

Abstract
Molecular‐targeted therapies directed against human epidermal growth factor receptor 2 (HER2) are evolving for various cancers. Neratinib is an irreversible pan‐HER tyrosine kinase inhibitor and has been approved by the FDA as an effective drug for HER2‐positive breast cancer. However, acquired resistance of various cancers to molecular‐targeted drugs is an issue of clinical concern, and emergence of resistance to neratinib is also considered inevitable. In this study, we established various types of neratinib‐resistant cell lines from HER2‐amplified breast and lung cancer cell lines using several drug exposure conditions. We analyzed the mechanisms of emergence of the resistance in these cell lines and explored effective strategies to overcome the resistance. Our results revealed that amplification of YES1, which is a member of the SRC family, was amplified in two neratinib‐resistant breast cancer cell lines and one lung cancer cell line. Knockdown of YES1 by siRNA and pharmacological inhibition of YES1 by dasatinib restored the sensitivity of the YES1‐amplified cell lines to neratinib in vitro. Combined treatment with dasatinib and neratinib inhibited tumor growth in vivo. This combination also induced downregulation of signaling molecules such as HER2, AKT and MAPK. Our current results indicate that YES1 plays an important role in the emergence of resistance to HER2‐targeted drugs, and that dasatinib enables such acquired resistance to neratinib to be overcome.
Keywords: breast cancer, drug resistance, lung cancer, neratinib, YES1
We found that YES1 played an important role in the emergence of resistance to neratinib. The combination therapy of dasatinib, which is a YES1 inhibitor, plus neratinib enabled the resistance to neratinib to be overcome.

1. INTRODUCTION
Human epidermal growth factor receptor 2 (HER2) is one of the receptor tyrosine kinases (RTK) and a member of the ErbB family of proteins. HER2 is activated by homodimerization, or by heterodimerization with other receptors of the ErbB family, such as epidermal growth factor receptor (EGFR), HER3 and HER4, and induces activation of downstream signaling pathways, such as the PI3K‐AKT and MAPK signaling pathways, promoting cell proliferation and survival.1 Thus, overexpression of HER2 is considered to be the oncogenic driver in several malignancies, including breast, lung, gastric, ovarian and esophageal cancers.2, 3, 4
There have been substantial advances in the development of HER2‐targeted therapies for HER2‐overexpressing cancers. HER2‐targeted drugs include trastuzumab, pertuzumab, lapatinib and trastuzumab emtansine (T‐DM1). However, acquired resistance to trastuzumab or lapatinib has been reported in cases of breast cancer.5, 6 Acquired resistance to trastuzumab in cases of gastric cancer has also been reported.7 Therefore, overcoming acquired resistance to HER2‐targeted therapies is a major clinical challenge in the treatment of HER2‐positive cancers.
Neratinib (HKI‐272) is an irreversible EGFR, HER2 and HER4 tyrosine kinase inhibitor (TKI),8 which has been reported to improve the overall survival in postoperative HER2‐positive breast cancer patients who have previously received trastuzumab‐based adjuvant therapy.9 Many trials of the efficacy of neratinib for other types of cancers are also ongoing. Previously, we reported the antitumor effect of neratinib against lung cancer harboring HER2 oncogene alterations.10 Moreover, it was reported that neratinib could be used to overcome trastuzumab resistance in cases of breast cancer.11 However, emergence of resistant cancer cells during the course of treatment with small‐molecule inhibitors is well known. Therefore, development of resistance to neratinib is strongly predicted.
In this study, we established neratinib‐resistant cells from the HER2‐amplified breast cancer cell line BT‐474 and the HER2‐amplified lung cancer cell line NCI‐H2170 (H2170). Then, we elucidated the mechanism of emergence of acquired resistance to neratinib and explored therapeutic strategies to overcome the resistance.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
The breast cancer cell line BT‐474 and the lung cancer cell line H2170 were used in this study. The cell lines were purchased from the ATCC. Neratinib‐resistant sublines were established from the BT‐474 and H2170 cell lines using the following procedures: cells were exposed to neratinib with stepwise escalation of the dose from 1 nmol/L to 2 μmol/L over 6 months (stepwise escalation method), or the cells were exposed to 2 µmol/L of neratinib from the start of the culture (high concentration method). Neratinib‐resistant sublines established by the stepwise escalation method were designated by the name of the parental cell line, followed by “‐NRS” and those established by the high concentration method were designated by the name of the parental cell line followed by “‐NRH”. If two neratinib‐resistant breast cancer sublines were established independently using the same method, the established cell lines were distinguished by adding a number to their names (eg, BT‐474‐NRS and BT‐474‐NRS2). All the cell lines were authenticated by short tandem repeat (STR) DNA analysis (Promega). All the breast cancer cell lines were cultured in DMEM containing 10% FBS and all the lung cancer cell lines were cultured in RPMI‐1640 medium containing 10% FBS under 5% CO2 at 37°C. Neratinib was purchased from Selleck Chemicals. Dasatinib was purchased from Bristol‐Myers Squibb.
