

Abstract
Ionizing radiation can damage DNA and, therefore, is a risk factor for cancer. Eker rats, which carry a heterozygous germline mutation in the tumor‐suppressor gene tuberous sclerosis complex 2 (Tsc2), are susceptible to radiation‐induced renal carcinogenesis. However, the molecular mechanisms involved in Tsc2 inactivation are unclear. We subjected Fischer 344 × Eker (Long Evans Tsc2 +/−) F1 hybrid rats to gamma‐irradiation (2 Gy) at gestational day 19 (GD19) or postnatal day 5 (PND5) and investigated the patterns of genomic alterations in the Tsc2 allele of renal tumors that developed at 1 year after irradiation (N = 24 tumors for GD19, N = 10 for PND5), in comparison with spontaneously developed tumors (N = 8 tumors). Gamma‐irradiation significantly increased the multiplicity of renal tumors. The frequency of LOH at the chromosome 10q12 region, including the Tsc2 locus, was 38%, 29% and 60% in renal carcinomas developed from the nonirradiated, GD19 and PND5 groups, respectively. Array comparative genomic hybridization analysis revealed that the LOH patterns on chromosome 10 in renal carcinomas were classified into chromosomal missegregation, mitotic recombination and chromosomal deletion types. LOH of the interstitial chromosomal deletion type was observed only in radiation‐associated carcinomas. Sequence analysis for the wild‐type Tsc2 allele in the LOH‐negative carcinomas identified deletions (nonirradiated: 26%; GD19: 21%) and base‐substitution mutations (GD19: 4%). Reduced expression of Tsc2 was also observed in the majority of the LOH‐negative carcinomas. Our results suggest that interstitial chromosomal deletion is a characteristic mutagenic event caused by ionizing radiation, and it may contribute to the assessment of radiation‐induced cancer risk.
Keywords: Eker rat, genomic signature, ionizing radiation, renal carcinoma, Tsc2
In this study, we used the Tsc2 heterozygous mutant Eker rat model to investigate the effects of radiation on the development of renal tumors and genomic alterations in those tumors. We found that in renal cancers of Eker rats, the tumor‐suppressor gene Tsc2 was inactivated by multiple mechanisms, including LOH with mitotic recombination or chromosomal deletion, point mutation, deletion and reduced Tsc2 expression. Importantly, our results suggest that interstitial chromosomal deletion is a characteristic mutagenic event caused by ionizing radiation, and it may be a useful indicator for more precise assessment of ionizing radiation‐induced cancer risk.

1. INTRODUCTION
Ionizing radiation is a mutagenic agent. Epidemiological studies of atomic‐bomb survivors and victims of the Chernobyl accident have demonstrated that exposure to ionizing radiation increases the risk of cancer.1, 2, 3, 4 Recent studies have suggested that computed tomography scans administered during childhood increase the risk of cancer development later in life.5, 6, 7 It has also been reported that long‐term cancer survivors who have received radiotherapy have a higher risk of secondary cancers compared with the general population.8, 9, 10, 11
Genomic alterations caused by the mutagenic effects of ionizing radiation can lead to cancer.12, 13, 14 Notably, amplification of several oncogenes, including C‐Myc, HER2 and CLIP2, has been more frequently observed in breast cancers of atomic bomb survivors and in papillary thyroid cancers of victims of the Chernobyl accident than in control populations.15, 16 In addition, a high frequency of deletions, balanced inversions and microsatellite alterations has been reported in radiotherapy‐induced secondary cancers.17, 18, 19 These radiation‐associated alterations in the cancer genome may be useful for estimating the risk of developing radiation‐induced cancer.
Identification of signatures of ionizing radiation in human cancers is difficult because cancer occurs as a result of the combined effects of radiation and other physical, chemical and biological factors, such as smoking, diet, ultraviolet radiation, viruses and hormones.20 Animal models are, therefore, important for investigating the contribution of a single factor to carcinogenesis. Characteristic patterns of LOH have been frequently observed in several solid tissues and tumors of mice exposed to ionizing radiation compared with nonirradiated tissues and spontaneous or chemical carcinogen‐induced tumors.21, 22, 23, 24 Eker rats spontaneously develop malignant adenocarcinoma via premalignant proliferative lesions of the renal proximal tubular epithelium (eg, phenotypically altered tubules, atypical hyperplasia and adenoma) with 100% incidence by 1 year of age.25 In these rats, a retrotransposon insertion was found in one allele of the gene tuberous sclerosis complex 2 (Tsc2), which maps to chromosome 10. Inactivation of the remaining wild‐type Tsc2 allele by spontaneous somatic mutation is responsible for renal carcinogenesis in Eker rats.26, 27, 28 Rat Tsc2, which shares 92% sequence similarity with human TSC2,29 encodes tuberin, which contains a GTPase‐activating protein (GAP) domain.30 Tsc2 can associate with GTP‐bound Rheb (a small GTPase and Ras homolog), thereby promoting GTP hydrolysis, which results in inhibition of mTOR activation.31 In cells deficient in TSC2, mTOR activation enhances the phosphorylation of the downstream substrates p70 S6 kinase and 4E‐BP1, which are involved in translation, thereby promoting cell proliferation and carcinogenesis.32, 33 A study using Eker rats demonstrated that exposure to ionizing radiation increases the incidence of renal tumors with a linear dose‐response relationship.34
We previously found that Eker rats are most susceptible to the development of renal tumors when irradiated with 2 Gy of gamma‐rays during the perinatal period.35, 36 The multiplicity of renal tumors developed at the age of 27 weeks was significantly increased in irradiated rats compared with nonirradiated rats. However, we could not perform molecular analyses owing to the small size of tumors. Thus, in this study, we extended the observation period from 27 to 52 weeks to obtain sufficiently large tumors to allow investigation of the patterns of genomic aberrations at the Tsc2 allele in spontaneous and radiation‐associated renal tumors of Eker rats and to identify radiation‐associated mutational events.
