

Abstract
Esophageal cancer is currently one of the most fatal cancers. However, there is no effective treatment. Increasing evidence suggests that interleukin (IL)‐33 has a significant role in tumor progression and metastasis. Currently, the underlying cellular and molecular mechanism of IL‐33 in promoting esophageal squamous cell carcinoma (ESCC) remains unclear. In this study, we investigated whether IL‐33 could induce the epithelial‐mesenchymal transition (EMT) in ESCC. Interleukin‐33 expression was examined in ESCC and corresponding adjacent normal tissues by immunohistochemistry and quantitative real‐time PCR experiments. Elevated IL‐33 levels were observed in ESCC tissues. Further in vitro experiments were undertaken to elucidate the effect of IL‐33 on migration and invasion in KYSE‐450 and Eca‐109 esophageal cancer cells. Knockdown of IL‐33 decreased the metastasis and invasion capacity in esophageal cancer cells, whereas IL‐33 overexpression showed the opposite effect. We then screened CCL2 which is a downstream molecule of IL‐33, and proved that IL‐33 could promote tumor development and metastasis by recruiting regulatory T cells (Tregs) through CCL2, and IL‐33 regulated the expression of CCL2 through transforming growth factor‐β in Treg cells. Knockdown of IL‐33 decreased the development of human ESCC xenografts in BALB/c nude mice. Collectively, we found that the IL‐33/nuclear factor‐κB/CCL2 pathway played an essential role in human ESCC progress. Hence, IL‐33 should be considered as an effective therapy target for ESCC.
Keywords: CCL2, esophageal squamous cell carcinoma, IL‐33, NF‐κB, Treg
Ying Yue et al. showed that IL‐33 could up‐regulate CCL2 expression and then recruit Treg cells through the NF‐κB pathway. Furthermore TGF‐β secreted by Tregs promotes the upregulation of IL‐33 in esophageal tumor cells. IL‐33 should be considered as an effective therapy target for esophageal cancer.
Abbreviations
- EMT
epithelial‐mesenchymal transition
- ESCC
esophageal squamous cell carcinoma
- IHC
immunohistochemistry
- IL‐33
interleukin‐33
- NF‐κB
nuclear factor‐κB
- TBS‐T
TBS containing 0.1% Tween‐20
- TGF‐β
transforming growth factor‐β
- Treg
regulatory T cell
1. BACKGROUND
Esophageal cancer is one of the most severe diseases that can influence human health both in developed and developing countries,1 especially in China. It was reported that ESCC accounted for 90% of total incidence of Chinese esophageal cancer cases.2 Due to the lack of early symptoms, many patients are diagnosed too late to be treated effectively.3 Studies found that the survival rate of advanced stage ESCC patients was no more than 10%.4, 5 Therefore, it is urgent to clarify the molecular pathogenesis of ESCC.
Interleukin‐33, a nuclear cytokine abundantly expressed both in endothelial and epithelial cells, is known as a natural ligand for ST2L and can be antagonized by soluble ST2.6, 7, 8, 9 Interleukin‐33 can activate many immune cells, such as Th2 cells.9 When there is cell injury or tissue damage, IL‐33 can be released as an alarm signal.10 It was reported that IL‐33 has different functions including tumor promotion or suppression. In ovarian cancer, the upregulation of IL‐33 dramatically promotes cancer cell proliferation, migration, and invasion.11 Hollande et al12 found that IL‐33 was essential and active for eosinophil‐mediated antitumor responses. Cui et al13 observed that the IL‐33/ST2 axis might be involved in the progression of human ESCC. However, the molecular mechanism of IL‐33 in ESCC development remains unclear. Transforming growth factor‐β is a well‐known immunosuppressor and proangiogenic factor that can regulate tumor proliferation and stemness.14 It exerts a dual function in tumorigenesis.15 Transforming growth factor‐β could inhibit tumorigenesis by regulating tumor growth and apoptosis and can also promote tumorigenesis by altering the tumor microenvironment to accelerate EMT.16 It was hypothesized that IL‐33 might induce EMT through the TGF‐β pathway.
Previous studies indicated that the IL‐33/ST2L axis could regulate inflammatory responses in many cancers, including ulcerative colitis, colitis‐associated cancer, and gastric cancer.17, 18 A previous study found that IL‐33 is restrictively expressed in murine squamous cell carcinoma cells, and it could activate FAK downstream to regulate CCL5 expression and tumor growth.19 In this study, we aimed to examine the expression and function of IL‐33 in ESCC and the underlying mechanism both in vitro and in vivo. Furthermore, the corresponding effect of IL‐33 in regulating cell proliferation, cell cycle, migration, invasion, and clonogenicity were also explored. Our results showed that TGF‐β can upregulate IL‐33 in ESCC, thus increasing the expression of CCL2 and inducing EMT through NF‐κB signaling. In summary, our study suggests that IL‐33 could be a potential therapeutic target for ESCC therapy.
2. MATERIALS AND METHODS
2.1. Patients and tissues
Specimens of patients who underwent cardiothoracic surgery at the First Affiliated Hospital of Zhengzhou University were collected between 2011 and 2014. All patients included signed the informed consent, and this collection was approved by the Ethics Committee Board in the First Affiliated Hospital of Zhengzhou University. Eighty‐seven ESCC and matched adjacent noncancerous tissues were collected independently. No patients had received preoperative chemotherapy or radiotherapy. After the surgical resection, all the tumor tissues were diagnosed as ESCC by standard protocol, 2 independent pathologists confirmed the pathological results separately. Patient's age, gender, tumor invasion level, lymph nodes metastasis, histological grade, and tumor stage were assessed and recorded. Details are shown in Table S1.
2.2. Cell lines
Human esophageal epithelial cell line HET‐1A and ESCC cell lines Eca‐109, KYSE‐450, KYSE‐70, EC9706, EC1, TE‐1, and TE‐7 were cultured in RPMI‐1640 (HyClone) medium, supplied with 10% FBS (HyClone), 100 μg/mL streptomycin, and 100 units/mL penicillin according to the standard protocol. All the cell lines were incubated at standard culture conditions (5% CO2, 37°C) in a sterile environment and were tested for IL‐33 expression.
2.3. Plasmid construction and transfection
KYSE‐450 was applied to stably overexpress IL‐33 by using pBpLV‐GFP + Puro vector plasmid (GenePharma). We qualified the level of IL‐33 by RT‐PCR and western blot analysis. Eca‐109 was used to knock down IL‐33. After transfection, cells were classified by a BD CantoII flow cytometer (Becton Dickinson). The successfully transfected cells were sorted by flow cytometry and were cultured.
2.4. Real‐time PCR
Total RNA were extracted by TRIzol reagent (Invitrogen). The RNA quality was assessed by NanoDrop 2000 (Thermo Fisher Scientific). The PrimeScript RT reagent kit (Takara) was used to obtain the cDNA with 1 μg RNA. Then we undertook RT‐PCR by using SYBR Premix ExTaq (Takara) in the Agilent Mx3005P system. Sequences of primers were showed in Table S2. Each experiment was repeated 3 times separately. Glyceraldehyde‐3‐phosphate dehydrogenase was used as control. Each target gene in our study was quantified with the 2−ΔΔCt method and was normalized to the level of GAPDH.
2.5. Immunohistochemistry
All resected slides were examined by 2 senior pathologists independently. Interleukin‐33 expression was scored by the proportion of positive stained cells and staining intensity. Staining intensity was grouped as: 0, negative; 1, yellowish; 2, light brown; and 3, dark brown. The scoring of immunoreactive tumor cells was set as follows: 0, <5%; 1, 5%‐25%; 2, 25%‐50%; 3, 50%‐75%; and 4, more than 75%. Immunoreactivity score was collected for each tissue by multiplying the intensity score with percentage.
2.6. Western blot analysis
Tissue samples and ESCC cells were lysed in ice‐cold RIPA buffer. Protein levels were assessed by BCA protein assay kit (Beyotime). The 12% SDS‐PAGE was used to separate equal amounts of protein before transferring protein to PVDF membranes. Then 5% nonfat dried milk was used in TBS‐T, then the membranes were incubated with specific primary Abs at 4°C. After washed in TBS‐T, membranes were incubated with secondary Abs. All the target bands were assessed by an enhanced chemiluminescence kit (Zhongshanjinqiao).
2.7. Cell proliferation
Proliferative ability of ESCC was assessed by using the CCK‐8 assay kit (Beyotime). Absorbance was measured at 450 nm at indicated time points by using a microplate reader.
2.8. Wound healing assays
Each well of the 6‐well plates was seeded with 2 × 105 cells, then the serum‐free medium was added until a confluent monolayer formed. A 200‐μL sterile pipette tip was used to scratch the confluent monolayers. Wound closure was examined by collecting digitized images at 0 and 24 hours.
2.9. Transwell migration and invasion assays
Generally, Transwell migration and invasion assays were carried out with 24‐well inserted plates (5 μm; Corning) according to the manufacturer's instructions. Cell suspension (2 × 105 cells/well) was added to the upper chamber, and 600 μL RMPI‐1640 containing no FBS was added to the lower chambers. After 24 hours, cells that attached to the lower surface were stained with 0.1% crystal violet for 15 minutes.
2.10. Isolation of Tregs
CD4+CD25+ Tregs were isolated from PBMC of healthy donors using a MoFlo XDP cytometer (Beckman Coulter). Cell sorting was carried out according to instructions. Positive sorting rate of cells after purification was confirmed over 90% by flow cytometry.
2.11. Cytometric bead array
Tumor cells were cultured for 72 hours and cell‐free supernatants were collected. Cytokine capture beads were added to the samples, and phycoerythrin detection reagent was added afterwards. Supernatants were incubated for 3 hours in the dark. Subsequently, bead pellets were collected and analyzed by using Cell Quest and CBC software.
2.12. Enzyme‐linked immunosorbent assay
According to the instructions, the level of CCL2 and IL‐33 were assessed by the ELISA kit purchased from BioLegend.
2.13. Animal model
A total of 10 female BALB/c nude mice (Vital River Laboratory Animal Technology) at age of 6 weeks were randomized into 2 groups (5 mice/group) and injected with tumor cells (1 × 106 cells in 100 μL PBS). We evaluated tumor length and width every 2 days. The tumor volume was calculated by the following formula: (length × width2)/2. In the Treg transplant animal experiment, 10 female NOD SCID mice were randomly divided into 2 groups. Both groups received an s.c. injection of 1 × 106 tumor cells. Tumor growth was assessed as above. Regulatory T cells (5 × 106) were transplanted through the tail vein 3 days before the mice were killed, then tumor tissues were collected. The proportion of infiltrated Tregs (CD4+CD25+FOXP3+) was analyzed by flow cytometry. All animal studies were approved by the Institutional Animal Care and Use committee of the First Affiliated Hospital of Zhengzhou University.
2.14. Statistical analysis
Student's t test or the χ2 test was used to compare data from different groups. We used the paired t test to analyze matched samples. Overall survival curves were plotted by the Kaplan‐Meier method. Analyses were undertaken using GraphPad Prism 7 software (GraphPad Software). P < .05 was considered as statistically significant difference in our study.
3. RESULTS
3.1. Levels of IL‐33 increased in ESCC tissues
First, the IL‐33 mRNA expression level was examined among 87 ESCC and matched normal tissues. The results indicated that the mRNA level of IL‐33 was significantly increased in ESCC tissues compared with normal tissues (P = .0006) (Figure 1A). The mRNA level of IL‐33 had a significant correlation with tumor stage (Figure 1B) and it was higher in poorly differentiated patients than in well differentiated groups (Figure 1C).
Figure 1.
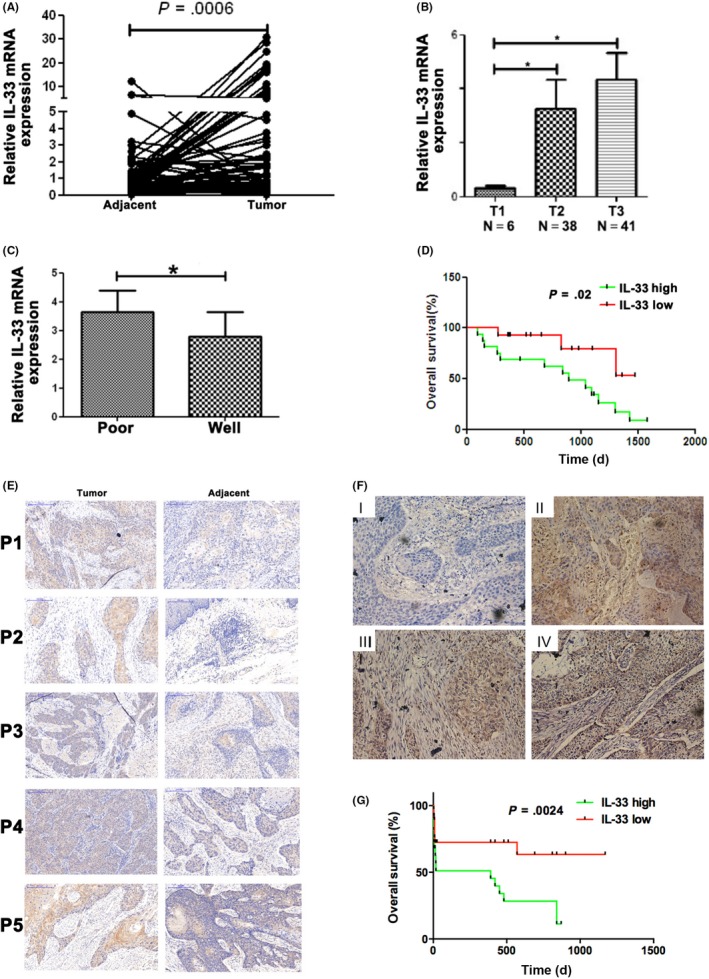
A,B, RT‐PCR analysis of interleukin (IL)‐33 mRNA expression in esophageal squamous cell carcinoma (ESCC) and paired noncancerous tissues (n = 87). C, RT‐PCR analysis of IL‐33 mRNA expression in poor and well differentiated tissues. D, Overall survival in ESCC patients with IL‐33‐low and IL‐33‐high mRNA expression. E, Representative images of immunohistochemical (IHC) staining for IL‐33 in ESCC tissues and adjacent normal tissues. F, Representative images of IHC staining for IL‐33 in different stages. G, Overall survival in ESCC patients with IL‐33‐low and IL‐33‐high protein expression. Mean ± SD of relative fold changes from triplicate experiments was plotted
The correlation of clinicopathological characteristics between IL‐33 level and patients are evaluated and described in Table S1. Among the 87 ESCC patients, 65 (74.7%) had higher expression of IL‐33 and 22 (25.3%) had a lower level. Moreover, IL‐33 level was significantly associated with invasive depth, differentiation degrees, TNM stage, and survival rates.
To confirm whether IL‐33 expression was related with poor prognosis in ESCC, survival analysis was carried out. According to the IL‐33 scores, 87 ESCC patients were divided into low or high expression groups (Figure 1D). Then IL‐33 protein expression was examined by IHC. We found that the percentage and intensity of IL‐33 in ESCC were obviously higher than in control groups (Figure 1E). Immunohistochemistry was used to represent the scoring method (Figure 1F,G). In the IL‐33‐high group, overall survival was lower than in the IL‐33‐low group (Figure 1G). Our results indicated that IL‐33 expression is closely correlated with tumor invasive depth, differentiation degree, TNM stage, and poor survival in ESCC patients.
3.2. Interleukin‐33 affects cell migration in vitro
The biological characteristics of IL‐33 were determined in HET‐1A and 7 other ESCC cell lines including Eca‐109, KYSE‐450, KYSE‐70, EC9706, EC9706 clone EC1, TE‐1, and TE‐7 by RT‐PCR and western blot analysis (Figure 2A,B). Eca‐109 has the highest IL‐33 mRNA expression, and KYSE‐450 has the lowest. Hence, for IL‐33‐overexpression experiments, KYSE‐450 cells were chosen, and for IL‐33‐knockdown experiments, Eca‐109 cells were chosen. Next, we applied the lentiviral system to obtain stable IL‐33 overexpression or knockdown cell lines to clarify the function of IL‐33.
Figure 2.
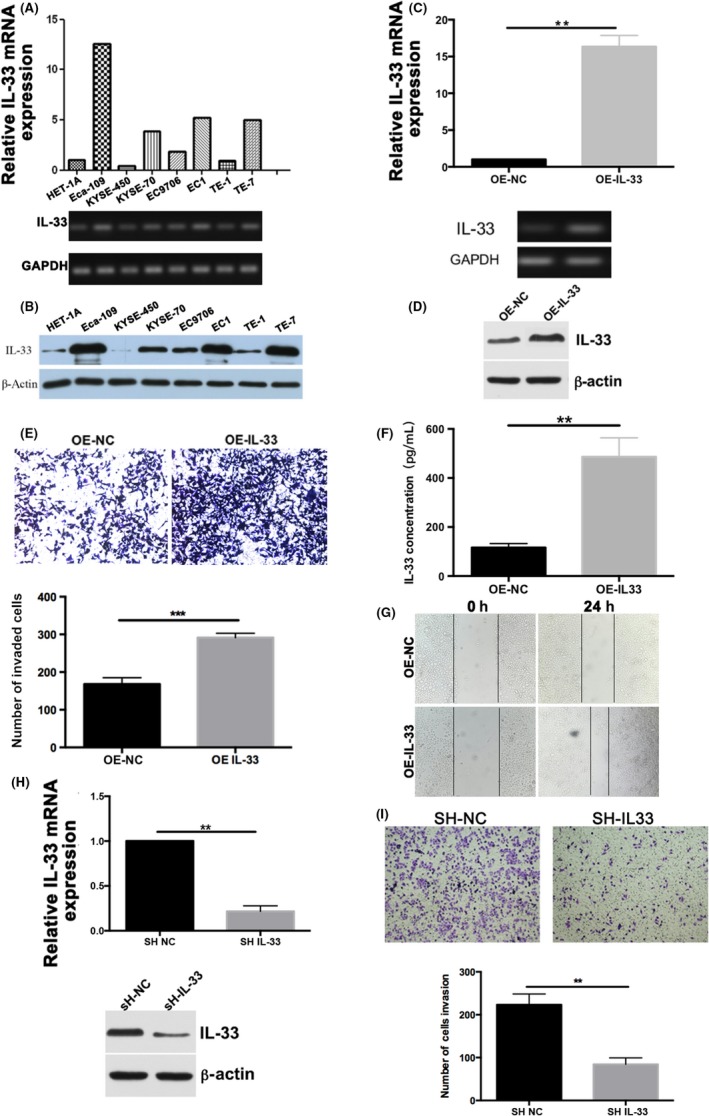
A, RT‐PCR analysis of interleukin (IL)‐33 expression in HET‐1A, Eca‐109, KYSE‐450, KYSE‐70, EC9706, EC9706 clone EC1, TE‐1, and TE‐7 cell lines. B, Western blot analysis for IL‐33 expression in esophageal squamous cell carcinoma (ESCC) cell lines. C,D, RT‐PCR and western blot analysis for IL‐33 overexpression (OE) in KYSE‐450 cells. E, Migratory and invasive capabilities of KYSE‐450 cells were evaluated using migration and invasion assays. F, ELISA analysis for IL‐33 overexpression in KYSE‐450 cells. G, Scratch healing was photographed under a microscope after 0 and 24 h. H, RT‐PCR and western blot analysis to detect the efficiency of IL‐33 knockdown in Eca‐109 cells. I, Transwell assay for ESCC cells with knockdown of IL‐33. Relative fold changes from triplicate experiments are plotted. NC, control
After transfection of IL‐33, the cells were sorted by flow cytometry. Interleukin‐33 increased in IL‐33‐overexpressing KYSE‐450 cells compared to control groups in both mRNA and protein levels (Figure 2C,D). Transwell analysis and ELISA also showed that the level of IL‐33 was higher than the control (Figure 2E,F). As shown in Figure 2G, IL‐33 overexpression significantly promoted the migration of ESCC in the wound healing assay. The mRNA and protein levels of IL‐33 were significantly and efficiently decreased in shIL‐33 Eca‐109 cells (Figure 2H). In addition, we found that the migration and invasion capacities of scrambled cells were remarkably greater than shIL‐33 cells (Figure 2I) in the Transwell analysis. The absorbance at 450 nm was not significantly elevated in the IL‐33 knockdown or overexpression groups compared to the control at 24, 48, or 72 hours (Figure S1A,B). In the proliferation assay, the number of cell clones formed in OE‐IL33 cells was similar (Figure S1C). Colony formation also showed that IL‐33 overexpression or knockdown did not affect cell proliferation (Figure S1C). The RT‐PCR, western blot analysis, and ELISA were carried out to detect the efficiency of IL‐33 knockdown (Figure S2A–C). After the knockdown of IL‐33, cell invasion ability was significantly reduced (Figure S2D). These results showed that IL‐33 played a vital role in cell migration and invasion, indicating that IL‐33 could be a potential target for antitumor therapy for ESCC.
3.3. Interleukin‐33 regulates CCL2 expression through NF‐κB signaling in ESCC
It was reported that various chemokines exert different effects on tumor growth, invasion, and metastasis. To understand how IL‐33 affects tumor progression, we investigated chemokine expression in the supernatants of tumor cells with either IL‐33 knockdown or overexpression by cytometric bead array. Results showed that IL‐33 overexpression significantly upregulated CCL2 (Figure 3A). Change of CCL2 expression was detected in ESCC cells using RT‐PCR and ELISA assays; we found that CCL2 expression was elevated with overexpression of IL‐33 (Figure 3B,C). I contrast, we utilized a lentiviral system to obtain IL‐33 knockdown cell lines. Downregulation of CCL2 level was observed in the shIL‐33 group compared with the empty retrovirus transfected group using RT‐PCR and ELISA assay (Figure 3D–F). The addition of IL‐33 recombinant protein could increase the CCL2 level (Figure 3G). Furthermore, when we knocked down the IL‐33 receptor ST2 and added exogenous recombinant IL‐33 in Eca‐109 cells, the CCL2 level did not change significantly (Figure 3G), indicating that IL‐33 might affect CCL2 expression by regulating IL‐33 receptor ST2. The NF‐κB signaling pathway is known to regulate initiation of tumorigenesis and tumor growth.20 To explore the underlying mechanism of IL‐33 on CCL2, the expression of CCL2 and NF‐κB was examined. Results revealed that the active form of NF‐κB (phosphorylated NF‐κB) and CCL2 were markedly decreased by knockdown of IL‐33 together with addition of NF‐κB inhibitor compared to the control group (Figure 3H). The same result was also achieved in the OE IL‐33 group (Figure S3). However, after knockdown of receptor ST2, the expression of CCL2 and NF‐κB was upregulated (Figure 3H). Additionally, both the expression of CCL2 and phosphorylated NF‐κB were decreased by knockdown of receptor ST2 and addition of IL‐33 (Figure 3H), indicating that IL‐33 might regulate NF‐κB signaling by binding to its receptor ST2 to affect the function of CCL2. Statistical analyses of the protein expression levels are shown in Figure S4A. Furthermore, RT‐PCR was carried out to analyze the CCL2 and IL‐33 gene levels in 35 ESCC patients and 76 ESCC patients from The Cancer Genome Atlas, the results showed that IL‐33 is significantly associated with CCL2 (Figure 3I). Therefore, we hypothesized that CCL2 could be a downstream target of IL‐33 in ESCC.
Figure 3.
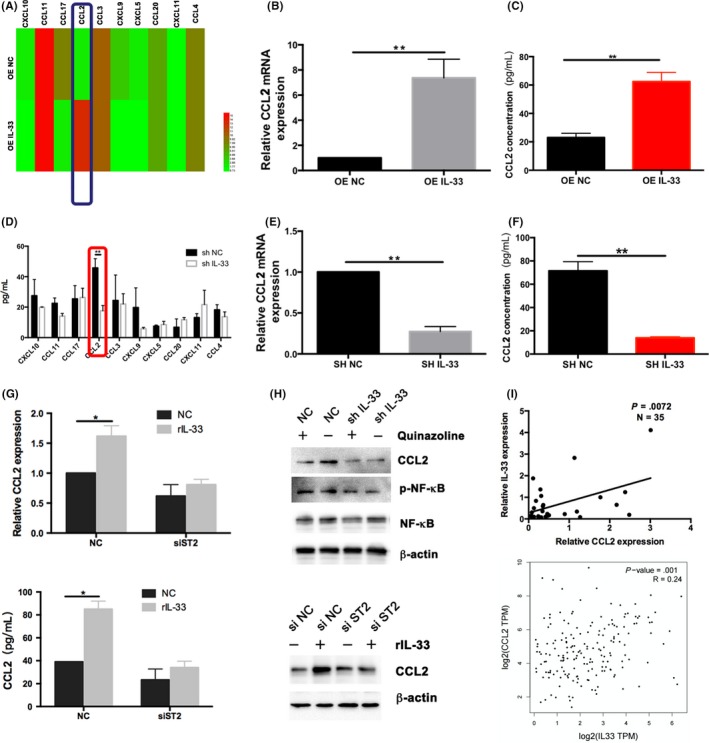
A, Interleukin (IL)‐33 regulates the expression of CCL2 in esophageal squamous cell carcinoma (ESCC) cells. A, Cytometric bead array (CBA) for changes of cytokines in KYSE‐450 cells overexpressing IL‐33 (OE‐IL‐33) and the control group (NC). B,C, RT‐PCR and ELISA to detect the change of CCL2 level. D, CBA for changes of cytokines in IL‐33 knockdown and control groups. E,F, RT‐PCR and ELISA to detect the change of CCL2 level with knockdown of IL‐33 in Eca‐109 cells. G, RT‐PCR and ELISA to detect the expression of CCL2 level with addition of IL‐33 and knockdown of IL‐33 receptor ST2. H, Western blotting revealed the protein level of active form of nuclear factor (NF)‐κB and CCL2 with knockdown of IL‐33 or its receptor. I, Association between CCL2 and IL‐33 gene levels in ESCC patients. Data are presented as mean ± SD
3.4. Interleukin‐33 regulates expression of CCL2 through TGF‐β
CCL2 has been shown to recruit Tregs.21 We used the Transwell assay to examine the chemotactic effect of IL‐33 and CCL2 on Tregs. Our results showed the proportion of CD4+ cells was higher in the OE‐IL‐33 group than in the control (Figure 4A). Regulatory T cells are involved in maintaining immune homeostasis and preventing autoimmunity, which can suppress immune responses through various mechanisms. The transcription factor Foxp3 is a major lineage regulator of Treg development and inhibitory activity.21 Interleukin‐33 was positively associated with Foxp3 at the mRNA level in 48 ESCC patients (Figure 4B). Addition of recombinant protein CCL2 could also exert a chemotactic effect on Tregs (Figure 4C). The migrated index was higher than the non‐CCL2 group. These results indicated that CCL2 might have a chemotactic effect on Treg cells.
Figure 4.
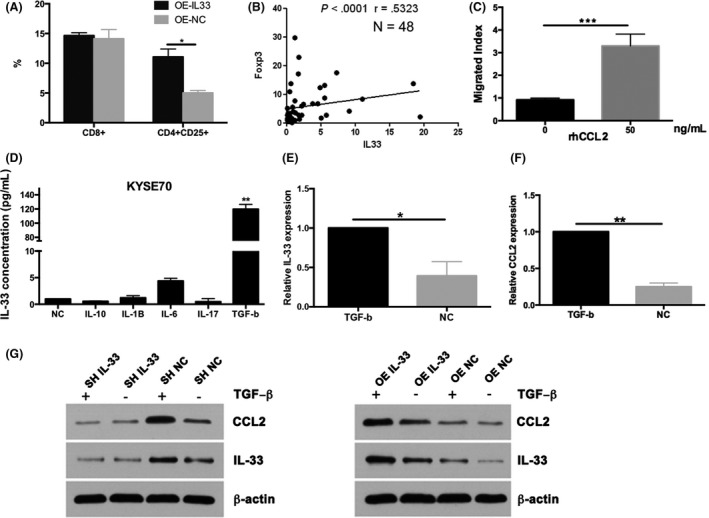
A, Transwell assays to detect the proportion of regulatory T cells (Tregs) with overexpression (OE) of interleukin (IL)‐33. B, Chemotactic effect of CCL2 to Tregs in 48 patients with esophageal squamous cell carcinoma by RT‐PCR and flow cytometry. C, Migrated index for addition of recombinant protein CCL2. D, ELISA analysis for IL‐33 with increased transforming growth factor‐β (TGF‐β) level. E,F, RT‐PCR for IL‐33 and CCL2 with addition of TGF‐β. G, Western blot analysis to observe the association between CCL2 and TGF‐β with IL‐33 overexpression or knockdown. Data are presented as mean ± SD
It was reported that TGF‐β can regulate CCL2 expression in cancer progression.22 Addition of TGF‐β recombinant protein could significantly increase the IL‐33 level when compared with control (Figure 4D). No significant difference was observed in terms of IL‐10, IL‐1β, IL‐6, or IL‐17. After adding TGF‐β to KYSE‐450 cells, IL‐33 and CCL2 mRNA was largely upregulated (Figure 4E,F). Then we investigated whether IL‐33 knockdown or overexpression could affect the CCL2 level with or without TGF‐β. Results showed that protein levels of CCL2 decreased significantly after knocking down IL‐33 or with addition of TGF‐β (Figure 4G). By adding exogenous TGF‐β and overexpressing IL‐33, CCL2 levels were upregulated (Figure 4G). Statistical analyses of the protein expression levels are shown in Figure S4B. It indicated that IL‐33 could regulate the expression of CCL2 though TGF‐β in the ESCC cells.
3.5. In vivo analysis of IL‐33 effects on tumor formation
To further investigate the impact of downregulating IL‐33 on tumor growth in vivo, we injected shIL‐33 cells or control cancer cells into nude mice. As shown in Figure 5A, control cells formed s.c. tumors earlier than the shIL‐33 group. In the IL‐33‐overexpressing group, we observed that s.c. tumors appeared earlier and were larger (Figure 5B). Tumor tissues from shIL‐33 cell‐injected mice had lower IL‐33 mRNA expression levels (Figure 5C). These findings suggested that knockdown of IL‐33 could reduce tumor burden and inhibit formation of tumors in vivo. The mRNA level of CCL2 was also decreased by knockdown of IL‐33 (Figure 5D). Furthermore, we injected IL‐33‐overexpressing KYSE‐450 cells into mice. As shown in Figure 5E,F, both IL‐33 and CCL2 mRNA levels were substantially increased. Epithelial‐mesenchymal transition plays crucial roles in the process of cancer metastasis,23 yet it remains unclear whether IL‐33 can promote EMT in ESCC. We analyzed EMT‐related genes from tumor tissues of nude mice and found that expression of N‐CAD, slug, VIM, and ZEB2 in shIL‐33 group was lower in the control group (Figure 5G).
Figure 5.
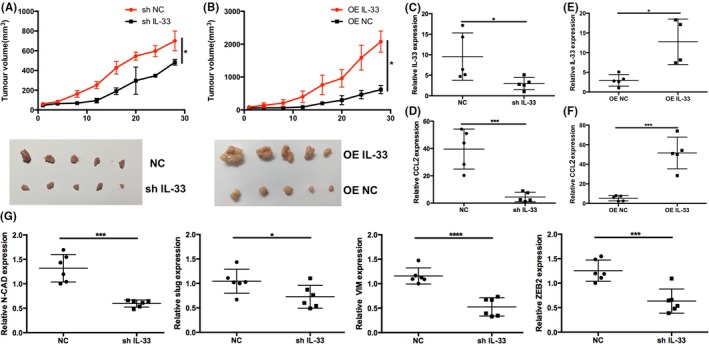
A,B, Tumor volumes in mice injected with shIL‐33 in Eca‐109 cells and overexpression (OE) of interleukin (IL)‐33 in KYSE‐450 cells (n = 5). C,D, RT‐PCR for IL‐33 and CCL2 with IL‐33 knockdown. E,F, RT‐PCR for IL‐33 and CCL2 with IL‐33 overexpression. G, RT‐PCR for detection of N‐CAD, slug, VIM, and ZEB2 with knockdown of IL‐33. Data are presented as mean ± SD
3.6. Chemotaxis of CCL2 cells on Tregs in vivo
The IL‐33 and CCL2 mRNA levels in 35 ESCC patients were further analyzed. At the same time, the proportion of Tregs was analyzed. We found that IL‐33 was positively associated with Tregs. To further investigate the chemotaxis of CCL2 on Tregs, Transwell assay was carried out and it was found that CCL2 had a chemotaxis effect on Treg cells. We injected control cells and shIL‐33 Eca‐109 cells into nude mice and transferred Tregs at day 20. The mice were killed at day 23. Flow cytometry analysis showed that the Treg proportion was lower (0.3%) in the shIL‐33 group compared with the control group (3.5%) (Figure 6A,B). In our xenograft mice model, similar chemotaxis effect was observed by IHC and RT‐PCR (Figure 6C,D). Our IHC results showed the expression of Foxp3 and CCL2 was downregulated by knockdown of IL‐33 (Figure 6C). We observed that, when IL‐33 was knocked down, the mRNA expression levels of Foxp3 and CCL2 were also decreased (Figure 6D), which indicated a positive correlation between Foxp3 and CCL2 in ESCC. We drew a schema showing the potential molecular mechanisms (Figure 6E).
Figure 6.
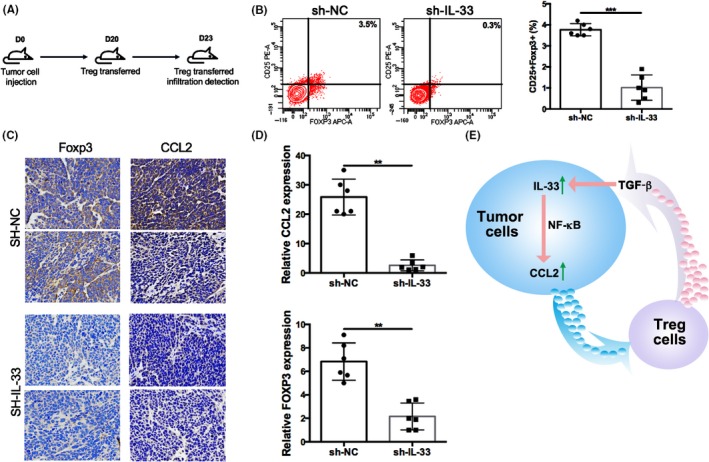
A, Esophageal squamous cell carcinoma tumor cell injection into nude mice (n = 5). D, day. B, Flow cytometry analysis shows the proportion of regulatory T cells (Tregs) in the shIL‐33 group and control group (NC). C, Immunohistochemical staining to detect the expression of Foxp3 and CCL2. D, RT‐PCR for Foxp3 and CCL2 with knockdown of interleukin (IL)‐33. E, Schema showing molecular mechanisms. Data are presented as mean ± SD
4. DISCUSSION
Esophageal squamous cell carcinoma is a fatal disease that remains difficult to diagnose and treat effectively, resulting in high rates of tumor metastasis, recurrence, and chemotolerance.15 Hence, it is urgent to get a deeper understanding. In our study, we examined the expression of IL‐33 of ESCC patients and observed a higher level of IL‐33 in tumor tissues and was closely related with worse clinical outcomes. Interleukin‐33 is positively associated with CCL2, and it could promote the expression of CCL2 through the NF‐κB pathway. Furthermore, we proved that IL‐33 could promote tumor development by regulating the expression of CCL2 as well as promoting metastasis by regulating EMT. These findings showed that IL‐33 might serve as a therapy target to suppress ESCC aggression and metastasis.
It was reported that IL‐33 could be secreted by necrotic epithelial cells,24, 25 and then bind to its cognate ST2 receptor, thus activating mitogen‐related protein kinase and NF‐κB.7, 26 Results from previous studies indicated that IL‐33 has a proinflammatory function and is considered as a “danger” signal.24, 27 Malik et al and Milovanovic et al9, 17 suggested that IL‐33 could be released into the tumor microenvironment to modulate tumor development, which indicates an inconsistent role in cancer development. It was reported that IL‐33 appeared at higher levels in colorectal cancer tissues.28, 29, 30 Here, we reported that IL‐33 was upregulated in ESCC tissue sections, suggesting that IL‐33 overexpression might be correlated with ESCC progress. Our results were confirmed by a recent study in murine squamous cell carcinoma cells,19 indicating that IL‐33 has a significant function in ESCC progression. In addition, functional analysis observed that IL‐33 was significantly related with invasive depth, degree of differentiation, and TNM stage and a poorer survival rate. Consistently, similar results have already been reported in colorectal cancer.30 A study from Yang et al31 also found that IL‐33 levels were significantly decreased both in adenocarcinoma and squamous cell carcinoma. Ding et al32 reported similar results in oral squamous cell carcinoma patients.
To further explore the biological mechanism of upregulated IL‐33 in ESCC, stable KYSE‐450‐OE‐IL‐33 and Eca‐109‐shIL‐33 cell lines were established. Our in vitro experiments showed that IL‐33 affected the invasion and migration of ESCC. Consistently, Tong et al11 reported that IL‐33 promoted the invasive and migratory capacity in human epithelial ovarian cancer cells. Zhang et al26 observed similar results in glioma cells. In our animal model, we examined the expression of metastasis‐related molecules by knockdown of IL‐33. Reportedly, CCL2 has the ability to promote invasion and migration in prostatic carcinoma and ovarian carcinoma.33, 34 Interleukin‐33 knockdown strongly downregulated CCL2, which could partly explain why IL‐33 knockdown impacted the reduced invasive capacity of ESCC cells. We found the expression of CCL2 was increased when exogenous IL‐33 recombinant protein was applied in ESCC cells. Gene expression of CCL2 in 76 patients was also measured as it was significantly associated with CCL2, which might be its downstream target. Furthermore, we knocked down ST2, an IL‐33 receptor, and the ability of IL‐33 to downregulate CCL2 was decreased. These results suggest that IL‐33 might promote the expression of CCL2 by binding to its receptor ST2.
Interleukin‐33 could recruit IL‐1R‐associated kinase 1 and 4 to the receptor complex in cytoplasmic region of ST2 by binding to the IL‐33 receptor, which triggers numerous signaling downstream, including NF‐κB, that might lead to inflammation.26 Activated NF‐κB enhances the expression of various proteins that could promote metastasis.24, 25, 28 In our results, after knockdown of IL‐33, the level of CCL2 was decreased when the NF‐κB pathway was inhibited. Furthermore, after knockdown of IL‐33 receptor ST2 together with applying recombinant protein IL‐33, we observed that IL‐33 could trigger the NF‐κB pathway and promote the expression of CCL2. We proved that IL‐33 could regulate CCL2 through the NF‐κB pathway by binding to its corresponding receptor. Zhang et al34 observed that CCL2 could increase tumor cell invasion and migration in prostate cancer. Furukawa et al reported similar results in ovarian cancer.35 In breast tumors, CCL2 expression was also associated with advanced disease status, tumor progression, and angiogenesis.22 Chang et al21 found that CCL2 produced by the glioma microenvironment was essential for the recruitment of T cells. In our experiment, we also found that the level of IL‐33 was associated with CCL2 in ESCC and might regulate the immunosuppression effect of CCL2 by recruiting Treg cells.
Epithelial‐mesenchymal transition plays crucial roles in the process of cancer development, which confers an aggressive phenotype to tumor cells.23 Most molecules were confirmed to participate in the process of EMT, including the downregulated intracellular adhesion molecule E‐cadherin and several increased markers from mesenchymal cells, such as fibronectin, vimentin, and N‐cadherin at the molecular level.36 Xu et al37 observed that IL‐33 could induce EMT, and promoted the pathogenesis of EMT of human kidney cells through p38 MAPK signaling. We assumed that IL‐33 might promote EMT and we found decreased EMT‐related gene expression including N‐CAD, ZEB2, VIM, and slug. In advanced stages, TGF‐β could induce EMT in cervical carcinoma cells.38 It was reported that TGF‐β could exert function as a potent pleiotropic cytokine by regulating cell and tissue development, differentiation, and homeostasis in mammals. Transforming growth factor‐β has a multitude of physiological functions, including promoting cancer progression and tumorigenesis.14, 39 In our study, IL‐33 depletion has a significant effect on CCL2 with addition of TGF‐β, which indicated that IL‐33 contributes to tumor immune escape by recruiting Treg cells with the presence of TGF‐β, thus promoting the occurrence and development of tumor.
In summary, our study shows that IL‐33 could promote tumor progress by upregulating CCL2 expression and recruiting Tregs through the NF‐κB pathway, and IL‐33 could promote ESCC metastasis in part by regulating EMT.
DISCLOSURE
The authors declare no potential conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
This study was supported by a grant from the National Natural Science Foundation of China (Grant No. U1804281). The authors acknowledge the assistance of their colleagues at the Department of Biotherapy Center of the First Affiliated Hospital of Zhengzhou University and the Seventh People's Hospital of Zhengzhou.
Yue Y, Lian J, Wang T, et al. Interleukin‐33‐nuclear factor‐κB‐CCL2 signaling pathway promotes progression of esophageal squamous cell carcinoma by directing regulatory T cells. Cancer Sci. 2020;111:795–806. 10.1111/cas.14293
Ying Yue, Jingyao Lian, and Tian Wang contributed equally to this work.
REFERENCES
- 1. Wu CC, Chen CJ. Esophageal carcinoma. N Engl J Med. 2015;372(15):1472. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87‐108. [DOI] [PubMed] [Google Scholar]
- 3. Tran GD, Sun XD, Abnet CC, et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian general population trial cohort in China. Int J Cancer. 2005;113(3):456‐463. [DOI] [PubMed] [Google Scholar]
- 4. Zhao J, He YT, Zheng RS, Zhang SW, Chen WQ. Analysis of esophageal cancer time trends in China, 1989–2008. Asian Pac J Cancer Prev. 2012;13(9):4613‐4617. [DOI] [PubMed] [Google Scholar]
- 5. Liang H, Fan JH, Qiao YL. Epidemiology, etiology, and prevention of esophageal squamous cell carcinoma in China. Cancer Biol Med. 2017;14(1):33‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liew FY, Girard JP, Turnquist HR. Interleukin‐33 in health and disease. Nat Rev Immunol. 2016;16(11):676‐689. [DOI] [PubMed] [Google Scholar]
- 7. Liu X, Hammel M, He Y, et al. Structural insights into the interaction of IL‐33 with its receptors. Proc Natl Acad Sci USA. 2013;110(37):14918‐14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mertz KD, Mager LF, Wasmer MH, et al. The IL‐33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology. 2016;5(1):e1062966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Milovanovic M, Volarevic V, Radosavljevic G, et al. IL‐33/ST2 axis in inflammation and immunopathology. Immunol Res. 2012;52(1–2):89‐99. [DOI] [PubMed] [Google Scholar]
- 10. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17(2):122‐131. [DOI] [PubMed] [Google Scholar]
- 11. Tong X, Barbour M, Hou K, et al. Interleukin‐33 predicts poor prognosis and promotes ovarian cancer cell growth and metastasis through regulating ERK and JNK signaling pathways. Mol Oncol. 2016;10(1):113‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hollande C, Boussier J, Ziai J, et al. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL‐33‐dependent eosinophil‐mediated control of tumor growth. Nat Immunol. 2019;20(3):257‐264. [DOI] [PubMed] [Google Scholar]
- 13. Cui G, Ren J, Xu G, Li Z, Zheng W, Yuan A. Cellular and clinicopathological features of the IL‐33/ST2 axis in human esophageal squamous cell carcinomas. Cancer Cell Int. 2018;18:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colak S, Ten Dijke P. Targeting TGF‐beta signaling in cancer. Trends Cancer. 2017;3(1):56‐71. [DOI] [PubMed] [Google Scholar]
- 15. Inman GJ. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev. 2011;21(1):93‐99. [DOI] [PubMed] [Google Scholar]
- 16. Seoane J, Gomis RR. TGF‐beta family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol. 2017;9(12):pii: a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malik A, Sharma D, Zhu Q, et al. IL‐33 regulates the IgA‐microbiota axis to restrain IL‐1alpha‐dependent colitis and tumorigenesis. J Clin Investig. 2016;126(12):4469‐4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun P, Ben Q, Tu S, Dong W, Qi X, Wu Y. Serum interleukin‐33 levels in patients with gastric cancer. Dig Dis Sci. 2011;56(12):3596‐3601. [DOI] [PubMed] [Google Scholar]
- 19. Serrels B, McGivern N, Canel M, et al. IL‐33 and ST2 mediate FAK‐dependent antitumor immune evasion through transcriptional networks. Sci Signal. 2017;10(508):eaan8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xia Y, Shen S, Verma IM. NF‐kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2(9):823‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang AL, Miska J, Wainwright DA, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid‐derived suppressor cells. Cancer Res. 2016;76(19):5671‐5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandal PK, Biswas S, Mandal G, et al. CCL2 conditionally determines CCL22‐dependent Th2‐accumulation during TGF‐beta‐induced breast cancer progression. Immunobiology. 2018;223(2):151‐161. [DOI] [PubMed] [Google Scholar]
- 23. Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci. 2014;35(9):479‐488. [DOI] [PubMed] [Google Scholar]
- 24. Gao X, Wang X, Yang Q, et al. Tumoral expression of IL‐33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. 2015;194(1):438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Davis C, Shah S, et al. IL‐33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 2017;56(1):272‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang JF, Wang P, Yan YJ, et al. IL33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2‐NF‐kappaB pathway. Oncol Rep. 2017;38(4):2033‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moulin D, Donze O, Talabot‐Ayer D, Mezin F, Palmer G, Gabay C. Interleukin (IL)‐33 induces the release of pro‐inflammatory mediators by mast cells. Cytokine. 2007;40(3):216‐225. [DOI] [PubMed] [Google Scholar]
- 28. Akimoto M, Maruyama R, Takamaru H, Ochiya T, Takenaga K. Soluble IL‐33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumour microenvironment. Nat Commun. 2016;7:13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akimoto M, Takenaga K. Role of the IL‐33/ST2L axis in colorectal cancer progression. Cell Immunol. 2018. [DOI] [PubMed] [Google Scholar]
- 30. Cui G, Qi H, Gundersen MD, et al. Dynamics of the IL‐33/ST2 network in the progression of human colorectal adenoma to sporadic colorectal cancer. Cancer Immunol Immunother. 2015;64(2):181‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang M, Feng Y, Yue C, et al. Lower expression level of IL‐33 is associated with poor prognosis of pulmonary adenocarcinoma. PLoS ONE. 2018;13(3):e0193428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding L, Ren J, Zhang D, et al. A novel stromal lncRNA signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via LncRNA‐CAF/interleukin‐33. Carcinogenesis. 2018;39(3):397‐406. [DOI] [PubMed] [Google Scholar]
- 33. Moisan F, Francisco EB, Brozovic A, et al. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol Oncol. 2014;8(7):1231‐1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C‐C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102(8):522‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furukawa S, Soeda S, Kiko Y, et al. MCP‐1 promotes invasion and adhesion of human ovarian cancer cells. Anticancer Res. 2013;33(11):4785‐4790. [PubMed] [Google Scholar]
- 36. Niknami Z, Eslamifar A, Emamirazavi A, Ebrahimi A, Shirkoohi R. The association of vimentin and fibronectin gene expression with epithelial‐mesenchymal transition and tumor malignancy in colorectal carcinoma. EXCLI J. 2017;16:1009‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Z, Zhao C, Wang Z, et al. Interleukin‐33 levels are elevated in chronic allograft dysfunction of kidney transplant recipients and promotes epithelial to mesenchymal transition of human kidney (HK‐2) cells. Gene. 2018;644:113‐121. [DOI] [PubMed] [Google Scholar]
- 38. Cheng K, Hao M. Metformin Inhibits TGF‐beta1‐induced epithelial‐to‐mesenchymal transition via PKM2 relative‐mTOR/p70s6k signaling pathway in cervical carcinoma cells. Int J Mol Sci. 2016;17(12):pii: E2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katsuno Y, Lamouille S, Derynck R. TGF‐beta signaling and epithelial‐mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25(1):76‐84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials