

Abstract
The SWI/SNF chromatin remodeling complex is composed of approximately 15 subunits, and approximately 20% of all cancers carry mutations in the genes encoding these subunits. Most of the genetic alterations in these genes are loss‐of‐function mutations. The identification of vulnerability based on synthetic lethality in cancers with SWI/SNF chromatin remodeling complex deficiency contributes to precision medicine. The SWI/SNF chromatin remodeling complex is involved in transcription, DNA repair, DNA replication, and chromosomal segregation. Cancers with deficiency in the SWI/SNF chromatin remodeling complex show increased vulnerability derived from the loss of these functions. Synthetic lethal targets have been identified based on vulnerabilities in the functions of the SWI/SNF chromatin remodeling complex. In this review article, we propose a precision medicine strategy using chemotherapeutic methods, such as molecular targeted therapy and immunotherapy, based on harnessing synthetic lethality in cancers with deficiency in the SWI/SNF chromatin remodeling complex.
Keywords: chromatin remodeling, epigenetics, molecular targeted therapy, precision medicine, synthetic lethality
The subunits of SWI/SNF chromatin remodeling complex are frequently deficient in various cancers. The identification of vulnerability based on synthetic lethality in cancers with SWI/SNF chromatin remodeling complex deficiency contributes to precision medicine. Synthetic lethal targets have been identified based on vulnerabilities in the functions of the SWI/SNF chromatin remodeling complex. In this review article, we propose a precision medicine strategy based on harnessing synthetic lethality in cancers with deficiency in the SWI/SNF chromatin remodeling complex.
![]()
1. THERAPEUTIC STRATEGY BASED ON SYNTHETIC LETHALITY FOR LOSS‐OF‐FUNCTION MUTATED CANCERS
Current precision medicine strategies for human cancers target activated proteins such as the tyrosine kinases EGFR, BRAF, and ALK‐fusion; these proteins are activated by gain‐of‐function genetic aberrations including gene mutation, amplification, and fusion (Figure 1).1, 2, 3 Activated oncogenes constitute a specific vulnerability of cancer cells. Inhibition of the synthesis or activity of these oncogenes results in cell death, specifically in cells expressing the activated oncogene; the dependence of cells on an oncogene for survival is defined as “oncogene addiction”.4 Only a fraction of cancers have an activated oncogene, whereas many cancers have other genetic aberrations such as loss‐of‐function (LOF) mutations. Certain LOF gene mutations of tumor suppressor genes confer druggable vulnerabilities on cancer cells. However, because genes with LOF mutations are inactivated, the LOF mutation gene product is not a target for inhibition (Figure 1). Harnessing synthetic lethality, such as that based on LOF gene mutations, has emerged as an attractive therapeutic strategy; however, to date, this strategy has not been widely successful. Synthetic lethality is defined by an interdependent relationship between 2 genes, which means that simultaneous loss of 2 genes, but not loss of either gene alone, leads to cell death (Figure 1).5, 6, 7 Cancer cells harboring a LOF gene mutation would therefore be vulnerable to inhibition of the synthetic lethal target.
Figure 1.
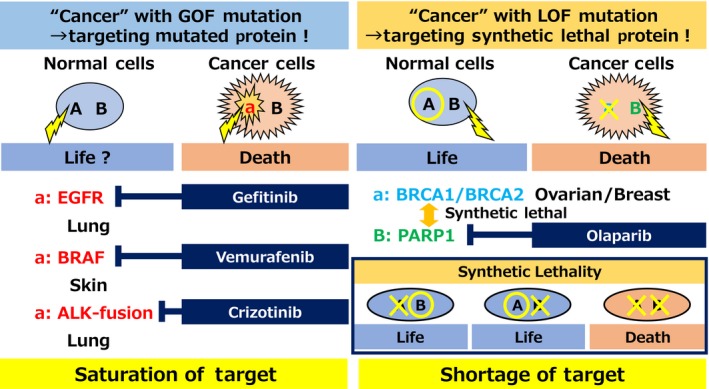
Precision medicine based on cancer mutations. PARP, poly(ADP‐ribose) polymerase
In this review, “synthetic lethal therapy” is defined as cancer therapy based on synthetic lethality. This strategy is based on the assumption that a cancer patient has a LOF mutation of “gene A” and gene A is synthetic lethal with gene B (Figure 2). In cancer cells, gene A is the LOF gene, and therapy with an inhibitor of B causes cell death based on synthetic lethality because of the simultaneous suppression of the function of both A and B. In normal cells, gene A is normal; therefore, inhibition of gene B does not affect the survival of normal cells. Synthetic lethal therapy is expected to have high selectivity against cancer cells and few side‐effects. Thus, the identification of cancer vulnerabilities associated with LOF gene mutations should lead to marked improvements in cancer therapy, as epitomized by the success of poly(ADP‐ribose) polymerase (PARP)1‐targeted therapy against hereditary breast and ovarian cancers harboring LOF mutations of the BRCA1 and BRCA2 genes.8, 9
Figure 2.
