

Abstract
Nearly one-hundred loci in the human genome have been associated with different forms of Charcot-Marie-Tooth disease (CMT) and related inherited neuropathies. Despite this wealth of gene targets, treatment options are still extremely limited, and clear “druggable” pathways are not obvious for many of these mutations. However, recent advances in gene therapies are beginning to circumvent this challenge. Each type of CMT is a monogenic disorder, and the cellular targets are usually well-defined and typically include peripheral neurons or Schwann cells. In addition, the genetic mechanism is often also clear, with loss-of-function mutations requiring restoration of gene expression, and gain-of-function or dominant-negative mutations requiring silencing of the mutant allele. These factors combine to make CMT a good target for developing genetic therapies. Here we will review the state of relatively established gene therapy approaches, including viral vector-mediated gene replacement and antisense oligonucleotides for exon skipping, altering splicing, and gene knockdown. We will also describe earlier stage approaches for allele-specific knockdown and CRIPSR/Cas9 gene editing. We will next describe how these various approaches have been deployed in clinical and preclinical studies. Finally, we will evaluate various forms of CMT as candidates for gene therapy based on the current understanding of their genetics, cellular/tissue targets, validated animal models, and availability of patient populations and natural history data.
Introduction:
After almost three decades of gene discovery, nearly 100 loci have been identified that underlie various forms of Charcot-Marie-Tooth disease (CMT) and similar forms of inherited peripheral neuropathy (Rossor et al., 2017; Timmerman et al., 2014). Despite this enormous increase in understanding the genetics of the disease, therapeutic options remain virtually non-existent. Mutations in the diverse genes and biological pathways associated with CMT can yield disease arising from loss-of-function, gain-of-function, dominant-negative or neomorphic mechanisms, thereby making the prospect of a single therapeutic approach to treat all, or even most, forms of CMT highly unlikely. Nevertheless, certain features of CMT disorders are advantageous for therapy development. Essentially all forms of CMT are monogenic, with a single driver mutation explaining disease, though penetrance and severity may vary with environment and genetic background/burden. CMT also has a well-defined population of target cells, typically peripheral myelinating Schwann cells and/or motor and sensory neurons. The combination of relatively well-defined and simple genetics and specific cellular targets makes CMT disorders excellent candidates for gene therapy approaches.
In this review, we will describe the current “Toolbox” of gene therapy strategies (Figure 1), highlighting examples where these have been successfully deployed in clinical or preclinical studies. We will then consider which forms of CMT may be the next good targets for developing gene therapy approaches and the steps that will need to be taken to successfully complete that development.
Figure 1.
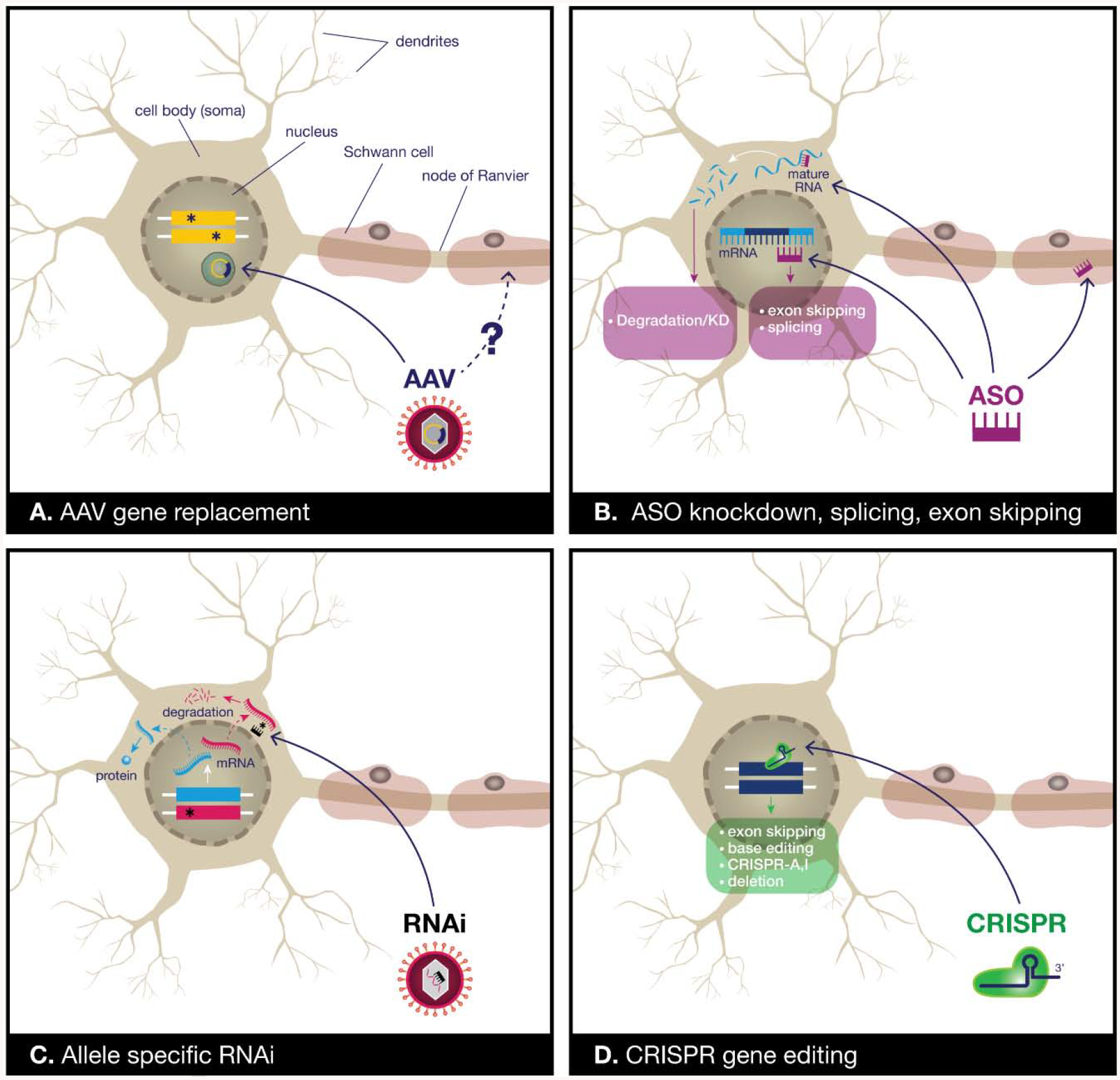
Our current and emerging gene therapy toolbox for CMT. A) Viral vectors for gene replacement can be used to restore gene expression, especially in recessive loss-of-function mutations. Adeno-associated virus (AAV) has been used extensively for this purpose, and the AAV9 serotype has good tropism for peripheral motor and sensory neurons, although the ability of AAVs to transduce Schwann cells is unclear. The viral payload persists in the nucleus as an extrachromosomal episome, providing long-term gene expression. B) Antisense oligonucleotides (ASOs) are small synthetic fragments of nucleic acid that can act in the nucleus to promote exon skipping or splicing of pre-mRNAs, or in the cytoplasm to promote degradation of mRNAs for gene knockdown. ASOs can enter neurons, but do not cross the blood-brain-barrier well and therefore need to be injected directly into the cerebrospinal fluid. However, they can also mediate gene knockdown in Schwann cells in preclinical studies. C) Allele-specific RNAi has been used in preclinical studies to treat a dominant mutation. RNAi sequences targeting mutant, but not wild-type mRNA are expressed using a microRNA shuttle driven by a U6 promoter. This construct can be delivered using an AAV. D) CRISPR-Cas9 approaches have produced promising preclinical results and can potentially be used to promote exon skipping, base-editing, transcriptional activation (CRISPR-A) or inactivation (CRISPR-I), and gene deletion. How to safely and effectively deliver the Cas9 and sgRNAs remains an area of active investigation.
Our gene therapy toolbox:
The best-validated tool in the gene therapy toolbox is gene replacement mediated by viral vectors. In particular, the emergence of Adeno-Associated Viruses (AAVs) as a safe and scalable choice for gene delivery has moved this field forward hugely in the past twenty years. AAVs include a variety of serotypes that differ in their capsid composition and thus their infectivity and the cell populations they transduce (their tropism). The AAV9 serotype has proven very effective for targeting motor and sensory neurons, the cell types of interest for many forms of CMT. Capsid modifications provide higher transduction efficiency in neurons following systemic delivery in animal studies (Chan et al., 2017). In particular, AAV-PHP.eB efficiently targeted dorsal root ganglion sensory neurons, the cardiac ganglion, and the enteric nervous system with intravenous delivery in adult mice, but the safety of this vector and its usefulness in humans is still being established. In addition to viral tropism, the route of delivery also influences the efficiency of transduction and the cell types being targeted. While intravenous systemic delivery is straightforward, the nervous system can be more efficiently transduced with delivery into the intrathecal space, and subpial delivery may be even more effective (Bravo-Hernandez et al., 2020; Meyer et al., 2015).
AAVs are capable of packaging inserts up to 4.6 kb of DNA, making them useful for many genes, but still limited for large constructs. AAVs are typically single stranded genomes, and gene expression from AAVs requires vector uncoating followed by conversion to double-stranded DNA, which can take several weeks to plateau (Ferrari et al., 1996; Hauck et al., 2004). As an alternative, modification generating self-complementary AAVs (scAAVs) creates a double stranded portion of the genome that serves to speed up scAAV expression after transduction, but further limits their capacity to ~2.3 kb. AAVs have the advantage of rarely integrating into the host genome, reducing their possible mutagenic effects. Instead, they persist as extrachromosomal episomes (Duan et al., 1998). In nondividing cells such as neurons, transduction leads to years-long expression. Theoretically, gene expression from an AAV vector should remain indefinitely, so long as the host cell remains intact (i.e. protected from disease and post-mitotic) and the promoter driving transgene expression is active. Indeed, numerous studies have reported sustained AAV-mediated gene expression for more than 1 year in mice, and up to 8–10 years in dogs and humans, which is essentially as long as AAV persistence has been measured experimentally (Buchlis et al., 2012; Elverman et al., 2017; Haidet et al., 2008; Jaen et al., 2017; Marshall et al., 2018; Nathwani et al., 2014; Pacak et al., 2007; Wang et al., 2012). Therefore, advantages of AAVs include relatively low immunogenicity, a nonintegrating genome, perdurance of expression, well-established capsid biology and tropism, and a proven clinical safety record.
For application to CMT, a disadvantage of AAVs is their poor transduction of Schwann cells. It is presently unclear if existing AAV serotypes can be used for efficiently delivering gene therapy payloads universally to Schwann cells, or if new capsid designs will be required. An alternative strategy is to use a different viral vector such as lentivirus for Schwann cell delivery, and this is described in detail in the accompanying review in this issue (Sargiannidou et al., 2019). Other vectors such as herpes virus are also being developed, but so far unproven. This review is focused on axonal forms of CMT, in which the target cells are the peripheral neurons.
In addition to virally mediated approaches, small synthetic nucleic acids, antisense oligonucleotides (ASOs), are also in common use. ASOs are modified to improve their bioavailability and pharmacokinetics in vivo, but they cross the blood-brain-barrier poorly, so targeting neurons often requires direct delivery to the nervous system, such as intrathecal injection. The conjugation of moieties such as peptides to promote targeting and internalization of ASOs by neurons may eventually overcome these issues (Hammond et al., 2016). ASOs are quite versatile in their actions. They have been used for gene knockdown, analogous to RNAi, in which the RNA-DNA duplex promotes RNase-H-mediated degradation of the transcript (Wu et al., 2004). ASOs are also used to enhance the correct splicing of genes and to promote exon skipping, when those approaches were genetically appropriate. In such applications, the ASO interacts with the pre-mRNA and splicing machinery, independent of RNase-H (Havens and Hastings, 2016). ASO-based treatments have the disadvantage of being less permanent than virally delivered therapies, with multiple doses required to maintain effects over the lifetime of an individual. However, this can also be viewed as a strength, allowing treatment to be stopped if negative effects emerge and allowing dosage to be optimized to patients’ responses over time. The action of ASOs can even be neutralized by dosing with a complementary “decoy” ASO (Rigo et al., 2014). ASOs also have the advantage of being chemically synthesized in a process that, as of now, is potentially more scalable and cost-effective than the production of viral vectors, although advancements in AAV manufacturing could someday close that gap, especially now that there are two FDA-approved AAV gene therapy products on the market and many more in the pipeline.
Viral-mediated gene replacement and ASOs are certainly the best-established approaches currently available, but other strategies are also emerging. These include virally delivered RNAi for gene knockdown, including the potential for allele-specific knockdown. This strategy is best applied to dominant mutations producing toxic gain-of-function (neomorphic) gene products or dominant-negative alleles where elimination of the mutant gene product alleviates the phenotype. This approach remains in the proof-of-concept preclinical stage, but the feasibility has been demonstrated in vivo. A similar emerging approach is CRISPR-based therapeutics. The potential for genome editing to permanently correct deleterious mutations at the chromosomal level is very exciting. To date, methods to correct mutations using homology directed repair are too inefficient to be viable therapeutically, but approaches such as CRISPR-A and CRISPR-I, where guide RNAs are used to specifically target a nuclease-dead CAS9 protein fused to a transcriptional activator (A) or inhibitor (I) are developing rapidly. Similarly, base-editing technology is also progressing rapidly. These approaches are reviewed elsewhere (Dominguez et al., 2016; Eid et al., 2018; Tadic et al., 2019; Vora et al., 2016), and here we will detail the use of CRISPR for exon skipping, which has been used in rigorous preclinical studies.
Examples of the successful application of these approaches – viral gene replacement, ASOs for knockdown, enhancing splicing and exon skipping, viral-vector-mediated RNAi for allele-specific knockdown, and CRISPR editing for exon skipping – in CMT and other genetic disorders are provided below. We will then address what forms of CMT may be candidates for these various approaches.
Examples of gene therapy success:
Spinal Muscular Atrophy:
Spinal muscular atrophy (SMA) represents a group of autosomal-recessive neurodegenerative disorders generally characterized by progressive muscle weakness and paralysis within proximal limb muscle resulting from the degeneration of lower motor neurons (Groen et al., 2018; Wang and Lunn, 2008). Its estimated incidence is 1 in 10,000 live births in European populations, with a high carrier frequency of ~1 in 54 (Sugarman et al., 2012; Verhaart et al., 2017a; Verhaart et al., 2017b). The clinical presentation of this disorder can vary, and is further classified, based on the age of onset and clinical progression, into SMA Type I-IV (D’Amico et al., 2011; Wang et al., 2007). About 60% of patients are diagnosed with SMA I, also known as Werdnig-Hoffmann disease, the most severe form of SMA and the leading genetic cause of infant death (D’Amico et al., 2011; Dubowitz, 1999; MacLeod et al., 1999). It presents very early in life, with most patients diagnosed at 6 months of age with an average survival of 2 years. Type II SMA is intermediate in severity, with onset before 18 months of age and patients never gaining the ability to walk. Types III & IV SMA are comparatively mild, with onsets after 18 months and in late adulthood, respectively (D’Amico et al., 2011; Wang et al., 2007). Patients with either Type III or IV experience mild muscle weakness and often retain the ability to walk (Wang et al., 2007).
All cases of SMA are caused by mutations within the survival motor neuron (SMN1) gene (Lefebvre et al., 1995). In humans, there is also an SMN2 gene, located within an inverted duplication adjacent to SMN1 on chromosome 5q13. The duplication contains four genes and varies from 0–4 copies per chromosome, making it prone to rearrangements and deletions (Tisdale and Pellizzoni, 2015). Indeed, variants within this region are often caused by unequal crossing over between the repeated units during paternal meiosis, increasing carrier frequency of this disease (Tisdale and Pellizzoni, 2015). The distal (telomeric) SMN1 and proximal (centromeric) SMN2 genes share 99.8% sequence homology and encode the same protein. SMN1 differs from SMN2 by only five nucleotide differences: one each within intron 6, exon 7, and exon 8, and two within intron 7 (Kashima and Manley, 2003; Monani et al., 1999). The nucleotide change within exon 8 falls within the 3’ untranslated region, while in exon 7, the C to T transition is in an exon splice enhancer site that, while translationally silent, alters splicing, and often results in the exclusion of exon 7 from SMN2 transcripts (Lorson et al., 1999; Monani et al., 1999). As a result, while SMN1 always yields full-length transcripts, SMN2 primarily produces a truncated transcript lacking exon 7 (~90%) and a greatly reduced number of full-length transcripts (~10%) (Cho and Dreyfuss, 2010; Lorson and Androphy, 2000). Considering exon 7 of the SMN genes encodes a functional domain that is responsible for self-oligomerization, SMNΔ7 protein oligomerizes less efficiently, is unstable, and rapidly degrades (Vitte et al., 2007). Altogether, these data from the human genetics of SMA indicate that the disease stems from a loss of SMN1 expression, and that effective therapies would either restore SMN1 or increase expression from whatever copies of SMN2 are present.
The first FDA-approved treatment for SMA, Nusinersen, is a modified 2’-O-methoxyethyl phosphorothioate antisense oligonucleotide (ASO) designed to increase the inclusion of exon 7 into SMN2 transcripts, and thus increase production of a functional, stable SMN protein, which can compensate for the disease-causing null and loss-of-function mutations in the SMN1 gene (Finkel et al., 2016; Finkel et al., 2017). This is accomplished by 10 arginine-serine dipeptide repeats within the ASO that drive the artificial recruitment of splicing enhancer factors to exon 7 of SMN2 (Finkel et al., 2016; Singh et al., 2006). Interim analysis of a phase III, double-blind, sham-procedure controlled clinical trial showed the post-onset benefit of Nusinersen for infantile-onset SMA patients who had two or more copies of the SMN2 gene (Finkel et al., 2016). Indeed, 40% of treated patients achieved critical developmental motor milestones compared to sham-procedure controls. This treatment was found to be so beneficial that all subjects within the placebo group were switched to Nusinersen. Similar effects of this SMN2-targeting ASO were identified when given to later-onset SMA patients with 2–4 copies of SMN2 (Mercuri et al., 2018). This study was stopped prematurely, based on a highly significant treatment effect based on the Hammersmith Functional Motor Scale – Expanded. Both studies imply that this treatment strategy may be therapeutically beneficial for all SMA patients across its phenotypic scale; however, both studies had non-responders, indicating possible variable effects.
Although highly efficacious, the effects of Nusinersen are transient, requiring patients to receive four intrathecal injections of the ASO within the first few weeks of treatment, and every 6 months thereafter (Finkel et al., 2017; Mendell et al., 2017; Parente and Corti, 2018). This type of injection is invasive and could cause adverse effects in patients. In response, an alternative gene therapy has been developed that involves a one-time intravenous delivery of SMN1 complementary DNA under the control of a hybrid cytomegalovirus enhancer-chicken beta-actin promoter with the use of a recombinant viral delivery vehicle: self-complementary adeno-associated viral vector (scAAV9) (Dominguez et al., 2011; Foust et al., 2009; Foust et al., 2010; Valori et al., 2010). This therapy is designed for rapid and sustained expression of SMN, not only in motor neurons, but also in other peripheral tissues that contribute to SMA symptoms, including the autonomic and enteric nervous systems, the heart, and the pancreas (Hamilton and Gillingwater, 2013; Sleigh et al., 2011). In a phase 1 clinical trial, 15 treated SMA patients exhibited longer survival, greater achievement of motor milestones, and significant improvements in motor function compared to historical cohorts (Mendell et al., 2017). Based on additional trials, phenotypic improvement can be achieved with a single, noninvasive, intravenous injection regardless of SMN2 copy number within each patient, and this AAV9-mediated gene therapy, Zolgensma, is now also FDA-approved, only the second such AAV-gene therapy treatment to be approved by the FDA.
Thus, SMA represents a neuromuscular disease in which motor neurons must be targeted and for which both ASO-based strategies for enhanced splicing and AAV9-based gene replacement have been successful and gained regulatory approval.
Duchenne’s Muscular Dystrophy:
Duchenne and Becker MDs are both X-linked, recessive forms of MD, together affecting 1 in 3,500 to 5,000 newborn males worldwide. Both disorders are caused by mutations in the dystrophin gene (DMD), the longest human gene, spanning 2.4 megabases of genomic DNA, 1% of the X chromosome (Kenwrick et al., 1987; Koenig et al., 1987). Although we are focusing on axonal CMTs, DMD offers a good case study because it is also a monogenic disorder and the target cell type (muscle) is also post-mitotic. It also highlights cutting-edge approaches, including ASO-mediated exon skipping, AAV-mediated gene replacement with engineered cDNAs, and preclinical studies using CRISPR-based therapeutics, but also challenges, including the complex gene structure and very large open reading frame.
DMD is a complicated transcription unit and contains at least seven independent, tissue-specific promoters and two polyadenylation sites, and produces 17 different isoforms that are named based their lengths and splicing patterns (Le Rumeur, 2015). The full-length isoform is expressed in all striated skeletal muscle and consists of an N-terminal domain that binds to actin filaments (encoded by exons 1–8), a central rod domain (exons 9–61), and a C-terminal cysteine-rich domain (exon 62–80) that help anchor dystrophin to the plasma membrane, where it interacts with many integral muscle proteins, including cytoskeletal actin, microtubules, and intermediate filaments to the extracellular matrix (Ervasti, 2007). Thus, dystrophin acts as scaffolding protein that is critical for sarcolemmal integrity. Any disruption of dystrophin’s scaffolding network can lead to either Duchenne or Becker MD.
Over 2/3 of patients with either Duchenne or Becker MD harbor mutations that delete one or more exons in DMD (Le Rumeur, 2015). Duchenne, the more severe of the two disorders, is caused by frameshift mutations that disrupt the open reading frame of DMD (Monaco et al., 1988). Patients with this form of MD exhibit severe, progressive muscle wasting causing the loss of ambulation within the first decade of life, and suffer from cardiomyopathy often leading to premature death (Kohler et al., 2005). Becker MD, meanwhile, is caused by in-frame deletions that result in a truncated but partially-functional dystrophin that often retains its N-terminal and C-terminal domains (Davies and Nowak, 2006; Rahimov and Kunkel, 2013). Patients with this disorder have a milder clinical pathology and normal lifespan (Bushby and Gardner-Medwin, 1993). Even individuals lacking more than 60% of the central rod domain are diagnosed with mild Becker muscular dystrophy. This implies that even a DMD gene with large in-frame deletions can yield partially functional dystrophin and ameliorate the severity of the disease. Thus, treatment development has been centered on altering splicing in an effort to restore the open reading frame within DMD genes affected by nonsense mutations. The goal is to turn a frameshift mutation into an in-frame deletion, in the hopes of converting severe forms of Duchenne MD to a milder Becker phenotype.
Towards this end, a series of ASOs designed to skip 20 different exons have been developed (exons 2, 8, 17, 19, 29, 40–46, 48–53, 55 and 59) and found to restore the reading frame of DMD, subsequently producing truncated dystrophin in various in vivo and in vitro models of Duchenne (Aartsma-Rus et al., 2003; Aartsma-Rus et al., 2004; Goyenvalle et al., 2004; McClorey et al., 2006; Surono et al., 2004; Takeshima et al., 2001; van Deutekom et al., 2001). Exondys 51 (eteplirsen), an ASO designed to skip exon 51 within DMD, is now approved for use in humans after successful clinical trials (Mendell et al., 2013). Specifically, this ASO binds to exon 51 in DMD and masks the exon inclusion signals that are used for splicing. Removal of exon 51 from an exon 45 to 50, 47 to 50, 48 to 50, 49 to 50, 50, 52 or 52 to 63 deleted transcript allows restoration of the open reading frame and synthesis of an internally-truncated, semi-functional dystrophin protein. By targeting exon 51, approximately 13% of patients with Duchenne could potentially be treated, the largest proportion of patients that could benefit from targeting a single dystrophin exon (Mendell et al., 2013). The Food and Drug administration accelerated approval for the use of Exondys 51, based on its ability to increase the expression of dystrophin in skeletal muscle in patients affected by Duchenne, although the extent of its efficacy and long-term effects on motor function are still being determined (Aartsma-Rus and Krieg, 2017).
Considering ASOs exhibit variable tissue transduction efficacies and transient effects, several exon-skipping approaches based on clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome editing are currently being developed (Juliano, 2016; Long et al., 2018; Ousterout et al., 2015b; Tabebordbar et al., 2016). This direct genome editing approach would only require a one-time treatment that would result in a permanent genomic modification and production of exon-deleted mRNA, which would, in turn, produce a semi-functional dystrophin protein. In a proof-of-concept study, AAV9-delivered CRISPR/Cas9 endonucleases and paired guide RNAs were shown to excise a nonsense mutation in exon 23 of the dmd gene in adult mdx mice, an established model of DMD (Tabebordbar et al., 2016). This restoration of the gene’s reading frame initiated the expression of dystrophin and partially recovered muscle function. Similar success was shown in a canine model of DMD (Amoasii et al., 2018). Over 3000 different frameshift mutations in DMD have been reported to cause Duchenne muscular dystrophy (Le Rumeur, 2015). Recent studies have shown that 60% of DMD mutations are clustered in two “hotspots” within the human DMD gene: one within exons 45 to 55 and another between exons 2 and 10 (Aartsma-Rus et al., 2009). Several research groups have developed a CRISPR/Cas9-based therapy that eliminates a 336-kb genomic region flanking exons 45 to 55 in the human DMD gene with the use of multiplexed single-guide RNAs (Ousterout et al., 2015a). This technique could restore dystrophin expression in human DMD myoblasts, yet it did exhibit off-target effects, including chromosomal rearrangements. To avoid the elimination of large genomic regions and in order to treat patients with mutations within exon 2–10, Long et al have recently developed a series of single guide RNAs designed to excise the 12 most common affected exons in DMD and induce the expression of dystrophin in both patient-derived iPSCs and derivative cardiomyocytes (Long et al., 2018). Remarkably, these single guides could even restore muscle force and contraction in three-dimensional engineered heart muscle culture derived from treated DMD patient-derived induced cardiomyocytes. While these results are promising in preclinical studies, the delivery of a CAS9 with an active nuclease using an AAV with persistent expression raises safety concerns and the possibility of off-target effects, particularly if the expression of the sgRNAs wanes.
An alternative gene therapy approach consists of a virally-delivered truncated dystrophin transgene with non-essential regions of the gene deleted. Known as ΔR4–R23/ΔCT, it is often referred to as micro-dystrophin (μ-dys). Remarkably, both intramuscular and intravenous administration of AAV vectors containing μ-dys expression cassettes significantly improve muscle membrane integrity and function in the mdx mouse and greatly extended the lifespan of dystrophin/utrophin double knockout mice (Gregorevic et al., 2004; Gregorevic et al., 2006; Harper et al., 2002; Liu et al., 2005; Yue et al., 2003; Yue et al., 2006). This therapy, now formally being tested in clinical trials for efficacy and safety (https://clinicaltrials.gov/ct2/show/NCT03375164), has a major advantage over exon-skipping methods as this treatment can treat any individual with Duchenne’s MD regardless of their distinct mutation in DMD.
Future personalized CRISPR/Cas9 based gene therapies may be developed, not just to convert a DMD mutation to a BMD mutation, but to correct the mutated DMD transcript. This way, a fully-functional dystrophin protein will be expressed, treating both Becker and Duchenne muscular dystrophies. This could potentially be accomplished by delivering a specific DNA donor template correcting the mutation along with an RNA guide designed to target the specific DMD mutations with Cas9. However, this approach is currently too inefficient to be considered clinically. Nonetheless, progress in DMD described above demonstrates the effectiveness of ASOs for exon skipping, the potential of CRISPR-based approaches, and the use of AAV-mediated delivery of truncated genes that can be packaged into viral particles. Of note, these approaches are all targeting muscle, not nerve, but both tissues are post-mitotic and should therefore permit lifelong AAV expression; thus the ability to correct the DMD gene dysfunction establishes important precedents for other diseases such as CMTs if the appropriate cell types can be targeted.
Hereditary Transthyretin amyloidosis:
Hereditary amyloid transthyretin amyloidosis (hATTR) is a rare dominant mutation that causes peripheral and autonomic neuropathy, usually progressing from distal sensorimotor neurons to more proximal neurons (Gertz et al., 2015). There are more than 130 known mutations in the transthyretin gene that have been linked to hATTR (Sekijima et al., 2018). Transthyretin monomers are intrinsically prone to misfolding, but are stabilized in tetrameric structures. Mutations in the transthyretin gene interfere with the tetramer formation and stabilization (Vieira and Saraiva, 2014); misfolded monomers polymerize, forming amyloid fibrils that deposit in the tissues and build up over time (Palaninathan, 2012).
Transthyretin is primarily produced in the liver for the transport of thyroxine and Vitamin A in the body (Vieira and Saraiva, 2014). Traditional treatment for hATTR is a liver transplant, removing the primary source of mutant transthyretin production, which can be life-changing for the patient, but is not always completely curative since transthyretin is also produced in the brain and retina (Banerjee, 2017). Pharmaceutical treatment for hATTR includes tafamidis and diflunisal, which stabilize the tetrameric structure of transthyretin (Gertz et al., 2019). Tafamidis has yet to be approved for use in the US and seems to have limited effectiveness, and only when used for early stages of the disease (Cortese et al., 2016; Lozeron et al., 2013). Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID), which has significant side effects associated with long term NSAID use (Azorin et al., 2017). Additionally, disease complications can make the use of NSAIDs unsafe.
Recently, two genetic approaches utilizing antisense oligonucleotides (ASO) and RNA interference (RNAi) technology have been approved by the FDA for the treatment of hATTR. Inotersen is an ASO produced by Akcea Therapeutics. The 20-oligonucleotide sequence binds selectively in the 3’ UTR end of transthyretin mRNA and initiates RNase H digestion of the mRNA (Ackermann et al., 2016). The ASO is not selective for the mutant mRNA, therefore it represses both wild-type and mutant protein expression (Ackermann et al., 2016). Preclinical testing in transgenic mice showed a significant decrease in transthyretin protein, with no evidence of cross reactivity with other mRNAs (Ackermann et al., 2016). Partisarin is an siRNA-based drug produced by Alnylam Pharmaceuticals. The small double-stranded RNA is packaged in a lipid complex as a delivery system for injection and transport to the liver, where it can specifically target hepatocytes-expressed transthyretin via RNAi (Suhr et al., 2015). The antisense strand of the siRNA sequence forms a complex with endogenous RNAi protein machinery (together called the RNA induced silencing complex, RISC), which selectively bind mRNA and digest it, reducing the amount of mRNA available for translation (Suhr et al., 2015). Partisarin significantly reduces the amount of transthyretin protein, but also has been associated with mild to moderate side-effects, most notably an allergic reaction to the lipid complex in which it is packaged (Adams et al., 2018). The success of both Inotersen and Partisarin at repressing transthyretin protein levels has provided patients living with hATTR treatment options that slow disease progression, while being less intrusive than liver transplantation.
Although hATTR does lead to peripheral neuropathy, the target organ for ASO knockdown is the liver, which sets it apart from most forms of CMT, where Schwann cells or peripheral motor and sensory neurons are the targets. As mentioned previously, gene therapy to target Schwann cells remains a challenge due to the lack of viral vectors with good tropism for this cell type (Sargiannidou et al., 2019). In the case of CMT1A, caused by gene duplication and overexpression of PMP22, ASO-mediated knockdown has been successful in preclinical studies using both mouse and rat models of the disease (Zhao et al., 2018). This and other approaches to regulate PMP22 levels are also described in more detail in the accompanying review by Pantera et al. in this issue (Pantera et al., 2020). Below we will provide examples of axonal neuropathies treated by gene therapy.
Giant Axonal Neuropathy:
The most advanced gene therapy specifically targeting an inherited peripheral neuropathy was developed for giant axonal neuropathy (GAN), a very rare form of CMT. GAN is an autosomal recessive disease characterized by progressive muscle weakness and premature death caused by enlarged axons with densely packed and disordered microtubules and intermediate filaments (Demir et al., 2005; Naldini, 2015). It is caused by loss-of-function mutations in GAN1, the gene that encodes gigaxonin, an E3 ubiquitin ligase adaptor involved in intermediate filament processing in neural cells (Bomont et al., 2000). Over 40 different mutations in GAN1 have been identified in patients with GAN (Houlden et al., 2007; Koop et al., 2007). They consist of in-frame deletion, insertion, missense, and nonsense mutations, all of which disrupt GAN1 function. However, both human and mouse heterozygous carriers of GAN1 mutations are unaffected by neuropathy or any other abnormalities, despite producing only half the normal amount of endogenous GAN1 (Mussche et al., 2013). This suggests that enhancing the expression of exogenous wild-type GAN1 in the nervous system may be of therapeutic benefit for patients with GAN. Indeed, proof-of-concept for an AAV-mediated GAN1 gene replacement strategy showed enhanced GAN1 expression eliminated intermediate filament aggregates in patient fibroblasts harboring different GAN1 mutations, and reduced neuronal intermediate filaments in the brain stem and spinal cord of Gan1 knockout mice (Mussche et al., 2013). The clinical benefit of GAN1 delivery in the mice was complicated by their comparatively mild phenotype, but the reversal of the enlarged axons, which are considered a hallmark of this disease, was accepted as proof-of-concept for correcting the pathology. Therapeutic safety and efficacy of scAAV9/JeT-GAN are currently being evaluated in clinical trials with the first patient having been treated in 2015 (https://clinicaltrials.gov/ct2/show/NCT02362438). This trial represents the first gene therapy trial for a peripheral neuropathy, and while GAN is rare, it sets an important precedent for the approach (Bailey et al., 2018).
CMT2D:
A second example of a gene therapy approach for an axonal neuropathy is the use of allele-specific knockdown to target dominant mutations in GARS as a treatment for CMT2D. In mouse studies, Gars mutations that cause peripheral neuropathy appear to do so through a neomorphic activity. This is supported by the lack of a dominant phenotype in heterozygous null mice, which do not produce a protein product (Seburn et al., 2006), and by the inability of 10–20 fold overexpression of the wild-type GARS gene to correct the neuropathy phenotype (Motley et al., 2011). Together, these results suggest that gene replacement would be ineffective (the transgenic overexpression did not produce benefit), whereas knockdown of the mutant gene product while leaving the wild-type allele intact would be beneficial (essentially the null heterozygous state). Importantly, GARS is an essential gene, so expression of the wild-type allele cannot be eliminated. Morelli et al. demonstrated the feasibility of such an approach in two mouse models of CMT2D: an existing allele (P278KY) that causes a dominant neuropathy, but which is not found in patients, and ΔETAQ, an allele causing a four amino acid internal deletion identified in a patient with severe, early-onset motor neuropathy (Morelli et al., 2019). RNAi sequences that specifically targeted either P278KY or ΔETAQ mRNAs were optimized in vitro for their ability to knock down the mutant target sequence without knocking down the wild type. These RNAi sequences were then incorporated into a miR-30 shuttle cassette with expression driven by a U6 promoter (Boudreau et al., 2011). In this way, the RNAi is produced in the cell through the miRNA processing pathway. This construct was then packaged into scAAV9 for in vivo delivery with tropism to motor and sensory neurons. In mice, both constructs were able to almost completely prevent neuropathy when delivered at birth, before the onset of symptoms at ~3 weeks-of-age. When delivered post-onset (at 5 or 9 weeks-of-age), there was still benefit, though much more modest, and the degree of benefit decreased as treatment was delayed. It appears that progression may have been slowed or stopped and innervation of intact NMJs was improved, even though reinnervation and regeneration was not observed. Both alleles of Gars targeted with RNAi differed from wild type by several base pairs (five for P278KY and twelve for ΔETAQ). Whether such an allele-specific knockdown approach would work for single base pair mutations will likely depend on the adjacent sequence, but even if specific sequences were found, it would also require separate tests of efficacy and safety for each unique RNAi sequence, creating a burdensome path to translation. An alternative strategy would be to produce a single vector that knocks down all GARS isoforms, mutant and wild-type, and in the same vector, to express an RNAi-resistant wild-type cDNA to restore GARS function. Such an approach has been used for alpha1 antitrypsin in the liver (Li et al., 2011). While the allele-specific targeting could potentially be achieved by ASOs or even CRISPR-based approaches, the knockdown-and-replace strategy would likely require a viral vector to deliver the RNAi and the wild-type cDNA together, and to maintain expression in transduced cells. Although the path to translation for CMT2D patients will require additional steps, the results of Morelli et al. establish the feasibility of this strategy (Morelli et al., 2019).
The lack of regeneration in the CMT2D preclinical studies with post-disease onset dosing is worth considering in this disease, and for CMT in general. With treatment after axon degeneration in the periphery, there was no evidence of regeneration, such as restoration of myelinated axon number or collateral sprouting at neuromuscular junctions by the remaining, intact motor nerve terminals (Morelli et al., 2019). Whether this failure of regeneration is because the axon degeneration is irreversible, because knockdown is incomplete, or because transduction efficiency in the spinal cord decreases with age, is unclear and will require additional research. In diseases like spinal muscular atrophy where there is loss of motor neuron cell bodies in the spinal cord, it is not surprising that the earlier treatment is delivered, the more effective it is, and that gene replacement does not lead to replacement of lost neurons. In cases of axonal dysfunction or degeneration without immediate death of the cell bodies, the regenerative capacity of the peripheral nervous system may allow regeneration, particularly if the root cause of the pathophysiology is addressed, such as through gene therapy. Without effective treatments, this remains hypothetical, but it can be tested in animal models with conditional induction or elimination of gene expression. For example, mice could be engineered such that a disease-associated mutation in Gars is flanked by loxP sites for Cre-mediated deletion. The mice could be maintained until axons begin to degenerate and the mutant gene could then be excised with an inducible Cre transgene. Such an experiment would lead to the complete deletion of mutant Gars in any given cell, eliminating the vagaries of viral transduction and extent of RNAi-mediated knockdown. This would reveal if axons are capable of regenerating once mutant Gars is eliminated, and define the time frame in which such regeneration is possible; reinnervation by motor neurons after extensive muscle atrophy may be ineffective, for example, even if the axon regenerates. Genetic experiments in model organisms could also address the cell autonomy of the pathogenic actions of these mutations. This more complete view of the disease and its potential for reversal would help in evaluating which patients would be mostly likely to benefit from a gene therapy treatment.
The next good CMT targets:
Given the toolbox of gene therapy approaches described and the lesson learned from the examples given, we will now consider which forms of CMT may be the next targets for gene therapy strategies (Table 1). To start preclinical studies, a good understanding of the genetics (LoF, GoF) and cellular targets is needed, as well as validated animal models of the disease. For an eventual clinical trial, a patient population with a defined natural history of their disease progression is also needed. The readiness for clinical trials in CMT is discussed in an accompanying review, including a discussion of biomarkers (Rossor et al., 2020). For CMT, some biomarkers may be subtype-specific, reflecting the particular disease mechanism associated with the gene or mutation, but others, such as circulating neurofilament levels, may apply to many forms of CMT, including the axonal forms (Sandelius et al., 2018).
Table 1.
The next good CMT targets for gene therapy development.
| Gene/disease | Mechanism/approach | Patients/natural history | Animal model |
|---|---|---|---|
| MFN2/CMT2A | LoF/dominant negative, increased MFN2 or MFN1 expression | Most common axonal neuropathy, natural history available | Transgenic mouse models, rat knockin? |
| GDAP1/CMT4A, CMT2K | LoF, gene replacement | Natural history available | Knockout mice |
| FIG4/CMT4J | Recessive LoF, gene replacement | Rare, natural history limited | Pale Tremor Mouse |
| IGHMBP2/CMT2S, SMARD1 | Recessive LoF, gene replacement | Rare, natural history limited, better for SMARD1 | Nmd mouse |
| CNTNAP1/CHN3, LCCS7 | Recessive LoF, gene replacement | Very rare, very limited natural history | Knockout and shambling mouse models |
CMT2A/MFN2:
Perhaps the best candidate for gene therapy is CMT2A, the most common form of axonal neuropathy, caused by mutations in Mitofusin2 (MFN2) (Zuchner et al., 2004). The genetics underlying CMT2A is somewhat confusing, with most mutations being dominant, but some appearing as recessives, and some producing additional symptoms such as optic atrophy and other CNS signs (Stuppia et al., 2015; Zuchner et al., 2006). MFN2 is involved in mitochondrial fusion, and without MFN2, there are more numerous, but smaller, fragmented mitochondria (Chen et al., 2003; Chen and Chan, 2005). To further complicate the genetics, MFN1 is a closely related gene that serves the same function. In the nervous system, MFN1 is expressed at quite low levels, possibly explaining why the nervous system is particularly vulnerable to MFN2 mutations, despite widespread MFN2 expression (Zhou et al., 2019). Mutant MFN2 in dominant alleles functions as a dominant negative, impairing the actions of the remaining intact MFN2 allele, and also disrupting the function of MFN1. Pharmacological strategies to improve the function of the remaining wild type MFNs and thus mitigate the negative effects of the mutant MFN2 are under development with promising results (Rocha et al., 2018). However, recent data in a mouse model of CMT2A suggests that gene therapy is also a potentially effective strategy (Zhou et al., 2019). The mouse model used expresses the pathogenic MFN2-R94Q allele, driven by the Thy1.1 promoter, which provides strong expression in a number of large projection neuron populations including motor neurons. These mice develop a marked phenotype, consistent with a dominant-negative genetic mechanism (both wild type Mfn2 alleles are still present), but importantly, this phenotype could be corrected with delivery of either MFN2 or MFN1 using AAV9. This is consistent with a dominant-negative action, where high level expression of wild type should be able to eventually outcompete the negative activities of the mutant protein. It is also consistent with the idea that low levels of MFN1 may contribute to the disease in the nervous system and raises the possibility that MFN1 (which is intact in CMT2A patients) could be elevated either pharmacologically or using strategies such as CRISPR-A in neurons.
Natural history data is available for CMT2A, as it is among the forms of neuropathy being tracked by the International Neuropathy Consortium (# NCT01193075). The animal models of CMT2A are also probably adequate for preclinical studies. In addition to the Thy1.1MFN2-R94Q transgenic mice mentioned above, other less aggressive models have also been developed, largely using transgenic expression of mutant forms of MFN2 (Bannerman et al., 2016; Cartoni et al., 2010; Detmer and Chan, 2007; Detmer et al., 2008; Misko et al., 2010). Rat models with human disease alleles knocked into the endogenous Mfn2 locus and valid axonal neuropathy phenotypes have also been described (Li et al., 2017), and these may ultimately be the best models for preclinical studies, since they avoid issues of overexpression or misexpression of the MFN2 gene. Passage Bio has announced in a press release that they are developing an MFN2 gene therapy using AAV delivery to treat CMT2A.
CMT4A/GDAP1:
Closely related to MFN2, but less common, are mutations in GDAP1 (Ganglioside-induced differentiation associated protein 1). GDAP1 mutations cause autosomal recessive CMT4A, but can also cause dominant CMT2K, and the clinical presentation can vary, with some cases including vocal cord paresis or reduced nerve conduction velocity (Azzedine et al., 2003; Baxter et al., 2002; Cuesta et al., 2002; Nelis et al., 2002). Thus, like MFN2, the inheritance and clinical presentation of GDAP1 mutations is variable. Also, like MFN2, GDAP1 is a mitochondrial protein, but in this case a mitochondrial fission protein (Bertholet et al., 2016; Niemann et al., 2005). Overall, the pathogenesis of GDAP1 mutations seems to be through loss of function, be it from recessive alleles or dominant-negative alleles. Consistent with that, knockout mice completely lacking the Gdap1 gene develop a neuropathy phenotype. Two independent strains of mice have been described with modestly differing age of onset and severity, but generally similar conclusions (Barneo-Munoz et al., 2015; Niemann et al., 2014). However, the phenotypes of both knockout mouse strains are milder and later onset than anticipated from the human disease. Nonetheless, these models may prove adequate for preclinical studies, or perhaps like Mfn2, a rat model would produce a more clinically relevant phenotype. In either case, the genetics support attempting a straightforward gene replacement for GDAP1 as well. GDAP1 expression in Schwann cells as well as neurons is important, and the deletion of the gene just in Schwann cells is sufficient to produce a neuropathy phenotype (Niemann et al., 2014), so both Schwann cells and neurons may need to be targeted. Possible adverse effects from overexpression have not been explored and may be most efficiently addressed using viral delivery of the gene in wild type mice as opposed to making transgenic strains driven by promoters producing expression patterns different than those that would result from the tropism of viral gene therapy vectors. CMT4A is also under study by the INC, and therefore some natural history data already exists.
Three rare, recessive forms of CMT are also nearing the readiness criteria of well-defined genetic mechanisms, good animal models, and available patient data. These include CMT4J, caused by mutations in FIG4; CMT2S (also spinal muscular atrophy with respiratory distress, SMARD1), caused by mutations in IGHMBP2, and a related disorder, congenital hypomyelinating neuropathy 3 (also lethal congenital contracture syndrome 7), caused by mutations in CNTNAP1.
CMT4J/FIG4:
CMT4J is caused by recessive mutations in FIG4, a PI(3,5)P2 phosphatase (Chow et al., 2007). A good animal model exists in the pale tremor mouse (plt). The cloning of plt led to the identification of FIG4 as a human disease gene (Chow et al., 2007). Using this mouse model, the genetics of FIG4 mutations are well-understood and the mutations are straightforward loss-of-function alleles. In both mice and humans, the loss of FIG4 results in vacuolization in motor neurons in the ventral spinal cord and sensory neurons in the dorsal root ganglia. The mice have vacuolization in other brain regions, which may be more severe than typical CMT4J pathology; however, complete LoF mutations in patients result in Yunis-Varon syndrome, which is more severe than CMT4J and involves additional organ systems, and thus may be more consistent with the plt mouse (Campeau et al., 2013). Eventually, motor and sensory neuron dysfunction and loss leads to severe neurological problems. The plt mice are lethal at ~1 month-of-age (on an F1 hybrid genetic background) and patients are highly variable, with some succumbing to disease as adolescents and other preserving function well into adulthood.
In mice, transgenic expression of FIG4 in neurons largely rescues the phenotype, whereas glial expression is less effective (Ferguson et al., 2012; Winters et al., 2011). This suggests a cell autonomous defect in neurons is largely responsible for the disease. Indeed, neuronal expression of FIG4 even rescued some aspects of demyelination in the mice, indicating non-cell autonomous effects are still driven by FIG4 in neurons. Nonetheless, whether correcting the heterogeneous demyelination seen in patients will require targeting Schwann cells as well as motor neurons is currently unknown and conditional knockout strategies in mice do suggest a role for Schwann cell-expressed Fig4 (Hu et al., 2018). Importantly, no adverse effects were observed in transgenic mice, possibly because FIG4 is in a complex with VAC14 and the kinase PIKFYVE (Lenk et al., 2011). The loss of FIG4 or other components of this complex destabilizes the complex and results in decreased protein levels of all other components. Therefore, a stoichiometric excess of FIG4 beyond other components of the complex may simply result in its degradation. Combined, this makes CMT4J a good gene therapy candidate with a good animal model, straightforward loss-of-function genetics making gene replacement logical, and a defined target of peripheral neurons, all supported by in vivo mouse studies that also showed no ill effects of overexpression. CMT4J is very rare and clinical documentation of its course is sparse. However, a foundation for the disease has been established (CureCMT4J.org) and a natural history study is recruiting patients (# NCT03810508).
CMT2S/IGHMBP2:
IGHMBP2 gene mutations also cause a recessive peripheral neuropathy as well as childhood motor neuron disease. Severe loss-of-function alleles cause spinal muscular atrophy with respiratory distress (SMARD1) (Grohmann et al., 2001), whereas weaker LoF alleles cause CMT2S, an axonal CMT (Cottenie et al., 2014). A good animal model for IGHMBP2 exists in the neuromuscular degeneration (nmd) mouse (Cox et al., 1998). Indeed, like FIG4, it was the identification of Ighmbp2 as the cause of nmd that led to its identification as a human disease gene, establishing the validity of the nmd mouse for SMARD1/CMT2S. The nmd allele is a splicing mutation that produces wild-type protein, but at much reduced levels. On their own, these mice may not recapitulate the full spectrum of disease presentations observed in patients, and with a typical lifespan less than 30 days on a C57BL/6 genetic background, they may represent the more severe end of the spectrum. A more detailed discussion of the genes and mouse models bridging peripheral neuropathy and motor neuron disease is provided in the accompanying review by Martin et al. (Martin et al., 2019).
The nmd mice have been used in preclinical gene therapy studies utilizing AAV9 to replace the IGHMBP2 gene (Nizzardo et al., 2015; Shababi et al., 2016). While these experiments in pre-onset mice were successful and encouraging, there was not an extensive test of the potential benefit of post-onset delivery of the therapy, which is a challenge given the aggressive phenotype of the mice. Furthermore, transgenic rescue of Ighmbp2 in neurons prevents the motor neuron disease in nmd, but reveals a dilated cardiomyopathy in the mice, raising the possibility that gene therapies would need to target other tissues and cell types beyond motor neurons (Maddatu et al., 2004). Case studies suggest involvement of other neuronal populations, such as the autonomic neurons of the gut, but the involvement of the heart or other organs in patients remains unclear (Tomaselli et al., 2018). While additional alleles of Ighmbp2 in animal models may be beneficial for preclinical studies, additional patient data to better establish the natural history are also needed. A trial to establish a genetic registry of pediatric motor neuron diseases including SMARD1 is underway (# NCT02532244), and limited natural history data is available from small scale and retrospective studies, but these are more focused on SMARD1 than on the milder CMT2S (Eckart et al., 2012; Viguier et al., 2019). While severe SMARD1 patients may lose motor neurons and require treatment as early as possible, the milder and slower progressing CMT2S patients may provide a longer window for therapeutic benefit from IGHMBP2 gene replacement. Genetically, the distinction of SMARD1 versus CMT2S appears to represent a spectrum of disease severity that corresponds to the severity of the loss of function of IGHMBP2. Therefore, the same gene replacement strategy would conceivably work for both diseases, but the differing clinical severity and progression could complicate natural history studies, trial designs, and outcome measures.
CHN3/CNTNAP1:
A third rare, recessive disorder is caused by mutations in CNTNAP1. Mutations in CNTNAP1 are associated with congenital hypomyelinating neuropathy type 3 (CHN3) and lethal congenital contracture syndrome type 7 (LCCS7) (Hengel et al., 2017; Lakhani et al., 2017; Laquerriere et al., 2014; Mehta et al., 2017; Nizon et al., 2017; Vallat et al., 2016). Both are severe, early-onset diseases of nerve conduction. CNTNAP1 encodes contactin-associated protein 1 (also called CASPR and a variety of other names). This protein is associated with a complex of cell adhesion molecules (most notably, Contactin) at the paranodal junctions of nodes of Ranvier (Rios et al., 2000). In its absence, the transverse bands observed by electron microscopy at the paranodes are disrupted (Hengel et al., 2017; Vallat et al., 2016). These structures are the septate-like junctions linking the Schwann cell processes to the axon and insuring proper ion channel localization at nodes, and thus enabling efficient nerve conduction in myelinated axons. The functions of CNTNAP1/CASPR are reviewed elsewhere (Bellen et al., 1998; Peles and Salzer, 2000; Rasband and Peles, 2015).
Although patients with CNTNAP1 mutations have a severe and early disease, the mutations appear to be straightforward loss-of-function alleles, again making them logical candidates for virus-mediated gene replacement. CNTNAP1 is reported to be specifically expressed in neurons and not in Schwann cells or oligodendrocytes (Einheber et al., 1997). This simplifies the gene replacement strategy, but the extent of CNS involvement beyond motor and sensory neurons may present a challenge. Since this disease largely results from defects in axonal conductance, it may also be more amenable to correction after the onset of symptoms, since the appropriate cell types are present, just not properly aligned through cell adhesion complexes. This possibility can be tested in preclinical studies. Several mouse alleles of Cntnap1 have been described, including engineered KO alleles (Bhat et al., 2001; Gollan et al., 2003), and several unpublished spontaneous alleles (the shambling mouse). In particular, shambling-5J has a pronounced phenotype, but survives for several months, improving its usefulness for post-onset studies and determining if the phenotype is indeed reversible after it develops. Thus, good animal models exist for preclinical studies, and a strategy for gene replacement seems sound, though additional work to confirm the cellular targets may be necessary. The rarity of the disease limits clinical characterization and natural history data, but like SMA, the relatively early and severe diseases associated with CNTNAP1 should make eventual clinical trials fairly short in duration to see effects and also increase the tolerance for risk, given the often fatal course of the disease if left untreated.
Other candidate disorders certainly exist. CMT2E, caused by dominant mutations in neurofilament light chain (NEFL), or Hereditary Sensory and Autonomic neuropathy 1 (HSAN1), caused by dominant mutations in SPTLC1 and −2, are both good candidates for allele-specific-knockdown approaches. In each disease, the mutant protein products contribute to the disease beyond simple loss of function and good animal models and patient data exist for both. The other tRNA synthetase-associated neuropathies are also potentially good candidates for allele-specific knockdown based on the success of that strategy for GARS mutations and working under the assumption that there is a shared mechanism underlying those diseases. However, those diseases are rarer and lack animal models at this time, increasing the challenge of developing such approaches. Patient availability and understanding of the genetics and pathogenic mechanisms may be limiting for many forms of CMT, but successes even in rare forms of the disease are important for setting precedents at these early stages of gene therapy implementation.
In summary, gene therapy is rapidly becoming more sophisticated and more accepted, with both viral vectors and ASOs now successfully approved for clinical use. The development of even newer technologies such as CRISPR-based therapeutics promises to further advance the capabilities of the field. There are successful clinical and preclinical studies in inherited peripheral neuropathies and related neuromuscular diseases that have sparked additional research and a search for new target genes. Several forms of CMT, including dominant and recessive diseases, are good candidates for the immediate development of gene therapy approaches. This will maintain the momentum of the field and provide additional information and precedent for gene therapy as a viable treatment strategy for these otherwise challenging diseases.
Highlights:
We describe the state-of-the-art for current gene therapy approaches, including viral vectors, antisense oligonucleotides, allele-specific approaches, and CRISPR strategies.
We highlight examples where gene therapy has been successfully applied to related neuromuscular diseases in preclinical studies and clinical trials.
We provide five disorders that are good candidates to be the next targets for gene therapy.
Acknowledgements:
We would like to thank Dr. Abigail Tadenev for helpful discussions and comments on the manuscript, and JAX Creative for assistance with graphics. This work is supported by grants from the NIH, including OD020351 (RWB) and NS105116 (RWB and SQH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Aartsma-Rus A, et al. , 2003. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet. 12, 907–14. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A, et al. , 2004. Antisense-induced multiexon skipping for Duchenne muscular dystrophy makes more sense. Am J Hum Genet. 74, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, et al. , 2009. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 30, 293–9. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A, Krieg AM, 2017. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 27, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann EJ, et al. , 2016. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid. 23, 148–157. [DOI] [PubMed] [Google Scholar]
- Adams D, et al. , 2018. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 379, 11–21. [DOI] [PubMed] [Google Scholar]
- Amoasii L, et al. , 2018. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 362, 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azorin SE, Cabib CE, Campistol JM, 2017. Diflunisal compassive use in transthyretin hereditary amyloid polyneuropathy: report of a first Spanish experience. Amyloid. 24, 105–106. [DOI] [PubMed] [Google Scholar]
- Azzedine H, et al. , 2003. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul Disord. 13, 341–6. [PubMed] [Google Scholar]
- Bailey RM, et al. , 2018. Development of Intrathecal AAV9 Gene Therapy for Giant Axonal Neuropathy. Mol Ther Methods Clin Dev. 9, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee DR, Lindsey E; Grogan Martha; Swiecicki Paul; Poterucha John; Heimbach Julie; Zeldenrust Steve; Gertz Morie; Edwards Brooks; Daly Richard; Klarich Kyle W; Dispenzieri Angela, 2017. Outcomes of Patients With Familial Transthyretin Amyloidosis After Liver Transplantation. Progress in transplantation. 27. [DOI] [PubMed] [Google Scholar]
- Bannerman P, et al. , 2016. Mice Hemizygous for a Pathogenic Mitofusin-2 Allele Exhibit Hind Limb/Foot Gait Deficits and Phenotypic Perturbations in Nerve and Muscle. PLoS One. 11, e0167573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneo-Munoz M, et al. , 2015. Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet. 11, e1005115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter RV, et al. , 2002. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 30, 21–2. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, et al. , 1998. Neurexin IV, caspr and paranodin--novel members of the neurexin family: encounters of axons and glia. Trends Neurosci. 21, 444–9. [DOI] [PubMed] [Google Scholar]
- Bertholet AM, et al. , 2016. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 90, 3–19. [DOI] [PubMed] [Google Scholar]
- Bhat MA, et al. , 2001. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 30, 369–83. [DOI] [PubMed] [Google Scholar]
- Bomont P, et al. , 2000. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 26, 370–4. [DOI] [PubMed] [Google Scholar]
- Boudreau R, et al. , 2011. Rapid cloning and validation of microRNA shuttle vectors : RNA Interference Techniques Vol. 1, Harper SQ, ^ Humana Press, New York, 19–37. [Google Scholar]
- Bravo-Hernandez M, et al. , 2020. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med. 26, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchlis G, et al. , 2012. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. 119, 3038–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushby KM, Gardner-Medwin D, 1993. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. 240, 98–104. [DOI] [PubMed] [Google Scholar]
- Campeau PM, et al. , 2013. Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet. 92, 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, et al. , 2010. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain : a journal of neurology. 133, 1460–9. [DOI] [PubMed] [Google Scholar]
- Chan KY, et al. , 2017. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. , 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2005. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 14 Spec No. 2, R283–9. [DOI] [PubMed] [Google Scholar]
- Cho S, Dreyfuss G, 2010. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, et al. , 2007. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 448, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese A, et al. , 2016. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 263, 916–924. [DOI] [PubMed] [Google Scholar]
- Cottenie E, et al. , 2014. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am J Hum Genet. 95, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox GA, Mahaffey CL, Frankel WN, 1998. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron. 21, 1327–37. [DOI] [PubMed] [Google Scholar]
- Cuesta A, et al. , 2002. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 30, 22–5. [DOI] [PubMed] [Google Scholar]
- D’Amico A, et al. , 2011. Spinal muscular atrophy. Orphanet J Rare Dis. 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KE, Nowak KJ, 2006. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 7, 762–73. [DOI] [PubMed] [Google Scholar]
- Demir E, et al. , 2005. Giant axonal neuropathy: clinical and genetic study in six cases. J Neurol Neurosurg Psychiatry. 76, 825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC, 2007. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 176, 405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, et al. , 2008. Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum Mol Genet. 17, 367–75. [DOI] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS, 2016. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 17, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez E, et al. , 2011. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 20, 681–93. [DOI] [PubMed] [Google Scholar]
- Duan D, et al. , 1998. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 72, 8568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz V, 1999. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 3, 49–51. [DOI] [PubMed] [Google Scholar]
- Eckart M, et al. , 2012. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics. 129, e148–56. [DOI] [PubMed] [Google Scholar]
- Eid A, Alshareef S, Mahfouz MM, 2018. CRISPR base editors: genome editing without double-stranded breaks. Biochem J. 475, 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, et al. , 1997. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol. 139, 1495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elverman M, et al. , 2017. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. 56, 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, 2007. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 1772, 108–17. [DOI] [PubMed] [Google Scholar]
- Ferguson CJ, et al. , 2012. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet. 21, 3525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari FK, et al. , 1996. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 70, 3227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, et al. , 2016. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 388, 3017–3026. [DOI] [PubMed] [Google Scholar]
- Finkel RS, et al. , 2017. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 377, 1723–1732. [DOI] [PubMed] [Google Scholar]
- Foust KD, et al. , 2009. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 27, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, et al. , 2010. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 28, 271–4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gertz MA, et al. , 2015. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol. 66, 2451–2466. [DOI] [PubMed] [Google Scholar]
- Gertz MA, et al. , 2019. Advances in the treatment of hereditary transthyretin amyloidosis: A review. Brain Behav. 9, e01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollan L, et al. , 2003. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 163, 1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyenvalle A, et al. , 2004. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 306, 1796–9. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, et al. , 2004. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 10, 828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, et al. , 2006. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 12, 787–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen EJN, Talbot K, Gillingwater TH, 2018. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 14, 214–224. [DOI] [PubMed] [Google Scholar]
- Grohmann K, et al. , 2001. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 29, 75–7. [DOI] [PubMed] [Google Scholar]
- Haidet AM, et al. , 2008. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A. 105, 4318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton G, Gillingwater TH, 2013. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med. 19, 40–50. [DOI] [PubMed] [Google Scholar]
- Hammond SM, et al. , 2016. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A. 113, 10962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, et al. , 2002. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 8, 253–61. [DOI] [PubMed] [Google Scholar]
- Hauck B, et al. , 2004. Intracellular viral processing, not single-stranded DNA accumulation, is crucial for recombinant adeno-associated virus transduction. J Virol. 78, 13678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens MA, Hastings ML, 2016. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 44, 6549–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H, et al. , 2017. CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurol Genet. 3, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, et al. , 2007. New mutations, genotype phenotype studies and manifesting carriers in giant axonal neuropathy. J Neurol Neurosurg Psychiatry. 78, 1267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, et al. , 2018. Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination. Ann Neurol. 83, 756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaen ML, et al. , 2017. Long-Term Efficacy and Safety of Insulin and Glucokinase Gene Therapy for Diabetes: 8-Year Follow-Up in Dogs. Mol Ther Methods Clin Dev. 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano RL, 2016. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima T, Manley JL, 2003. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 34, 460–3. [DOI] [PubMed] [Google Scholar]
- Kenwrick S, et al. , 1987. Molecular analysis of the Duchenne muscular dystrophy region using pulsed field gel electrophoresis. Cell. 48, 351–7. [DOI] [PubMed] [Google Scholar]
- Koenig M, et al. , 1987. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 50, 509–17. [DOI] [PubMed] [Google Scholar]
- Kohler M, et al. , 2005. Quality of life, physical disability, and respiratory impairment in Duchenne muscular dystrophy. Am J Respir Crit Care Med. 172, 1032–6. [DOI] [PubMed] [Google Scholar]
- Koop O, et al. , 2007. Genotype-phenotype analysis in patients with giant axonal neuropathy (GAN). Neuromuscul Disord. 17, 624–30. [DOI] [PubMed] [Google Scholar]
- Lakhani S, et al. , 2017. Identification of a novel CNTNAP1 mutation causing arthrogryposis multiplex congenita with cerebral and cerebellar atrophy. Eur J Med Genet. 60, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquerriere A, et al. , 2014. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum Mol Genet. 23, 2279–89. [DOI] [PubMed] [Google Scholar]
- Le Rumeur E, 2015. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 15, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, et al. , 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 80, 155–65. [DOI] [PubMed] [Google Scholar]
- Lenk GM, et al. , 2011. Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet. 7, e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, et al. , 2011. Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc Natl Acad Sci U S A. 108, 14258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. , 2017. A RAT MODEL OF CMT2A DEVELOPS A PROGRESSIVE NEUROPATHY. Journal of the Peripheral Nervous System. 22, 331. [Google Scholar]
- Liu M, et al. , 2005. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther. 11, 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, et al. , 2018. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv. 4, eaap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, et al. , 1999. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 96, 6307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ, 2000. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 9, 259–65. [DOI] [PubMed] [Google Scholar]
- Lozeron P, et al. , 2013. Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol. 20, 1539–45. [DOI] [PubMed] [Google Scholar]
- MacLeod MJ, et al. , 1999. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 3, 65–72. [DOI] [PubMed] [Google Scholar]
- Maddatu TP, et al. , 2004. Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum Mol Genet. 13, 1105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall MS, et al. , 2018. Long-Term Improvement of Neurological Signs and Metabolic Dysfunction in a Mouse Model of Krabbe’s Disease after Global Gene Therapy. Mol Ther. 26, 874–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PB, et al. , 2019. Overlapping spectrums: The clinicogenetic commonalities between Charcot-Marie-Tooth and other neurodegenerative diseases. Brain Res. 146532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClorey G, et al. , 2006. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 13, 1373–81. [DOI] [PubMed] [Google Scholar]
- Mehta P, et al. , 2017. Novel mutation in CNTNAP1 results in congenital hypomyelinating neuropathy. Muscle Nerve. 55, 761–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, et al. , 2013. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 74, 637–47. [DOI] [PubMed] [Google Scholar]
- Mendell JR, et al. , 2017. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Mercuri E, et al. , 2018. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. 378, 625–635. [DOI] [PubMed] [Google Scholar]
- Meyer K, et al. , 2015. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther. 23, 477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko A, et al. , 2010. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 30, 4232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco AP, et al. , 1988. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 2, 90–95. [DOI] [PubMed] [Google Scholar]
- Monani UR, et al. , 1999. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 8, 1177–83. [DOI] [PubMed] [Google Scholar]
- Morelli KH, et al. , 2019. Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J Clin Invest. 129, 5568–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley WW, et al. , 2011. Charcot-Marie-Tooth-Linked Mutant GARS Is Toxic to Peripheral Neurons Independent of Wild-Type GARS Levels. PLoS Genet. 7, e1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussche S, et al. , 2013. Restoration of cytoskeleton homeostasis after gigaxonin gene transfer for giant axonal neuropathy. Hum Gene Ther. 24, 209–19. [DOI] [PubMed] [Google Scholar]
- Naldini L, 2015. Gene therapy returns to centre stage. Nature. 526, 351–60. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, et al. , 2014. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 371, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelis E, et al. , 2002. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 59, 1865–72. [DOI] [PubMed] [Google Scholar]
- Niemann A, et al. , 2005. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 170, 1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann A, et al. , 2014. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain. 137, 668–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizon M, et al. , 2017. Two novel variants in CNTNAP1 in two siblings presenting with congenital hypotonia and hypomyelinating neuropathy. Eur J Hum Genet. 25, 150–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizzardo M, et al. , 2015. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci Adv. 1, e1500078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout DG, et al. , 2015a. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 6, 6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout DG, et al. , 2015b. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther. 23, 523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak CA, et al. , 2007. Long-term skeletal muscle protection after gene transfer in a mouse model of LGMD-2D. Mol Ther. 15, 1775–81. [DOI] [PubMed] [Google Scholar]
- Palaninathan SK, 2012. Nearly 200 X-ray crystal structures of transthyretin: what do they tell us about this protein and the design of drugs for TTR amyloidoses? Current medicinal chemistry. 19. [DOI] [PubMed] [Google Scholar]
- Pantera H, Shy ME, Svaren J, 2020. Regulating PMP22 expression as a dosage sensitive neuropathy gene. Brain Res. 1726, 146491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parente V, Corti S, 2018. Advances in spinal muscular atrophy therapeutics. Ther Adv Neurol Disord. 11, 1756285618754501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Salzer JL, 2000. Molecular domains of myelinated axons. Curr Opin Neurobiol. 10, 558–65. [DOI] [PubMed] [Google Scholar]
- Rahimov F, Kunkel LM, 2013. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. 201, 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, 2015. The Nodes of Ranvier: Molecular Assembly and Maintenance. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo F, et al. , 2014. Pharmacology of a central nervous system delivered 2’-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 350, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, et al. , 2000. Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci. 20, 8354–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha AG, et al. , 2018. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science. 360, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor AM, et al. , 2017. Peripheral neuropathy in complex inherited diseases: an approach to diagnosis. J Neurol Neurosurg Psychiatry. 88, 846–863. [DOI] [PubMed] [Google Scholar]
- Rossor AM, Shy ME, Reilly MM, 2020. Are we prepared for clinical trials in Charcot-Marie-Tooth disease? Brain Res. 1729, 146625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelius A, et al. , 2018. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology. 90, e518–e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargiannidou I, Kagiava A, Kleopa KA, 2019. Gene therapy approaches targeting Schwann cells for demyelinating neuropathies. Brain Res. 146572. [DOI] [PubMed] [Google Scholar]
- Seburn KL, et al. , 2006. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 51, 715–26. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, et al. , 2018. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 13, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M, et al. , 2016. Rescue of a Mouse Model of Spinal Muscular Atrophy With Respiratory Distress Type 1 by AAV9-IGHMBP2 Is Dose Dependent. Mol Ther. 24, 855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, et al. , 2006. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 26, 1333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Gillingwater TH, Talbot K, 2011. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis Model Mech. 4, 457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuppia G, et al. , 2015. MFN2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci. 356, 7–18. [DOI] [PubMed] [Google Scholar]
- Sugarman EA, et al. , 2012. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 20, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr OB, et al. , 2015. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 10, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surono A, et al. , 2004. Chimeric RNA/ethylene-bridged nucleic acids promote dystrophin expression in myocytes of duchenne muscular dystrophy by inducing skipping of the nonsense mutation-encoding exon. Hum Gene Ther. 15, 749–57. [DOI] [PubMed] [Google Scholar]
- Tabebordbar M, et al. , 2016. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 351, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadic V, et al. , 2019. CRISPR/Cas9-based epigenome editing: An overview of dCas9-based tools with special emphasis on off-target activity. Methods. 164–165, 109–119. [DOI] [PubMed] [Google Scholar]
- Takeshima Y, et al. , 2001. Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain Dev. 23, 788–90. [DOI] [PubMed] [Google Scholar]
- Timmerman V, Strickland AV, Zuchner S, 2014. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes (Basel). 5, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale S, Pellizzoni L, 2015. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 35, 8691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli PJ, et al. , 2018. IGHMBP2 mutation associated with organ-specific autonomic dysfunction. Neuromuscul Disord. 28, 1012–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat JM, et al. , 2016. Contactin-Associated Protein 1 (CNTNAP1) Mutations Induce Characteristic Lesions of the Paranodal Region. J Neuropathol Exp Neurol. 75, 1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valori CF, et al. , 2010. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2, 35ra42. [DOI] [PubMed] [Google Scholar]
- van Deutekom JC, et al. , 2001. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 10, 1547–54. [DOI] [PubMed] [Google Scholar]
- Verhaart IEC, et al. , 2017a. A multi-source approach to determine SMA incidence and research ready population. J Neurol. 264, 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaart IEC, et al. , 2017b. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 12, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira M, Saraiva MJ, 2014. Transthyretin: a multifaceted protein. Biomol Concepts. 5, 45–54. [DOI] [PubMed] [Google Scholar]
- Viguier A, et al. , 2019. Spinal muscular atrophy with respiratory distress type 1: A multicenter retrospective study. Neuromuscul Disord. 29, 114–126. [DOI] [PubMed] [Google Scholar]
- Vitte J, et al. , 2007. Refined characterization of the expression and stability of the SMN gene products. Am J Pathol. 171, 1269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora S, et al. , 2016. Next stop for the CRISPR revolution: RNA-guided epigenetic regulators. FEBS J. 283, 3181–93. [DOI] [PubMed] [Google Scholar]
- Wang CH, et al. , 2007. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 22, 1027–49. [DOI] [PubMed] [Google Scholar]
- Wang CH, Lunn MR, 2008. Spinal muscular atrophy: advances in research and consensus on care of patients. Curr Treat Options Neurol. 10, 420–8. [DOI] [PubMed] [Google Scholar]
- Wang Z, et al. , 2012. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 20, 1501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters JJ, et al. , 2011. Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. J Neurosci. 31, 17736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, et al. , 2004. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 279, 17181–9. [DOI] [PubMed] [Google Scholar]
- Yue Y, et al. , 2003. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 108, 1626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu M, Duan D, 2006. C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 14, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HT, et al. , 2018. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest. 128, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, et al. , 2019. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest. 130, 1756–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, et al. , 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 36, 449–51. [DOI] [PubMed] [Google Scholar]
- Zuchner S, et al. , 2006. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 59, 276–81. [DOI] [PubMed] [Google Scholar]