

Abstract
The ECM protein EFEMP1 (fibulin‐3) is associated with all types of solid tumor through its cell context‐dependent dual function. A variant of fibulin‐3 was engineered by truncation and mutation to alleviate its oncogenic function, specifically the proinvasive role in glioblastoma multiforme (GBM) cells at stem‐like state. ZR30 is an in vitro synthesized 39‐kDa protein of human fibulin‐3 variant. It has a therapeutic effect in intracranial xenograft models of human GBM, through suppression of epidermal growth factor receptor/AKT and NOTCH1/AKT signaling in GBM cells and extracellular MMP2 activation. Glioblastoma multiforme is highly vascular, with leaky blood vessels formed by tumor cells expressing endothelial cell markers, including CD31. Here we studied GBM intracranial xenografts, 2 weeks after intratumoral injection of ZR30 or PBS, by CD31 immunohistochemistry. We found a 70% reduction of blood vessel density in ZR30‐treated xenografts compared with that of PBS‐treated ones. Matrigel plug assays showed the effect of ZR30 on suppressing angiogenesis. We further studied the effect of ZR30 on genes involved in endothelial transdifferentiation (ETD), in 7 primary cultures derived from 3 GBMs under different culture conditions. Two GBM cultures formed mesh structures with upregulation of ETD genes shortly after culture in Matrigel Matrix, and ZR30 suppressed both. ZR30 also downregulated ETD genes in two GBM cultures with high expression of these genes. In conclusion, multifaceted tumor suppression effects of human fibulin‐3 variant include both suppression of angiogenesis and vasculogenic mimicry in GBM.
Keywords: extracellular compartment, malignant glioma, novel cancer therapeutic, syngeneic primary culture, vasculogenic mimicry
Vascularization in glioblastoma multiforme intracranial xenograft was reduced by intratumoral injection of ZR30, an in vitro synthesized 39‐kDa protein of human fibulin‐3 variant.
![]()
1. INTRODUCTION
Malignant gliomas are the most dangerous of primary brain tumors. Glioblastoma multiforme (GBM) is the most common, most aggressive, and highest grade of malignant glioma, with an average median survival of only 12‐14 months.1 Radical surgical resection followed by adjuvant radiotherapy/chemotherapy is standard treatment for malignant gliomas, however, tumor recurrence occurs in nearly all instances. Human fibulin‐3 variant (h‐fibulin‐3‐v), a novel cancer therapeutic agent, has been developed in our laboratory after recognizing that its parental protein EFEMP1 has a role in cancer evolution with a cell context‐dependent dual function.2, 3, 4 Human fibulin‐3 variant functions in the extracellular compartment through multifaceted tumor suppression mechanisms. As such, it would effectively inhibit tumor cells from reforming tumor mass, and would be less susceptible to therapeutic escape leading to recurrence. These mechanisms include suppressing tumor growth and invasion by its influence on intracellular growth signals of different tumor subpopulations, eg, epidermal growth factor receptor (EGFR)/AKT in glioma cells in proliferation state, and NOTCH1/AKT in glioma cells in stem‐like state.
ZR30 is an in vitro synthesized protein version of h‐fibulin‐3‐v without signal peptide that has shown therapeutic efficacy in a human GBM intracranial (i.c.) xenograft model with a survival end‐point following even a single intratumoral administration.5 The same multifaceted tumor suppression mechanisms of ZR30 were found in treated GBM cell lines and primary cultures, as also shown by induction of h‐fibulin‐3‐v expression in GBM cells.4, 5 Initially, EFEMP1 was reported to have a role in antiangiogenesis.6 The cell context‐dependent dual function of EFEMP1 in cancer appears to complicate its role in tumor vascularization. A provascularization function of EFEMP1 was implicated in ovarian cancer with lymph node metastasis,7 and proven in a highly metastatic cell line of human pancreatic adenocarcinoma,8 whereas antitumor vascularization function was also proven in a highly proliferative cell line of GBM.9 Both were shown in s.c. xenograft models by overexpression of ectopic EFEMP1. Here we sought the effect of ZR30 on tumor vascularization using a clinically relevant intracranial GBM xenograft model, with a focus on angiogenesis and the vascularization potential of GBM cells.
Tumor cells express a vascular phenotype through a process called endothelial transdifferentiation (ETD). It is characterized by upregulation of genes in embryonic development that program hematopoietic and vascular development.10 Tumor vascularization includes both angiogenesis and pathologic vascularization. The latter refers to leaky blood vessels formed by tumor cells, known as vasculogenic mimicry (VM) that supports rapidly growing tumors. Although most of the blood vessels in tumor are derived from angiogenesis, VM is an important part of tumor vascularization associated with cancer progression, including GBM.11, 12, 13, 14, 15, 16, 17 None of the antiangiogenic therapies currently in clinical use, which mainly target vascular endothelial growth factor (VEGF) and its receptor KDR, has shown a significant improvement in overall survival of GBM patients.18 Agents inhibiting VM might not only improve antiangiogenic therapies through synergistic and/or additive effects, when coupled with current antiangiogenic therapies, but could significantly improve survival as well.
2. MATERIALS AND METHODS
2.1. Ethics statement
The use of fresh GBM tissue to establish primary cultures in this study was approved by the institutional review board of the University of California (UC) Irvine, with informed consent obtained by the Tissue Bank of the Department of Pathology and Laboratory Medicine, UC Irvine College of Health Sciences. The use of nude mice for i.c. human GBM xenograft models in this study was approved by the Institutional Animal Care and Use Committee (IACUC) of UC Irvine (2006‐2689) and the First Affiliated Hospital of Soochow University (2017‐175). The use of nude mice for the Matrigel plug in vivo angiogenesis assay in this study was approved by the IACUC of Sun Yat‐sen University Cancer Center (L102012018120H). Their care was in accordance with institution guidelines.
2.2. Human GBM primary cultures
Human GBM‐derived syngeneic primary cultures used in this study were established in the Brain Tumor Research Laboratory, UC Irvine, following methods detailed previously.19 In brief, single cells of GBM specimens were equally divided into 2 or 3 portions to culture in 2 (namely SA and NS) or 3 (namely SA, NS, and EC) conditions. In the SA condition, cells were cultured with DMEM/F12 medium supplemented with 10% FBS in collagen‐coated (initial culture) or regular culture dishes (subsequent passages). In the NS condition, cells were cultured with DMEM/F12 medium supplemented with epidermal growth factor (EGF, 20 ng/mL), basic fibroblast growth factor (bFGF, 10 ng/mL), and 5% B27 (Invitrogen), initially in low‐binding or 1% agar‐coated dishes (for 3‐4 weeks), then in adherent conditions in fibronectin (1 μg/cm2)‐coated dishes, and back and forth for at least 2 rounds of adherent and spheroid cultures. In the EC condition, adherent cultures were maintained in endothelial cell growth medium (PromoCell, http://www.promocell.com/) in fibronectin (1 μg/cm2)‐coated dishes. The NS‐conditioned cells were cultured in fibronectin‐coated dishes prior to in vitro and in vivo analyses. All cells were cultured in a 37°C humid chamber containing 5% CO2.
Cells used in this study were validated to be free of mycoplasma by analyzing culture medium with MycoAlert PLUS Mycoplasma from Lonza and/or PCR‐based Venor GeM Mycoplasma Detection Kit (Sigma‐Aldrich). Identification of mutations in TP53, PTEN, IDH1, and IDH2 and DNA copy number variation of PTEN and EGFR were carried out using cDNA and DNA samples of GBM primary cultures, following PCR‐based mutation assay with sequencing and comparative quantitative PCR, respectively, as described previously.3, 19 The genetic identity profiles of 16 short tandem repeats were determined based on DNA samples by IDEXX RADIL, with results shown in Table 1.
Table 1.
Gene abnormality and short tandem repeat (STR) profiles of syngeneic glioblastoma multiforme (GBM) primary cultures
| Tumor | Histology | Clinical data | Established cultures | Culture condition | Mutation, deletion, or amplification of genes | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP53 | IDH1 | IDH2 | PTEN | EGFR | |||||
| GBM51 | GBM | 61 y, F, recurrent | 51A | NS | wt | wt | wt | wt | High amp |
| 51B | SA | wt | wt | wt | wt | No amp | |||
| STR: AMEL(X), CSF1PO(10, 12), D13S317(11, 12), D16S539(11, 13), D18S51(12, 15), D21S11(29), D3S1358(13, 15), D5S818(10, 11), D7S820(9), D8S1179(14), FGA(20), Penta D(11, 12), Penta E(5, 10), TH01(8, 9), TPOX(8), vWA(14) | |||||||||
| GBM98 | GBM | 70 y, F, de novo | 98A | NS | L257V | wt | wt | Homozygous p.Q298 trunc | No amp |
| 98B | SA | L257V | wt | wt | Homozygous p.Q298 trunc | No amp | |||
| 98E | EC | L257V | wt | wt | Homozygous p.Q298 trunc, heterozygous pR55 trunc | Low amp | |||
| STR: AMEL(X), CSF1PO(9, 10), D13S317(11), D16S539(12, 13), D18S51(17, 19), D21S11(30, 31.2), D3S1358(16, 17), D5S818(11, 12), D7S820(8, 11), D8S1179(12, 13), FGA(20), Penta D(8, 14), Penta E(13, 15), TH01(6, 9), TPOX(8, 10), vWA(16) | |||||||||
| GBM97 | GBM‐O | 45 y, M secondary | 97A | NS | wt | wt | wt | Homozygous deletion | Low amp |
| 97B | SA | wt | wt | wt | Homozygous deletion | Low amp | |||
| STR: AMEL(X, Y), CSF1PO(12, 13), D13S317(11), D16S539(12, 13), D18S51(14, 16), D21S11(31, 32.2), D3S1358(15, 18), D5S818(11, 12), D7S820(8, 11), D8S1179(13), FGA(24, 26), Penta D(9, 14), Penta E(14, 15), TH01(6, 8), TPOX(8, 9),vWA(15, 17) | |||||||||
amp, amplification; EC, adherent cultures maintained in endothelial cell growth medium in fibronectin (1 μg/cm2)‐coated dishes; F, female; M, male; NS, cells cultured with DMEM/F12 medium supplemented with epidermal growth factor (20 ng/mL), basic fibroblast growth factor (10 ng/mL), and 5% B27 (Invitrogen), initially in low‐binding or 1% agar‐coated dishes (for 3‐4 weeks), then in adherent conditions in fibronectin (1 μg/cm2)‐coated dishes, and back and forth for at least 2 rounds of adherent and spheroid cultures; SA, cells cultured with DMEM/F12 medium supplemented with 10% FBS in collagen‐coated (initial culture) or regular culture dishes (subsequent passages); trunc, truncated.
2.3. Human fibulin‐3 variant protein
ZR30 used in this study was provided by Ziren Research. It was made by an in vitro cell‐free system based on the sequence of the h‐fibulin‐3‐v, excluding the signal peptide, as detailed previously.5
2.4. Intracranial human GBM xenografts and immunohistochemistry
Each BALB/c nude mouse (SPF level, 5‐6 weeks old, female; Shanghai SLAC Laboratory Animal Co.) was implanted with a guide cannula in the right frontal lobe, and then 3 μL glioma cells (9:1 mixture of 51A‐GFP and 51B‐RFP, a total of 1 × 105 cells) was injected through the guide cannula, as described previously.5 Nineteen days after cell implantation, 5 μL PBS or ZR30 (180 ng/μL) was injected through the guide cannula into the cell implantation site. Two weeks after treatment, right hemispheres were removed from both groups of mice and processed for immunohistochemistry using primary Ab of anti‐CD31 (ab182981; Abcam), as detailed previously.17 Blood vessel density in xenografts was determined by positive signals counted by ImageJ based on images of a 20× lens, 5 images per section, 2 sections per mouse brain (frontal and hind parts), and 2‐3 brains per group.
2.5. Matrigel plug in vivo angiogenesis assay
BALB/c nude mice were divided into 2‐3 groups (2‐3 mice per group), s.c. injected on both sides with 1 mL Matrigel (~10 mg/mL) with or without the addition of human VEGF‐165 (100 ng/mL; Invitrogen) and/or ZR30 (100 or 300 ng/mL). Seven days later, mice were killed and Matrigel plugs were carefully removed, fixed in 4% formaldehyde, paraffin‐embedded, then sectioned (4 μm) for Masson’s trichrome staining and CD31 immunohistochemistry. The experiment was repeated with addition of bFGF (300 ng/mL; R&D Systems) on top of the addition of VEGF‐165 and/or ZR30.
2.6. Matrigel‐based tubular network assay
Corning Matrigel matrix (~10 mg/mL) was melted at 4°C overnight, added to ice‐cold 24‐well plates (0.2 mL/well) or 96‐well plates (0.05‐0.08 mL/well) and solidified by incubating at 37°C for more than 1 hour. The GBM cells were suspended in human endothelial basal medium (H1168B; Cell Biologics) supplemented with 2% FBS or DMEM/F12 with the addition of 10% FBS, then added in duplicates or triplicates to Matrigel‐coated 24‐ or 96‐well plates, in concentrations of 1‐2 × 105/well or 5 × 104/well, respectively. Images were taken periodically (every 1 or 2 hours) under an inverted microscope for up to 24 hours, and analyzed using the Fiji distribution of ImageJ20 at version ImageJ 2.0.0‐rc‐15/1.49k; Java 1.6.0_65 (available as a Life‐Line version from https://imagej.net/Downloads).
2.7. Real‐time PCR quantification of gene expressions
Diluted cDNA samples in 10 mmol/L Tris.HCL (pH 7.5) were used in standard‐based real‐time PCR, using a 2‐step PCR program (95°C for 3 seconds, 65°C for 30 seconds) with fast 96‐well reaction plates (Applied Biosystems) and FastStart DNA MasterPLUS SYBR Green I (Roche). Each run of real‐time PCR includes a serial of 10‐fold diluted standard composed of PCR‐amplified fragment of each gene in equal ratio with other genes including internal control genes. Information regarding PCR primers and amplicons are shown in Table 2.
Table 2.
Real‐time PCR primer sequences and amplicon melting temperature
| Gene symbol | 5′ sequence | 3′ sequence | Melting (°C) |
|---|---|---|---|
| ACTB | TCCTTCCTGGGCATGGAGT | TGATCTTCATTGTGCTGGGT | 88.5 |
| CDH5 | AATCACGATAACACGGCCAAC | CATATCCTCGCAGAAGGTGAAC | 89.9 |
| DLL4 | ATTGCCACGGAGGTATAAGG | CTTGGCAAGTTCACAGTCTG | 87.2 |
| GAPDH | CAATATGATTCCACCCATGG | GGTTCACACCCATGACGAAC | 89.3 |
| GATA2 | CTCTACCACAAGATGAATGG | GGCCTGTTAACATTGTGCAG | 89.6 |
| LMO2 | TTCAGAGGAACCAGTGGATG | TGAGATAGTCTCTCCGGCAGA | 91.7 |
| NANOG | CAAACAGGTGAAGACCTGGT | CAGGTTGAATTGTTCCAGGT | 85.7 |
| NOTCH4 | TCTCAAGGCACTGAAGCCAA | CTTCCATCTCAGATTCCTGG | 89.2 |
| PECAM1 | CCAAGGTGAAAGACTGAACC | GACTATCTGGACTGTGTTGC | 84.8 |
| POU3F2 | AATGAATGTCACAAGGAGATGG | ACATAGTGGTAAGGTTCAGTGG | 86.5 |
| POU5F1 | GGCTGGGTTGATCCTCG | CCTCCGGGTTTTGCTCC | 92.0 |
| SALL2 | GTGTCTTTGTCATTGTCTGG | ACTAGGTTAGGAAGCTCTGG | 84.7 |
| SOX2 | GAACACCAATCCCATCCAC | ACTTCCTGCAAAGCTCCTAC | 82.1 |
| TUBA1A | CAACCATGCGTGAGTGCATC | GAAGGTGTTGAAGGAATCATC | 88.8 |
| VIM | GAGGAGAGCAGGATTTCTCTG | ATCGTGATGCTGAGAAGTTTCG | 82.0 |
2.8. Statistical analysis
One‐way ANOVA with post‐hoc pairwise comparisons was used to determine ZR30’s effect on VEGF‐stimulated angiogenesis in Matrigel plugs and gene expression. Poisson regression was utilized to analyze the effect of ZR30 on each cell line’s mesh numbers formed in Matrigel from 2 independent experiments, which were adjusted as fixed effect. The equal‐variance t test was used to compare blood vessel density in PBS‐ and ZR30‐treated xenografts. Gene expression was calculated as 10 000 × (absolute ratio to ACTB). All comparisons used 2‐sided tests at an alpha = 0.05 significance level using SAS version 9.4 (SAS Institute) and Excel 2013 (Microsoft).
3. RESULTS
3.1. ZR30 suppresses tumor vascularization in human GBM i.c. xenografts
As shown previously,19 i.c. implantation of a mixture of 100 000 cells of 51A and 51B (9:1) formed i.c. xenografts with the tumor mass largely composed of 51B cells that expressed highly proangiogenic genes or proteins, whereas the peritumoral zone was largely composed of 51A cells that expressed highly proinvasive genes or proteins. We used this i.c. human GBM xenograft model to examine the effect of intratumoral injection of ZR30 on tumor vascularization during the course of tumor development, which was 19‐33 days following cell implantation. Immunohistochemistry of endothelial cell marker CD31 showed positive staining of blood vessels in PBS‐treated xenografts (Figure 1A). Comparison of blood vessel density (BVD) in xenografts showed a significant two‐thirds reduction in ZR30‐treated xenograft compared with that of PBS‐treated controls (Figure 1B).
Figure 1.
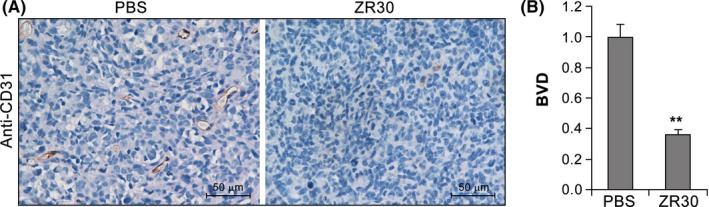
ZR30 inhibits blood vessel formation in intracranial xenografts of human glioblastoma multiforme primary cultures. A, Representative pictures of CD31 immunohistochemistry on PBS‐ or ZR30‐treated xenografts. B, Comparison of blood vessel density (BVD) in ZR30‐ and PBS‐treated xenografts in 3 and 2 mice, respectively. The average of BVD in PBS‐treated xenografts was set at unity. Columns, mean (PBS, n = 20; ZR30, n = 30); bars, SEM. P < .05; ** P < .005
Immunohistochemistry of glial fibrillary acidic protein (GFAP) revealed negative staining of GFAP in central tumor mass of both PBS and ZR30‐treated xenografts. Because 51A cells express GFAP, whereas 51B cells do not,19 GFAP‐negative tumor mass is consistent with our prior findings that 51B formed tumor masses in syngeneic GBM i.c. xenograft models of 51A and 51B (9:1). Thus, ZR30‐mediated suppression of BVD is within 51B‐formed tumor mass.
3.2. ZR30 suppresses angiogenesis in Matrigel plugs in vivo
To examine the effect of ZR30 on angiogenesis, we undertook an in vivo Matrigel plug assay. As shown in Figure 2A, 1 week after s.c. injection of liquefied Matrigel without addition of VEGF‐165 (negative control), the surface of Matrigel plugs did not show blood vessels, which was in great contrast with the addition of VEGF‐165 (positive control). The Matrigel plugs containing both VEGF‐165 and ZR30 were slightly pinkish with no vessels on the surface. The assay was repeated with 2 doses of ZR30, and vessels inside the Matrigel plugs were imaged after Masson’s trichrome staining (Figure 2B). Relative vessel density in Matrigel plugs of negative (group 1), positive (2), and treatment groups (3 and 4) were compared. As shown in Figure 2C, vessel density in Matrigel plugs of positive control was significantly higher by an average of 5.2‐fold, in comparison with that of negative controls. Vessel density in treatment groups were significantly reduced in comparison with that of the positive control in a dose‐dependent manner. Comparison of vessel density between negative control and high‐dose treatment groups showed a lack of statistical significance (P = .14), indicating ZR30‐mediated abolition of blood vessel formation in Matrigel plugs induced by VEGF‐165.
Figure 2.
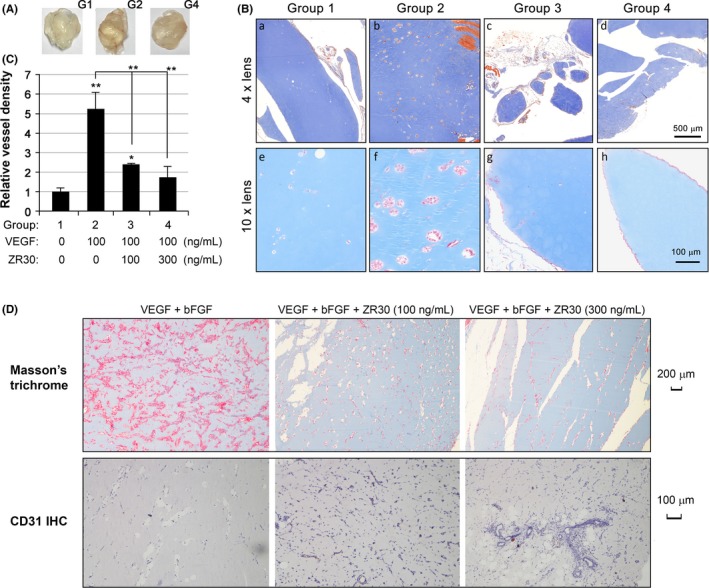
ZR30 inhibits angiogenesis in vivo. A, Representative pictures of Matrigel plugs removed from mice 1 week after s.c. injection from negative control (G1), positive control (G2), and ZR30 treatment (G4) groups of mice. B, Representative pictures of Masson’s trichrome stained sections of Matrigel plugs. C, Comparison of vessel density in 4 groups of Matrigel plugs. Columns, mean (n = 3); bars, SD. *P < .05; **P < .005. D, Masson’s trichrome and CD31 immunohistochemistry (IHC) of Matrigel plugs containing VEGF‐165 (100 ng/mL) and basic fibroblast growth factor (bFGF, 300 ng/mL), with or without ZR30 (100 or 300 ng/mL)
Masson’s trichrome is often used to stain connective tissue. Nuclei and other basophilic structures, such as collagen fibers, are stained blue; cytoplasm, muscle, erythrocytes, and keratin are stained bright red. It was used to visualize and quantify angiogenesis in Matrigel plugs.21 In plugs of positive control containing VEGF‐165, we found vessel structures with bright red staining inside from Masson’s trichrome, but no CD31‐positive cells. The H&E staining confirmed vessels containing red blood cells in the positive control. This suggests that VEGF‐165 can induce angiogenesis in the designed conditions, but is not able to stabilize blood vessel without other growth factors, such as bFGF, which was used by others in their Matrigel in vivo angiogenesis assays.6
To verify the effect of ZR30 on inhibition of angiogenesis, the same in vivo angiogenesis assay was carried out with modification by addition of bFGF to liquefied Matrigel in positive control and treatment groups. As shown in Figure 2D, in positive control plugs, we found massive numbers of structures with irregular shape stained bright red along, with some areas that were stained blue by Masson’s trichrome; however, no CD31‐positive cells were observed on immunohistochemistry. Our explanation of this phenomenon is that bFGF functions to induce and maintain functional blood vessels. Together with VEGF, bFGF induced massive amounts of angiogenesis in Matrigel plugs, and consequent depletion of bFGF might result in massive vessel degeneration.
In this modified Matrigel plug assay, we found ZR30 significantly suppressed massive areas of positive Masson’s trichrome staining, with visibility of vessels lined by CD31‐positive cells (Figure 2D, middle and right panels). Our explanation for this contradicting phenomenon is that dramatically less positive Masson’s trichrome staining in plugs of treated cells indicated low levels of angiogenesis and consequently low consumption of bFGF in vessel maintenance, which allowed detection of CD31‐positive cells and vessels in these plugs at the time removed for analysis. Results shown in Figure 2D confirmed our speculation that bFGF is required to maintain the viability of vessels following angiogenesis. Overall data from Matrigel angiogenesis assays using different angiogenesis‐inducing agents consistently indicated the antiangiogenesis function of ZR30.
3.3. ZR30 suppresses Matrigel matrix induction of ETD gene expression
Repeated Matrigel‐based tubular network assay was carried out using primary cultures (51B, 97B, and 98B) that showed formation of mesh structures shortly (4‐6 hours) after placement on solidified Matrigel matrix in both culture mediums (human endothelial basal medium with addition of 2% FBS and DMEM/F12 with addition of 10% FBS). Real‐time PCR was used to quantify expression levels of genes before and after formation of mesh structures, using cDNA samples converted from total RNA of cells cultured in SA conditions (SA‐2D) for 25 hours and Matrigel matrix (Matrigel‐3D) for 6 hours under the same seeding density. Genes of pluripotent embryonic stem cell, hematopoietic and vascular development, VM (NOTCH4)10 and GBM cell pluripotency (POU3F2, SOX2, SALL2, and OLIG2)22 were examined, together with genes involved in housekeeping and epithelial‐mesenchymal transition (EMT). The expression level of genes was shown as an absolute ratio to ACTB times 10 000, as shown in Table 3. The 3 housekeeping genes and the gene involved in EMT (VIM) were highly expressed. Embryonic, pluripotency, and some hematopoietic vascular genes (NANOG, POU5F1, POU3F2, SALL2, and NOTCH4) were moderately expressed (more than 1 of 1000 copies of ACTB), and some other hematopoietic vascular genes (CDH5, PECAM1, GATA2, LMO2, and DLL4) had low expression (less than 1 of 1000 copies of ACTB). Other examined developmental and hematopoietic vascular genes (SOX2, OLIG2, VWF, and TEK) were not, or were rarely expressed (less than 1 of 10 000 copies of ACTB).
Table 3.
Differential gene expressions in glioblastoma multiforme primary cultures in SA‐2D and Matrigel‐3D conditions
| Function | Housekeeping | EMT | Embryonic, pluripotency | Hematopoietic, vascular development | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | GAPDH | TUBA1A | VIM | NANOG | SALL2 | POU5F1 | POU3F2 | NOTCH4 | CDH5 | PECAM1 | GATA2 | LMO2 | DLL4 |
| 51B | |||||||||||||
| 2D | 2301 | 3651 | 11 733 | 83 | 64 | 92 | 22 | 26 | 3.1 | 5.3 | 1.0 | 7.0 | 0.05 |
| (119) | (124) | (1981) | (32) | (15) | (41) | (5.0) | (4.4) | (0.4) | (0.6) | (0.03) | (3.3) | (0.05) | |
| 3D | 1193 | 3689 | 2344 | 744 | 278 | 905 | 64 | 285 | 26 | 18 | 3.4 | 26 | 0.39 |
| (502) | (75) | (685) | (116) | (14) | (81) | (1.4) | (31) | (1.0) | (0.6) | (2.0) | (4.3) | (0.39) | |
| 3D/2D | 0.5 | 1.0 | 0.2 | 9.0 | 4.4 | 9.9 | 2.9 | 11.1 | 8.3 | 3.4 | 3.6 | 3.7 | 7.9 |
| 98B | |||||||||||||
| 2D | 4652 | 1713 | 7677 | 313 | 91 | 129 | 352 | 41 | 2.8 | 6.6 | 0.8 | 8.2 | 0.11 |
| (1535) | (266) | (2193) | (162) | (26) | (8.6) | (23) | (2.5) | (0.1) | (1.5) | (0.01) | (1.7) | (0.11) | |
| 3D | 1908 | 2105 | 5000 | 1064 | 275 | 975 | 295 | 357 | 20 | 31 | 1.7 | 21 | 0.17 |
| (66) | (48) | (147) | (69) | (4.2) | (61) | (38) | (38) | (3.8) | (5.3) | (0.1) | (4.0) | (0.04) | |
| 3D vs 2D | 0.4 | 1.2 | 0.7 | 3.4 | 3.0 | 7.6 | 0.8 | 8.8 | 6.9 | 4.8 | 2.3 | 2.6 | 1.5 |
| 97B | |||||||||||||
| 2D | 11 017 | 3576 | 10 726 | 474 | 1653 | 137 | 1399 | 50 | 54 | 7.6 | 1.6 | 7.8 | 0.31 |
| (746) | (48) | (722) | (143) | (96) | (31) | (25) | (1.1) | (14) | (0.6) | (0.4) | (1.9) | (0.09) | |
| 3D | 7927 | 2207 | 2393 | 573 | 648 | 194 | 705 | 51 | 54 | 14.4 | 1.1 | 4.7 | 2.0 |
| (2702) | (620) | (643) | (146) | (125) | (49) | (103) | (11) | (7.5) | (2.0) | (0.03) | (1.4) | (0.3) | |
| 3D vs 2D | 0.7 | 0.6 | 0.2 | 1.2 | 0.4 | 1.4 | 0.5 | 1.0 | 1.0 | 1.9 | 0.7 | 0.6 | 6.5 |
Gene expressions are shown as mean (SEM) of ratio to ACTB times 10 000 in cells cultured in 24‐well plates (200 000/well) coated with Matrigel matrix for 6 h (referred to as 3D), or uncoated for 25 h (referred to as 2D). Bold text indicates >2‐fold change in gene expression between 3D and 2D conditions.
Abbreviation: EMT, epithelial‐mesenchymal transition.
As shown in Table 3, comparison of these gene expression levels between Matrigel‐3D and SA‐2D conditions showed little change in TUBA1A in all 3 GBM primary cultures. Approximately 50% reduction of GAPDH in both 51B and 98B, not in 97B, and 70%‐80% reduction of VIM in 51B and 97B, not in 98B, were seen between Matrigel‐3D and SA‐2D conditions. In contrast, 2‐ to 10‐fold increase of embryonic, pluripotency, hematopoietic, and vascular development genes (NANOG, SALL2, POU5F1, NOTCH4, CDH5, PECAM1, GATA2, and LMO2) were observed in both 51B and 98B, and additionally 3‐fold increase in POU3F2 and 8‐fold increase in DLL4 in 51B. However, none of these Matrigel matrix‐mediated gene upregulations were seen in 97B, except a 6.5‐fold increase of DLL4. These embryonic, pluripotency, hematopoietic, and vascular development genes are ETD genes; the results of our study on the effect of ZR30 on their expression is described below.
As shown in Figure 3, addition of ZR30 in the medium of the 3D‐Matrigel culture significantly reduced the number of mesh structures in a dose‐dependent manner. It also suppressed most of the upregulated ETD genes, including SALL2, POU5F1, NOTCH4, and CDH5 in 51B and 98B, and NANOG, POU3F2, and PECAM1 in 51B (Figure 4). It was noted that NANOG and POU3F2 had lower basal expression levels in 51B, 26% and 6% compared with 98B, respectively. There are high basal levels of ETD genes in 97B, where SALL2 and CDH5 levels were approximately 20‐fold higher compared with that in 51B and 98B, and POU3F2 levels were more than 40‐fold higher compared with that in 51B. Although 97B cells formed mesh structures in Matrigel matrix and ZR30 significantly reduced the number of mesh structures by one third (P < .05), no effect of ZR30 was observed on the regulation of ETD genes in 97B, except for a 25% downregulation of CDH5, after 6 h of culture on Matrigel matrix.
Figure 3.
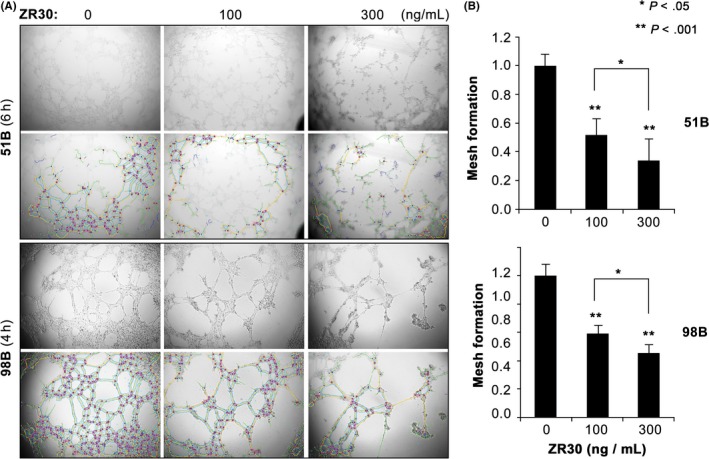
ZR30’s dose‐dependent suppression of mesh formation in Matrigel matrix. A, Representative images of 51B and 98B (rows 1 and 3) after plating 50 000 cells in 0.1 mL DMEM/F12 supplemented with 10% FBS with or without ZR30, on solidified Matrigel in 96‐well plates. Meshes were superimposed by ImageJ (rows 2 and 4) and the numbers were counted. B, Relative mesh counts normalized to no‐treatment control. Columns are least square means and bars are SE from data obtained in 2 independent experiments with 2‐3 replicates
Figure 4.
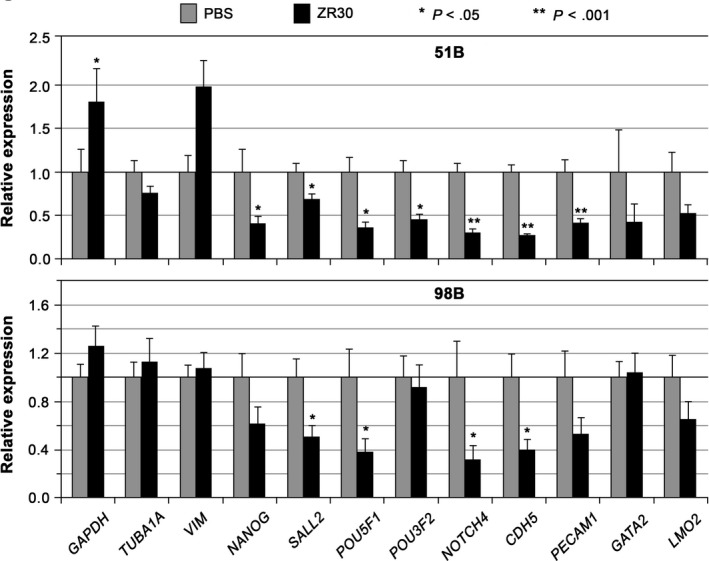
ZR30 downregulates endothelial transdifferentiation genes upregulated after 6 h of culture in Matrigel matrix. Gene expression from cells cultured in medium with or without addition of ZR30 (20 ng for 51B, 100 ng for 98B) in 3D‐Matrigel were compared for effect of ZR30. Columns, geometric means of relative expression (n = 2‐4); bars, SEM
We further examined mesh formation in Matrigel matrix by 51A, 97A, and 98A, the syngeneic primary cultures of 51B, 97B, and 98B, respectively (see Table 1), which were established under NS conditions. No formation of mesh structures was observed for 51A or 97A up to 24 hours. Significantly fewer (approximately one third) meshes were formed by 98A, compared with 98B, 4 hours after placement on the Matrigel matrix. As shown in Figures 3 and 4, ZR30‐mediated suppression of ETD gene upregulation in 98B correlated with its suppression of mesh formation in Matrigel matrix. However, after 6 hours of culture of 51A and 98A cells on Matrigel matrix in medium with/without addition of ZR30, these ETD gene expression levels were not significantly altered.
3.4. ZR30 downregulates ETD genes highly expressed in GBM primary cultures
The above data shows ZR30’s inhibition of Matrigel matrix‐induced ETD gene expression in 51B and 98B cells, especially in cells with low basal expression levels. We then examined the effect of ZR30 on ETD genes in cells with high basal expression levels after a relatively long‐term treatment in conditions that established the cultures. As shown in Figure 5, 4 ETD genes (NANOG, POU5F1, NOTCH4, and CDH5) were significantly downregulated in 97B cells following 25 h of treatment by ZR30 in a dose‐dependent manner.
Figure 5.
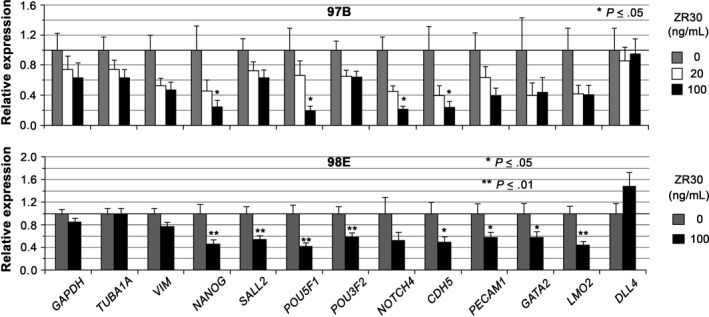
ZR30 downregulates endothelial transdifferentiation genes highly expressed in glioblastoma multiforme cells under their in vitro culture conditions. Relative expression levels in 97B and 98E, normalized to ACTB, and average expression levels of all genes in each culture of no‐treatment were set at unity. For 97B cells, columns are geometric means of relative expression in 25 h of culture with or without ZR30 treatment (n = 2); bars, SEM. For 98E cells, columns are geometric means of relative expression at 1, 2, and 3 wk of culture with or without ZR30 treatment (n = 5‐6); bars, SEM
Hematopoietic gene DDL4 was barely expressed in all SA‐conditioned GBM cells (see Table 1). In contrast, it was moderately expressed (10 000 × [absolute ratio to ACTB] = 22 ± 9.6) in 98E, a syngeneic culture of 98B and 98A established in EC conditions, at a level comparable with embryonic, pluripotency, and hematopoietic developmental genes in GBM primary cultures (see Table 3). Other hematopoietic vascular genes (NOTCH4, PECAM1, and CDH5) were also expressed at a higher level in 98E, compared to 98B. As shown in Figure 5, 8 ETD genes (NANOG, SALL2, POU5F1, POU3F2, CDH5, PECAM1, GATA2, and LMO2) were significantly downregulated by ZR30 in 98E. The effect of ZR30 on suppressing ETD gene expression in 98E cells was shown after 1 week, which was maintained through continuous treatment of cells in the following 2 weekly passages, with further reduction by 50% in PECAM1, 40% in NANOG, and 20%‐30% in SALL2, POU5F1, POU3F2, and CDH5. A 50% reduction in NOTCH1 expression was only shown in 98E after 3 weeks of treatment, and not after a 1 or 2 weeks of treatment.
3.5. ZR30 does not affect GBM cell growth in vitro
Data shown above indicated the transcriptional downregulation of ETD genes in GBM cells by ZR30. We examined its effect on GBM cell proliferation under in vitro culture conditions, by CCK‐8 cell proliferation and Trypan blue exclusion assays. The CCK‐8 assay of GBM cell growth from 1000 cells/well in 96‐well plates for 6‐14 days showed that ZR30 does not suppress 97B, 97A, or 98A cell growth in vitro, with only a slight inhibition on the growth of slow‐growing GBM cells of 97A and 98A during the third week in culture (Figure 6A). We then determined effect of ZR30 on GBM cells over 3 weeks culture of 2 weekly passages, with counting of viable cells by exclusion of Trypan blue. Cell counting was carried out weekly, and doubling time was calculated. As shown in Figure 6B, 98A and 98B cell growth speed remained similar over 3 weeks of culture, whereas 98E and 97A cell growth speed was slowed down in the third week of culture (passage 2). ZR30 did not affect growth speed of all 4 analyzed GBM primary cultures following 1‐3 weeks of treatment.
Figure 6.
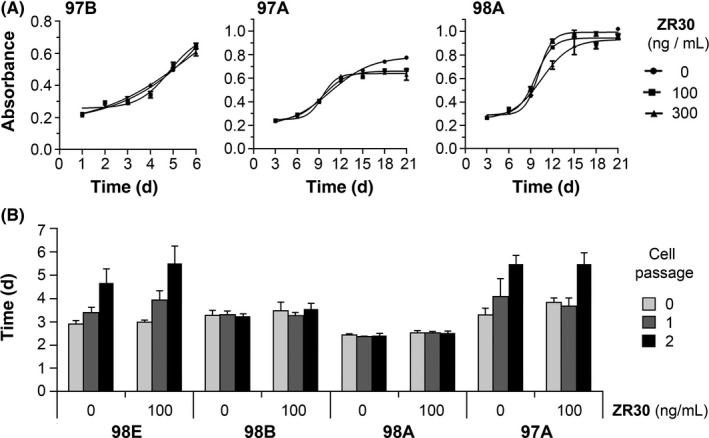
Effect of ZR30 on glioma cell growth. A, CCK‐8 colorimetric assay for the determination of cell growth, measured daily (for 97B) or every 3 days (for 97A and 98A) for absorbance at 450 nm by cells (1000 cells/well in 96‐well plate with 5 replicates in 0.1 mL culture medium with or without ZR30), which were changed after CCK‐8 assay. B, Cell doubling time calculated based on the number of viable cell with exclusion of Trypan blue (0.02%), counted 1 week after seeding 20 000 cells/well in a 12‐well plate with 3 replicates in 1 mL culture medium with or without ZR30. The assay was repeated on cells with or without 2 weekly passages
4. DISCUSSION
Immunohistochemistry analysis following 2 weeks of treatment of GBM i.c. xenografts by ZR30 or PBS showed suppression effects of ZR30 on BVD. Results from in vivo angiogenesis assays using 2 Matrigel plug formulas consistently showed a positive effect of ZR30 in suppression of angiogenesis. As the Ab used for immunohistochemistry recognizes CD31 of both human and mouse, we could not conclude whether ZR30‐induced reduction of BVD in human i.c. GBM xenografts is due to its suppressive effect on angiogenesis, VM, or both.
However, analysis of ZR30’s effect on ETD gene expression showed suppression of most of the upregulated ETD genes in 2 SA‐conditioned cultures (51B and 98B) shortly after placing in Matrigel matrix. ZR30 also caused suppression of ETD gene expression in an EC‐conditioned GBM primary culture (98E) with high expression of vascular marker genes (eg, DLL4), as well as an SA‐conditioned GBM primary culture (97B) with high basal expression levels. The effect of ZR30 on downregulation of ETD‐genes is consistent with suppression of VM.
The GBM subpopulation of cells in 98E harbors a new mutation in the PTEN gene (see Table 1), indicating a later evolved subpopulation with vascular features. Results of this study suggest that tumor cells in a vascular state, as well as those in the proliferation state with plasticity to switch into a vascular state, could have arisen at the advanced stages of cancer evolution, through some of the known selection pressure forces, including an ever‐changing tumor microenvironment, overgrowth, hypoxia, and low pH. The involvement of hypoxia‐related genes in VM10 is consistent with the finding that VM‐forming tumor cells reside within the tumor mass core.
Our current approach in exploring the role of ZR30 on inhibition of vascularization in GBM could not address whether ZR30 could destabilize the existing blood vessels in GBM. The animal experiments with nude mice forming i.c. xenografts showed rapid body weight reduction 1 week after treatment; on average, the reduction was 10% in both PBS and ZR30 treatment groups. This confirms that the treatment was given at the time of the tumors’ aggressive growth. The marked reduction of BVD (70% reduction compared with control) by ZR30 in GBM i.c. xenografts at the stage of aggressive growth supports ZR30’s effect in suppressing new blood vessel formation and possibly destabilizing newly formed blood vessels. This is an intriguing question for a future study.
Both PI3K/AKT signaling and MMP2 have been shown to be related regulators in the formation of vascular‐like networks.23, 24 We have shown ZR30’s effect on inhibition of extracellular activation of MMP2 and attenuation of phosphorylation of AKT in both types of glioma cells with high expression of NOTCH1 or EGFR.5 Along with suppression of GBM cell ETD, these findings suggest at least 3 convergent, but independent, mechanisms of action of ZR30 in suppression of GBM vascularization.
In the last 2 decades, attempts at “starving cancer to death” by targeting tumor blood vessels have failed to improve overall survival.25 The reasons include: (i) failure to see cancer’s capability to change from a high proliferation to high invasion state; and (ii) targeting VEGF and its receptor KDR works on suppressing angiogenesis by normal endothelial cells, but not tumor cells, which have undergone ETD to form VM or vessel cooption, where tumor cells migrate along the preexisting vessels of the host organ.26 ZR30’s dual function in suppression of angiogenesis and VM is unique in targeting tumor vascularization. Hence, ZR30 as a novel cancer therapeutic, directly deliverable into the tumor site and functioning in the extracellular compartment, could effectively treat GBM through its multifaceted and unique mechanisms of action, including suppression of tumor cell growth and invasive features and its supporting vascular microenvironment.
DISCLOSURE
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This work was supported, in part, by a grant provided by the National Natural Science Foundation of China (Grant No. 81672484, 81372685, and 81572475), the Musella Foundation for Brain Tumor Research and Information, generous gifts from the Stern Family and Jasmin Jahanshahi Fire Safety Foundation via MAKAN, the National Basic Research Program of China (973, No. 2015CB755505), the Guangzhou Science Technology Project (No. 201508020125), and the Science and Technology Planning Project of Guangdong Province (No. 2016A020213004).
Ke C, Luo J‐r, Cen Z‐w, et al. Dual antivascular function of human fibulin‐3 variant, a potential new drug discovery strategy for glioblastoma. Cancer Sci. 2020;111:940–950. 10.1111/cas.14300
Ke and Luo are equal contributing authors.
Contributor Information
Zhong‐ping Chen, Email: chenzp57@mail.sysu.edu.cn.
Yi‐Hong Zhou, Email: yihongz@uci.edu.
REFERENCES
- 1. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017;19:v1‐v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu Y, Ke C, Ru N, et al. Cell context‐dependent dual effects of EFEMP1 stabilizes subpopulation equilibrium in responding to changes of in vivo growth environment. Oncotarget. 2015;6:30762‐30772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou YH, Afrasiabi K, Linskey ME. Extracellular control of chromosomal instability and maintenance of intra‐tumoral heterogeneity. J Cancer Metastas Treat. 2018;4:15. [Google Scholar]
- 4. Zhou YH, Hu Y, Yu L, et al. Weaponizing human EGF‐containing fibulin‐like extracellular matrix protein 1 (EFEMP1) for 21st century cancer therapeutics. Oncoscience. 2016;3:208‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y, Hu Y, Liu C, et al. Human fibulin‐3 protein variant expresses anti‐cancer effects in the malignant glioma extracellular compartment in intracranial xenograft models. Oncotarget. 2017;8:106311‐106323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albig AR, Neil JR, Schiemann WP. Fibulins 3 and 5 antagonize tumor angiogenesis in vivo. Cancer Res. 2006;66:2621‐2629. [DOI] [PubMed] [Google Scholar]
- 7. Chen J, Wei D, Zhao Y, Liu X, Zhang J. Overexpression of EFEMP1 correlates with tumor progression and poor prognosis in human ovarian carcinoma. PLoS ONE. 2013;8:e78783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seeliger H, Camaj P, Ischenko I, et al. EFEMP1 expression promotes in vivo tumor growth in human pancreatic adenocarcinoma. Mol Cancer Res. 2009;7:189‐198. [DOI] [PubMed] [Google Scholar]
- 9. Hu Y, Pioli PD, Siegel E, et al. EFEMP1 suppresses malignant glioma growth and exerts its action within the tumor extracellular compartment. Mol Cancer. 2011;10:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726‐2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagenblast E, Soto M, Gutierrez‐Angel S, et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature. 2015;520:358‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williamson SC, Metcalf RL, Trapani F, et al. Vasculogenic mimicry in small cell lung cancer. Nat Commun. 2016;7:13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang JP, Liao YD, Mai DM, et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: a meta‐analysis. Angiogenesis. 2016;19:191‐200. [DOI] [PubMed] [Google Scholar]
- 14. Wang SY, Ke YQ, Lu GH, et al. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J Neurooncol. 2013;112:339‐345. [DOI] [PubMed] [Google Scholar]
- 15. Jhaveri N, Chen TC, Hofman FM. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016;380:545‐551. [DOI] [PubMed] [Google Scholar]
- 16. Li C, Chen YS, Zhang QP, et al. Vasculogenic mimicry persists during glioblastoma xenograft growth. Glioma. 2018;1:16‐21. [Google Scholar]
- 17. Mei X, Chen YS, Chen FR, Xi SY, Chen ZP. Glioblastoma stem cell differentiation into endothelial cells evidenced through live‐cell imaging. Neuro Oncol. 2017;19:1109‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arbab AS, Jain M, Achyut BR. Vascular mimicry: The next big glioblastoma target. Biochem Physiol. 2015;4:e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou YH, Chen Y, Hu Y, et al. The role of EGFR double minutes in modulating the response of malignant gliomas to radiotherapy. Oncotarget. 2017;8:80853‐80868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Passaniti A, Taylor RM, Pili R, et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest. 1992;67:519‐528. [PubMed] [Google Scholar]
- 22. Suva ML, Rheinbay E, Gillespie SM, et al. Reconstructing and reprogramming the tumor‐propagating potential of glioblastoma stem‐like cells. Cell. 2014;157:580‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hess AR, Seftor EA, Seftor RE, Hendrix MJ. Phosphoinositide 3‐kinase regulates membrane Type 1‐matrix metalloproteinase (MMP) and MMP‐2 activity during melanoma cell vasculogenic mimicry. Cancer Res. 2003;63:4757‐4762. [PubMed] [Google Scholar]
- 24. Chiablaem K, Lirdprapamongkol K, Keeratichamroen S, Surarit R, Svasti J. Curcumin suppresses vasculogenic mimicry capacity of hepatocellular carcinoma cells through STAT3 and PI3K/AKT inhibition. Anticancer Res. 2014;34:1857‐1864. [PubMed] [Google Scholar]
- 25. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donnem T, Hu J, Ferguson M, et al. Vessel co‐option in primary human tumors and metastases: an obstacle to effective anti‐angiogenic treatment? Cancer Med. 2013;2:427‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]