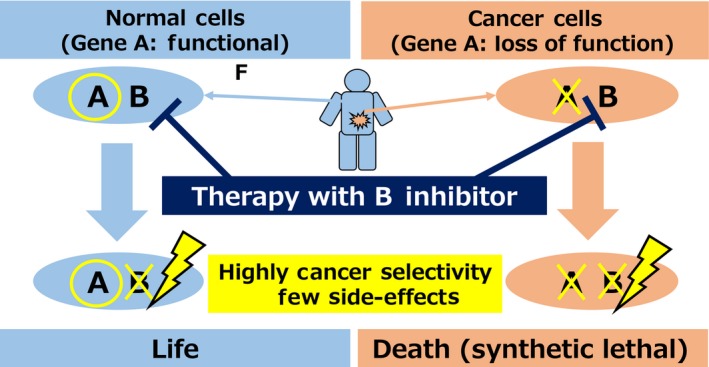
Synthetic lethal therapy: cancer therapy based on synthetic lethality
2. ROLE OF CHROMATIN REGULATING FACTORS
Chromatin regulating factors are largely divided into 2 groups: chromatin remodeling complexes and histone modifying factors (Figure 3). Chromatin remodeling complexes use the energy of ATP hydrolysis and maintain chromatin structure by opening or closing chromatin through sliding, ejecting, repositioning, or inserting nucleosomes, which are histone octamers composed of histones H2A, H2B, H3, and H4.10 Histone modifying factors maintain the interaction between DNA and histones through histone methylation/demethylation, acetylation/deacetylation, and ubiquitylation/deubiquitylation. These chromatin regulating factors control binding of various functional proteins, such as transcription factors, DNA replication factors, DNA repair factors, and chromosome segregation factors to chromatin by remodeling and modifying chromatin structure. Therefore, chromatin regulating factors contribute to the regulation of transcription, DNA repair, DNA replication, and chromosomal segregation.
Figure 3.
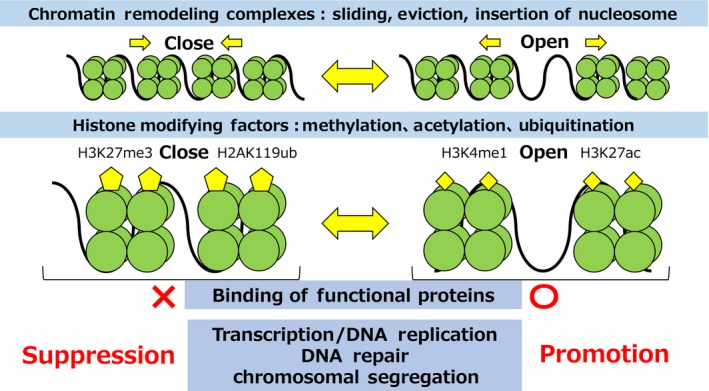
Role of chromatin regulators
3. GENETIC ABNORMALITY OF SWI/SNF CHROMATIN REMODELING GENES IN VARIOUS CANCERS
Recent advances in genome‐wide sequencing technologies have contributed to the identification of most gene mutations associated with cancer. Comprehensive genome studies identified mutations in genes involved in chromatin regulation in approximately 50% of cancers.11, 12, 13, 14 Most of the mutations in chromatin regulating genes are LOF mutations such as deleterious missense mutations, frameshift mutations, and chromosomal deletions. Mutations in SWI/SNF chromatin remodeling genes occur with high frequency in cancer; they are detected in approximately 20% of all cancer patients.15, 16 The SWI/SNF chromatin remodeling complex is composed of 15 subunits, and it is classified into 3 complexes comprising different subunits (Figure 4): the BRG1/BRM‐associated factor (BAF) complex, the polybromo‐associated BAF (PBAF) complex, and the noncanonical BAF (ncBAF) complex.17, 18 The function of the SWI/SNF chromatin remodeling complex relies on the catalytic activities of the SWI/SNF2‐like ATPase and helicase domains. It is composed of accessory subunits harboring chromatin‐binding motifs, such as a bromodomain. Specifically, LOF genetic aberrations of SMARCA4, ARID1A, ARID2, PBRM1, and SMARCB1 are common in various cancers such as lung cancer, ovarian clear cell carcinoma, skin cancer, renal clear cell carcinoma, and rhabdoid tumors, respectively. Gain‐of‐function genetic aberrations of SS18 by fusion of SS18 and SSXs (SSX1, SSX2, or SSX4) are observed in all synovial sarcoma patients.19 Most SWI/SNF chromatin remodeling genes, except the SS18‐SSX fusion, cause LOF genetic aberrations in cancer. Therefore, the development of therapies based on synthetic lethality would be a promising therapeutic strategy. In this review, we introduce a synthetic lethal therapy strategy for the treatment of cancers with genetic aberrations of SWI/SNF chromatin remodeling genes (Figures 4 and 5).
Figure 4.
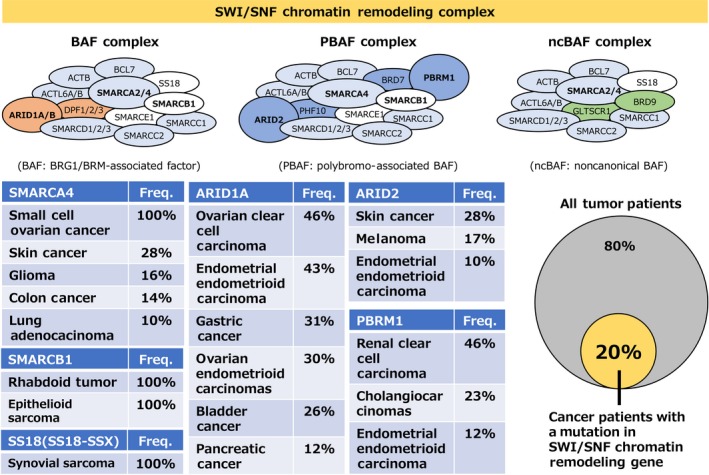
Genetic abnormality of SWI/SNF chromatin remodeling components in various cancers
Figure 5.
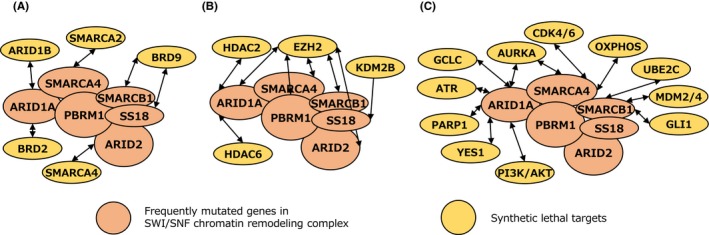
Synthetic lethal targets in cancers deficient in the SWI/SNF chromatin remodeling complex. A, Synthetic lethal targets based on targeting the interaction between 2 subunits in the SWI/SNF chromatin remodeling complex. B, Synthetic lethal targets based on targeting the competitor with the SWI/SNF chromatin remodeling complex. C, Synthetic lethal targets based on targeting the function of the SWI/SNF chromatin remodeling complex
4. SYNTHETIC LETHAL TARGETS BASED ON TARGETING THE INTERACTION BETWEEN 2 SUBUNITS IN THE SWI/SNF CHROMATIN REMODELING COMPLEX
The SWI/SNF chromatin remodeling complex is a large complex composed of many different subunits, including functional subunits, such as catalytic, structural, and DNA binding subunits. Therefore, cancers deficient in the SWI/SNF chromatin remodeling complex are vulnerable because the interactions between subunits can be targeted for inactivation (Figure 5A). SMARCA4 and its paralog SMARCA2 have an ATPase domain that supplies energy for chromatin remodeling activity. Therefore, the ATPase function of the SWI/SNF chromatin remodeling complex requires either SMARCA4 or SMARCA2. According to the mutually exclusive paralogs SMARCA4 and SMARCA2, the SWI/SNF chromatin remodeling complex exists as a SMARCA4‐containing complex or a SMARCA2‐containing complex. Therefore, loss of SMARCA4 or SMARCA2 leads to functional deficiency of the SMARCA4‐containing complex or SMARCA2‐containing complex, respectively. Work from our group showed that SMARCA4‐deficient lung adenocarcinoma cells are sensitive to siRNA‐mediated suppression of SMARCA2.20 This was supported by follow‐up studies.21, 22 Conversely, SMARCA2‐deficient esophageal squamous carcinoma cells are sensitive to inhibition of SMARCA4.23 These observations suggest that the genetic relationship between SMARCA4 and SMARCA2 is a synthetic lethal relationship. The development of SMARCA4 and SMARCA2 inhibitors would be promising as a synthetic lethal therapy strategy for SMARCA2‐deficient cancers and SMARCA4‐deficient cancers, respectively. SMARCA2 and SMARCA4 proteins are potential druggable targets because they possess ATPase and bromo domains. The development of inhibitors of SMARCA4 and SMARCA2 based on protein degradation could provide promising therapeutic opportunities (WO2016138114; Genentech and Constellation).
Similar to the synthetic lethal relationship between the SMARCA4 and SMARCA2 paralogous pair, ARID1A and its paralog ARID1B form a synthetic lethal pair. ARID1A and ARID1B are mutually exclusive components of the BAF complex. ARID1A‐deficient ovarian clear cell carcinoma cells are sensitive to siRNA‐mediated suppression of ARID1B. Loss of both ARID1A and ARID1B causes collapse of the BAF complex and leads to loss of functional activity of the BAF complex.24 Therefore, inhibition of ARID1B function is a promising strategy for ARID1A‐deficient cancers. However, because ARID1B does not contain functional domains, such as a catalytic domain that could serve as a target for drug development, it would be difficult to develop an inhibitor against ARID1B. A modified approach to the inhibition of ARID1B may be the use of BRD2 inhibitors because inhibition of BRD2 represses the transcriptional expression of ARID1B. In fact, ARID1A‐deficient cancer cells are more sensitive to BRD2 inhibitors than ARID1A‐proficient cancer cells.25 Therefore, BRD2 inhibitors would be promising for the treatment of ARID1A‐deficient cancers because of induction of synthetic lethality through suppression of ARID1B.
SMARCB1 and SS18 are components of the BAF complex. SMARCB1 is genetically aberrant because of LOF gene mutation or chromosomal deletion in pediatric and juvenile cancers such as rhabdoid tumor and epithelioid sarcoma.26, 27 SS18 is genetically aberrant because of fusion with SSXs (SSX1, SSX2, or SSX4) in synovial sarcoma. The SS18‐SSX fusion protein is incorporated into the BAF complex, resulting in loss of the SMARCB1 protein from the BAF complex.19, 28 This suggests that the BAF complex containing the SS18‐SSX fusion protein is deficient in SMARCB1 function. SMARCB1‐deficient cancer cells and SS18‐SSX fusion cancer cells are synthetic lethal because of inhibition of a subunit of the ncBAF complex, such as BRD9.18, 29, 30 Thus, BAF complex‐deficient cancers, such as SMARCB1‐deficient cancers, depend on the function of the residual ncBAF complex. A BRD9 inhibitor would therefore be a promising strategy for SMARCB1‐deficient cancers.
Comprehensive analysis of synthetic lethality between 2 factors in all subunits of the SWI/SNF chromatin remodeling complex showed that the SMARCA4‐ARID2, SMARCA4‐ACTB, and SMARCC1‐SMARCC2 pairs are significant for synthetic lethality.31 Considering synthetic lethality among these pairs, the SMARCA4 and ARID2 relationship would be a potential target for synthetic lethal therapy because SMARCA4 and ARID2 are frequently mutated in cancer.
5. SYNTHETIC LETHAL TARGETS BASED ON TARGETING THE COMPETITOR WITH THE SWI/SNF CHROMATIN REMODELING COMPLEX
Transcription is regulated by the balance between the promotion and repression of gene expression by various chromatin regulating factors. Therefore, cancers deficient in the SWI/SNF chromatin remodeling complex are vulnerable because of the abrogation of transcriptional balance (Figure 5B). The SWI/SNF chromatin remodeling complex promotes gene expression by inducing chromatin relaxation at the promoter and enhancer regions. In contrast, polycomb repressive complex 2 (PRC2), which is composed of the methyltransferase EZH2 and the noncatalytic regulatory subunits SUZ12 and EED, represses gene expression by promoting chromatin compaction through histone H3 K27 trimethylation (H3K27me3) at the promoter and enhancer regions. The SWI/SNF chromatin remodeling complex and PRC2 thus interact in a competitive manner to promote and repress transcription.32, 33 In cancer cells deficient in SMARCA4, SMARCB1, ARID1A, and PBRM1 of the SWI/SNF chromatin remodeling complex, inhibition of EZH2 causes synthetic lethality.32, 34, 35, 36 This competitive relationship suggests that deficiency in the SWI/SNF chromatin remodeling complex promotes PRC2 activity through loss of the complex's restraint; cancer cells deficient in SWI/SNF chromatin remodeling complex thus depend on PRC2 activity.37, 38 An inhibitor of EZH2 would be promising for SWI/SNF chromatin remodeling complex‐deficient cancers. Several EZH2 inhibitors have been developed by various companies. The EZH2 inhibitor tazemetostat is currently in clinical trials39; it has shown limited clinical activity in SMARCB1‐deficient epithelioid sarcoma (NCT02601950).
Although the SWI/SNF chromatin remodeling complex promotes the transcription of many genes, it also represses the transcription of certain genes. The histone deacetylase HDAC6 gene is repressed by the SWI/SNF chromatin remodeling complex. ARID1A deficiency upregulates HDAC6 expression through derepression caused by deficiency in the SWI/SNF chromatin remodeling complex. ARID1A‐deficient cancers depend on the activation of HDAC6, and inhibition of HDAC6 causes synthetic lethality in ARID1A‐deficient cancers.40 The HDAC6 inhibitors would be promising agents for the treatment of ARID1A‐deficient cancers.
6. SYNTHETIC LETHAL TARGETS BASED ON TARGETING THE FUNCTION OF THE SWI/SNF CHROMATIN REMODELING COMPLEX
The SWI/SNF chromatin remodeling complex functions in transcription, DNA repair, DNA replication, and chromosomal segregation. Therefore, cancers deficient in the SWI/SNF chromatin remodeling complex are vulnerable because of the abrogation of these cellular functions (Figure 5C). Regarding DNA repair, the SWI/SNF chromatin remodeling complex is involved in DNA double‐strand break repair and DNA damage checkpoint regulation. The PARP1 inhibitor olaparib is approved for BRCA1/2‐deficient ovarian and breast cancers, which are deficient in homologous recombination repair, a DNA double‐strand break repair mechanism. In ARID1A‐deficient cancers, which are deficient in homologous recombination‐mediated DNA double‐strand break repair, treatment with a PARP inhibitor causes synthetic lethality.41
In the SWI/SNF chromatin remodeling complex, the BAF complex is required for chromatin binding of topoisomerase II (TOP2), which is necessary for DNA replication and chromosomal segregation. ARID1A mediates the physical interaction between the BAF complex and TOP2.42 Thus, TOP2 dysfunction in BAF complex‐deficient tumors, such as ARID1A‐deficient cancers, results in aberrant DNA replication and chromosomal segregation. Based on the abnormality of those cellular functions, BAF complex‐deficient cancer cells are selectively sensitive to inhibitors of the cell cycle regulators cyclin‐dependent kinase (CDK)4/CDK6, the DNA replication checkpoint factor ATR, and the chromosomal segregation factor Aurora kinase A.43, 44, 45, 46, 47
The SWI/SNF chromatin remodeling complex is involved in the regulation of several metabolic pathways. The energy supply in cancer cells is derived from ATP generated by the glycolytic pathway. However, SMARCA4‐deficient lung cancer cells depend on energy supplied by the oxidative phosphorylation pathway rather than the glycolytic pathway, and are therefore sensitive to inhibition of oxidative phosphorylation.48 In addition, the cells are exposed to oxidative stress, such as that caused by reactive oxygen species (ROS), which damage DNA and proteins. Excessive generation of ROS leads to induction of cell death. However, the intracellular antioxidant system can effectively mediate resistance to oxidative stress by suppressing ROS through the antioxidant metabolite glutathione (GSH). Thus, the balance between oxidative stress and the antioxidant metabolite GSH maintains cellular homeostasis. We recently showed that impairment of the GSH metabolic pathway is a vulnerability of ARID1A‐deficient cancer cells.49 Glutathione is a tripeptide metabolite synthesized from cysteine, glutamate, and glycine. ARID1A positively regulates the transcription of SLC7A11, which encodes a protein required for the maintenance of intracellular cysteine.49 Deficiency in ARID1A results in SLC7A11 downregulation, which decreases intracellular cysteine. The low basal level of GSH in ARID1A‐deficient cancer cells leads to vulnerability. Inhibition of GSH by the GSH inhibitor APR‐246 or buthionine sulfoximine, an inhibitor of the glutamate cysteine ligase catalytic subunit (which catalyzes the synthesis of GSH), causes excessive increase of ROS and leads to synthetic lethality in ARID1A‐deficient cancer cells.49 These data indicate that inhibitors of metabolic pathways, such as the oxidative phosphorylation pathway and the GSH metabolic pathway, are promising therapeutic agents for cancers with deficiencies in the SWI/SNF chromatin remodeling complex because of vulnerabilities derived from metabolic pathway deficiencies.
7. EFFICIENT STRATEGIES OTHER THAN SYNTHETIC LETHAL ONES FOR THE THERAPY OF SWI/SNF CHROMATIN REMODELING COMPLEX‐DEFICIENT CANCERS
ARID1A and PBRM1 are components of different complexes, namely, the BAF complex and PBAF complex, respectively. Therefore, immunotherapies against ARID1A‐deficient cancers and PBRM1‐deficient cancers should be based on different mechanisms. Cancers with a genetic aberration in mismatch repair are characterized by hypermutation frequency and should respond to immune checkpoint inhibition using programmed death‐ligand 1 (PD‐L1) and programmed death‐1 (PD‐1) Abs because of increased antigen presentation.50 The ARID1A‐containing BAF complex interacts with mismatch repair factors and positively regulates mismatch repair. Therefore, ARID1A‐deficient cancer cells have a high mutation rate because of the deficiency in mismatch repair.51 Cancer immunotherapy would be a promising strategy for ARID1A‐deficient cancers. In addition to mismatch repair deficiency, the activity of the interferon‐γ signaling pathway modulates the sensitivity to cancer immunotherapy.52 The PBRM1‐containing PBAF complex functions in the repression of genes involved in promoting interferon‐γ signaling pathway activity.53 PBRM1‐deficient renal clear cell carcinoma would benefit from cancer immunotherapy because the interferon‐γ signaling pathway is activated in PBRM1‐deficient cancer cells. Cancer immunotherapy is a promising strategy for cancers deficient in the SWI/SNF chromatin remodeling complex.
The effect of SWI/SNF chromatin remodeling complex deficiency on the response to chemotherapy using cytotoxic anticancer agents remains unclear. Ovarian clear cell carcinoma has the highest rate of ARID1A mutation among cancers. ARID1A‐deficient ovarian clear cell carcinoma cells are selectively sensitive only to gemcitabine among the standard chemotherapeutic agents for ovarian clear cell carcinoma that are currently available.54 The first‐line standard chemotherapy for ovarian clear cell carcinoma is combination therapy with paclitaxel and carboplatin. The use of gemcitabine is limited to tumors showing relapse after paclitaxel and carboplatin therapy. However, gemcitabine shows promise for use as a first‐choice agent for optimized therapy in ARID1A‐deficient ovarian clear cell carcinoma in the future.
8. CONCLUDING REMARKS
The SWI/SNF chromatin remodeling complex is composed of many subunits, and the encoding genes are frequently mutated in various cancers. Whether an inhibitor of a synthetic lethal target in cancers deficient in one gene could be effective for cancers with a deficiency in another gene, including genes encoding the subunits of the SWI/SNF chromatin remodeling complex, remains unknown. However, because the SWI/SNF chromatin remodeling complex is divided into the BAF, PBAF, and ncBAF complexes, expanding the indications for an inhibitor of a synthetic lethal target might not be simple. Frequently mutated genes tend to be characteristic of different tumor types, such as SMARCA4 in lung adenocarcinoma (10% frequency), ARID1A in ovarian clear cell carcinoma (50% frequency), PBRM1 in renal clear cell carcinoma (40% frequency), and SMARCB1 in rhabdoid tumors (100% frequency). Mutation of each gene is important for carcinogenesis in every tumor type. The mechanism underlying the effect of cancer‐specific gene mutations can be elucidated by determining the functional relationship based on synthetic lethality. ARID1A mutation occurs in the early stage of precancerous endometriosis and clear cell adenofibroma.55, 56 In addition, ROS could contribute to cancer transformation by promoting gene mutation or by stimulating cellular signaling.57, 58, 59, 60 The basal level of ROS increases as a result of a decrease of GSH in ARID1A‐deficient ovarian cancer cells.49 These findings suggest that ARID1A abrogation is involved in oncogenesis by contributing to a dysregulated balance between ROS and GSH homeostasis. The clinical success rate of therapy based on biomarkers of LOF mutations in SWI/SNF chromatin remodeling genes could be improved by identifying promising drug targets using the concept of synthetic lethality. Many synthetic lethal targets for cancers with SWI/SNF chromatin remodeling complex deficiency have been identified. Inhibitors of these synthetic lethal targets have been developed, many of which are currently under clinical trials or approved for clinical use (Table 1). It is expected that these promising drugs will be approved for clinical application in cancers with SWI/SNF chromatin remodeling complex deficiency in the near future.
Table 1.
Candidate drugs against synthetic lethal targets in cancers deficient in the SWI/SNF chromatin remodeling complex
| Synthetic lethal target | Genetic abnormality | Candidate drug |
|---|---|---|
| ARID1B | ARID1A | – |
| ATR | ARID1A | VX‐970/AZD6738 |
| AURKA | ARID1A/SMARCA4 | MLN8273 |
| BRD2 | ARID1A | I‐BET‐762 |
| BRD9 | SMARCB1/SS18‐SSX | – |
| CDK4/CDK6 | SMARCA4 + SMARCA2 | Palbociclib/ribociclib/abemaciclib |
| EZH2 | ARID1A/SMARCA4/SMARCB1/PBRM1 | Tazemetostat/valemetostat |
| GCLC | ARID1A | BSO |
| GSH | ARID1A | APR‐246 |
| GLI1 | SMARCB1 | LDE225 |
| HDAC2 | ARID1A | Volinostat |
| HDAC6 | ARID1A | ACY‐125 |
| KDM2B | SS18‐SSX | – |
| MDM2/MDM4 | SMARCB1 | ATSP‐7041/idasanutlin |
| OXPHOS | SMARCA4 | IACS‐010759 |
| PARP1 | ARID1A | Olaparib/rucaparib |
| PD‐1/PD‐L1 | ARID1A/PBRM1 | Nivolumab |
| PI3K/AKT | ARID1A | BKM120 |
| SMARCA2 | SMARCA4 | – |
| SMARCA4 | SMARCA2/ARID2 | – |
| SS18‐SSX | SS18‐SSX | – |
| UBE2C | SMARCB1 | Bortezomib/MLN2238 |
| YES1 | ARID1A | BMS‐354825/SKI‐606 |
DISCLOSURE
The authors have no conflict of interest to declare.
ACKNOWLEDGMENTS
We thank Dr Takashi Kohno for valuable guidance in writing this paper.
Sasaki M, Ogiwara H. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex‐deficient cancers. Cancer Sci. 2020;111:774–782. 10.1111/cas.14311
REFERENCES
- 1. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;13(11):772‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendelsohn J. Personalizing oncology: perspectives and prospects. J Clin Oncol. 2013;31(15):1904‐1911. [DOI] [PubMed] [Google Scholar]
- 3. Yoh K, Seto T, Satouchi M, et al. Vandetanib in patients with previously treated RET‐rearranged advanced non‐small‐cell lung cancer (LURET): an open‐label, multicentre phase 2 trial. Lancet Respir Med. 2016;5(1):42-50. [DOI] [PubMed] [Google Scholar]
- 4. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. [DOI] [PubMed] [Google Scholar]
- 5. Iglehart JD, Silver DP. Synthetic lethality–a new direction in cancer‐drug development. N Engl J Med. 2009;361(2):189‐191. [DOI] [PubMed] [Google Scholar]
- 6. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725‐1735. [DOI] [PubMed] [Google Scholar]
- 7. Fang B. Development of synthetic lethality anticancer therapeutics. J Med Chem. 2014;57(19):7859‐7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110‐120. [DOI] [PubMed] [Google Scholar]
- 9. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 10. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273‐304. [DOI] [PubMed] [Google Scholar]
- 11. Beck S, Bernstein BE, Campbell RM, et al. A blueprint for an international cancer epigenome consortium. A report from the AACR Cancer Epigenome Task Force. Cancer Res. 2012;72(24):6319‐6324. [DOI] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Research N , Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet. 2013;45(10):1113‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polak P, Karlic R, Koren A, et al. Cell‐of‐origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518(7539):360‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol. 2019;21(2):152‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015;1(5):e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mashtalir N, D'Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 2018;175(5):1272–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Michel BC, D'Avino AR, Cassel SH, et al. A non‐canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20(12):1410‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18‐SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153(1):71‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oike T, Ogiwara H, Tominaga Y, et al. A synthetic lethality‐based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73(17):5508‐5518. [DOI] [PubMed] [Google Scholar]
- 21. Hoffman GR, Rahal R, Buxton F, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1‐deficient cancers. Proc Natl Acad Sci U S A. 2014;111(8):3128‐3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson BG, Helming KC, Wang X, et al. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34(6):1136‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ehrenhofer‐Wolfer K, Puchner T, Schwarz C, et al. SMARCA2‐deficiency confers sensitivity to targeted inhibition of SMARCA4 in esophageal squamous cell carcinoma cell lines. Sci Rep. 2019;9(1):11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helming KC, Wang X, Wilson BG, et al. ARID1B is a specific vulnerability in ARID1A‐mutant cancers. Nat Med. 2014;20(3):251‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berns K, Caumanns JJ, Hijmans EM, et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene. 2018;37(33):4611‐4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chun HE, Lim EL, Heravi‐Moussavi A, et al. Genome‐wide profiles of extra‐cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell. 2016;29(3):394‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203‐206. [DOI] [PubMed] [Google Scholar]
- 28. McBride MJ, Pulice JL, Beird HC, et al. The SS18‐SSX fusion oncoprotein Hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell. 2018;33(6):1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang X, Wang S, Troisi EC, et al. BRD9 defines a SWI/SNF sub‐complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun. 2019;10(1):1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife. 2018;7:e41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schick S, Rendeiro AF, Runggatscher K, et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet. 2019;51(9):1399‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poynter ST, Kadoch C. Polycomb and trithorax opposition in development and disease. Wiley Interdiscip Rev Dev Biol. 2016;5(6):659‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bitler BG, Aird KM, Garipov A, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A‐mutated cancers. Nat Med. 2015;21(3):231‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim KH, Kim W, Howard TP, et al. SWI/SNF‐mutant cancers depend on catalytic and non‐catalytic activity of EZH2. Nat Med. 2015;21(12):1491‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922‐7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Lee RS, Alver BH, et al. SMARCB1‐mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet. 2017;49(2):289‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakayama RT, Pulice JL, Valencia AM, et al. SMARCB1 is required for widespread BAF complex‐mediated activation of enhancers and bivalent promoters. Nat Genet. 2017;49(11):1613‐1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B‐cell non‐Hodgkin lymphoma and advanced solid tumours: a first‐in‐human, open‐label, phase 1 study. Lancet Oncol. 2018;19(5):649‐659. [DOI] [PubMed] [Google Scholar]
- 40. Bitler BG, Wu S, Park PH, et al. ARID1A‐mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19(8):962‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dykhuizen EC, Hargreaves DC, Miller EL, et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature. 2013;497(7451):624‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williamson CT, Miller R, Pemberton HN, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tagal V, Wei S, Zhang W, et al. SMARCA4‐inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX‐680 in non‐small cell lung cancers. Nat Commun. 2017;8:14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xue Y, Meehan B, Macdonald E, et al. CDK4/6 inhibitors target SMARCA4‐determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Commun. 2019;10(1):558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xue Y, Meehan B, Fu Z, et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non‐small cell lung cancer. Nat Commun. 2019;10(1):557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu C, Lyu J, Yang EJ, Liu Y, Zhang B, Shim JS. Targeting AURKA‐CDC25C axis to induce synthetic lethality in ARID1A‐deficient colorectal cancer cells. Nat Commun. 2018;9(1):3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deribe YL, Sun Y, Terranova C, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med. 2018;24(10):1627. [DOI] [PubMed] [Google Scholar]
- 49. Ogiwara H, Takahashi K, Sasaki M, et al. Targeting the vulnerability of glutathione metabolism in ARID1A‐deficient cancers. Cancer Cell. 2019;35(2):177–190. [DOI] [PubMed] [Google Scholar]
- 50. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shen J, Ju Z, Zhao W, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miao D, Margolis CA, Gao W, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359(6377):801‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuroda T, Ogiwara H, Sasaki M, et al. Therapeutic preferability of gemcitabine for ARID1A‐deficient ovarian clear cell carcinoma. Gynecol Oncol. 2019. [DOI] [PubMed] [Google Scholar]
- 55. Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer‐associated mutations in endometriosis without cancer. N Engl J Med. 2017;376(19):1835‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guan B, Mao TL, Panuganti PK, et al. Mutation and loss of expression of ARID1A in uterine low‐grade endometrioid carcinoma. Am J Surg Pathol. 2011;35(5):625‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oberley LW. Free radicals and diabetes. Free Radic Biol Med. 1988;5(2):113‐124. [DOI] [PubMed] [Google Scholar]
- 58. Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, Heintz NH. Redox‐dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal. 2006;8(9–10):1447‐1459. [DOI] [PubMed] [Google Scholar]
- 59. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1‐15. [DOI] [PubMed] [Google Scholar]
- 60. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931‐947. [DOI] [PubMed] [Google Scholar]