2.2. Cell viability assay
The antitumor effects of the drugs were determined by MTS assay in vitro. Cells (3000/well) were seeded on a 96‐well plate, and then serial dilutions of the drugs were added 24 hours later. The cells were incubated in the presence of the drugs for 72 hours under 5% CO2 at 37°C. Cell viability was determined by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). The optical densities of the samples were measured at 492 nm using a Multiskan FC Microplate Photometer (Thermo Fisher Scientific). The antitumor effect of each drug was expressed as the inhibitory concentration at 50% (IC50). The assays were repeated three times. Data were expressed as the mean ± standard error (SE).
2.3. Western blotting
Cells were washed in ice‐cold PBS and lysed with lysis buffer, a mixture of RIPA buffer, phosphatase inhibitor cocktails 2 and 3 (Sigma‐Aldrich) and Complete Mini (Roche). The cell lysate was collected after centrifugation at 20 000 g for 15 minutes at 4°C. Protein was quantified using the DC Protein Assay Kit (Bio‐Rad Laboratories), fractionated on SDS‐PAGE and blotted onto a membrane using the Trans‐Blot Turbo Transfer System (Bio‐Rad Laboratories). After the membrane was blocked with 5% skim milk in TBS‐containing 0.05% Tween 20 (T‐TBS) for 1 hour, it was probed with the primary antibody overnight at 4°C, followed by incubation with the secondary antibody for 1 hour at 25°C. Proteins were detected using ECL Prime Western Blotting Detection Reagent (General Electric Company) and by scanning the membrane using ImageQuant LAS 4000 (General Electric Company). The primary antibodies used were as follows: phospho‐HER2‐Tyr877, HER2, phospho‐AKT‐Ser473, AKT, phospho‐MAPK‐Tyr202/204, MAPK, phospho‐SRC family‐Tyr416, YES1, cleaved PARP (Cell Signaling Technology) and Actin (used as the loading control) (Merck Millipore). The secondary antibody was HRP‐conjugated anti–mouse or anti–rabbit IgG (Santa Cruz Biotechnology).
2.4. Copy number assay
Genomic DNA was extracted from the cell lines using the DNeasy Blood & Tissue Kit (Qiagen). The copy number was determined with the StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific) using Taqman copy number assays (Thermo Fisher Scientific). TaqMan RNase P Control (Thermo Fisher Scientific) was used as the reference gene. The relative copy number in each sample was determined by comparing the ratio of the Ct value of the target gene to that of the reference gene in each sample with the ratio in standard genomic DNA (Merck), after validating that the efficiencies of the PCR reactions of both the target and reference genes were equal. The gene copy number in standard genomic DNA was set at 2. Samples were analyzed in triplicate. The assays were repeated three times. Data were expressed as mean ± SE.
2.5. Gene expression assay
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) and reverse‐transcribed with the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative RT‐PCR (qRT‐PCR) was performed on the StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific) using TaqMan Gene Expression Assays (Thermo Fisher Scientific). The gene expression level was calculated using the delta‐delta CT method. GAPDH was used as the endogenous control. The assays were repeated three times. Data were expressed as mean ± SE.
2.6. siRNA transfection
siRNA specific for YES1 (Silencer1 Select Validated siRNA #4390824) and the non‐targeting control (Silencer1 Select Negative Control No. 2 siRNA #4390846) were purchased from Thermo Fisher Scientific. Each siRNA (5 nmol/L of the dose) was transfected to the cells using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific). After the siRNA transfection, the cells were incubated for 72 hours under 5% CO2 at 37°C.
2.7. Co–immunoprecipitation
Protein lysates were immunoprecipitated with anti–HER2 antibody (Cell Signaling Technology) or normal mouse IgG (Santa Cruz) using Dynabeads Protein G (Thermo Fisher Scientific), in accordance with the manufacturer's instructions.
2.8. Xenograft mouse model
Six‐week‐old BALB/c‐nu/nu female mice were purchased from Charles River Laboratories (Yokohama, Japan). All mice were provided with sterilized food and water and housed in a barrier facility under a 12:12‐hour light‐dark cycle. BT‐474‐NRS2 (107 cells) was suspended in 100 μL of DMEM with Matrigel Basement Membrane Matrix (Corning) mixture (1:1 ratio) and injected subcutaneously into the backs of the mice. Tumors were measured using digital calipers, and the tumor volumes were calculated using the formula: volume = 1/2 × [(shortest diameter)2 × (the longest diameter)]. When the tumor volumes exceeded approximately 150 mm3, the mice were randomly allocated to one of four groups: a control group, a neratinib (10 mg/kg/d) group, a dasatinib (15 mg/kg/d) group and a neratinib (10 mg/kg/d) plus dasatinib (15 mg/kg/d) group (n = 6 per group). The drugs were suspended in 0.5 w/v (%) methyl cellulose and administered by oral gavage five times a week for 4 weeks. The tumor volumes were measured twice a week. Data were expressed as mean ± SE. The protocol was approved by the Animal Care and Use Committee, Okayama University (Permit Number: OKU‐2019328).
2.9. Statistical analysis
All the statistical analyses in this study were performed using EZR version 1.40 (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R version 3.5.2 (The R Foundation for Statistical Computing, Vienna, Austria).12 Specifically, the software is a modified version of R commander designed to add statistical functions frequently used in biostatistics. Differences were compared using one‐way analysis of variance (multiple groups) or the t test (two groups). P < 0.05 was considered as being indicative of statistical significance.
3. RESULTS
3.1. Establishment of neratinib‐resistant cell lines
The breast cancer cell line BT‐474 and lung cancer cell line H2170 with HER2 amplification were exposed to neratinib using two different methods: the stepwise escalation method (NRS series) and the high concentration method (NRH series). The parent cell lines are known to be highly sensitive to neratinib. As a result, we obtained six neratinib‐resistant cell lines: BT‐474‐NRS, BT‐474‐NRS2, BT‐474‐NRH, BT‐474‐NRH2, H2170‐NRS and H2170‐NRH.
The IC50 values of neratinib are shown in Table 1. The IC50 values of neratinib for these six resistant cell lines were at least 100‐fold the values for the parental cell lines.
Table 1.
IC50 values for neratinib and the methods of drug exposure in neratinib‐resistant cell lines
| Cancer type | Cell line | Method of drug exposure |
Neratinib IC50 Mean ± standard error (μmol/L) |
|---|---|---|---|
| Breast | BT‐474 | — | 0.0028 ± 0.00019 |
| BT‐474‐NRS | Stepwise | 0.97 ± 0.18 | |
| BT‐474‐NRS2 | Stepwise | 1.45 ± 0.25 | |
| BT‐474‐NRH | High | 1.69 ± 0.35 | |
| BT‐474‐NRH2 | High | 1.44 ± 0.26 | |
| Lung | H2170 | — | 0.029 ± 0.0059 |
| H2170‐NRS | Stepwise | >10 | |
| H2170‐NRH | High | 3.96 ± 0.49 |
‐, not applicable.
3.2. Status of human epidermal growth factor receptor 2 and human epidermal growth factor receptor 2‐related signaling in the neratinib‐resistant cell lines
We evaluated the expression and phosphorylation levels of the HER2‐related signaling molecules (Figure 1A). Both BT‐474‐NRS and BT‐474‐NRS2 showed elevated phosphorylation levels of HER2 and AKT. BT‐474‐NRH, BT‐474‐NRH2, H2170‐NRS and H2170‐NRH showed downregulated expression of HER2. Then, we evaluated the copy number and mRNA expression of HER2 (Figure S1); the copy number and mRNA expression of HER2 seemed to be decreased, as compared to those in the parental cell lines, in H2170‐NRS and H2170‐NRH.
Figure 1.
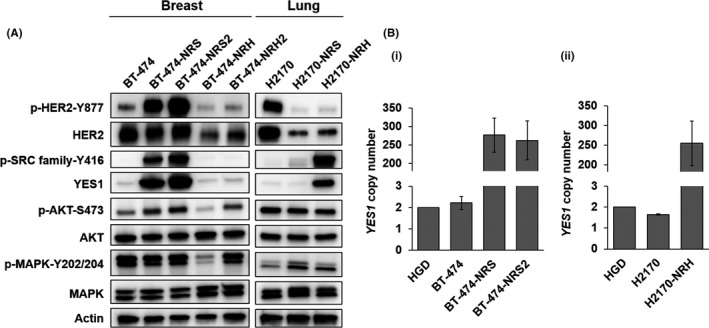
YES1, a member of the SRC family, is amplified and overexpressed in neratinib‐resistant cell lines. A, Protein expression and phosphorylation profile of neratinib‐resistant breast cancer and lung cancer cell lines. B, Copy number assay of YES1 in (i) BT‐474, BT‐474‐NRS, BT‐474‐NRS2, (ii) H2170 and H2170‐NRH. Human genomic DNA (HGD) was used as the control (two copies). The assay was repeated three times. Data are shown as mean ± standard error
3.3. YES1 upregulation in neratinib‐resistant cell lines
We previously reported that YES1, a member of the SRC family, plays an important role in the emergence of acquired resistance of HER2‐positive breast cancer to trastuzumab and lapatinib.13 Therefore, we investigated the expression levels of YES1. Both the expression levels of YES1 and the phosphorylation levels of the SRC family proteins were upregulated in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH (Figure 1A). We also determined the copy number of YES1, which revealed gain of the copy number in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH (Figure 1B).
3.4. Effect of YES1 knockdown in the neratinib‐resistant cell lines
To clarify the effect of YES1 in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH, we performed YES1 knockdown using siRNA and confirmed that the antitumor effect of neratinib was restored in the resistant cell lines after YES1 knockdown (Figure 2A). We performed Western blot analysis to investigate the effect of YES1 knockdown on the HER2‐related signaling pathways. Phosphorylation of the SRC family proteins was suppressed by YES1 knockdown in the resistant cell lines. Furthermore, a greater degree of inhibition of the phosphorylation of HER2, AKT and MAPK was observed with the combination of YES1 knockdown and neratinib than with either alone (Figure 2B). Furthermore, phosphorylation of HER2 was also suppressed by YES1 knockdown. To identify the interaction between HER2 and YES1, we performed a co–immunoprecipitation (IP) assay, which revealed that YES1 was bound to HER2 in BT‐474‐NRS and BT‐474‐NRS2 (Figure S2).
Figure 2.
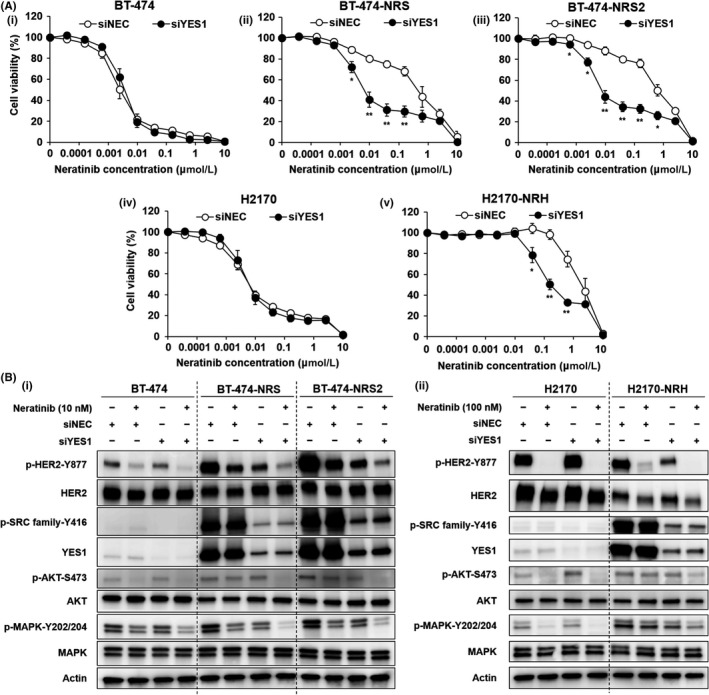
YES1 knockdown overcomes neratinib resistance. A, MTS assay to assess the sensitivity to neratinib of (i) BT‐474, (ii) BT‐474‐NRS, (iii) BT‐474‐NRS2, (iv) H2170 and (v) H2170‐NRH after YES1 knockdown. Cells were transfected with siRNA followed by treatment with neratinib for 72 h. The assay was repeated three times. Data are shown as mean ± SE; *P < 0.05, **P < 0.01 vs negative control (siNEC). B, Effect of YES1 knockdown on the protein expression and phosphorylation profiles in the presence/absence of neratinib treatment in (i) BT‐474, BT‐474‐NRS, BT‐474‐NRS2, (ii) H2170 and H2170‐NRH. The cells were treated with neratinib for 2 h after the siRNA transfection
3.5. Effect of dasatinib in the neratinib‐resistant cell lines
We focused on the SRC inhibitor dasatinib because YES1, one of the members of the SRC family proteins, was upregulated in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH. We used the combination of neratinib plus dasatinib to overcome the resistance to neratinib. The combination of neratinib and dasatinib exerted remarkable antitumor effects against the resistant cell lines (Figure 3A). We also performed western blot analysis to elucidate the effect of the combination therapy. In the resistant cell lines, phosphorylations of HER2, AKT and MAPK were inhibited to a greater extent by the combination of neratinib plus dasatinib than by either drug alone, similar to the case for the combination of YES1 knockdown plus neratinib (Figure 3B). We evaluated the presence of apoptosis by analyzing the amount of cleaved PARP, an apoptosis marker, and revealed that apoptosis was also induced by the combination of neratinib plus dasatinib (Figure 3C).
Figure 3.
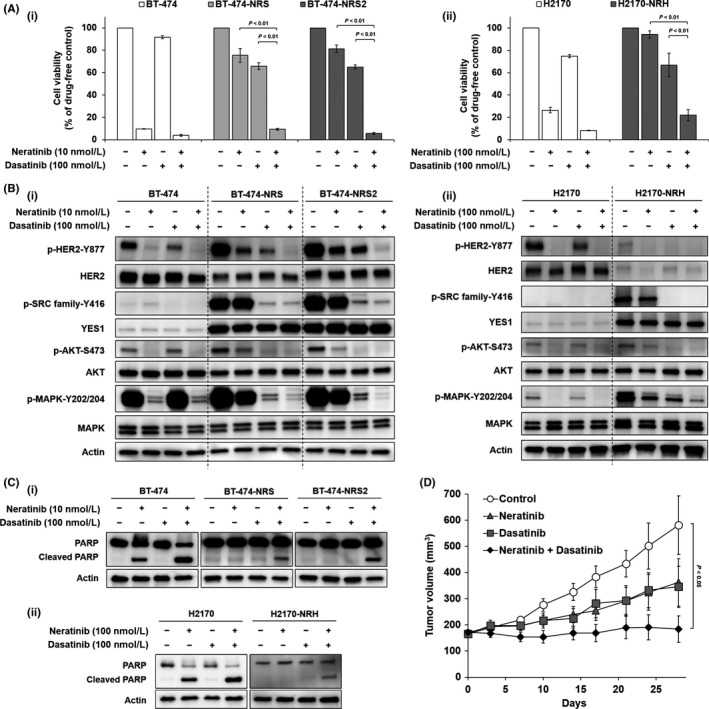
Dasatinib, an SRC family inhibitor, enables the resistance to neratinib to be overcome. A, MTS assay to assess the effect of neratinib alone, dasatinib alone and the combination of neratinib plus dasatinib on (i) BT‐474, BT‐474‐NRS, BT‐474‐NRS2, (ii) H2170 and H2170‐NRH. The cells were treated with the drugs for 72 h. The assay was repeated three times. Data are shown as mean ± standard error. B, Effect of neratinib alone, dasatinib alone and the combination of neratinib plus dasatinib on protein phosphorylation in (i) BT‐474, BT‐474‐NRS, BT‐474‐NRS2, (ii) H2170 and H2170‐NRH. The cells were treated with the drugs for 2 h. C, Apoptosis assay by detection of PARP cleavage in (i) BT‐474, BT‐474‐NRS, BT‐474‐NRS2, (ii) H2170 and H2170‐NRH. The cells were treated with neratinib alone, dasatinib alone or the combination of neratinib plus dasatinib for 48 h. D, Growth of BT‐474‐NRS2 in BALB/c nude mice. The mice were given neratinib (10 mg/kg) alone, dasatinib (15 mg/kg) alone or the combination of neratinib plus dasatinib by oral gavage five times a week. Data are shown as mean ± SE (n = 6)
Furthermore, based on the results of in vitro assays, we performed an in vivo assay to examine the effect of the combination of neratinib plus dasatinib using BT‐474‐NRS2. The tumor growth was significantly suppressed in the combination therapy group as compared to the control group (Figure 3D). No significant differences were observed in the body weights of the treated mice (Figure S3).
4. DISCUSSION
In this study, we established six neratinib‐resistant cell lines from two HER2‐amplified cell lines. Two methods were used to establish the resistance, the stepwise escalation method and the high concentration method, based on our previous report that the condition of EGFR‐TKI exposure influenced the mechanism of emergence of acquired resistance in lung cancer cell lines.14 Clinically, the reported mean trough plasma concentration of neratinib in patients after once‐daily oral administration of the drug at the dose of 240 mg is 52‐59 ng/mL (0.093‐0.106 μmol/L).15 The IC50 values of neratinib for neratinib‐resistant cell lines were sufficiently high. Therefore, we considered that the neratinib‐resistant cell lines established by us were sufficiently resistant to neratinib.
Activation of CYP3A4 has been reported as one of the mechanisms underlying the development of resistance to neratinib, and resistance to neratinib conferred cross‐resistance to trastuzumab, lapatinib and afatinib.16 In our study, we found amplification of YES1 in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH. YES1 is a member of the SRC family of proteins, which are non–receptor tyrosine kinases.17 The SRC family proteins include c‐SRC, YES1, FYN, LYN, FGR, BLK, HCK, LCK and FRK, which are key regulators of signal transduction.18 Our previous study showed that amplification of YES1 was associated with resistance to HER2 inhibitors, such as trastuzumab and lapatinib, in breast cancer13 and afatinib in gastric cancer.19 Moreover, YES1 amplification was also found to be associated with resistance to EGFR inhibitors in patients with lung cancer.20 Therefore, our results suggest that YES1 amplification is a mechanism of resistance to not only neratinib, but also to general inhibitors of the ErbB family.
We focused on dasatinib (BMS‐354825), which is a dual SRC/ABL kinase inhibitor,21 as a useful drug to overcome resistance to neratinib. In clinical settings, dasatinib is used to treat chronic myelogenous leukemia and Philadelphia chromosome‐positive acute lymphoblastic leukemia, targeting the BCR‐ABL fusion protein.22 Dasatinib has also been reported to inhibit YES1.23 In the current study, the combination of dasatinib plus neratinib exerted a marked antitumor effect against YES1‐amplified neratinib‐resistant cells, both in vitro and in vivo. Furthermore, we reported that this combination therapy was also effective against trastuzumab and trastuzumab/lapatinib‐resistant cells harboring YES1 amplification.13 These results suggest that combined dasatinib plus neratinib treatment should be considered in clinical cases where reassessment of the tumor cells after they acquire resistance to HER2‐targeted therapies reveals amplification of YES1.
Human epidermal growth factor receptor 2 was more phosphorylated in BT‐474‐NRS and BT‐474‐NRS2 compared to the parental BT‐474, whereas HER2 expression was decreased in H2170‐NRH compared to the parental H2170. Despite this difference, the combination therapy of neratinib plus inhibition of YES1 exerted a remarkable antitumor effect in all these cell lines. Phosphorylation of AKT and MAPK was slightly suppressed by a single agent of neratinib in BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH, as shown in Figure 3B. Their phosphorylation might have been reduced due to the suppression of not only HER2 but also EGFR and/or HER4, because neratinib is a pan‐HER inhibitor. It is highly possible that BT‐474‐NRS and BT‐474‐NRS2 depend on HER2 because the phosphorylation of HER2 was promoted compared to the parental BT‐474. In contrast, EGFR and/or HER4 pathway might be activated rather than HER2 in H2170‐NRH because the expression and phosphorylation of HER2 was decreased in H2170‐NRH compared to the parental H2170. Although whether or not BT‐474‐NRS, BT‐474‐NRS2 and H2170‐NRH still depend on HER2 has not been clarified, inhibition of YES1 results in overcoming the neratinib resistance in the cells harboring YES1 amplification regardless of HER2 expression status.
We revealed previously that cancer stem cell (CSC)‐like features and epithelial‐to‐mesenchymal transition (EMT) were involved in the resistance of lung cancer to ErbB family‐TKI.14, 24, 25 Therefore, we also investigated the expression levels of the CSC markers and EMT markers in the resistant cell lines. ALDH1A1 is a putative CSC marker, and we found overexpression of ALDH1A1 in BT‐474‐NRH and BT‐474‐NRH2 (Figure S4). N‐cadherin and vimentin, which are EMT markers, were not detected in any of the resistant cell lines (data not shown). CSC‐like features were also found in the neratinib‐resistant cell lines. Development of a therapeutic strategy against cells with CSC‐like features is one of the tasks for the future.
Expression of HER2 protein was downregulated in BT‐474‐NRH, BT‐474‐NRH2, H2170‐NRS and H2170‐NRH. Clinically, loss of HER2 amplification has been reported after treatment with trastuzumab in patients with breast and gastric cancer.26, 27, 28 Moreover, there are some reports from vitro studies which suggest downregulated expression of HER2 protein in a neratinib‐resistant breast cancer cell line and a decrease of the copy number of HER2 in an afatinib‐resistant lung cancer cell line.16, 24 Reduction of HER2 protein expression may be one of the factors contributing to the resistance to HER2 inhibitors, because it results in a decrease in the amount of the drug target.
Phosphorylation of HER2 was suppressed by a single agent of YES1 knockdown or dasatinib treatment. This is consistent with our observation in a previous study.13 According to other reports, this phenomenon was also observed with saracatinib (AZD‐0530) (a SRC/ABL inhibitor) treatment in trastuzumab‐resistant breast cancer and gastric cancer.29, 30 It has been reported that SRC interacts with HER2.17 Therefore, we proposed a hypothesis that YES1 may also interact with HER2. We performed co–IP assay to clarify the interaction of YES1 with HER2 and demonstrated that YES1 bound directly with HER2. Thus, YES1 may bind to and activate HER2. Further studies are necessary for obtaining a better understanding.
In conclusion, we found that YES1 played an important role in the emergence of resistance to HER2‐targeted drugs. Furthermore, the combination therapy of dasatinib plus neratinib enabled the resistance to neratinib to be overcome. These findings are expected to contribute to improvement of the therapeutic outcomes in patients with breast cancer or lung cancer harboring HER2 amplification.
DISCLOSURE
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank Dr Takehiro Matsubara (Biobank, Okayama University Hospital, Okayama, Japan) and Ms Fumiko Isobe (Department of General Thoracic Surgery and Breast and Endocrinological Surgery, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan) for their technical support. This work was supported by the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number 19H00408 [to T. Takeda] and 19K09070 [to T. Shien]).
Takeda T, Yamamoto H, Suzawa K, et al. YES1 activation induces acquired resistance to neratinib in HER2‐amplified breast and lung cancers. Cancer Sci. 2020;111:849–856. 10.1111/cas.14289
REFERENCES
- 1. Zhang H, Berezov A, Wang Q, et al. Review series ErbB receptors: from oncogenes to targeted cancer therapies. Molecules. 2007;117:2051‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER‐2/neu proto‐oncogene in human breast and ovarian cancer. Science. 1989;244:707‐712. [DOI] [PubMed] [Google Scholar]
- 3. Meert AP, Martin B, Paesmans M, et al. The role of HER‐2/neu expression on the survival of patients with lung cancer: a systematic review of the literature. Br J Cancer. 2003;89:959‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagaraja V, Eslick GD. HER2 expression in gastric and oesophageal cancer: a metaanalytic review. J Gastrointest Oncol. 2015;6:143‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2‐targeted therapies in HER2 gene‐amplified breast cancer. Ann Oncol. 2012;17:3007‐3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mohd MSN, Crown J, Hennessy BT. Overcoming resistance and restoring sensitivity to HER2‐targeted therapies in breast cancer. Ann Oncol. 2012;23:3007‐3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roukos DH. Editorial: Targeting gastric cancer with trastuzumab: new clinical practice and innovative developments to overcome resistance. Ann Surg Oncol. 2010;17:14‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wissner A, Mansour TS. The development of HKI‐272 and related compounds for the treatment of cancer. Arch Pharm (Weinheim). 2008;341:465‐477. [DOI] [PubMed] [Google Scholar]
- 9. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab‐based adjuvant therapy in HER2‐positive breast cancer (ExteNET): 5‐year analysis of a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18:1688‐1700. [DOI] [PubMed] [Google Scholar]
- 10. Ogoshi Y, Shien K, Yoshioka T, et al. Anti–tumor effect of neratinib against lung cancer cells harboring HER2 oncogene alterations. Oncol Lett. 2019;17:2729‐2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Canonici A, Gijsen M, Mullooly M, et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget. 2013;4:1592‐1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanda Y. Investigation of the freely available easy‐to‐use software “EZR” for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeda T, Yamamoto H, Kanzaki H, et al. Yes1 signaling mediates the resistance to Trastuzumab/Lap atinib in breast cancer. PLoS ONE. 2017;12:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell‐like properties in cancer cells. Cancer Res. 2013;73:3051‐3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burstein HJ, Sun Y, Dirix LY, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2‐positive breast cancer. J Clin Oncol. 2010;28:1301‐1307. [DOI] [PubMed] [Google Scholar]
- 16. Breslin S, Lowry MC, O'Driscoll L. Neratinib resistance and cross‐resistance to other HER2‐targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br J Cancer. 2017;116:620‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513‐609. [DOI] [PubMed] [Google Scholar]
- 18. Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906‐7909. [DOI] [PubMed] [Google Scholar]
- 19. Yoshioka T, Shien K, Takeda T, et al. Acquired resistance mechanisms to afatinib in HER2‐amplified gastric cancer cells. Cancer Sci. 2019;110:2549‐2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan PD, Narzisi G, Jayaprakash AD, et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc Natl Acad Sci U S A. 2018;115:E6030‐E6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lombardo LJ, Lee FY, Chen P, et al. Discovery of N‐(2‐chloro‐6‐methylphenyl)‐2‐(6‐(4‐(2‐hydroxyethyl)‐ piperazin‐1‐yl)‐2‐methylpyrimidin‐4‐ylamino)thiazole‐5‐carboxamide (BMS‐354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658‐6661. [DOI] [PubMed] [Google Scholar]
- 22. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib‐resistant Philadelphia chromosome‐positive leukemias. N Engl J Med. 2006;354:2531‐2541. [DOI] [PubMed] [Google Scholar]
- 23. Montero JC, Seoane S, Ocaña A, Pandiella A. Inhibition of Src family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. 2011;17:5546‐5552. [DOI] [PubMed] [Google Scholar]
- 24. Torigoe H, Shien K, Takeda T, et al. Therapeutic strategies for afatinib‐resistant lung cancer harboring HER2 alterations. Cancer Sci. 2018;109:1493‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hashida S, Yamamoto H, Shien K, et al. Acquisition of cancer stem cell‐like properties in non‐small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015;106:1377‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pietrantonio F, Caporale M, Morano F, et al. HER2 loss in HER2‐positive gastric or gastroesophageal cancer after trastuzumab therapy: implication for further clinical research. Int J Cancer. 2016;139:2859‐2864. [DOI] [PubMed] [Google Scholar]
- 27. Guarneri V, Dieci MV, Barbieri E, et al. Loss of HER2 positivity and prognosis after neoadjuvant therapy in HER2‐positive breast cancer patients. Ann Oncol. 2013;24:2990‐2994. [DOI] [PubMed] [Google Scholar]
- 28. Wang RX, Chen S, Jin X, Chen CM, Shao ZM. Weekly paclitaxel plus carboplatin with or without trastuzumab as neoadjuvant chemotherapy for HER2‐positive breast cancer: loss of HER2 amplification and its impact on response and prognosis. Breast Cancer Res Treat. 2017;161:259‐267. [DOI] [PubMed] [Google Scholar]
- 29. Zhang S, Huang WC, Li P, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han S, Meng Y, Tong Q, et al. The ErbB2‐targeting antibody trastuzumab and the small‐molecule SRC inhibitor saracatinib synergistically inhibit ErbB2‐overexpressing gastric cancer. MAbs. 2014;6:403‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials