the two N-terminal linkers in close proximity of the CTD results in a situation where the CTDs are further apart in the free form of the protein. LC8 binding would then be necessary to bring the CTDs in close proximity and to stimulate the activity of the replication complex. Consistent with this model, our minigenome assay has demonstrated that abrogation of the LC8-RavP interaction leads to a severe attenuation in polymerase activity. We propose that RavP movement along the nucleocapsid is a rate-limiting step in viral transcription and replication, and LC8 facilitates this process by restricting the CTDs to a more ideal “walking” conformation (Fig. 6). However, another possibility is that a proximal orientation of the CTDs could help RavP to release the RNA from adjacent RavN molecules to provide RavL access to the RNA. Future work could distinguish between the possibilities by identifying whether the affinity between RavN and the viral RNA is affected by the presence of RavP free, and bound to LC8.
Fig. 6. Models depicting how LC8 binding facilitates polymerase activity.

In the absence of LC8 (right), the CTDs (green ovals) occupy both distal and proximal conformations, leading to non-ideal movement of the polymerase along the RavNRNA complex (maroon). When RavP is bound to LC8 (left), CTDs are forced into a proximal conformation, facilitating polymerase movement along the RavNRNA complex and leading to increased polymerase efficiency, evinced by the minigenome assay comparison of RavP-WT to RavP-AAA (Fig. 5D).
In support of our dimer elongation hypothesis, it is interesting to note that an analogous pair of related viral proteins, the VP35s of the Ebola and Marburg viruses, show a strikingly similar pattern in their LC8 binding sites. The Ebola and Marburg viruses are members of the Filoviridae family, and are in the same Mononegavirales order as RAV and VSV. The VP35 protein expressed in the Filoviridae family serves analogous functions to P, including suppression of the host immune response and bridging the viral polymerase and nucleoprotein to facilitate viral transcription and replication (50). While the Ebola virus VP35 is known to bind LC8 (2), the Marburg virus version does not contain the TQT motif necessary for LC8 binding (Fig. 5E). Additionally, the LC8-binding site occurs in precisely the region occupied by the Marburg virus’s longer multimerization domain. This pair of examples may suggest that, while RavP and Ebola VP35 can perform the same functions as VSVP and Marburg VP35 when bound to LC8, they have some additional viral functions in their free form. We propose that the lengthening of the dimerization domain, and subsequent restrictions in the CTD, allows RavP to switch between selected conformations within the ensemble, affecting various downstream functions. The high conservation of the LC8 motif within the Lyssavirus genus, implies that LC8 performs an important function not operating in other genera.
Previous work on 53BP1, an essential protein in the double strand break repair pathway, has demonstrated the importance of an LC8 binding site adjacent to a self-association domain (51). While the self-association domain is sufficient for dimerization without LC8, LC8 binding is necessary for efficient localization and double strand break repair. The same concept is demonstrated with the essential Nup159 subunit of the nuclear pore, where multiple LC8 sites adjacent to a dimeric coiled- coil are necessary to augment the dimerization effect, and both the coiled-coil and LC8 are required for optimal function (52). These results parallel our own, where LC8 binding extends the dimerization domain resulting in conformational restriction of the folded CTD domains necessary for replication.
Summary
Structural, dynamics, thermodynamics, and minigenome data presented here suggest that LC8 binding results in a structural ensemble of RavP-LC8 that is highly biased towards conformations where the CTDs are oriented on the same side of the protein, as in VSVP, for efficient transcription and replication. This model lays the groundwork for future innovative investigations into LC8-linked, viral infection cycles.
Materials and Methods
Bacterial expression constructs and protein production
RavP constructs containing the N-terminal region (residues 1–152, RavP-N), the C-terminal region (residues 140–297, RavP-C) and full-length RavP (residues 1–297, RavP-FL) were generated using PCR, and then cloned into a modified pET24d expression vector with an N-terminal His6 tag followed by a tobacco etch virus (TEV) protease cut site. LC8 from Drosophila melangoster was also sub-cloned into a pET24d expression vector with N-terminal His6 tag and TEV cutting site. RavP-FL constructs with the TQT residues mutated to AAA (RavP-AAA), or the Cys at residue 261 mutated to a Ser (RavP-C261S), were generated using a QuikChange Lightning mutagenesis kit (Agilent, Santa Clara, California). DNA sequences were verified by automated sequencing. The recombinant vectors were transformed into BL21 Rosetta (DE3) cell lines for protein expression. Bacterial culturing is common in the Barbar lab, and was performed as described in (37).
Paramagnetic labelling
Purified RavP-C261S was labelled with MTSL (Toronto Research Chemicals, Toronto, Canada) at the sole remaining Cysteine, C297, as described in (53). Briefly, SEC-purified, 100 μM RavP-C261S was incubated with 10 mM DTT for 2 hours at 25 °C to ensure that C297 was reduced. The sample was then buffer exchanged using a HiTrap Desalting column (GE Healthcare) into an MTSL labelling buffer composed of 50 mM Tris, 100 mM sodium chloride, 1 mM sodium azide, pH 7.5. A stock of 50 mM MTSL in ethanol was added to RavP-C261S at a 15X molar excess (1.5 mM MTSL in 100 μM RavP-C261S), and labelling proceeded in the dark at 4 °C for 16 hours. Labeled samples were then buffer exchanged into NMR buffer (50 mM sodium phosphate, 100 mM sodium chloride, 1 mM sodium azide, 5 mM ethylenediaminetetraacetic acid, pH 6.5) to remove any excess MTSL label, and concentrated to 150 μM RavP-C261S. LC8-bound samples were made by adding 1.8 mM LC8 in NMR buffer directly to pre-labelled RavP-C261S at a molar ratio of 1.2:1 LC8:RavP-C261S. After paramagnetic NMR spectra were collected, samples were reduced by the addition of 2 mM Ascorbate (100 mM stock in NMR buffer), followed by a 2 hour incubation period to ensure complete reduction before diamagnetic spectra were collected.
Eukaryotic cell culture and microscopy
HeLa Kyoto cells stably expressing LC8-GFP (54) were a kind gift from I. Poser and A. Hyman (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany). The cells were cultured in «DMEM medium with 10% (v/v) fetal calf serum and with 1% (v/v) penicillin/streptomycin.
pTIT plasmids (55) encoding RavL from RAV strain SAD-L16 (0.5 μg), RavN from CVS-11 (1.5 μg), and RavP-WT/RavP-AAA from CVS-11 (1.0 μg) were transfected into HeLa-LC8-GFP cells (2*106 cells) using Lipofectamine 2000 (Invitrogen, Carlsbad, California). These plasmids were cotransfected with 0.5 μg of a plasmid encoding the T7 RNA polymerase, as previously described (25).
The challenge virus standard (CVS, French CVS 1151) strain of rabies virus was grown in N2A cells. HeLa-LC8-GFP cells (2*106 cells) were infected with CVS at an MOI of 10, and collected for fluorescent imaging at 48 hours post-infection.
We used the minigenome system as previously described (39), with slight modifications. BSR cells stably expressing the T7 polymerase (BSR-T7) were transfected with pRL-TK (0.1 μg), pDI-Luc (0.8 μg), and pTIT plasmids encoding RavL or RavL-AAA (0.33 μg), RavN (1 μg), and RavP-WT/RavP-AAA(0.5 μg). Forty-eight hours after transfection, firefly and Renilla luciferase activities were measured using a dual-luciferase assay kit as described by the manufacturer (Promega, Madison, Wisconsin). The Renilla luciferase gene is constitutively expressed at low levels, and provides an internal control to account for differences in cell expression rates and concentrations. Therefore, our luminescence readings are a ratio of Firefly/Renilla luciferase activity. Experiments were performed in triplicate on two biological replicates.
In IFNα-dependent signaling assays, HEK-293T cells were transfected with pRL-TK (0.75 μg), pISRE-Luc (2.5 μg), and pCDNA3.1 encoding RavP-WT, RavP-AAA, or an empty vector control (2.5 μg) (56). Twenty-four hours after transfection, activated cells were treated with 2,000 U/mL of human recombinant IFNα for six hours at 37 °C, while non-activated controls were not. Cells were then harvested, and firefly and Renilla luciferase activities were measured using a dual-luciferase kit (Promega). Values displayed in Figure 5C are a normalized ratio of activated to non-activated cells, and are the result of two biological replicates performed in triplicate.
Cells were fixed for 10 min with 4% paraformaldehyde (PFA) and permeabilized for 10 min with 0.1% TritonX-100 in PBS. Cells were incubated for 1 hour at RT with the rabbit polyclonal anti-RavP antibody (Dilution of 1/1000) previously described (57), then washed and incubated for 1 hour with Alexa fluor conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, Massachusetts). Following washing, cells were mounted with ImmunoMount (Thermo Fisher Scientific) containing DAPI. Images were captured using a Leica SP8 confocal microscope.
Isothermal titration calorimetry
ITC experiments for the interactions of LC8 with RavP-N, RavP-C, and RavP-FL were performed using a Microcal (North Hampton, MA) VP-ITC microcalorimeter at 25 °C in buffer composed of 50 mM sodium phosphate, 50 mM sodium chloride, 1 mM sodium azide, 5 mM β-mercaptoethanol, pH 7.5. In all experiments, an initial 2 μl injection was followed by 26 injections of 10 μl LC8 (300 μM) into 25 μM RavP constructs in the sample cell. Protein samples and buffer were degassed before data collection. The data were processed using Origin 7.0 (Malvern Panalytical, Malvern, UK) and fit to a single-site binding model. The recorded data are the averages of three independent experiments, with uncertainty reported as the standard deviation from the mean or the error in the fit, whichever was greater.
Circular dichroism
Spectra were recorded on a JASCO 720 spectropolarimeter using a 1 mm cell at a concentration of 10 μM in 20 mM sodium phosphate, 1 mM sodium azide, pH 7.5, at 25 °C. Spectra for samples of LC8-bound proteins were collected using a 1:1 molar concentration of binding partners. Difference spectra were obtained by subtracting the 10 μM LC8 spectrum from the spectra of the bound form.
SEC-MALLS
Size-exclusion chromatography (SEC) combined with multi-angle laser light scattering (MALLS) and refractometry was performed at 20 °C with a flow rate of 0.5 mL min−1 on a Superdex 200 HR 10/300 GL column (GE Healthcare) equilibrated with 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 50 mM arginine, 50 mM glutamate and 0.2 mM TCEP buffer supplemented with a protease inhibitor cocktail (Complete EDTA-free, Roche). MALLS detection was performed with a DAWN-HELEOS II detector (Wyatt Technology, Santa Barbara, California) using a 690 nm laser light source. The protein concentration was measured with an Optilab T-rEX detector (Wyatt Technology) using a refractive-index increment, dn/dc, of 0.185 mL g−1. Weight-averaged molar masses (MW) were calculated with ASTRA (Wyatt Technology) (58).
NMR measurements
NMR measurements were collected at 25 °C using 150 μM 13C/15N labeled RavP-FL, RavP-N, or RavP-C in a buffer containing 50 mM sodium phosphate (pH 6.5), 100 mM sodium chloride, 1 mM sodium azide, 5 mM β-mercaptoethanol, 5 mM ethylenediaminetetraacetic acid, a protease inhibitor mixture (Roche Applied Science), 2,2-dimethylsilapentane-5-sulfonic acid for 1H chemical shifts referencing, and 10% D2O (vol/vol). Experiments for the LC8-bound samples were collected using a 1:1 molar ratio of 150 μM LC8:RavP construct. Band selective excitation short transient (BEST) 1H-15N TROSY-HSQC (59) spectra were compared before and after 3D experiments to verify sample stability. Additionally, samples were checked for degradation using sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Backbone resonance assignments for constructs were determined using triple resonance experiments, including HNcoCACB, HBHANH, HBHAcoNH, HNCO, HNCA, HNcoCA, HNcaNNH, HNcocaNNH (60), and hCCcoNHs (61). These experiments were carried out on a Bruker 700-MHz spectrometer. Backbone resonance assignments for RavP-FL were determined from comparison of HSQC spectra of the constructs with BEST 1H-15N TROSY-HSQC spectra of RavP-FL, and verified using a BEST TROSY-HNCO experiment collected on a Bruker 800-MHz spectrometer.
Longitudinal (T1) and transverse (T2) relaxation times were determined at 800-MHz using standard HSQC-based pulse sequences (62). T1 measurements involved eight time points that ranged from 20 ms to 1200 ms, and were collected as an interleaved experiment. The T2 experiments had nine time points ranging from 16.96 ms to 271.36 ms. All T1 and T2 experiments used a recovery delay of 2 s. Two-dimensional TROSY-based steady-state 1H-15N NOE experiments were collected using a simultaneous recovery delay and saturation period of 6 s. Additionally, a three-dimensional HNCO-based 1H-15N NOE experiment was used to measure heteronuclear NOE values for peaks that overlapped in the two-dimensional experiment.
Phase modulated CLEAN chemical exchange (CLEANEX-PM) experiments were run using Fast-HSQC detection on an 800-MHz Spectrometer at 25 °C, pH 6.5, and with a mixing time of 50 ms (36). HNCO-based CLEANEX spectra were also collected for RavP-FL samples under the same conditions. A recovery delay of 1.7 s was used for the HSQC-based experiment, and 2 s for the HNCO-based CLEANEX (61). All dynamics data were collected at 800-MHz.
Residual dipolar couplings were collected at 800-MHz for linker residues using the amide RDCs by TROSY spectroscopy (ARTSY) method, with a 1H dephasing duration of 10.75 ms (63). This method utilizes a ratio of interleaved reference and attenuated spectra to determine the 1JNH and 1DNH values, rather than traditional splitting experiments, and therefore bypasses some of the issues with extreme peak broadening in the upfield component of the split peaks. Free and LC8-bound RavP samples were run both in standard NMR buffer, as well as in pf1 phage aligned solution (Asla Biotech, Riga, Latvia). A 50 mg/mL stock of Pf1 phage was added into samples to a final concentration of 10 mg/ml, where significant 2H splitting was seen.
PRE data were collected at 800-MHz using a 1H-15N HSQC-TROSY pulse sequence, and data are presented as a ratio of the oxidized to reduced peak intensities. Spectra were compared to unlabeled spectra to verify that the MTSL label was not impacting RavP structure.
NMR data analysis
All spectra were processed with either TopSpin (Bruker BioSpin, Billerica, Massachusetts) or NMRPipe (64), and analyzed using Sparky (65). Cα, Cβ, and C’ chemical shifts were compared to random coil values at the specified temperature and pH to determine the secondary structure (66, 67).
T1 and T2 relaxation experiments were analyzed using Sparky (65). Errors for 1H-15N NOE and peak intensity ratios were calculated from the intensities of the baseline noise. Errors for RDCs were calculated as described in (63). Reduced spectral density mapping was performed on the NMR relaxation data as previously described (35, 68) to yield values for spectral densities J(0), J(ωN), and J(0.87ωH).
Small-angle x-ray scattering
SAXS data were collected at the BioSAXS beamline BM29 (Grenoble) at the European Synchrotron utilizing an in-line analytical SEC S200 column (10/300 GL, GE Healthcare). SAXS measurements were performed with a Pilatus 1M detector at a distance of 2.867 m allowing a q range of 0.03 to 4.94 nm with a wavelength of 0.09919 nm. The scattering from the buffer before and after each sample measurement was used for background subtraction. Data were analyzed using Primus (ATSAS) (69) and ScÅtter (Bioisis) program suites (70). The volume of correlation method was used to determine the molecular mass (71). The radius of gyration (Rg) was calculated using both the Guinier approximation and the pair distribution function. SAXS datasets were scaled and averaged to produce one unique I(q) curve.
Model generation and molecular dynamics simulation
A combined atomistic coarse-grained and classical explicit solvent molecular dynamics simulations approach was used to generate ensembles of conformers suitable for fitting the SAXS data for both the free and bound complex. Models of the LC8-RavP complex were built with the rigid body SAXS modeling software CORAL (69) using the structures of the DD and CTD domains (22, 27), as well as a representative LC8-peptide structure. As CORAL builds the regions for which no high-resolution structure are available as beads, the resulting model was then processed to generate an all-atom model. The all atom model of the LC8-RavP complex was then used as input for molecular dynamics simulations. To generate the starting model for RavP, LC8 was deleted from the model of the complex. Models of RavP and LC8-RavP were then simulated in GROMACS 5 (72) using either an atomistic coarse-grained structure-based model (73) or explicit solvent classical molecular dynamics simulations (MDS). In the case of the structure-based model MDS, a time step of 0.0005 time units was used and the simulation was coupled to a temperature bath via Langevin dynamics. A single 100 ns trajectory was obtained for both RavP and LC8-RavP, and snapshots were extracted every 50 ps to create two ensembles with 2000 models each. In the case of classical MDS, we generated multiple trajectories for an aggregated simulation time of ~ 250 ns for RavP and ~ 400 ns for LC8-RavP. MDS was performed using charmm22* (74) to simulate collapsed states, and the amber03ws and amber99SBws forcefields (75) to reproduce the properties of intrinsically disordered proteins (Fig. S6). At the beginning of each simulation, the protein was immersed in a box of SPC/E water, with a minimum distance of 1.0 nm between protein atoms and the edges of the box. The genion tool was used to add 150 mM NaCl (72). Long range electrostatics were treated with the particle-mesh Ewald summation (76). Bond lengths were constrained using the P-LINCS algorithm. The integration time step was 5 fs. The v-rescale thermostat and the Parrinello–Rahman barostat were used to maintain a temperature of 300 K and a pressure of 1 atm. Each system was energy minimized using 1,000 steps of steepest descent and equilibrated for 500 ps with restrained protein heavy atoms prior to production simulations. Radius of gyration versus time, RMSF and interdomain distances were calculated using GROMACS routines. Snapshots were extracted every 200 ps from each trajectory, leading to the generation of ~ 1250 models of RavP and ~ 2000 models of LC8-RavP.
For each model from the RavP and LC8-RavP ensembles, theoretical SAXS patterns were calculated with CRYSOL (77) and ensemble optimization fitting was performed with GAJOE (78). The optimum selected ensemble size and relative weights of the models were determined automatically by GAJOE. Goodness of fit was demonstrated by the low χ values using the ensemble optimization method (χEOM).
By comparison with other methods for constructing ensembles (e.g. Ensemble (79) or Asteroids (80)), simulations of trajectories can provide an easier way to generate all-atom models of physically accessible conformers for multimeric molecules or multi-molecular complexes. The possible drawback with classical MDS that intrinsically disordered proteins are too compact can be avoided by combining different methods and different force fields, in particular as was done here, by using force fields specifically devised for disordered proteins (75). Clearly, our different trajectories started from an initial conformation that has an Rg value larger or equivalent to the experimental Rg value, and although some led to more compact conformations (those using Charmm force fields), others led to larger conformers (those using SBM or amber force field; Fig S6). Extracting conformers from these different trajectories generates ensembles with great diversity of compactness. Modeling SAXS data with ensembles generated by SBM or MDS simulations has been reported for different systems (e.g. (81–83).
Structure prediction and ensemble visualization
Secondary structure predictions were obtained via PSIPRED v3.3 (31) and Agadir (33). Regions of order and disorder were predicted using DIS0PRED3 (84) and IUPred2A (85). Domain boundaries were predicted using DomPred (31). Ensemble analysis of the models generated from the SAXS profile was carried out using the Ensemblator program (38, 86), which aligns structures based on common regions, and describes regions of variability using a variety of complementary methods. Here we use the locally overlaid dipole residual (LODR) function to get information on residue-level backbone similarity from a pairwise comparison of all models in the ensemble. The LODR analyzes differences in local structure by aligning each dipeptide within the protein and calculating the RMSD for the backbone of the next amino acid within the protein sequence.
Phylogenetics
Phosphoprotein sequences for viruses in the Rhabdoviridae family were obtained from the UniProt server. Alignments and trees were generated with Clustal Omega (87), by comparing representative phosphoproteins for viruses within different genera in the Rhabdoviridae family. Representative viruses were lettuce necrotic yellow virus (Cytorhabdovirus), orchid fleck virus (Dichorhavirus), rabies virus (Lyssavirus), viral hemorrhagic septicemia virus (Novirhabdovirus), perch rhabdovirus (Perhabdovirus), Drosophila melanogaster sigmavirus (sigmavirus), spring viremia of carp virus (Sprivivirus), Tibrogargan virus (Tibrovirus), Durham virus (Tupavirus), vesicular stomatitis Indian virus (Vesiculovirus). Phosphoproteins from three genera that did not show a similar domain architecture were excluded from the analysis (Ephemerovirus, Varicosavirus, and Nucleorhabdovirus). Sequence conservation analysis for phosphoproteins related to RavP was carried out using the ConSurf server (88), with the final conservation coloring based on 48 unique sequences with a maximum percent identity of 85%. Conservation scores were then mapped onto a representative protomer from within the SAXS ensemble.
Supplementary Material
Highlights.
The nearly 100% lethality rate for the rabies virus has been linked to interactions between the rabies virus phosphoprotein (RavP) and the host hub protein LC8.
NMR, SAXS, and MDS were used to build a structural ensemble for both RavP and the RavP-LC8 complex.
Interactions between RavP and LC8 are very tight due to the bivalency effect, and LC8 binding restricts the mobility of RavP’s C-terminal domain.
Cell-based studies show that LC8 interactions are essential for efficient transcription/replication.
Acknowledgements
N.J. was supported in part by the Chateaubriand Fellowship of the Office for Science & Technology of the Embassy of France in the United States. This work was supported by National Institutes of Health Grant GM 084276 to E.B.. The authors also acknowledge Helena Kovacs and Rainer Kuemmerle at Bruker BioSpin for help with NMR data collection, Nathalie Scrima for technical support, Grant Larson for figure development assistance, and Andrew Brereton, Nikolaus Loening, and Patrick Reardon for insightful conversations and data interpretation. Support to facilities includes the Oregon State University NMR Facility funded in part by the National Institutes of Health, HEI Grant 1S10OD018518, and by the M. J. Murdock Charitable Trust grant # 2014162. This work was also supported by the Fondation de la Recherche Médicale (Equipe FRM DEQ20170336754) and used the platforms of the Grenoble Instruct Center (ISBG: UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
Abbreviations
- CD
circular dichroism
- CTD
C-terminal domain
- DD
dimerization domain
- EOM
ensemble optimization method
- IDP
intrinsically disordered protein
- HSQC
heteronuclear single quantum coherence
- IFN
interferon
- ITC
isothermal titration calorimetry
- LODR
locally overlaid dipole residuals
- MALLS
multi-angle laser light scattering
- MDS
molecular dynamics simulations
- NMR
nuclear magnetic resonance
- PRE
paramagnetic relaxation enhancement
- RAV
rabies virus
- RavL
rabies polymerase
- RavN
rabies nucleoprotein
- RavP
rabies phosphoprotein
- RDC
residual dipolar coupling
- SAXS
small angle X-ray scattering
- SBM
structure-based modelling
- SEC
size exclusion chromatography
- STAT1
signal transducer and activator of transcription
- TEV
tobacco etch virus
- VSVP
vesicular stomatitis virus phosphoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raux H, Flamand A, Blondel D (2000) Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74(21): 10212–10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kubota T, et al. (2009) Ebolavirus VP35 Interacts with the Cytoplasmic Dynein Light Chain 8. J Virol 83(13):6952–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayappa KD, et al. (2015) Human immunodeficiency virus type 1 employs the cellular dynein light chain 1 protein for reverse transcription through interaction with its integrase protein. J Virol 89(7): 3497–3 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbar E (2008) Dynein Light Chain LC8 Is a Dimerization Hub Essential in Diverse Protein Networks†. Biochemistry 47(2):503–508. [DOI] [PubMed] [Google Scholar]
- 5.Rapali P, et al. (2011) DYNLL/LC8: a light chain subunit of the dynein motor complex and beyond. FEBS J 278(17):2980–2996. [DOI] [PubMed] [Google Scholar]
- 6.Jespersen N, et al. (2019) Systematic identification of recognition motifs for the hub protein LC8. Life Sci alliance 2(4):e201900366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nyarko A, Barbar E (2011) Light chain-dependent self-association of dynein intermediate chain. J Biol Chem 286(2):1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapali P, et al. (2011) Directed evolution reveals the binding motif preference of the LC8/DYNLL hub protein and predicts large numbers of novel binders in the human proteome. PLoS One 6(4). doi: 10.1371/journal.pone.0018818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan GS, Preuss MAR, Williams JC, Schnell MJ (2007) The dynein light chain 8 binding motif of rabies virus phosphoprotein promotes efficient viral transcription. Proc Natl Acad Sci US A 104(17):7229–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kienzle TE (2007) Rabies (Chelsea House).
- 11.Chenik M, Chebli K, Gaudin Y, Blondel D (1994) In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J Gen Virol 75(11):2889–2896. [DOI] [PubMed] [Google Scholar]
- 12.Chenik M, Schnell M, Conzelmann KK, Blondel D (1998) Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72(3): 1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidy A, Chelbi-Alix M, Blondel D (2005) Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79(22):14411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albertini AAV, et al. (2006) Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313(5785):360–363. [DOI] [PubMed] [Google Scholar]
- 15.Makokha M, Hare M, Li M, Hays T, Barbar E (2002) Interactions of cytoplasmic dynein light chains Tctex-1 and LC8 with the intermediate chain IC74. Biochemistry 41(13):4302–4311. [DOI] [PubMed] [Google Scholar]
- 16.Jurado S, et al. (2012) ATM substrate Chk2-interacting Zn 2+ finger (ASCIZ) is a bi-functional transcriptional activator and feedback sensor in the regulation of dynein light chain (DYNLL1) expression. J Biol Chem 287(5):3156–3 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo KWH, et al. (2005) The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J Biol Chem 280(9):8172–8179. [DOI] [PubMed] [Google Scholar]
- 18.Benison G, Karplus PA, Barbar E (2007) Structure and Dynamics of LC8 Complexes with KXTQT-Motif Peptides: Swallow and Dynein Intermediate Chain Compete for a Common Site. J Mol Biol 371(2): 457–468. [DOI] [PubMed] [Google Scholar]
- 19.Erdős G, et al. (2017) Novel linear motif filtering protocol reveals the role of the LC8 dynein light chain in the Hippo pathway. PLoS Comput Biol 13(12):e1005885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clardy J, Liang J, Jaffrey SR, Guo W, Snyder SH (1999) Structure of the PIN/LC8 dimer with a bound peptide. Nat Struct Biol 6(8):735–740. [DOI] [PubMed] [Google Scholar]
- 21.Clark S, Nyarko A, Lohr F, Karplus PA, Barbar E (2016) The Anchored Flexibility Model in LC8 Motif Recognition: Insights from the Chica Complex. Biochemistry 55(1): 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov I, Crepin T, Jamin M, Ruigrok RWH (2010) Structure of the dimerization domain of the rabies virus phosphoprotein. J Virol 84(7):3707–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leyrat C, et al. (2012) Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J Mol Biol 423(2):182–197. [DOI] [PubMed] [Google Scholar]
- 24.Poisson N, et al. (2001) Molecular basis for the interaction between rabies virus phosphoprotein P and the dynein light chain LC8: Dissociation of dynein-binding properties and transcriptional functionality of P J Gen Virol 82(11):2691–2696. [DOI] [PubMed] [Google Scholar]
- 25.Nikolic J, et al. (2017) Negri bodies are viral factories with properties of liquid organelles. Nat Commun 8(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiltzer L, et al. (2014) Interaction of rabies virus P-protein with STAT proteins is critical to lethal rabies disease. J Infect Dis 209(11): 1744–1753. [DOI] [PubMed] [Google Scholar]
- 27.Mavrakis M, McCarthy AA, Roche S, Blondel D, Ruigrok RWH (2004) Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J Mol Biol 343 (4): 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerard FCA, et al. (2009) Modular Organization of Rabies Virus Phosphoprotein. J Mol Biol 388(5):978–996. [DOI] [PubMed] [Google Scholar]
- 29.Kidane AI, et al. (2013) Structural features of LC8-induced self-association of swallow. Biochemistry 52(35):6011–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radnai L, et al. (2010) Affinity, Avidity, and Kinetics of Target Sequence Binding to LC8 Dynein Light Chain Isoforms. J Biol Chem 285(49):38649–38657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292(2):195–202. [DOI] [PubMed] [Google Scholar]
- 32.Yabukarski F, et al. (2016) Ensemble Structure of the Highly Flexible Complex Formed between Vesicular Stomatitis Virus Unassembled Nucleoprotein and its Phosphoprotein Chaperone. J Mol Biol 428(13):2671–2694. [DOI] [PubMed] [Google Scholar]
- 33.Muñoz V, Serrano L (1994) Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Mol Biol 1(6):399–409. [DOI] [PubMed] [Google Scholar]
- 34.Frueh DP (2014) Practical aspects of NMR signal assignment in larger and challenging proteins. Prog Nucl Magn Reson Spectrosc 78:47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrow N, Zhang O, Szabo A, Torchia D, Kay L (1995) Spectral density function mapping using 15N relaxation data exclusively. JBiomol NMR 6(2): 153–162. [DOI] [PubMed] [Google Scholar]
- 36.Hwang TL, van Zijl PCM, Mori S (1998) Accurate quantitation of water-amide proton exchange rates using the Phase-Modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme RID B-8680–2008. J Biomol NMR 11(2):221–226. [DOI] [PubMed] [Google Scholar]
- 37.Clark S, et al. (2018) Multivalency regulates activity in an intrinsically disordered transcription factor. Elife 7. doi: 10.7554/eLife.36258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brereton AE, Karplus PA (2018) Ensemblator v3: Robust atom-level comparative analyses and classification of protein structure ensembles. Protein Sci 27(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fouquet B, et al. (2015) Focal Adhesion Kinase Is Involved in Rabies Virus Infection through Its Interaction with Viral Phosphoprotein P. J Virol 89(3): 1640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer A, Nolden T, Nemitz S, Perlson E, Finke S (2015) A Dynein Light Chain 1 Binding Motif in Rabies Virus Polymerase L Protein Plays a Role in Microtubule Reorganization and Viral Primary Transcription. J Virol 89(18):9591–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, et al. (2016) Rabies virus phosphoprotein interacts with ribosomal protein L9 and affects rabies virus replication. Virology 488:216–224. [DOI] [PubMed] [Google Scholar]
- 42.Chelbi-Alix MK, Vidy A, El Bougrini J, Blondel D (2006) Rabies viral mechanisms to escape the IFN system: the viral protein P interferes with IRF-3, Stat1, and PML nuclear bodies. J Interferon Cytokine Res 26(5):271–280. [DOI] [PubMed] [Google Scholar]
- 43.Oksayan S, et al. (2012) A novel nuclear trafficking module regulates the nucleocytoplasmic localization of the rabies virus interferon antagonist, P protein. J Biol Chem 287(33):28112–28121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, et al. (2017) BECN1-dependent CASP2 incomplete autophagy induction by binding to rabies virus phosphoprotein. Autophagy:00–00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kammouni W, et al. (2015) Rabies virus phosphoprotein interacts with mitochondrial Complex I and induces mitochondrial dysfunction and oxidative stress. J Neurovirol 21(4):370–382. [DOI] [PubMed] [Google Scholar]
- 46.Blondel D, et al. (2002) Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21(52):7957–7970. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, et al. (2016) The co-chaperone Cdc37 regulates the rabies virus phosphoprotein stability by targeting to Hsp90AA1 machinery. Sci Rep 6(March):27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curran J (1998) A role for the Sendai virus P protein trimer in RNA synthesis. J Virol 72(5):4274–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribeiro E de A, et al. (2009) Binding of rabies virus polymerase cofactor to recombinant circular nucleoprotein-RNA complexes. J Mol Biol 394(3):558–575. [DOI] [PubMed] [Google Scholar]
- 50.Takamatsu Y, Kolesnikova L, Becker S (2018) Ebola virus proteins NP, VP35, and VP24 are essential and sufficient to mediate nucleocapsid transport. Proc Natl Acad Sci U S A 115(5): 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker JR, et al. (2018) The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat Commun 9(1):5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaik M, et al. (2015) Structural basis for assembly and function of the Nup82 complex in the nuclear pore scaffold. J Cell Biol 208(3):283–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venditti V, Fawzi NL (2018) Probing the Atomic Structure of Transient Protein Contacts by Paramagnetic Relaxation Enhancement Solution NMR, pp 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poser I, et al. (2008) BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods 5(5):409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finke S, Conzelmann KK (1999) Virus promoters determine interference by defective RNAs: selective amplification of mini-RNA vectors and rescue from cDNA by a 3’ copy-back ambisense rabies virus. J Virol 73(5):3818–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lieu KG, et al. (2013) The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J Virol 87(14):8261–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lahaye X, et al. (2009) Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J Virol 83(16):7948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wyatt (1998) Submicrometer Particle Sizing by Multiangle Light Scattering following Fractionation. J Colloid Interface Sci 197(1):9–20. [DOI] [PubMed] [Google Scholar]
- 59.Favier A, Brutscher B (2011) Recovering lost magnetization: Polarization enhancement in biomolecular NMR. J Biomol NMR 49( 1): 9–15. [DOI] [PubMed] [Google Scholar]
- 60.Yoshimura Y, Kulminskaya NV, Mulder FAA (2015) Easy and unambiguous sequential assignments of intrinsically disordered proteins by correlating the backbone 15N or 13C’ chemical shifts of multiple contiguous residues in highly resolved 3D spectra. J Biomol NMR 61(2): 109–121. [DOI] [PubMed] [Google Scholar]
- 61.Grzesiek S, Bax A (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc 114(16):6291–6293. [Google Scholar]
- 62.Farrow NA, et al. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33(19):5984–6003. [DOI] [PubMed] [Google Scholar]
- 63.Fitzkee NC, Bax A (2010) Facile measurement of 1H-15N residual dipolar couplings in larger perdeuterated proteins. J Biomol NMR 48(2):65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delaglio F, et al. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6(3):277–293. [DOI] [PubMed] [Google Scholar]
- 65.Lee W, Tonelli M, Markley JL (2015) NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31(8): 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kjaergaard M, Brander S, Poulsen FM (2011) Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. JBiomolNMR 49(2):139–149. [DOI] [PubMed] [Google Scholar]
- 67.Kjaergaard M, Poulsen FM (2011) Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J Biomol NMR 50(2): 157–165. [DOI] [PubMed] [Google Scholar]
- 68.Barbar E, Hare M, Makokha M, Barany G, Woodward C (2001) NMR-detected order in core residues of denatured bovine pancreatic trypsin inhibitor. Biochemistry 40(32):9734–42. [DOI] [PubMed] [Google Scholar]
- 69.Franke D, et al. (2017) ATSAS 2.8 : a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 50(4):1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Petoukhov MV, et al. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45(2):342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rambo RP, Tainer JA (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496(7446):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4(3):435–447. [DOI] [PubMed] [Google Scholar]
- 73.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN (2010) SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res 38(Web Server issue):W657–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piana S, Lindorff-Larsen K, Shaw DE (2011) How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys J 100(9):L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Best RB, Zheng W, Mittal J (2014) Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J Chem Theory Comput 10(11):5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Essmann U, et al. (1995) A smooth particle mesh Ewald method. JChem Phys 103(19):8577–8593. [Google Scholar]
- 77.Svergun D, Barberato C, Koch MHJ, IUCr (1995) CRYSOL – a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J Appl Crystallogr 28(6):768–773. [Google Scholar]
- 78.Tria G, Mertens HDT, Kachala M, Svergun DI (2015) Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2(2):207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choy W-Y, Forman-Kay JD (2001) Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J Mol Biol 308(5): 1011–1032. [DOI] [PubMed] [Google Scholar]
- 80.Nodet G, et al. (2009) Quantitative Description of Backbone Conformational Sampling of Unfolded Proteins at Amino Acid Resolution from NMR Residual Dipolar Couplings. J Am Chem Soc 131(49): 17908–17918. [DOI] [PubMed] [Google Scholar]
- 81.Jamros MA, et al. (2012) Substrate-Specific Reorganization of the Conformational Ensemble of CSK Implicates Novel Modes of Kinase Function. PLoS Comput Biol 8(9):e1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chumnarnsilpa S, Robinson RC, Grimes JM, Leyrat C (2015) Calcium-controlled conformational choreography in the N-terminal half of adseverin. Nat Commun 6(1):8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM (2014) Drastic changes in conformational dynamics of the antiterminator M2–1 regulate transcription efficiency in Pneumovirinae. Elife 3. doi: 10.7554/eLife.02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones DT, Cozzetto D (2015) DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31(6):857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mészáros B, Erdos G, Dosztanyi Z (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46(W1):W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clark SA, Tronrud DE, Andrew Karplus P (2015) Residue-level global and local ensemble-ensemble comparisons of protein domains. Protein Sci 24(9):1528–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sievers F, et al. (2014) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7(1):539–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ashkenazy H, et al. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.