2. MATERIALS AND METHODS
2.1. Experimental animals
A total of 59 F1 hybrid rats produced by mating Fischer 344 female rats with Long Evans male rats carrying the heterozygous Eker mutation (Tsc2 +/−) were used in this study. Among them, 13 F1 hybrid rats were used as the control group, while the remaining 26 and 20 F1 hybrid rats were used as gamma‐irradiated at gestational day 19 (GD19) and at postnatal day 5 (PND5) groups, respectively. All experimental protocols in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the National Institute of Radiological Sciences (NIRS; approval no. 07‐1075) and were performed in strict accordance with the NIRS Guide for Care and Use of Laboratory Animals.
2.2. Irradiation and sample preparation
Pregnant rats were either not irradiated (controls) or subjected to whole‐body gamma‐irradiation (2 Gy) at GD19, or the pups were not irradiated or irradiated at PND5. The irradiation was conducted using a Gammacell 40 irradiator containing Cs‐137 source (Atomic Energy of Canada) with a dose rate of 0.7 Gy/min. When the offspring reached 52 weeks of age, both kidneys were removed and fixed in 10% neutral buffered formalin at room temperature for 24 hours and then cut into five transverse sections per kidney. These sections were embedded in paraffin and further sectioned to a thickness of 4 μm for histopathological examination. Renal tumors (>2 mm diameter) removed during dissection were frozen in liquid nitrogen. Genomic DNA and total RNA were isolated from frozen renal tumors using an AllPrep DNA/RNA/Protein Mini Kit (Qiagen).
2.3. Multiplicity of renal proliferative lesions
H&E‐stained kidney sections from nonirradiated rats and from rats irradiated at GD19 or PND5 were used to count the multiplicity of renal proliferative lesions. The renal lesions were classified into three histological types: (a) phenotypically altered tubule; (b) atypical hyperplasia; or (c) adenoma or adenocarcinoma.
2.4. LOH analysis
LOH analysis for the Tsc2 locus and the 10 microsatellite loci spanning rat chromosome 10 were performed as described35 using genomic DNA isolated from 8 tumor samples of nonirradiated rats and 24 and 10 tumor samples of rats irradiated at GD19 and PND5, respectively. PCR for the 10 microsatellite loci was carried out in 25 μL reaction volume containing 50 ng genomic DNA, 0.2 mM dNTP, 1.4 mM MgCl2, 0.2 μM of each primer and 0.625 U rTaq polymerase. The primer sequences and PCR conditions are listed in Table S1. The PCR products were resolved on an acrylamide gel using capillary electrophoresis (HAD‐GT12, eGene, Qiagen).
2.5. Array comparative genomic hybridization analysis
Array comparative genomic hybridization (CGH) analysis was performed using the Agilent Rat Genome CGH Microarray Kit (Agilent Technologies). Fluorescent labeling of DNA, microarray hybridization and post–hybridization washing were conducted as per the manufacturer's protocol. The array images were scanned using an Agilent Microarray Scanner (G2565CA), and signal intensities were measured with Feature Extraction software ver. 10.5.1. The obtained data were imported into Agilent Genomic Workbench 6.5 and analyzed using the Aberration Detection Method 2 algorithm.
2.6. Sequence analysis
Reverse transcription was performed to generate cDNA using 5 μg of total RNA, ReverTra Ace (Toyobo) and random primers. Then, the cDNA was amplified using four primer pairs covering the coding region of Tsc2. PCR was carried out in 25 μL reaction volume containing 0.2 μM of each primer, 0.2 mM dNTP, 1.5 mM MgCl2, 0.625 U AmpliTaq Gold polymerase (Perkin Elmer Japan) and 1 μL cDNA. The primer sequences and PCR conditions are listed in Table S1. PCR products were resolved by 2% agarose gel electrophoresis and then sequenced directly using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). To identify the mutations, the cDNA sequences were compared with the rat Tsc2 gene sequence registered in the GenBank database (accession: NM_012680.3).
2.7. Quantitative RT‐PCR
Quantitative RT‐PCR was performed on an Mx3000P real‐time PCR system (Agilent Technologies) using SYBR Premix Ex Taq (Takara Bio Inc). Analysis of the cDNA sequence revealed a splice‐site variant lacking exon 32 that was predominantly expressed in almost all tumors without LOH or mutation in Tsc2 (18 of 19 cases). We therefore examined the expression of this variant as a Tsc2 wild‐allele‐derived transcript. The expression of the housekeeping gene Actin was used to normalize the data. The primer sequences and PCR conditions are listed in Table S1. Relative gene expression was calculated using the method.24 A mean value of Tsc2 expression derived from the kidneys of 6 Eker rats with Tsc2 +/+ (52 weeks of age) was used as a reference value.
2.8. Immunohistochemistry
Fixed sections were deparaffinized through a graded ethanol series, rehydrated, and washed with TBS containing 0.1% (w/v) Tween 20. Sections were then incubated overnight at 4°C with the following primary antibodies at the indicated dilution: (a) phospho‐Akt1 (Ser473) (D9E) rabbit mAb (1:25) (Cell Signaling Technology); (b) phospho‐mTOR (Ser2448) (49F9) rabbit mAb (1:50) (Cell Signaling Technology); (c) phospho‐S6 ribosomal protein (Ser235/236) (91B2) rabbit mAb (1:75) (Cell Signaling Technology); and (d) phospho‐4E‐BP1 (Thr37/46) (236B4) rabbit mAb (1:400) (Cell Signaling Technology). Bound antibodies were visualized with a Vectastain Elite ABC Kit (Vector Laboratories) using a biotinylated secondary antibody against rabbit IgG and diaminobenzidine as a chromogen. All sections were then counterstained with hematoxylin.
2.9. Statistical analysis
Statistical analysis was performed with the Wilcoxon rank sum test to assess the significance of differences in the multiplicity of renal lesions between groups. The differences were considered significant at values of P < 0.05.
3. RESULTS
3.1. Multiplicity of renal proliferative lesions in Eker rats exposed to ionizing radiation
Representative histopathological images of renal lesions from irradiated Tsc2 +/− F1 rats are shown in Figure 1. Lesion histology did not differ between the nonirradiated and irradiated groups. The multiplicities of renal lesions for each group are shown in Table 1. Compared with the nonirradiated group, the multiplicity of phenotypically altered tubules was significantly greater in males irradiated at PND5 (P < 0.05). The multiplicity of atypical hyperplasias was significantly greater in both males and females irradiated at GD19 and PND5 (P < 0.05). The multiplicity of renal tumors (adenoma and adenocarcinoma) was significantly greater in males and females irradiated at GD19 and PND5 (P < 0.05). These results indicated that ionizing radiation has a carcinogenic effect in the Tsc2 +/− rats and increases the total number of renal tumors at 52 weeks of age by hastening the advance of renal carcinogenesis.
Figure 1.
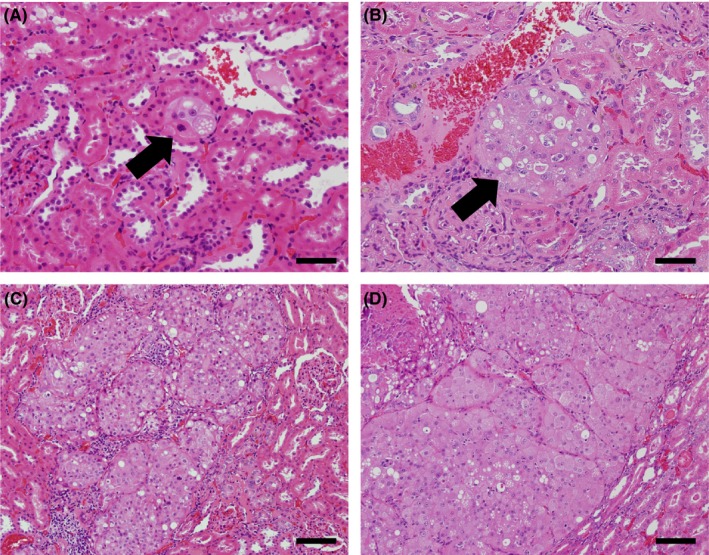
Representative histopathological identification of lesions in the kidneys of gamma‐irradiated Tsc2 +/− F1 rats. H&E‐stained sections revealed phenotypically altered tubules (A), atypical hyperplasia (B), adenoma (C) and adenocarcinoma (D). A phenotypically altered tubule and atypical hyperplasia are indicated by arrows. Scale bars, in (A) and (B), 50 μm; (C) and (D), 100 μm
Table 1.
Multiplicity of renal proliferative lesions in Tsc2 +/− F1 rats with or without exposure to gamma radiation
| N | Phenotypically altered tubule | Atypical hyperplasia | Adenoma and adenocarcinoma | |
|---|---|---|---|---|
| Multiplicity (number of lesions in five kidney sections/animal) | ||||
| Male | ||||
| Control (0 Gy) | 8 | 30.1 ± 10.0a | 4.1 ± 3.4 | 2.9 ± 1.5a |
| GD19 (2 Gy) | 8 | 33.4 ± 15.0a | 25.8 ± 12.7*,a | 7.6 ± 1.6*,a |
| PND5 (2 Gy) | 9 | 73.6 ± 20.6*,† | 54.0 ± 12.6*,†,a | 6.2 ± 2.8*,a |
| Female | ||||
| Control (0 Gy) | 5 | 50.8 ± 18.6 | 3.4 ± 2.3 | 0.4 ± 0.5 |
| GD19 (2 Gy) | 18 | 48.1 ± 21.5 | 9.7 ± 5.0* | 3.3 ± 1.7* |
| PND5 (2 Gy) | 11 | 66.0 ± 14.8† | 18.8 ± 5.0*,† | 2.2 ± 1.3* |
| Male and female | ||||
| Control (0 Gy) | 13 | 38.1 ± 16.8 | 3.8 ± 2.9 | 1.9 ± 1.7 |
| GD19 (2 Gy) | 26 | 43.5 ± 21.4 | 14.6 ± 10.9* | 4.6 ± 2.6* |
| PND5 (2 Gy) | 20 | 69.4 ± 17.6*,† | 34.7 ± 20.1*,† | 4.0 ± 2.9* |
Data represent the mean ± SD.
GD, gestational day; PND, postnatal day.
P <0 .05, Control vs gestational day (GD19) or postnatal day 5 (PND5); † P <0 .05, GD19 vs PND5; a P < 0.05, male vs female.
3.2. Frequency and pattern of LOH on chromosome 10
As shown in Figure 2A, the frequency of LOH at the Tsc2 locus in spontaneous tumors and tumors developed after irradiation at GD19 and PND5 was 38% (3 of 8), 29% (7 of 24) and 60% (6 of 10), respectively. Three distinct LOH patterns were detected: (a) LOH spanning from locus D10MIT14 to locus D10RAT7 (covering the entire chromosome 10) (pattern a); (b) LOH spanning from the Tsc2 locus to the distal locus (D10RAT7) (pattern b); and (c) LOH ranging from the Tsc2 locus to the center of the chromosome (D10RAT32) (pattern c). Interestingly, pattern c was observed only in irradiated groups, whereas patterns a and b were observed in all experimental groups. To evaluate the pattern of genomic copy‐number aberrations in the tumors, 1 tumor sample of each LOH type was chosen from each group (nonirradiated: 2, irradiated at GD19: 3, PND5: 3) for array CGH analysis (Figure 2B). Tumor samples with continuous LOH patterns a and b had normal copy numbers throughout chromosome 10, indicating that the LOH was caused by chromosomal missegregation or mitotic recombination. In contrast, genomic copy‐number losses spanning from chromosome 10q12 to q24 were detected in the tumor samples exhibiting pattern c from groups GD19 and PND5, indicating that this LOH was caused by interstitial deletion. Notably, this interstitial chromosomal deletion was not found when we examined LOH in an additional 11 spontaneous renal tumors from fixed kidney sections (data not shown). These results suggested that there is a quality difference in the LOH patterns between spontaneous and radiation‐associated tumors and that the LOH with interstitial chromosomal deletion is a signature for radiation‐associated tumors.
Figure 2.
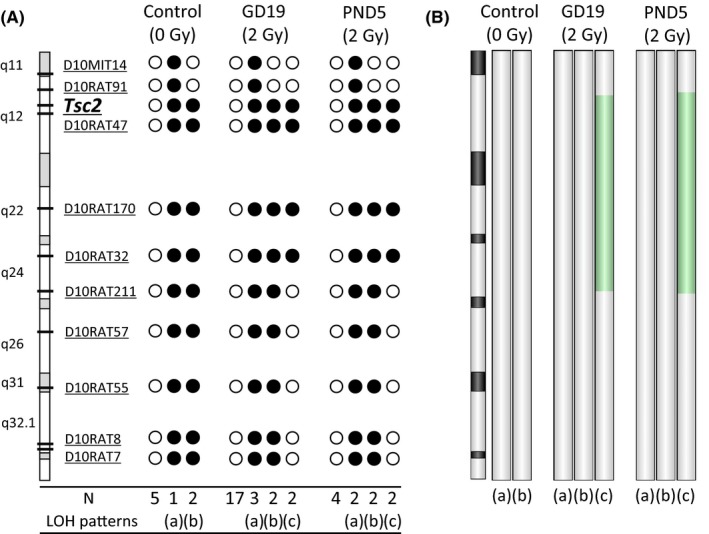
LOH patterns reveal different mutational events in spontaneous and radiation‐associated renal tumors from Tsc2 +/− F1 rats. A, Polymorphic markers and their positions are shown in the chromosome schematic. Closed circles (●) represent LOH. N, number of cases that have the same LOH pattern. B, Changes in DNA copy number revealed by array comparative genomic hybridization (CGH) analysis. Deleted regions detected by array CGH analysis are shown in green
3.3. Tsc2 sequence analysis
Sequence analysis of Tsc2 in LOH‐negative tumor samples (5, 17 and 4 samples from nonirradiated control, GD19 and PND5 groups, respectively) was performed to identify the mutations occurring in the wild‐type Tsc2 allele. As shown in Table 2 and Figure 3, two and six mutations were identified in the tumors of the nonirradiated and GD19 groups, respectively. Among them, seven mutations were deletions and the eighth was a base substitution. One sample (TK718) had two deletions in the Tsc2 gene. Five of the eight mutations were predicted to produce aberrant truncated proteins owing to a frameshift. These results suggested that there is no qualitative difference in the patterns of the Tsc2 mutations between spontaneous and radiation‐associated tumors and that deletion is the major type of mutation that causes the inactivation of Tsc2 function in tumors.
Table 2.
Mutations in Tsc2 in spontaneous and radiation‐associated renal tumors
| Number | Group | Sample ID | DNA | Exon | Protein alteration | Size of deletion (bp) |
|---|---|---|---|---|---|---|
| 1 | Control (0 Gy) | TK711 | c.1120_1257del | 12 | p.Asn374_Pro419del | 138 |
| 2 | TK727 | c.4387delC | 34 | p.Arg1463Glu, fs17 | 1 | |
| 3 | GD19 (2 Gy) | TK756 | c.444_445delCA | 5 | p.Asp148Glu, fs44 | 2 |
| 4 | TK718 | c.1802_2541del | 17 | p.Leu604Thr, fs962 | 740 | |
| 5 | TK718 | c.4735_4846del | 37 | p.Gly1579Ser, fs11 | 112 | |
| 6 | TK709 | c.3351delG | 29 | p.Gln1118Ser, fs73 | 1 | |
| 7 | TK719 | c.4943T > A | 38 | p.Val1648Asp | n.a. | |
| 8 | TK708 | c.4996_5074del | 39 | p.Gly1666_Asn1722del | 79 |
del, deletion; GD19, gestational day 19; fs, frameshift; n.a., not applicable.
Figure 3.
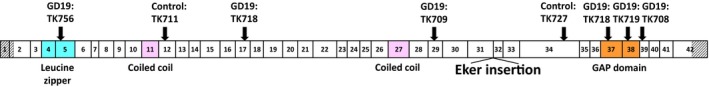
Distribution of Tsc2 mutations in spontaneous and radiation‐associated rat renal tumors. A schematic depicts the structure of rat Tsc2, including 42 numbered exons and the locations of the functional domains (colored). Hatched boxes indicate the 5′ and 3′ UTR. Arrows indicate the positions of the identified mutations
3.4. Expression of Tsc2
To examine the downregulation of Tsc2 in the LOH‐negative or Tsc2 mutation‐negative tumors, expression of a remaining Tsc2 wild‐allele‐derived splicing variant was analyzed in LOH‐negative or Tsc2 mutation‐negative renal tumor samples (3, 12 and 4 samples from nonirradiated control, GD19 and PND5 groups, respectively) as well as normal kidney tissues (Figure 4). The expression of Tsc2 was less than half of the average level of normal kidney tissues in 67% (2 of 3), 58% (7 of 12) and 100% (4 of 4) of the tumors of the nonirradiated, GD19 and PND5 groups, respectively. These results suggested that Tsc2 expression is downregulated in the majority of tumors without LOH or mutation in Tsc2, suggesting that inactivation of Tsc2 may involve an epigenetic mechanism.
Figure 4.
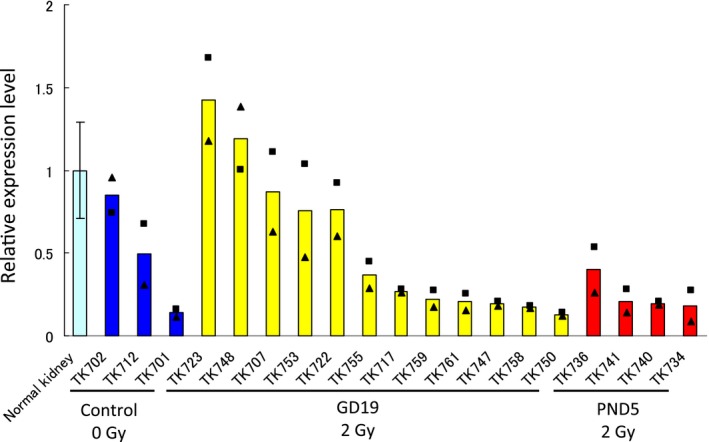
Quantitative RT‐PCR assessment of Tsc2 expression in normal rat kidney tissues, spontaneous renal tumors and radiation‐associated renal tumors. The data for normal kidney tissues (the mean ± SD of 6 samples) and for renal tumors (the average of two independent experiments, ■ and ▲) are shown. The data for tumors were normalized to the average expression value for normal kidney tissues
3.5. Activation of the mTOR pathway
To examine the activation of the mTOR pathway in the LOH‐negative renal tumors, immunohistochemical staining of several regulators in the mTOR pathway was assessed. Representative images of phospho‐Akt1 (Ser473), phospho‐mTOR (Ser2448), phospho‐4E‐BP1 (Thr37/46), and phospho‐S6 ribosomal protein (Ser235/236) staining in renal adenocarcinomas are shown in Figure S1. As expected, positive staining for these regulators was observed in all tumors, indicating that the mTOR pathway was activated in the LOH‐negative renal tumors.
4. DISCUSSION
The present study aimed to investigate genomic aberrations at the Tsc2 allele in radiation‐associated renal tumors of Tsc2 +/− rats. We showed that: (a) the multiplicity of renal tumors that had developed in irradiated rats by 52 weeks of age significantly increased compared with that of nonirradiated rats; (b) LOH at the Tsc2 locus and mutations of Tsc2 were identified in the renal tumors; (c) interstitial chromosomal deletion on chromosome 10, including the Tsc2 locus, was found only in the tumors of irradiated rats; and (d) Tsc2 expression in the majority of tumors was lower than that of normal kidney tissues.
The multiplicity of atypical hyperplasia and renal tumors was significantly increased in the irradiated groups compared with the nonirradiated control group (Table 1). Consistent with our previous study,35, 36 this result indicates that the actively growing kidney during the perinatal period is susceptible to radiation‐induced renal carcinogenesis. However, in our previous observations of nonirradiated rats at 27 weeks of age, female rats were more predisposed to renal preneoplastic lesions than were males, whereas no difference in the multiplicity of neoplastic lesions was observed between male and female rats.35, 36 Consistent with this finding, estrogen has been shown to promote the development of preneoplastic lesions in kidneys of female Eker rats and to inhibit the progression of human renal carcinoma cells through activation of estrogen receptor β.37, 38 In the present study, by extending the observation period from 27 to 52 weeks, we found that the multiplicity of neoplastic lesions was more prevalent in male rats than in female rats at 52 weeks of age. In humans, a similar gender difference is observed in kidney cancer, where the cancer incidence is twofold higher in men than in women.39 In this regard, it has been speculated that androgen promotes tumorigenesis and progression in kidneys, whereas estrogen inhibits tumorigenesis/progression.39
LOH is a major mechanism for inactivation of tumor‐suppressor genes in cancer. In this study, LOH at the Tsc2 locus was detected in both spontaneous (38%) and radiation‐associated tumor samples (29‐60%) (Figure 2A.). The frequency of LOH did not differ significantly between the tumors. Consistent with our results, other studies using Eker rats have reported that the frequencies of LOH at the Tsc2 locus are 44%‐60% in spontaneous40, 41 and 29% in radiation‐induced renal tumors.42
Interestingly, the interstitial LOH pattern has also been reported in some radiation‐induced tumors, including mice models of medulloblastoma in heterozygous Ptch1 23, 24 and of intestinal tumors in heterozygous Apc. 43, 44 A high frequency of the interstitial LOH pattern at the tumor‐suppressor Ikaros locus has also been reported in radiation‐induced thymic lymphomas in mice, compared with spontaneously developed or alkylating agent‐induced lymphomas.21 Furthermore, using mouse kidney cell lines, it has been reported that the interstitial LOH pattern is induced as a delayed effect of gamma, proton and iron‐ion radiation exposure.45 In those studies, however, it was not clear whether the LOH was caused by chromosomal missegregation, mitotic recombination or deletion. Therefore, in the present study we examined the copy number of the Tsc2 locus by array CGH analysis and found that LOH of the interstitial deletion type was observed only in tumors of irradiated groups (Figure 2B). Because the interstitial deletion is likely the result of the misrejoining of two DNA double‐strand breaks induced by ionizing radiation within the same chromosome,46 these data indicate that interstitial chromosomal deletion is a radiation‐specific mutational event.
Kobayashi et al reported Tsc2 mutations in 33% (7 of 21) of spontaneous and 33% (3 of 9) of ethylnitrosourea‐induced renal tumors without LOH in Eker rats.29 They also reported that deletion, duplication or base‐substitution mutations in Tsc2 were observed in spontaneous tumors, whereas only base‐substitution mutations were observed in ethylnitrosourea‐induced tumors. In our study, sequence analysis of Tsc2 in LOH‐negative tumor samples identified two deletions in spontaneous tumors and five deletions and one base substitution in radiation‐associated tumors (Table 2). These mutations were predicted to produce truncated proteins, except for 1 sample, which produced an aberrant protein. It should be noted that, in the present study, small deletions were found in the tumors in both nonirradiated and irradiated groups. In addition, no difference in the size of the deletions was observed between the groups. Given that small deletions are also frequently observed as spontaneous mutations in other models,47, 48, 49, 50 our data suggest that small deletions are induced by ionizing radiation but not a specific mutational event for radiation exposure.
Approximately 50% of the tumor samples we assessed had no Tsc2 mutations. This result was unexpected because tumorigenesis in this model is associated with either the loss or the inactivation of a wild‐type allele according to Knudson's two‐hit theory.51 However, we found that Tsc2 expression was downregulated in the majority of tumors (Figure 4). Activation of the Tsc2‐mTOR pathway in these tumors was also confirmed by immunohistochemistry (Figure S1). These results indicate the involvement of epigenetic alterations in the inactivation of the Tsc2 wild‐type allele. In fact, promoter DNA hypermethylation and silencing of TSC2 have been reported in human oral squamous cell cancer and acute leukemia.52, 53
Concerning the difference in the dependency of age‐at‐irradiation, high LOH frequency was observed in the PND5 group (60%, 6 of 10) compared with that in the GD19 group (29%, 7 of 24) (Figure 2). In contrast, the Tsc2 mutation frequency in the GD19 group (29%, 5 of 17) was higher than that in the PND 5 group (0%, 0 of 4) (Table 2). Although, in both cases, no statistically significant difference was observed between the groups, radiation exposure to actively growing kidney cells at perinatal stage (GD19) and subsequent perturbation of normal kidney development35 may be involved in the increase in the Tsc2 gene mutations and other mechanisms, such as the promoter DNA hypermethylation, resulting in the decrease of LOH frequency, in the GD19 group.
In conclusion, our study identified the genomic mutational events for the inactivation of the tumor‐suppressor gene Tsc2 in renal carcinogenesis of Eker rats exposed to ionizing radiation. We also identified interstitial chromosomal deletions in the tumors as a mutagenic event induced by radiation exposure. Identification of radiation‐associated signatures will inform the assessment of cancer risk caused by ionizing radiation.
DISCLOSURE
The authors declare that there are no conflicts of interest.
Supporting information
ACKNOWLEDGEMENTS
The authors thank the Laboratory Animal and Genome Sciences Section, NIRS, National Institutes for Quantum and Radiological Science and Technology (QST), for animal facility management. This work was supported in part by a Grant‐in‐Aid for Scientific Research (A) (15H01834) from the Japan Society for the Promotion of Science. The animals used in this experiment are part of the J‐SHARE (Japan Storehouse of Animal Radiobiology Experiments) project under construction by the NIRS, QST.
Inoue T, Kokubo T, Daino K, et al. Interstitial chromosomal deletion of the tuberous sclerosis complex 2 locus is a signature for radiation‐associated renal tumors in Eker rats. Cancer Sci. 2020;111:840–848. 10.1111/cas.14307
Contributor Information
Toshiaki Kokubo, Email: kokubo.toshiaki@qst.go.jp.
Shizuko Kakinuma, Email: kakinuma.shizuko@qst.go.jp.
REFERENCES
- 1. Ozasa K. Epidemiological research on radiation‐induced cancer in atomic bomb survivors. J Radiat Res. 2016;57(suppl 1):i112‐i117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grant EJ, Brenner A, Sugiyama H, et al. Solid cancer incidence among the life span study of atomic bomb survivors: 1958–2009. Radiat Res. 2017;187:513‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cardis E, Kesminiene A, Ivanov V, et al. Risk of thyroid cancer after exposure to 131I in childhood. J Natl Cancer Inst. 2005;97:724‐732. [DOI] [PubMed] [Google Scholar]
- 4. Zablotska LB, Ron B, Rozhko AV, et al. Thyroid cancer risk in Belarus among children and adolescents exposed to radioiodine after the Chornobyl accident. Br J Cancer. 2011;104:181‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brenner DJ, Hall EJ. Computed tomography–an increasing source of radiation exposure. N Engl J Med. 2007;357:2277‐2284. [DOI] [PubMed] [Google Scholar]
- 6. Mathews JD, Forsythe AV, Brady Z, et al. Cancer risk in 680 000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. BMJ. 2013;346:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pearce MS, Salotti JA, Little MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet. 2015;380:499‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newhauser WD, de Gonzalez AB, Schulte R, Lee C. A review of radiotherapy‐induced late effects research after advanced technology treatments. Front Oncol. 2016;6:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Friedman DL, Whitton J, Leisenring W, et al. Subsequent neoplasms in 5‐year survivors of childhood cancer: the childhood cancer survivor study. J Natl Cancer Inst. 2010;102:1083‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Gonzalez AB, Gilbert E, Curtis R, et al. Second solid cancers after radiotherapy: a systematic review of the epidemiological studies of the radiation dose‐response relationship. Int J Radiat Oncol Biol Phys. 2013;86:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newhauser WD, Durante M. Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer. 2011;11:438‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mukherjee D, Coates PJ, Lorimore SA, Wright EG. Responses to ionizing radiation mediated by inflammatory mechanisms. J Pathol. 2014;232:289‐299. [DOI] [PubMed] [Google Scholar]
- 13. Sokolov M, Neumann R. Global gene expression alterations as a crucial constituent of human cell response to low doses of ionizing radiation exposure. Int J Mol Sci. 2015;17:55‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szumiel I. Ionizing radiation‐induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol 2015;91:1‐12. [DOI] [PubMed] [Google Scholar]
- 15. Miura S, Nakashima M, Ito M, et al. Significance of HER2 and C‐MYC oncogene amplifications in breast cancer in atomic bomb survivors. Cancer. 2008;112:2143‐2151. [DOI] [PubMed] [Google Scholar]
- 16. Hess J, Thomas G, Braselmann H, et al. Gain of chromosome band 7q11 in papillary thyroid carcinomas of young patients is associated with exposure to low‐dose irradiation. Proc Natl Acad Sci USA. 2011;108:9595‐9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonin‐Laurent N, Gibaud A, Huygue M, et al. Specific TP53 mutation pattern in radiation‐induced sarcomas. Carcinogenesis. 2006;27:1266‐1272. [DOI] [PubMed] [Google Scholar]
- 18. Behrens C, Travis LB, Wistuba II, et al. Molecular changes in second primary lung and breast cancers after therapy for Hodgkin's disease. Cancer Epidemiol Biomarkers Prev. 2000;9:1027‐1035. [PubMed] [Google Scholar]
- 19. Behjati S, Gundem G, Wedge DC, et al. Mutational signatures of ionizing radiation in second malignancies. Nat Commun. 2016;7:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimada Y, Nishimura M, Kakinuma S, et al. Combined effect of ionizing radiation and alkylating agents on cancer induction. Genes Environ. 2007;29:29‐37. [Google Scholar]
- 21. Shimada Y, Nishimura M, Kakinuma S, et al. Radiation‐associated loss of heterozygosity at the Znfn1a1 (Ikaros) locus on chromosome 11 in murine thymic lymphomas. Radiat Res. 2000;154:293‐300. [DOI] [PubMed] [Google Scholar]
- 22. Ponomareva ON, Rose JA, Lasarev M, Rasey J, Turker MS. Tissue‐specific deletion and discontinuous loss of heterozygosity are signatures for the mutagenic effects of ionizing radiation in solid tissues. Cancer Res. 2002;62:1518‐1523. [PubMed] [Google Scholar]
- 23. Tsuruoka C, Blyth BJ, Morioka T, et al. Sensitive detection of radiation‐induced medulloblastomas after acute or protracted gamma‐ray exposures in Ptch1 heterozygous mice using a radiation‐specific molecular signature. Radiat Res. 2016;186:407‐414. [DOI] [PubMed] [Google Scholar]
- 24. Ishida Y, Takabatake T, Kakinuma S, et al. Genomic and gene expression signatures of radiation in medulloblastomas after low‐dose irradiation in Ptch1 heterozygous mice. Carcinogenesis. 2010;31:1694‐1701. [DOI] [PubMed] [Google Scholar]
- 25. Hino O, Kobayashi T, Momose S, Kikuchi Y, Adachi H, Okimoto K. Renal carcinogenesis: genotype, phenotype and dramatype. Cancer Sci. 2003;94:142‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG. Predisposition to renal carcinoma in the Eker rat is determined by germ‐line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci USA. 1994;91:11413‐11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kobayashi T, Hirayama Y, Kobayashi E, Kubo Y, Hino O. A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat Genet. 1995;9:70‐74. [DOI] [PubMed] [Google Scholar]
- 28. Kobayashi T, Urakami S, Cheadle JP, et al. Identification of a leader exon and a core promoter for the rat tuberous sclerosis 2 (Tsc2) gene and structural comparison with the human homolog. Mamm Genome. 1997;8:554‐558. [DOI] [PubMed] [Google Scholar]
- 29. Kobayashi T, Urakami S, Hirayama Y, et al. Intragenic Tsc2 somatic mutations as Knudson's second hit in spontaneous and chemically induced renal carcinomas in the Eker rat model. Jpn J Cancer Res. 1997;88:254‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573‐576. [DOI] [PubMed] [Google Scholar]
- 31. Inoki K, Guan KL. Tuberous sclerosis complex, implication from a rare genetic disease to common cancer treatment. Hum Mol Genet. 2009;18:R94‐R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2016;25:545‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex‐2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3‐kinase/Akt pathway. Mol Cell. 2002;10:151‐162. [DOI] [PubMed] [Google Scholar]
- 34. Hino O, Klein‐Szanto AJ, Freed JJ, et al. Spontaneous and radiation‐induced renal tumors in the Eker rat model of dominantly inherited cancer. Proc Natl Acad Sci USA. 1993;90:327‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kokubo T, Kakinuma S, Kobayashi T, et al. Age dependence of radiation‐induced renal cell carcinomas in an Eker rat model. Cancer Sci. 2010;101 616‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morioka T, Blyth BJ, Imaoka T, et al. Establishing the Japan‐Store house of animal radiobiology experiments (J‐SHARE), a large‐scale necropsy and histopathology archive providing international access to important radiobiology data. Int J Radiat Biol. 2019;95:1372‐1377. [DOI] [PubMed] [Google Scholar]
- 37. Wolf DC, Goldsworthy TL, Donner EM, Harden R, Fitzpatrick B, Everitt JI. Estrogen treatment enhances hereditary renal tumor development in Eker rats. Carcinogenesis. 1998;19:2043‐2047. [DOI] [PubMed] [Google Scholar]
- 38. Yu C, Ho J, Huang Y, et al. Estrogen inhibits renal cell carcinoma cell progression through estrogen receptor‐b activation. PLoS ONE. 2013;8:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lucca I, Klatte T, Fajkovic H, Martino MD, Shariat SF. Gender differences in incidence and outcomes of urothelial and kidney cancer. Nat Rev Urol. 2015;12:585‐592. [DOI] [PubMed] [Google Scholar]
- 40. Yeung RS, Xiao GH, Everitt JI, Jin F, Walker CL. Allelic loss at the tuberous sclerosis 2 locus in spontaneous tumors in the Eker rat. Mol Carcinog. 1995;14:28‐36. [DOI] [PubMed] [Google Scholar]
- 41. Kubo Y, Mitani H, Hino O. Allelic loss at the predisposing gene locus in spontaneous and chemically induced renal cell carcinomas in the Eker rat. Cancer Res. 1994;54:2633‐2635. [PubMed] [Google Scholar]
- 42. Hino O, Mitani H, Sakaurai J. ‘Second hit’ of Tsc2 gene in radiation induced renal tumors of Eker rat model. Int Congr Ser. 2002;1236:163‐174. [Google Scholar]
- 43. Haines J, Dunford R, Moody J, Ellender M, Cox R, Silver A. Loss of heterozygosity in spontaneous and X‐ray‐induced intestinal tumors arising in F1 hybrid min mice: evidence for sequential loss of APC+ and Dpc4 in tumour development. Genes. 2000;28:387‐394. [DOI] [PubMed] [Google Scholar]
- 44. Ellender M, Harrison JD, Meijne E, et al. Intestinal tumours induced in ApcMin/+mice by X‐rays and neutrons. Int J Radiat Biol. 2011;87:385‐399. [DOI] [PubMed] [Google Scholar]
- 45. Turker MS, Grygoryev D, Lasarev M, et al. Simulated space radiation‐induced mutants in the mouse kidney display widespread genomic change. PLoS ONE. 2017;12:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yatagai F. Mutations induced by heavy charged particles. Biol Sci Space. 2004;18:224‐234. [DOI] [PubMed] [Google Scholar]
- 47. Stankowski LF Jr, Hsie AW. Quantitative and molecular analyses of radiation‐induced mutation in AS52 cells. Radiat Res. 1986;105:37‐48. [PubMed] [Google Scholar]
- 48. Tindall KR, Stankowski LF Jr. Molecular analysis of spontaneous mutations at the gpt locus in Chinese hamster ovary (AS52) cells. Mutat Res. 1989;220:241‐253. [DOI] [PubMed] [Google Scholar]
- 49. Ono T, Ikehata H, Pithani VP, et al. Spontaneous mutations in digestive tract of old mice show tissue‐specific patterns of genomic instability. Cancer Res. 2004;64:6919‐6923. [DOI] [PubMed] [Google Scholar]
- 50. Nakayama T, Sawai T, Masuda I, et al. Tissue‐specific and time‐dependent clonal expansion of ENU‐induced mutant cells in gpt delta mice. Environ Mol Mutagen. 2017;58:592‐606. [DOI] [PubMed] [Google Scholar]
- 51. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chakraborty S, Mohiyuddin SA, Gopinath K, Kumar A. Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer. 2008;8:163‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu Z, Wang M, Wang L, et al. Aberrant expression of TSC2 gene in the newly diagnosed acute leukemia. Leuk Res. 2009;33:891‐897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials