

Abstract
Charge detection mass spectrometry (CDMS) is mainly utilized to determine the mass of intact molecules. Previous applications of CDMS have determined the mass-to-charge ratio and charge of large polymers, DNA molecules, and native protein complexes, from which corresponding mass values could be assigned. Recent advances have demonstrated CDMS using an Orbitrap mass analyzer yields reliable assignment of integer charge states that enables individual ion mass spectrometry (I2MS) and spectral output directly into the mass domain. Here, I2MS analysis was extended to isotopically resolved fragment ions from intact proteoforms for the first time. With a radically different bias for ion readout, I2MS identified low abundance fragment ions containing many hundreds of residues that were undetectable by standard Orbitrap measurements, leading to a doubling in the sequence coverage of triosephosphate isomerase. Thus, MS/MS with detection of individual ions (MS/I2MS) provides far greater ability to detect high mass fragment ions and exhibits strong complementarity to traditional spectral readout in this, its first application to top-down mass spectrometry.
Keywords: Charge Detection Mass Spectrometry, Individual Ion Mass Spectrometry, MS/MS, Tandem MS, Top Down Mass Spectrometry, Orbitrap, Charge Detection
Graphical Abstract
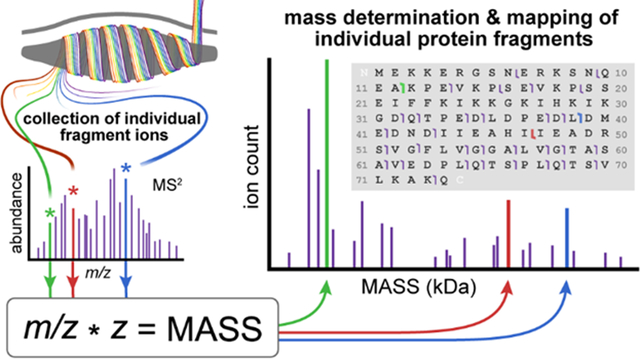
Introduction
In past years, charge detection mass spectrometry (CDMS) has been associated with the charge assignment and mass determination of large polymers, DNA molecules, and protein complexes. Complexes have ranged from native protein assemblies with a molecular weight of a few hundred kiloDaltons to much larger viruses and bacteriophages in the megaDalton regime1, 2. Traditionally, these experiments have been performed on specialized, custom-built instruments specifically designed for charge detection of individual ions in an electrostatic linear ion trap analyzer3–5.
Recent developments have revealed that the charge of an ion can be accurately assigned using the commercially available Orbitrap analyzer; two recent reports showed detection of individual ions, with one having charge assignment with uncertainty of ±3–4 unit charges6 and relying on prior m/z knowledge for species separation, while the other approach performs direct charge assignment on 6+ to 70+ ions with >96% reliability7. The latter was accomplished utilizing the summation of ion signal as a function of time (Selective Temporal Overview of Resonant Ions; STORI plot analysis)8 coupled with the Orbitrap analyzer’s ability to measure ion frequencies with high accuracy9. The end result showed that high resolution mass spectra can be produced by determining the correct quantized charge value of the ion signals7. Orbitrap-based individual ion mass spectrometry (I2MS) has therefore expanded traditional CDMS applications from the mass determination of large, heterogeneous complexes to accurate mass assignment of >500 proteoforms in a complex mixtures7. Furthermore, because I2MS analysis can take place on standard Orbitrap platforms, all the features of these instruments can be exploited, including dissociation techniques such as higher-energy collisional dissociation (HCD).
Here, we expand the application of I2MS from measurements of intact proteoforms and their complexes to establish a new option for analyzing top-down tandem mass spectra, individual ion MS/MS analysis (MS/I2MS). Utilizing the capability of a Q-Exactive Plus for HCD, we demonstrate that it is possible to isolate multiple ions by m/z, fragment them in the HCD cell, and readout individual fragment ion signals within the Orbitrap analyzer, accurately determine their charge, and create a true mass spectrum of resulting fragment ions. Upon comparison with traditional readout of the same population of fragment ions for a 40 kDa protein, we show an increase of 48% in obtainable sequence coverage with the addition of MS/I2MS. This expansion in coverage is accompanied by a 3-fold increase in the median mass values of identified fragment ions from ~6.5 kDa to ~18.5 kDa for the traditional and new MS/I2MS readouts, respectively.
Experimental Section
Assembly and purification of pJL1-SUMO-TPI.
An 1132 bp linear DNA construct encoding the SUMO-TPI fusion was synthesized by Integrated DNA Technologies (Skokie, IL). The pJL1 backbone DNA was amplified via PCR using primers pJL1BB_F: 5’-ATGTATATCTC CTTCTTAAAG-3’ and pJL1BB_R: 5’-GTCGACCGGCTGCTAACAAAG-3’. Fifty ng each of insert and backbone PCR product were incubated with Gibson assembly reagents as previously described to assemble plasmid pJL1-TPI-SUMO10. Assembly reaction products were transformed into E. coli strain DH5α, and individual colonies were screened via colony PCR using primers TPIcPCR_F: 5’- GGTGTGATTGCGTGCATCGG-3’ and pJL1cPCR_R: 5’-CTCGCTCAGGCGCAATCACG-3’ to identify a strain carrying a correctly-assembled plasmid. For use in cell-free protein synthesis (CFPS) reactions, pJL1-SUMO-TPI DNA was purified from 100 mL of saturated overnight culture using Hi-Speed® Plasmid Maxi Kit (Qiagen; Venlo, The Netherlands).
Cell-free synthesis of SUMO-TPI.
Cell-free extracts were prepared as previously described from a derivative of recoded E. coli strain C321.ΔA.75911 modified to endogenously express T7 RNA polymerase. To achieve cell-free protein synthesis of SUMO-TPI, extracts were combined with purified pJL1-SUMO-TPI DNA and other reagents to assemble 15 μL reactions as previously described11. For applications requiring purified SUMO-TPI, after completion of the synthesis reactions crude reaction volume was incubated with Ni-NTA agarose (Qiagen; Venlo, Netherlands) and SUMO-TPI was purified following manufacturer protocol. An illustration of the N terminal His/SUMO-tagged TPI is shown within the top inset in Figure 1a.
Figure 1.

(a) Intact mass spectrum of SUMO-tagged Triosephosphate Isomerase (TPI, ~40 kDa) with the top inset illustrating a TPI construct diagram and bottom (red outlined) inset illustrating the isolated +39 charge state used to produce the subsequent (b) MS/I2MS spectrum. Four isotopic distributions of matched fragment ions (B97, B155, Y208, and Y341) demonstrate several of the 59 assigned fragment ions. Assigned charge (z) vs. m/z spectrum (c) for the deconvolution of the complex m/z spectrum into the mass domain. The inset illustrates the correct charge assignment for one isotopically resolved fragment ion.
Sample Preparation and MS Analyses.
SUMO-tagged TPI (~40 kDa) was desalted using an Amicon Ultra-0.5 mL centrifugal filter and prepared in 1 μM concentrations for subsequent analysis. The TPI sample was denatured in 60:40 water:acetonitrile with 0.2% acetic acid. Samples were electrosprayed with a custom nano-electrospray source (spray voltage +0.8 to 1.6 kV) and analyzed with a Q-Exactive Plus (Thermo Fisher Scientific; Bremen, Germany) mass spectrometer12, 13.
Ensemble and I2MS data were acquired on the same instrument and under comparable ion source and fragmentation conditions. All MS/MS data were produced from the isolated +39 charge state of TPI with a higher-energy collisional dissociation (HCD) using 16 normalized collisional energy (NCE) units. Ensemble data were collected with a traditional central electrode (CE) voltage of −5 kV and individual ion data were acquired with an optimized CE voltage of −1 kV to promote ion survival for accurate charge assignment. In addition, a corresponding ensemble measurement acquired with a reduced central electrode voltage of −1 kV for comparative purposes can be found in the Supplemental Information (Supplemental Figure 5; Supplemental Table 3). A 2 second acquisition time was used for all MS/MS scan events.
I2MS Data Collection and Processing.
The I2MS charge assignment and ion mass determination process has been previously described7, 14. A brief workflow of the MS/I2MS process is shown in Supplemental Figure 1. Briefly, an average of ~700 individual ions were collected per acquisition event. The m/z of individual ions were determined through normal Fourier-transform Mass Spectrometry (FTMS) analysis, while the charge of the ion was simultaneously measured as the summation of its ion signal as a function of time (STORI plot)14. Once the quantized charge and m/z values of the ion were assigned, the mass of the species was calculated and utilized to create a true mass spectrum.
A fragment search against the precursor amino acid sequence was carried out using TDValidator (Proteinaceous; Evanston, IL) on spectra containing ~2 million ions. To acquire 2 million ions for each data set 3,205 I2MS spectra (~100 min. of total acquisition time) were collected and 25 ensemble spectra (~ 1 min. of total acquisition time; Automatic Gain Control = 1×106 charges/scan) were averaged for subsequent processing. Fragment ions were identified by matching their isotopic distributions to theoretical isotopic distributions generated using the Mercury15 and Averagine16 algorithms. All fragment ions were identified within a ±20 ppm max tolerance of their theoretical values. Other search metrics included a charge range of 1–20 and a S/N cutoff of 2. To make ions searchable in TDValidator, neutral mass MS/I2MS spectra were transformed into theoretical +1 (M+H) distributions. To calculate P-scores, matched and unmatched fragment monoisotopic masses were uploaded to ProSight Lite (http://prosightlite.northwestern.edu/),17 with 20 ppm tolerances. Monoisotopic masses were calculated for ensemble data with Xtract software within the Xcalibur program (Thermo Fisher Scientific, Version 4.0.27.10). A simple script was produced to format MS/I2MS mass bin distributions into centroided peaks that were passed into Xtract software to produce monoisotopic masses.
Results and Discussion
Traditional MS/MS analysis of larger protein ions using collisional dissociation techniques produce well known fragmentation patterns, typically with the predominance of fragment B and Y ions <10 kDa18, 19. However, fragment production becomes scarce toward the center of the protein sequence, particularly for larger mass proteins20, 21. Supplemental Figure 2 illustrates this trend with a typical top-down fragmentation spectrum recorded using the standard MS/MS readout for the 39+ charge state of a key protein in glycolysis, triosephosphate isomerase (TPI). The graphical fragment map in Supplemental Figure 2 mainly shows protein cleavages towards the termini of the protein. The lack of identified fragment ions of high mass hinders the overall characterization by yielding too little MS/MS information regarding the center composition of the proteoform22. This stands as a significant challenge for top-down mass spectrometry to provide precise localization of diverse post-translational modifications, polymorphisms, or mutations23, 24.
The optimized spectrum of the intact TPI construct along with the isolated +39 charge state (red inset) and resultant MS/I2MS spectrum are shown in Figure 1a and b, respectively. While the intact SUMO-tagged TPI mass is 39,729.3 Da, the TPI fragment mass spectrum contains well resolved isotopic distributions ranging from 9 – 39 kDa. The four insets in Figure 1b clearly demonstrate a few of the matched fragment isotopic distributions utilized to produce the corresponding graphical fragment map shown in Figure 2a (blue markers). The fragment map has 59 fragment ions matched to theoretical isotopic distributions. The size of the matched fragment ions ranged from 92 to 362 amino acid residues. Respectable P-scores of 1.1 × 10−12 and 7.1 × 10−18 were determined for MS/I2MS and ensemble MS/MS data. The slightly lower P-score for the MS/I2MS data is attributed to a larger number of unmatched fragments upon processing through Xtract software. This higher number of unmatched fragments is attributed to an increased sensitivity to pick up internal fragment species and the current limitation of utilizing Xtract software on mass, not m/z, results. However, when both measurements are combined the overall P-score improves to 1.5 × 10−19. A more robust method to measure confidence and scoring will be tailored to I2MS technology as it matures. A complete list of matched fragment ions, assigned fragment charges, and their ppm mass errors for both ensemble and MS/I2MS techniques is included in the accompanying Supplemental Information (Supplemental Figure 3 and 4; Supplemental Tables 1 and 2). Figure 1c, a plot of assigned individual ion charge (z) as a function of m/z, illustrates the complex transformation that occurs upon transitioning from the m/z to mass domain through I2MS analysis. Each dot corresponds to an individual isotope ion of a complicated mix of fragments and the various charge states that are created upon precursor fragmentation. The inset provides an example of an isotopic distribution, where a charge of +15 was assigned. Using this information, a robust comparison of identified fragment ions via traditional ensemble MS/MS analysis can be completed.
Figure 2.

(a) Overlaid ensemble (red) and MS/I2MS (blue) graphical fragment maps. (b) Box and whisker plots comparing the masses of ions observed in traditional (ensemble) MS2 spectrum (left; 34 matched fragment ions) and MS/I2MS spectrum (right; 59 total matched fragment ions). (c) Plot of fragment ion relative intensity as a function of molecular weight for ensemble (red) and MS/I2MS techniques (blue).
Fragments identified via traditional ensemble and MS/I2MS analysis show biases toward different sections of the TPI protein sequence. Figure 2a demonstrates these differences with overlaid fragmentation maps for ensemble (red) and MS/I2MS (blue) analysis. The ensemble (traditional MS/MS) fragment map is composed of 34 fragments with 50% of fragment ions containing less than 50 amino acids. All 59 fragment ions in the MS/I2MS fragment map counterpart were over 90 residues in length with 69% of matched fragments containing over 150 amino acids. The difference in fragment identifications between the two techniques is highly visible in Figure 2b with the box and whisker plot. First quartile/ median/ third quartile values for identified fragment ions by ensemble and MS/I2MS techniques were 1,810/6,613/15,419 kDa and 16,597/18,736/31,766 Da, respectively. In addition, the median fragment charge states for ions identified by ensemble versus I2MS techniques were +11 and +20, respectively. The average charge state for the MS/I2MS fragment ions was determined by averaging the charge calculated for each individual ion depicted in Supplemental Figure 4. These large differences in detected fragment sizes can be attributed to the many sensitivity advantages of MS/I2MS analysis. Sensitivity gains from I2MS processing include high precision information pertaining to exact m/z placement of the ion, removal of interfering noise signals, direct determination of ion stability, and an inherent lowering of spectral peak density within the mass (not m/z) domain.
Differences in identified fragments between ensemble and individual ion mass spectrometry techniques can be attributed to several factors. First, the charge detection capabilities of the Orbitrap analyzer have previously been demonstrated to be limited to ions with a +6 charge and above7. Ions with charges below that threshold cannot be separated from instrumental noise. As a result, fragments under ~6 kDa will not be detected using MS/I2MS analysis. Second, MS/I2MS processing removes all non-stable ion signals and eliminates interfering noise signals. As a result, the clean I2MS spectral baseline helps extend the intra-spectral dynamic range for fragment mass identifications.
Extending the dynamic range of fragment ion detection is essential to identifying low abundance fragments. In m/z space, the ion signal of larger protein fragments is distributed across many isotope and charge state channels, leading to a lower average signal than for small protein fragments. For this reason, the relative abundance of molecular weight fragment peaks over 10 kDa must be much higher than fragments with molecular weights around 2 – 3 kDa. Figure 2c illustrates that to identify higher mass fragment ions in ensemble measurements (red squares) their relative abundance must be much higher than their lower mass counterparts. However, MS/I2MS analysis consolidates multiple low S:N fragment charge state distributions into one higher S:N mass distribution and reduces spectral noise. As a result, MS/I2MS analysis increases identification of larger mass fragment ions regardless of relative abundance. As Figure 2c demonstrates, MS/I2MS analysis detects and matches fragment signals with relatively low abundance over 10 kDa (blue triangles). These factors shift the fragment ion size distributions typically seen in ensemble MS/MS experiments to higher mass fragment distributions, ultimately increasing the identification of cleavage sites toward the middle of the protein.
Upon combining traditional MS/MS and MS/I2MS techniques, robust identifications of fragments containing only a few to hundreds of residues can be readily assigned to characterize proteins of interest. This complementary analysis holds promise for increased coverage of large proteins and their complexes which can reach molecular weight values well in excess of 100 kDa. With this in mind, this publication only investigates one charge state of one protein and further exploration of this technique applied to MS/MS analysis is clearly needed. We are confident that future studies with MS/I2MS will open up new avenues for the exploration of proteoforms and their complexes through tandem MS.
Conclusion
Individual ion mass spectrometry is capable of producing true mass spectra for fragment ions. Here, the inherent sensitivity of individual ion analysis shifted the detection bias from small terminal fragments to the identification of larger fragment ions containing hundreds of amino acids. The addition of MS/I2MS to the MS/MS analysis toolbox adds a potentially complementary tool for increasing fragmentation coverage and yielding more complete proteoform characterization.
Supplementary Material
Figure S1: Schematic of general MS/I2MS workflow, Figure S2: −5 kV ensemble MS/MS spectrum and fragment map, Figure S3: Mass error histograms, Figure S4: Histograms for assigned charges for fragments, Figure S5: −1 kV ensemble MS/MS spectrum and fragment map, Table S1: −5 kV ensemble MS/MS matched fragment list, Table S2: MS/I2MS matched fragment list, Table S3: −1 kV ensemble MS/MS matched fragment list
Acknowledgements
This work was funded by the Intensifying Innovation program from Thermo Fisher Scientific and was carried out in collaboration with the National Resource for Translational and Developmental Proteomics under Grant P41 GM108569 from the National Institute of General Medical Sciences, National Institutes of Health with additional support from the Sherman Fairchild Foundation. JOK received additional funding from the American Society for Mass Spectrometry from a 2019 Post-Doctoral Career Development Award. LFS is a Gilliam Fellow of the Howard Hughes Medical Institute. We would like to thank Ryan Fellers and Bryan Early for their software development support.
Footnotes
Conflict of Interest
MWS is an employee of Thermo Fisher Scientific.
References
- 1.Keifer DZ, Motwani T, Teschke CM & Jarrold MF Measurement of the accurate mass of a 50 MDa infectious virus. 30, 1957–1962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keifer DZ, Pierson EE & Jarrold MF Charge detection mass spectrometry: weighing heavier things. Analyst 142, 1654–1671 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Elliott AG, Harper CC, Lin H-W & Williams ER Mass, mobility and MSn measurements of single ions using charge detection mass spectrometry. Analyst 142, 2760–2769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner WH A Gated Electrostatic Ion Trap To Repetitiously Measure the Charge and m/z of Large Electrospray Ions. Analytical Chemistry 69, 4162–4168 (1997). [Google Scholar]
- 5.Keifer DZ, Shinholt DL & Jarrold MF Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Analytical Chemistry 87, 10330–10337 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Wörner TP et al. Resolving heterogeneous high-mass macromolecular machineries by Orbitrap-based single particle charge detection mass spectrometry. BioRxiv 717413 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Kafader JO et al. Multiplexed Single Ion Mass Spectrometry Improves Measurement of Proteoforms and Their Complexes. BioRxiv 715425 (2019). [Google Scholar]
- 8.Kafader JO et al. STORI Plots Enable Accurate Tracking of Individual Ion Signals. Journal of The American Society for Mass Spectrometry 30, 2200–2203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kafader JO et al. Measurement of Individual Ions Sharply Increases the Resolution of Orbitrap Mass Spectra of Proteins. Analytical Chemistry 91, 2776–2783 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Martin RW et al. Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nature Communications 9, 1203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belov ME et al. From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Analytical Chemistry 85, 11163–11173 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Skinner OS et al. An informatic framework for decoding protein complexes by top-down mass spectrometry. Nature Methods 13, 237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kafader JO et al. STORI Plots Enable Accurate Tracking of Individual Ion Signals. Journal of The American Society for Mass Spectrometry (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockwood AL, Van Orden SL & Smith RD Rapid Calculation of Isotope Distributions. Analytical Chemistry 67, 2699–2704 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Senko MW, Beu SC & McLaffertycor FW Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. Journal of the American Society for Mass Spectrometry 6, 229–233 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Fellers RT et al. ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 15, 1235–1238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toby TK, Fornelli L & Kelleher NL Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annual Review of Analytical Chemistry 9, 499–519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catherman AD, Skinner OS & Kelleher NL Top Down Proteomics: Facts and Perspectives. Biochemical and biophysical research communications 445, 683–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skinner OS et al. Top-down characterization of endogenous protein complexes with native proteomics. Nature Chemical Biology 14, 36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haverland NA et al. Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. Journal of The American Society for Mass Spectrometry 28, 1203–1215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schachner LF et al. Standard Proteoforms and Their Complexes for Native Mass Spectrometry. Journal of The American Society for Mass Spectrometry 30, 1190–1198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han X, Jin M, Breuker K & McLafferty FW Extending Top-Down Mass Spectrometry to Proteins with Masses Greater Than 200 Kilodaltons. 314, 109–112 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Chait BT Mass Spectrometry: Bottom-Up or Top-Down? 314, 65–66 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Schematic of general MS/I2MS workflow, Figure S2: −5 kV ensemble MS/MS spectrum and fragment map, Figure S3: Mass error histograms, Figure S4: Histograms for assigned charges for fragments, Figure S5: −1 kV ensemble MS/MS spectrum and fragment map, Table S1: −5 kV ensemble MS/MS matched fragment list, Table S2: MS/I2MS matched fragment list, Table S3: −1 kV ensemble MS/MS matched fragment list