

Abstract
Migraineurs can show brain hyperexcitability and oxidative stress that may promote headache. Since hyperexcitability can enhance oxidative stress which promotes hyperexcitability, ending this feed-back loop may reduce migraine. Neocortical spreading depression, an anrefeimal model of migraine begins with hyperexcitability and triggers oxidative stress in the neocortical area involved and in the trigeminal system, which is important to pain pathway nociceptive activation in migraine. Additionally, oxidative stress causes increased trigeminal ganglion calcitonin gene-related peptide release and oxidative stress can reduce spreading depression threshold. Insulin-like growth factor-1 significantly protects against spreading depression in vitro by reducing oxidative stress and it is effective against spreading depression after intranasal delivery to animals. Here, we used adult male rats and extend this work to study the trigeminal system where insulin-like growth factor-1 receptors are highly expressed. Recurrent neocortical spreading depression significantly increased surrogate markers of trigeminal activation - immunostaining for trigeminal ganglion oxidative stress, calcitonin gene related peptide levels and c-fos in the trigeminocervical complex versus sham. These effects were significantly reduced by intranasal delivery of insulin-like growth factor-1 a day before recurrent neocortical spreading depression. Furthermore, intranasal treatment with insulin-like growth factor-1 significantly reduced naïve levels of trigeminal ganglion calcitonin gene related peptide versus sham with no impact on blood glucose levels. Intranasal delivery of insulin-like growth factor-1 not only mitigates neocortical spreading depression, a cause of migraine hyperexcitability modeled in animals, but also when neocortical spreading depression is triggered by suprathreshold stimuli, insulin-like growth factor-1 effectively reduces nociceptive activation in the trigeminal system.
Keywords: migraine, intranasal delivery, CGRP, trigeminal ganglion, trigeminal nucleus, oxidative stress
Graphical Abstract
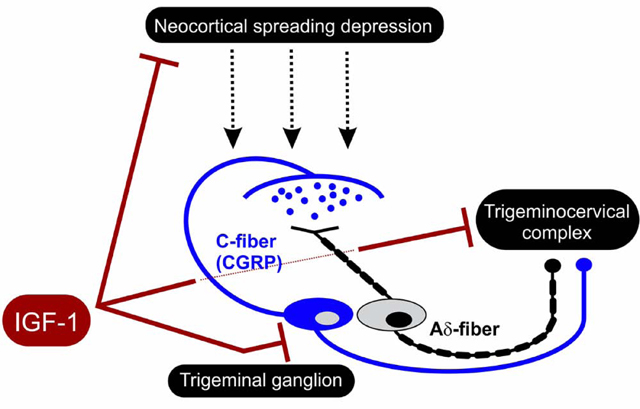
1. Introduction
Central nervous system aberrant electrical activity that includes hyperexcitability can be seen in migraine patients (Goadsby et al., 2017; Dodick, 2018; Vigano et al., 2019) along with increased levels of oxidative stress that are seen both during and between migraine attacks (Yilmaz et al., 2007; Tuncel et al., 2008; Aytac et al., 2014; Yigit et al., 2018). Since hyperexcitability can enhance oxidative stress and oxidative stress promotes hyperexcitability, unraveling this vicious cycle especially as it relates to the trigeminal system (i.e., the trigeminal ganglion and trigeminocervical complex) may lead to novel therapeutics for migraine (Borkum, 2016; Goadsby et al., 2017). Shatillo and colleagues (Shatillo et al., 2013) show that neocortical spreading depression, the most likely cause of migraine aura and perhaps a cause of related pain (Moskowitz et al., 1993; Zhang et al., 2011), triggers oxidative stress not only in the neocortical area involved in spreading depression, but also in the trigeminal system, which is important to pain pathway activation in migraine (Shatillo et al., 2013). They also note that oxidative stress can increase calcitonin gene-related peptide (CGRP) release in dorsal root ganglion neurons (Shatillo et al., 2013). Anti-CGRP biologics designed to block migraine pain are now available and are making important improvements to migraine treatment. However, they do not prevent the initiation of migraine (Hansen et al., 2011; Messlinger et al., 2012) and more therapeutic options are still needed because some migraineurs experience limited response to current medications (Mitsikostas and Reuter, 2017). Thus, there is an unmet need for therapeutics to treat frequent migraine sufferers more effectively.
Increased physical and intellectual activity (termed environmental enrichment) can reduce subsequent neurological disease in animals and humans. Environmental enrichment is known to reduce susceptibility to neocortical spreading depression in animal models (Pusic et al., 2014). Likewise, increased physical activity reduces susceptibility to migraine in humans (Irby et al., 2016; Lemmens et al., 2019). Improved understanding of the bases for this widespread preconditioning neuroprotection from environmental enrichment may lead to new therapeutics for brain diseases such as migraine. For example, insulin-like growth factor-1 (IGF-1) levels increase with environmental enrichment (Ciucci et al., 2007) and IGF-1 enters brain with increased sensory stimulation (Nishijima et al., 2010). In vitro modeling of migraine using spreading depression in hippocampal slice cultures shows that IGF-1 treatment significantly protects against spreading depression (Grinberg et al., 2012; Grinberg et al., 2013). This effect involves abrogation of microglial oxidative stress, a factor that otherwise can trigger the hyperexcitability burst needed to promote spreading depression (Grinberg et al., 2012; Grinberg et al., 2013). Also, IGF-1 significantly protects against spreading depression in vivo after intranasal administration [i.e., which includes direct nose-to-brain delivery (Grinberg et al., 2017)] in adult rats.
Here we extend this work to show that intranasal delivery of IGF-1 also significantly prevents nociceptive activation of the trigeminal system from recurrent spreading depression. This work appeared in preliminary form (Won and Kraig, 2019a; Won and Kraig, 2019b).
2. Results
2.1. Impact of spreading depression and intranasal IGF-1
We first confirmed that our recurrent spreading depression model could trigger a significant activation of the trigeminal ganglion and trigeminocervical complex. We did not examine the impact of a single spreading depression since this is often not sufficiently robust activation in anesthetized animals to probe for experimental changes (Grinberg et al., 2017; Wang et al., 2016). Results are shown in Figure 1. Consistent with results previously reported (Grinberg et al., 2017), microinjection of less than 10 nl of 0.5 M KCl was sufficient to reproducibly evoke SD. Recurrent spreading depression induced a significant (p < 0.001, power 1.00) increase in malondialdehyde immunostaining in the trigeminal ganglion. Specific natural logarithm ratios (spreading depression / sham) were 2.00 ± 0.190 (n = 5/group) which reflects a 639% increase versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Similarly, recurrent spreading depression induced a significant (p < 0.001, power = 1.00) increase in CGRP immunostaining in the trigeminal ganglion. Specific natural logarithm ratios (spreading depression / sham) were 1.60 ± 0.31 (n = 5/group) which reflects a 395% increase versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Finally, recurrent spreading depression evoked a significant (p < 0.001, power = 1.00) increase in c-fos positive cells. Specific natural logarithm ratios (spreading depression/sham c-fos positive cells) were 1.04 ± 0.20 (n = 5–7/group) which reflects a 183% increase versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Accordingly, we evoked 8–9 spreading depressions for all experimental animals in this study treated with nasal IGF-1 or vehicle (Figure 2).
Figure 1. Spreading depression-induced activation in the trigeminal ganglion and trigeminocervical complex.

(A) Recurrent 0.5M KCl (≤ 10 nl) KCl microinjections triggered 7–9 spreading depressions over the 90 minute recording period (8.4 ± 0.4 spreading depressions). (B) No spreading depressions occurred from similar 0.5M NaCl micro-injections. (C-E) Spreading depression induced trigeminal ganglion increased immunostaining for malondialdehyde. Representative images show malondialdehyde immunostaining images after recurrent SD (C) and sham (D) in the V1 area of the trigeminal ganglion. Scale bar = 25 μm. (E) Natural logarithm ratios (SD/sham) showed that recurrent SD caused a significantly (***p < 0.001) increase in malondialdehyde immunostaining which reflects a 639% increase compared to no difference (ND) in ratios (i.e., Ln = 0). (F-H) Similarly, trigeminal ganglion CGRP immunostaining was increased after recurrent SD. Images show representative CGRP immunostaining after recurrent SD (F) and sham (G) in the trigeminal ganglion. Scale bar = 25 μm. (H) Natural logarithm ratios (SD/sham) showed recurrent SD caused a significant (***p < 0.001) increase in CGRP immunostaining which reflects a 395% increase compared to no difference (ND) in ratios (i.e., Ln = 0). (I-K) Trigeminocervical complex c-fos immunostaining was also increased after recurrent SD. Representative images show c-fos immunostaining within the superficial laminae I and II (red area of interest) of the trigeminocervical complex at −4.5 mm from obex after (I) spreading depression (SD) or (J) sham. Scale bar = 200 μm. (K) Natural logarithm ratios (SD/sham) show that recurrent SD caused a significant (***p < 0.001) increase in c-fos positive cells which reflects a 183% compared to no difference (ND).
Figure 2. Representative neocortical spreading depression responses from KCl stimulation.

Records show typical large extracellular voltage deflections of spreading depression. Intranasal administration of IGF-1 (or vehicle) was delivered. Twenty-four hours later, neocortical spreading depression was induced every 10 minutes for 90 minutes via pressurized episodic micro-injections of nl quantities of 0.5 M KCl into the rostral neocortex with confirmation of spreading depression established via electrophysiological microelectrode recordings from caudal neocortex. Representative neocortical spreading depression slow electrical changes are shown. 10 mV scale bar is negative down. Immediately after spreading depression stimulation, tissues were harvested for determination of the degree of trigeminal system activation in the trigeminal ganglion and trigeminocervical complex.
Second, we confirmed that IGF-1 receptors are widely distributed in the trigeminal ganglion (Figure 3), consistent with evidence shown previously by others (Wang et al., 2014).
Figure 3. Trigeminal ganglion IGF-1 receptor distribution.

Representative images show that IGF-1 can have direct ligand-receptor interactions via its cognate receptor that is found throughout the adult rat trigeminal ganglion [(A) V1, ophthalmic division; (B) V2, maxillary division; and (C) V3, mandibular division]. Scale bar = 50 μm.
Third, parental administration of IGF-1 is FDA approved for treatment of short stature due to primary IGF-1 deficiency in children (Ipsen Biopharmaceuticals, 2016). In this patient population, a subset of children receiving high doses of systemic IGF-1 exhibit hypoglycemia and headache (Ipsen Biopharmaceuticals, 2016). Accordingly, we probed for any impact of intranasal IGF-1 on blood glucose levels. No evidence of hypoglycemia after intranasal delivery of IGF-1 was seen (Figure 4). Specific time-based blood glucose levels (n = 8/group at 0 hour and n = 4/group otherwise) for succinate buffer (sham) and IGF-1 treated groups were, respectively: 0hour: 132 ± 11 and 124 ± 7; 0.3-hour: 213 ± 14 and 222 ± 13; 1.0-hour: 167 ± 19 and 146 ± 12; 3.0-hour: 129 ± 8 and 119 ± 8; 6.0-hour: 134 ± 6 and 134 ± 3; 24-hour: 143 ± 9 and 147 ± 16 mg/dl. No significant (p > 0.21–1.00) difference between matched time pairs (t-test) was seen. The systematic elevation of blood glucoses seen at 0.3 hours was significant (ANOVA, p < 0.001, power = 1.00), albeit transient over initial level, increases that most likely reflect the expected side effect of exposure to isoflurane anesthesia for the 20 minutes needed for nasal administration (Zuurbier et al., 2008). The lowest IGF-1 glucose-related level seen was 114 mg/dl [compared to control (132 mg/dl; time 0)], which is within the normal range for Wistar rats [85–132 mg/dl (Kohn and Clifford, 2002)]. While the glucose level that defines hypoglycemia is variable, a drop of 1 mM is not considered hypoglycemic.
Figure 4. Intranasal treatment with IGF-1 did not cause hypoglycemia.

Serum glucose was measured in non-fasted rats under brief isoflurane anesthesia after intranasal delivery of vehicle (red) or IGF-1 (black) compared to immediately before treatments (light gray). As expected, glucose rose at the conclusion of intranasal treatments due to continued isoflurane exposure. However, no significant differences were seen between treatment pairs (t-test) or 0 time (light gray) versus other values when compared via ANOVA plus post hoc Holm-Sidak except at the 0.3 hour point (***p < 0.001). The lowest IGF-1 glucose-related level seen was 119 mg/dl [compared to control (128 mg/dl; time 0)], which is within the normal range for Wistar rats [85–132 mg/dl (Kohn and Clifford, 2002)]. Dotted lines show normal glucose range. While the glucose level that defines hypoglycemia is variable, a drop of 1 mM is not considered hypoglycemic. Furthermore, no IGF-1 glucose level was hypoglycemic. Results shown as mean ± SEM.
Fourth, up to 5% of patients treated systemically with IGF-1 might complain of headache. Accordingly, we used CGRP immunostaining as a metric for trigeminal ganglion nociceptive activation potentially associated with headache (Figure 5). Results demonstrated for the first time that intranasal IGF-1 did not cause an increase in trigeminal ganglion CGRP levels consistent with what might be expected from headache (Durham, 2006), but instead showed a highly significant decrease in CGRP levels in naïve animals (p < 0.001, power = 1.00, n = 7/group). Specific natural logarithm ratios (IGF-1/sham) were −1.66 ± 0.32 which reflects an 81% decrease in CGRP levels.
Figure 5. Intranasal delivery of IGF-1 significantly reduced CGRP levels in the trigeminal ganglion of naïve animals.

Representative images show CGRP immunostaining of the V1 area of the trigeminal ganglion after treatment with (A) IGF-1 or (B) vehicle. (C) Natural logarithm ratios of immunostaining levels after intranasal treatment [naïve (IGF-1/sham)] show that IGF-1 significantly (***p < 0.001) reduced CGRP levels which reflects an 81% reduction versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Scale bar = 25 μm.
Treatment with succinate alone (i.e., log-ratios of succinate/control) showed no significant impact on baseline CGRP levels compared to control animals. Specific logarithm ratios were 0.022 ± 0.19 (p = 0.916, power = 0.05, n = 7/group). For these naïve animal CGRP measurements and the measurements of CGRP and malondialdehyde in the trigeminal ganglion after neocortical spreading depression the p value is based on comparison of the log-ratio of IGF-1/sham immunostaining integrated intensity versus 0. For c-fos positive cells an analogous log-ratio strategy was employed for measurements in the trigeminocervical complex.
2.2. Intranasal IGF-1 inhibits trigeminal ganglion and trigeminocervical complex activation after recurrent spreading depression
Next, we determined how intranasal IGF-1 treatment impacted trigeminal ganglion activation after recurrent spreading depression. Spreading depression was triggered every nine-ten minutes for up to nine spreading depressions. Consistent with the results of Shatillo and coworkers (Shatillo et al., 2013), recurrent spreading depression triggered oxidative stress in the trigeminal ganglion. However, oxidative stress measured via malondialdehyde immunostaining in the trigeminal ganglion following recurrent spreading depression, was significantly (p < 0.001, power 1.00) reduced by pretreatment with IGF-1 (Figure 6). Specific natural logarithm ratios (IGF1/sham) were −1.76 ± 0.62 (n = 5/group) which reflect an 83% decrease in malondialdehyde levels. Similarly, pretreatment with intranasal IGF-1 caused a significant (p < 0.001, power = 1.00) reduction in CGRP levels in the trigeminal ganglion after recurrent spreading depression (Figure 6). Specific log-ratio values were −0.90 ± 0.15 (n = 5/group) which reflect a 59% decrease in CGRP levels. However, no significant (p = 0.347, power = 0.136) changes in blood CGRP levels were found after pretreatment with IGF-1 (or vehicle) followed by recurrent spreading depression. Specific values were 12.1 ± 3.4 pg/ml, with two samples below the detection limit (n = 6/group; vehicle pretreatment) and 7.7 ± 1.6 pg/ml with three samples below the detection limit (n = 6/group; IGF-1 pretreatment).
Figure 6. Intranasal delivery of IGF-1 significantly reduced spreading depression-induced activation in the trigeminal ganglion and trigeminocervical complex.

Representative images show malondialdehyde immunostaining of the V1 area of the trigeminal ganglion after pretreatment with (A) IGF-1 or (B) vehicle which was followed a day later by 90 minutes of recurrent spreading depression. (C) Natural logarithm ratio levels of immunostaining after intranasal treatment (IGF-1/sham) showed that IGF-1 significantly (***p < 0.001) reduced malondialdehyde levels which reflects an 83% reduction versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Scale bar = 25 μm. (D-F) IGF-1 pretreatment reduced immunostaining for CGRP. Representative images show CGRP immunostaining of the V1 area of the trigeminal ganglion after pre-treatment with (D) IGF-1 or (E) vehicle which was followed a day later by 90 minutes of recurrent spreading depression. (F) Natural log ratio levels of immunostaining after intranasal treatment (IGF-1/sham) show that IGF-1 significantly (***p < 0.001) reduced CGRP levels which reflects a 59% reduction versus comparison to no difference (ND) in ratios (i.e., Ln = 0). Scale bar = 25 μm. (G-H) IGF-1 pretreatment reduced immunostaining for c-fos. Representative images show c-fos immunostaining at −4.5 mm caudal to the obex within (red area of interest) laminae I and II of the trigeminocervical complex after pre-treatment with (G) IGF-1 or (H) succinate buffer which was followed a day later by 90 minutes of recurrent spreading depression. Scale bar = 200 μm. (I) Natural logarithm ratios (IGF-1/sham) show that nasal IGF-1 significantly (***p < 0.001) reduced c-fos positive cells which reflects a 45% reduction versus comparison to no difference (ND) in ratios (i.e., Ln = 0).
Finally, recurrent spreading depression activates neurons in the superficial laminae of the caudal trigeminal nucleus and upper cervical spinal cord (i.e., trigeminocervical complex) (Goadsby et al., 2017; Kunkler and Kraig, 2003; Moskowitz et al., 1993) (Figure 1). Accordingly, we used trigeminocervical complex activation from spreading depression to test the impact of pretreatment with intranasal delivery of IGF-1 (Figure 6). Results show that intranasal pretreatment with IGF-1 caused a significant (p < 0.001, power = 0.97) reduction of the natural logarithm ratio of c-fos positive cells (IGF-1/sham) in the trigeminocervical complex superficial laminae at −4.5 mm from the obex. Specific natural logarithms were −0.60 ± 0.06 (n =7/group) which reflects a 45% reduction in c-fos positive cells from spreading depression by nasal IGF-1.
3. Discussion
3.1. Summary
This study shows that intranasal IGF-1 is an effective means to inhibit trigeminal system activation associated with migraine modeled in rats using spreading depression. In this initial proof-of-principle study we used only males. Future work will include females.
3.2. Intranasal delivery of IGF-1
Intranasal delivery of therapeutics is increasingly recognized to be a safe and effective means to treat neurological disorders (Djupesland et al., 2014). Intranasal IGF-1 quickly enters the brain within minutes through olfactory, and importantly, trigeminal nerve routes and influences brain function (Grinberg et al., 2017).
IGF-1 passes into the brain along the olfactory and trigeminal nerves to the cerebrospinal fluid where it then appears to widely enter brain after bulk flow movement through the cerebrospinal fluid space and into the brain parenchyma via flow from perivascular spaces (Lochhead et al., 2015; Thorne et al., 2004). It is notable that, following intranasal delivery, the highest levels of tracer are seen in the trigeminal ganglion. The levels in the trigeminal ganglion are ~10-fold greater than levels in the olfactory bulb within 30 minutes of administration (Thorne et al., 2004). Many have questioned the translational utility of extrapolating intranasal delivery data from rats to humans. This concern stems from differences between rodents and humans in intranasal structure and olfactory mucosa size where delivered agents would be expected to cross. Perhaps this focus needs to be expanded to examine the potential role of the trigeminal nerves as a passage way from nose to brain. Potentially, focusing on the trigeminal route into the brain via intranasal delivery of therapeutics may be especially well-suited to developing migraine therapeutics since the trigeminal ganglion is the “first pain-pathway related stop” along intranasal-related passage way to the brain.
3.3. IGF-1 and oxidative stress
Delineation of mechanisms by which IGF-1 reduces oxidative stress in the trigeminal ganglion is beyond the scope of this work. However, some direction can be provided from the existing literature and our results here to suggest potential mechanisms that may differ between the central nervous system and the trigeminal ganglion. Differences in the sources of oxidative stress between brain and the trigeminal ganglion may lead to novel information about how nasal IGF-1 can mitigate oxidative stress in preclinically modeled migraine, and by extension potentially migraine seen in patients. Spreading depression evoked by hyperexcitability from recurrent electrical stimulation in hippocampal brain slices requires microglia and their activation to promote release of the pro-inflammatory cytokine tumor necrosis factor alpha and reactive oxygen species, which together help to initiate spreading depression (Grinberg et al., 2012; Grinberg et al., 2013; Pusic et al., 2014). Conversely, treatment with IGF-1 abrogates this microglial oxidative stress and inflammatory activation and in so doing inhibits initiation of spreading depression (Grinberg et al., 2012; Grinberg et al., 2013). Hippocampal brain slice culture responses frequently parallel similar responses in vivo (Kunkler et al., 2005) Thus, we hypothesize that the role of microglia shown to occur in hippocampal brain slices may well extend to conditions in vivo, including that in neocortex.
In contrast, oxidative stress in the trigeminal ganglion after recurrent neocortical spreading depression involves cells that morphologically resemble neurons with increased malondialdehyde, a product of lipid peroxidation (Shatillo et al., 2013). This enhanced production of malondialdehyde in the trigeminal ganglion is so robust that it can be detected in the blood of migraine patients (Yilmaz et al., 2007; Tuncel et al., 2008; Aytac et al., 2014; Yigit et al., 2018) suggesting its use as a potential biomarker for migraine treatment. Activated neurons generate reactive oxygen species, which can result in oxidative stress if reactive oxygen species generation exceeds anti-oxidant levels. Given that mitochondria are concentrated in neurons, malondialdehyde can cause mitochondrial dysfunction that can directly promote production of reactive oxygen species (Long et al., 2009). Notably, intranasal IGF-1 reduced trigeminal ganglion malondialdehyde levels by 83% (Figure 6). Furthermore, using brain slice cultures in vitro, oxidative stress from menadione exposure, a mitochondrial inhibitor, is significantly reduced by IGF-1 (Grinberg et al., 2012; Grinberg et al., 2013). In fact, IGF-1 reduces naïve culture levels of reactive oxygen species significantly below that of controls [see Figure 4 (Grinberg et al., 2012)]. This leads us to hypothesize that a principle means by which intranasal IGF-1 may inhibit malondialdehyde levels in the trigeminal ganglion after recurrent spreading depression is likely to involve effects on mitochondrial function.
3.4. IGF-I interactions with CGRP
Oxidative stress triggers increased levels of CGRP in the trigeminal ganglion. For example, infusion of glyceryl trinitrate, a nitric oxide donor which enhances oxidative stress in brain (Li, et al., 2016), results in a significant increase in the number of CGRP-immunopositive cells in rat trigeminal ganglia (Dieterle, et al., 2011). Additionally, spreading depression generates oxidative stress (Shatillo et al., 2013) and induces CGRP protein expression in trigeminal ganglia (Chen et al., 2017). Ex vivo trigeminal ganglion studies show increase in oxidative stress is associated with increase in CGRP gene and peptide expression and can be inhibited by treatment with an antioxidant (Raddant et al., 2014).
CGRP, in turn, plays a key role in migraine pathophysiology (Goadsby et al., 2017; Iyengar et al., 2019). Intravenous CGRP causes headache in migraineurs (Lassen et al., 2002). Also, healthy individuals demonstrate headache following CGRP infusion, which can be prevented by a CGRP receptor antagonist (Petersen et al., 2005). Agents that interfere with released CGRP function are effective against migraine (Edvinsson et al., 2018; Iyengar et al., 2019). In animals, intraperitoneal CGRP injection causes migraine-like photophobia that is reversed by an anti-CGRP antibody (Mason et al., 2017). Also, local subcutaneous injection of CGRP in the periorbital area of mice elicits delayed periorbital allodynia that is inhibited by local and intraperitoneal injection of a CGRP antagonist (De Logu et al., 2019). Finally, anti-CGRP agents reduce migraine-like behavior and/or trigeminocervical c-fos levels in animal migraine models (Filiz et al., 2019; Ramachandran et al., 2014).
Importantly, neuropeptides such as CGRP are synthesized in cell bodies and move to synaptic terminals for release (Schwartz, 2000). Once released, newly formed neuropeptide must come from the cell body to replenish synaptic levels for release (Schwartz, 2000). Accordingly, cellular levels of CGRP may directly reflect amounts of CGRP that are released with cellular activation such as from trigeminal ganglion activation. If so, then the IGF-1-related changes in trigeminal ganglion support a potential pain relieving ability of IGF-1 in migraine. This conclusion is supported by the findings here that nasal IGF-1 triggered a significant decline in CGRP levels in naïve animals and had a similar significant impact on lowering trigeminal ganglion CGRP after recurrent spreading depression. This interpretation is bolstered by the fact that direct administration of IGF-1 to brain is anti-nociceptive (Bitar et al., 1996).
Oxidative stress, including that from mitochondrial dysfunction, may also be a key stimulus for increased CGRP production. Others have shown that trigeminal ganglion CGRP levels and related c-fos activation in the trigeminocervical complex are elevated with various preclinical models of migraine and these changes can be prevented by therapeutic treatments designed to treat migraine (Filiz et al., 2019; Greco et al., 2016; Sixt et al., 2009) similar to the results reported here. A potentially important consideration in this regard is data reported by the Burstein laboratory. Melo-Carillo and co-workers show that anti-CGRP agents selectively block Aδ trigeminal electrical activity and related high threshold trigeminal nucleus neurons (Melo-Carrillo et al., 2017). In contrast, C-fibers, which contain CGRP and activate wide dynamic range neurons in the trigeminal nucleus, are not inhibited. The authors suggest this partial impact on the trigeminal system may account for the partial efficacy seen in treatment of migraine patients with anti-CGRP agents (Melo-Carrillo et al., 2017). Intranasal IGF-1 treatment may have more widespread effects because of its more global impact on migraine-related pathophysiology.
Perhaps the most important potential impact of IGF-1 on migraine pathophysiology will come from its ability to reduce oxidative stress. Reactive oxygen species promote trigeminal system nociceptive activation via TRPA1 channel activation (Shatillo et al., 2013). Increased TRPA1 signaling promotes spreading depression, suggesting that TRPA1 channel blockade may be a potential target for improved migraine treatment due to an ability to reduce oxidative stress (Jiang et al., 2019). Most existing prophylactic migraine agents show some degree of anti-oxidant effect (Borkum, 2016) providing further support for targeting oxidative stress in migraine treatment. However, while TRPA1 channels appear to be a promising target for therapeutics development, considerable hurdles remain (Skerratt, 2017). In contrast, intranasal IGF-1 showed blockade of oxidative stress, like that hoped for with TRPA1 channel blockade, including an 83% reduction in trigeminal ganglion malondialdehyde levels after recurrent spreading depression.
3.5. Conclusion
Increased production of IGF-1 is a naturally occurring adaptive response to environmental enrichment in animals, which is protective against preclinical modeled migraine. As a result, it may have a high benefit/risk ratio as a therapeutic for brain, especially when delivered phasically, consistent with the basic tenet of environmental enrichment-based science [i.e., physiological stress followed by an adaptation period, cycled repeatedly (Kraig et al., 2010)] if it can be translated to the human condition. IGF-1 seems particularly well-suited for intranasal delivery for migraine because of the likely dominant entry pathway via the trigeminal pathway to the brain. As a result, intranasal IGF-1 delivery not only reduces brain susceptibility to preclinical migraine hyperexcitability (inferred from reduced susceptibility to spreading depression) but also surrogates of trigeminal system hyper-activity (Chen et al., 2017) [i.e., trigeminal ganglion oxidative stress (malondialdehyde) and CGRP as well as trigeminocervical c-fos]. Perhaps intranasal IGF-1 will mitigate the depletion of antioxidants seen in migraine that leads to worsening of this disorder (Yilmaz et al., 2007; Tuncel et al., 2008; Aytac et al., 2014; Yigit et al., 2018).
Experimental Procedures
4.1. Animals
Adult (250–450 g) male Wistar rats (n = 90 total) (Charles River Laboratories, Wilmington, MA) were used in this study and initially housed two/cage until after surgery when animals were housed with one animal/cage.
Housing included use of static micro isolator cages with corn cob bedding, Enviro-dri nesting material (Shepard Specialty Papers, Watertown, TN) and nestlets (Ancare Corporation, Bellmore, NY) for enrichment. Rats were maintained in a 12-hour light-dark cycle with controlled humidity and temperature in our Central Animal Facility. Rats had free access to food and water throughout experiments and were observed daily for evidence of normal feeding, grooming and ambulatory activity. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Chicago, were conducted in accordance with the Guidelines of the National Institutes of Health Guide for Care and Use of Laboratory Animals (2011) and were patterned after ARRIVE guidelines.
Animals were removed from the central facility for intranasal treatment and surgical procedures as described below in the investigators’ nearby lab. Once awake after treatment and initial surgery (see below), they were returned to the central animal facility. The next day, animals were again brought to the investigators’ lab for induction of spreading depression followed by harvesting of brains. Experiments were performed during the mid-portion of the light cycle. Rats were randomly divided into control and experimental groups and sample sizes were based on our previous published studies that show a significant change and Power of always greater than 0.8 and most often equal to 1.00. All treatments and data analyses were completed under blinded conditions. Routine chemicals were purchased from Sigma or ThermoFisher if not otherwise specified below.
4.2. Intranasal treatment
Intranasal delivery of 150 μg human recombinant IGF-1 [(hIGF-1), #191-G1; R&D Systems, Minneapolis, MN] in 50 μl sodium succinate buffer followed our previously described approach (Grinberg et al., 2017). Briefly, animals were anesthetized with inhalational 5% isoflurane (Butler Schein Animal Health, Dublin, Ohio) in oxygen with spontaneous respirations in an anesthesia box. Once anesthetized, animals were placed in the recumbent position on a Plexiglas plate fitted with a Kopf rat nose cone (model 906; Tujunga, CA). There, a 10 μl pipettor was used to deliver 5 μl of hIGF-1 [or vehicle (succinate buffer)] every two minutes to alternating nostrils for a total of 50 μl. Arterial oxygen tension was continuously monitored with a cutaneous oximeter (2500A VET; Nonin Medical, Plymouth, MN) and ranged from 95–100 mm Hg here and in all experiments. Animals were maintained at 37 ± 0.5 °C using an overhead infra-red heating lamp coupled to a rectal thermometer and related temperature regulation unit (TC5500; Digi-Sense, Vernon Hills, IL). Rats were observed daily after treatments. No adverse effects (i.e., altered feeding, grooming or ambulatory behavior) were seen from intranasal treatments in this work or our prior experiments that involved treatment with IGF-1 for up to two weeks (Grinberg et al., 2017).
4.3. Blood glucose analyses
Blood glucose was monitored in a subset of animals that were not used for other analyses. Immediately before intranasal treatments and while anesthetized, blood glucose was sampled at time zero using a sterile, 27 gauge hypodermic needle to puncture the tip of the tail. A second drop of blood from punctures was used for glucose measurements (Contour Next EZ; Ascensia Diabetes Care, US, Inc.; Parsippany, NJ). After intranasal treatments (hIGF-1 or vehicle), animals were briefly re-anesthetized and maintained as above for each blood draw done over time (i.e., 0.3, 3, 6, and 24 hours).
4.4. Spreading depression
For determination of whether recurrent spreading depression (versus sham) triggered increased trigeminal ganglion activation (i.e., malondialdehyde and CGRP levels) as well as previously demonstrated trigeminocervical c-fos activation (Moskowitz et al., 1993) animals were prepared as described below for initial surgery which was followed the next day by recurrent spreading depression for 90 minutes. For sham controls, 0.5M NaCl was injected instead of 0.5M KCl.
Induction of recurrent spreading depression followed previously published aseptic techniques from our laboratory with modifications (Grinberg et al., 2017; Kraig et al., 1991; Kunkler and Kraig, 2003; Moskowitz et al., 1993; Pusic et al., 2014). These prior reports include the rationale for using inhalational isoflurane for study of spreading depression in rats (Pusic et al., 2014). Animals were anesthetized with 5% inhalational isoflurane in oxygen using an anesthesia box. Once anesthetized, animals were transferred to a table top stereotaxic unit with animals held in place via blunt, soft-tipped ear bars and a custom-fitted nose cone with soft nose bar for delivery of isoflurane (3% during surgery). Anesthetic gas was removed by nose cone vacuum to prevent room contamination. At this point eyes were treated with Artificial Tears™ (#17478–062-35; Akorn, Lake Forest, IL). The animal head was then shaved with electric clippers (ChroMini®; Wahl UK, LTD, Heme, Kent, England) and cleansed with Betadine Solution (Purdue Products, L.P.; Stamford, CT). Next, 0.05 ml of 0.25% Bupivacaine (Hospira, Inc., Lake Forest, IL) was injected subcutaneously to either side of what would become a small left of midline incision to expose the underlying skull.
After five minutes, a left of midline incision ~ 1.5 cm was created and the underlying bone was scraped free of connective tissue. Skull hemostasis was achieved using Bone Wax (#CPB31A; CP Medical Inc., Portland, OR). Next, small < 2 mm diameter craniotomies were drilled with saline cooling using a Dremel tool and #106 engraving cutter bit (Dremel, Racine WI) at 3 mm anterior to bregma and 2 mm lateral to the midline (for subsequent KCl injections to induce spreading depression) and 6 mm caudal to bregma and 4.5 mm lateral to the midline (for subsequent spreading depression recordings). The incision area was cleansed with Betadine Solution and the incision was closed with Autoclips® (3427631; Clay Adams, Becton Dickinson Primary Care Diagnostics, Becton Dickinson and Company, Sparks, MD). The closed head wound was wiped with Betadine Solution before returning animals to their individual cages within the Central Animal Facility.
One day after intranasal treatment and surgery or simply surgery for KCl versus NaCl intracerebral nano-injections, animals were again anesthetized with 5% isoflurane in oxygen followed by subcutaneous Bupivacaine (0.25%) injection (0.05 ml to either side of the head incision site. Five minutes later, the wound clips were removed, skin spread open and the surgical area wiped with Betadine Solution. Animals were then transferred to a modified stereotaxic unit with nose cone delivery of isoflurane (which was progressively reduced to 1.5% with released anesthetic removed from the lab by vacuum placed in the nose cone). Eyes were coated with Artificial Tears™ to reduce potential nociceptive activation from anesthesia air current. To further reduce potential facial nociceptive activation, ear bars were replaced with clamps made from one cm wide 5 ml syringe tube walls cut to form somewhat less than a half circle. The inside curve of these pieces was fitted with soft foam rubber for placement adjacent to rat head at the level of the ears with the outside of the curved pieces attached perpendicularly to an ear bar rod that fits into the traditional stereotaxic ear bar holders. Similarly, the nose clamp was fitted with a piece of foam rubber to reduce potential nociceptive activation of trigeminal afferents from restraint. Animal temperature was regulated to 37 ± 0.5 °C via a heater water bath and circulated warm water (#NO-1199900328/Q20; Pharmacia Biotech, Gebrüder, Germany) via copper tubing placed beneath the animal. Temperature was monitored with a rectal thermometer probe and thermometer (Digi-Sense). Arterial oxygen tension was continuously monitored with a pulse oximeter as described above. Adequacy and consistency of anesthesia in spontaneously breathing animals was monitored by noting: 1) arterial oxygen tension that was kept between 95100 mm Hg; 2) pink skin color in appendages; 3) regular respirations; and 4) absence of withdrawal to hind paw pinch [Guide for the Care and Use of Laboratory Animals (2011); (Tremoleda et al.,2012)]. As previously noted, we recognize that volatile anesthesia in spontaneously breathing animals will lead to a modest rise in arterial carbon dioxide tension which could inhibit initiation of spreading depression (Grinberg et al. 2017; Kraig et al., 1991). However, this hypercarbia would create a systematic confound that would be equally applied to all animals/groups.
For spreading depression induction, a 1.2 mm diameter glass tube with an 8 μm tip diameter (#6010; A-M Corporation, Everett WA) was filled with 0.5 M KCl and attached to a PicoSpritzer-II electronic valve system holder (Parker Hannifin, Hollis, NH) with the tip positioned 750 μm below the pial surface at the anterior craniotomy site using an M1 micromanipulator (Narishige, International, Amytiville, NY). KCl injections (every 9–10 minutes over 90 minute period) were completed with 20 pounds of pressure for a period of time needed to reliably eject 10–30 nl of KCl, volumes that approximated the amount of KCl needed to trigger spreading depression a day after IGF-1 treatment in our previous study (Grinberg et al., 2017). At the conclusion of experiments KCl micro-injection volume was confirmed by ejecting a bolus into 3In-One™ light machine oil (WD-40 Company, San Diego, CA) contained in the depression of a depression well microscope slide (#48333–002; VWR Scientific Products, Buffalo Grove, IL). The diameter of the injectate was then measured with a compound microscope to estimate the volume injected into brain. We recognize that injection volume in oil may not exactly reflect amount released in vivo (Nicholson, 2001). However, any variances would be a systematic error and therefore have no impact on our relative estimates here. A remote reference electrode made from a blunted tip Pasteur pipette filled with 150 mM saline and 3.5% agar was placed on left temporalis muscle.
For recording spreading depression, 4 μm tip diameter micropipettes were fashioned for 1.2 mm diameter glass tubes (#6030; A-M) and driven 750 μm below the pial surface at the caudal craniotomy site using a Canberra micromanipulator (Narishige). All electrode placements were performed under microscopic observation (Wild M-8 stereomicroscope; Leica Micro Systems, Inc. Buffalo Grove, IL) with standard LED flashlight illumination. The craniotomy surface was warmed (37 ± 0.5 °C) with normal saline flowing at ~ 2 ml/min via an in-line heater and controller (#SH27B; TC-344 C; Warner Instruments, Hamden CT).
Electrophysiological signals to confirm the large defining interstitial direct current negative shift of spreading depression were measured with an A1 Axoprobe amplifier system (Axon Instruments, Burlington, CA) and digitally stored via an analog-digital converter (1440A, Axon) board as well as the time point of KCl injections. This digitized data was stored as permanent files within and external to the lab in a secure facility.
4.5. Immunostaining
At the conclusion of spreading depression, inhalational isoflurane was raised to 5% for a few minutes and then animals were more deeply anesthetized by intraperitoneal injection of ketamine/Xylazine into the lower right abdominal quadrant. Rats were euthanized by cardiac perfusion fixation by first injecting with 0.1 mL heparin (10,000 U/mL; Sagent Pharmaceuticals, Schaumburg, IL) followed by 250 ml of 150 mM sodium chloride and then by 250 ml 4% paraformaldehyde in 0.1M phosphate buffered saline [(PBS), pH 7.2] with both flowing under 120 mm Hg regulated pressure. Afterward, brains plus attached cervical spinal cord as well as trigeminal ganglia were removed and post-fixed for 24 hours followed by immersion in 20% sucrose in 0.1 M PBS at 4 °C. Days later, neural specimens were frozen (−30 °C) in 2-methyl butane (#O3551–4; ThermoFisher) and stored at −80 °C in sealed containers.
The ipsilateral (left) to neocortical spreading depression trigeminal ganglion of each animal was cut into 20 μm thick, consecutive, longitudinal sections using a cryostat (#3050S; Leica, Buffalo Grove, IL) and mounted as one section per gelatin coated slide. Sections containing the V1 (ophthalmic) division of the trigeminal ganglion were identified and three sections, at least 40 μm apart, from each animal were selected for malondialdehyde, a marker of oxidative stress (Shatillo et al., 2013), and CGRP immunohistochemistry.
Sections for malondialdehyde immunohistochemistry of the trigeminal ganglion were blocked in 10% normal donkey serum containing 0.3% Triton X-100 (diluent buffer) for 1 hour at room temperature and then incubated overnight in primary antibody (Table 1) in diluent buffer at 4°C. Sections were rinsed with PBS and incubated in 2o antibody (Table 1) for 1 hour at room temperature.
Table 1.
Description of antibodies used for immunohistochemistry
| Name | Catalog# (RRID#) |
Host | Dilution | Detects | Supplier | |
|---|---|---|---|---|---|---|
| 1° Ab | c-fos | ABE457 (AB_2631318) |
rabbit | 1/10,000 | N-terminus of c-fos | Millipore, Temecula, CA |
| CGRP | C8198 (AB_259091) |
rabbit | 1/1,000 | rat CGRP | Sigma, St. Louis, MO |
|
| MDA | ab6463 (AB_305484) |
rabbit | 1/500 | rat MDA | AbCam, Cambridge, MA |
|
| IGF-1Rβ | 3027 (AB_2122378) |
rabbit | 1/100 | C-terminus of IGF-1Rβ |
Cell Signaling Technology, Danvers, MA |
|
| Name | Catalog# | Against | Dilution | Supplier | ||
| 2° Ab | Alexa 594 (goat) |
ab150084 | rabbit | 1/500 (CGRP) 1/700 (c-fos, MDA) |
AbCam, Cambridge, MA |
|
For CGRP and IGF-1 receptor beta (IGF-1Rβ immunostaining of the trigeminal ganglion, sections were pre-treated with cold acetone (−20 °C) for 10 minutes, rinsed with PBS and then incubated in PBS containing 0.1% Triton X-100 (PBS-T) for 15 minutes. The sections were blocked in 5% normal goat serum/PBS for 1 hour at room temperature before overnight incubation at 4°C in rabbit polyclonal antibody (Table 1) diluted in PBS-T containing 1% bovine serum albumin and 3% normal goat serum. Sections were then incubated in 2° antibody (Table 1) diluted in PBS-T containing 1% bovine serum albumin for 1 hour at room temperature.
For c-fos immunostaining of the trigeminocervical complex, the brainstem/cervical spinal cord was cut into 40 μm thick coronal sections using a cryostat. Laterality was controlled for by marking the right, contralateral (to spreading depression side) ventral aspect of the brainstem/cervical spinal cord. Six consecutive sections were collected every 1.5 mm from the obex (0 mm) caudally through cervical spinal cord (i.e., −1.5 mm, −3.0 mm, −4.5 mm, −6.0 mm) as previously described (Kunkler and Kraig, 2003; Moskowitz et al., 1993). For each animal, three sections were randomly selected from each brainstem/spinal cord level and processed for c-fos expression. c-fos immunohistochemistry was performed on free floating sections by incubating in 10% normal donkey serum containing 0.3% Triton X-100 for 1 hour at room temperature and then overnight in rabbit anti-c-fos (Table 1) in diluent buffer at 4°C. Sections were rinsed with PBS and incubated in 2° antibody (Table 1) for 1 hour at room temperature. The sections were rinsed with PBS and mounted on gelatin coated slides.
Slides containing immunostained sections were coverslipped with Prolong™ Gold Antifade mounting media (#P36930; Life Technologies, Eugene, OR). The specificity of the immunolabeling was verified by omission of the primary antibody, using only secondary antibody staining.
Characterization of the antibodies used in this study is provided as follows. Preabsorption of the rabbit anti-CGRP antibody with 10 μM rat CGRP abolished specific tissue staining (manufacturer’s data sheet). In addition, this antibody does not cross react with substance P, VIP, NPY, calcitonin or somatostatin. The specificity of the rabbit anti-IGF-1Rβ antibody has been demonstrated in IGF-1Rβ knockout mice (Epaud et al., 2012). The IGF-1Rβ antibody has been validated by the manufacturer via western blot and does not cross react with the insulin receptor. The c-fos antibody used was evaluated by western blot by the manufacturer and showed an appropriate molecular weight (~60/56 kDa). The malondialdehyde antibody used was tested by ELISA by the manufacturer.
4.6. Computer-based digital image quantification
We used computer-based and blinded semi-quantitative digital quantification of immunostaining metrics to test the ability of intranasal IGF-1 to impact trigeminocervical complex activation. To help reduce inter-experiment variability, immunostaining was performed on paired samples (e.g., sham and experimental) sections for all markers (malondialdehyde, CGRP and c-fos).
Imaging was performed using a Photometrics self-calibrating digital camera QuantEM512SC camera (500 × 500 pixels; Photometrics, Tucson, AZ) run on MetaMorph software (v. 7.0.4; Molecular Devices, Sunnyvale, CA) under blinded conditions. Power for both the camera/computer system and ultraviolet light illumination was stabilized for uniform behavior using regulated power supplies (APC-Pro1500; Schneider Electric, West Kingston, RI). The acquisition system also used an electronic shutter (Sutter Lambda SC; Novato, CA), Prior Lumen 200 watt Xeon lamp; Rockford, MA) running on an DMRE2 inverted microscope (Leica) at 20× gain for malondialdehyde and CGRP images and 5× gain for c-fos images. Image exposure, camera gain and thresholding were uniformly set [i.e., equivalently for experimental and sham groups for each imaging metric (i.e., malondialdehyde, CGRP and c-fos)]. Resultant images were digitally stored as TIFF files for subsequent analyses that continued under blinded conditions where image integrated fluorescence intensity was registered using MetaMorph software for malondialdehyde and CGRP.
For c-fos image analyses, the 5x digitally stored TIFF files were electronically enlarged to facilitate cell counting to 6,400 × 6,400 pixels and adjusted for brightness/contrast equivalently between experimental group pairs using Image J (v. 1.43i; National Institutes of Health public access). c-fos labeled nuclei were manually counted in paired samples by an observer blinded to experimental conditions. Manual cell counts were tabulated using the cell counter function in ImageJ. Only intensely stained, round or oval-shaped nuclei were counted in laminae I-II [obex through −6.0 mm, recording the average of n = 3 sections (technical replicates) at each anatomical level per animal (n ≥ 5 biological replicates/group)].
4.7. CGRP plasma assays
Blood was collected via cardiac puncture using a 21 gauge syringe needle and transferred into a BD Vacutainer K3 EDTA blood collection tube (366450, Becton, Dickinson & Co., Franklin Lakes, NJ). Samples were spun 1,500 g × 10 min at 4°C. The plasma was transferred to sterile 2.0 ml, screw cap, polypropylene tubes and stored at −80°C until analysis. Plasma samples were sent to Cayman Chemical Company (Ann Arbor, Michigan) for analysis of CGRP content using ELISA methods. According to the vendor, the detection limit is >10 pg/ml.
4.8. Statistical methods
We used a verified method of quantifying immunostaining log-ratios (experimental / sham) as a further means to reduce potential run-to-run variations in immunostaining for malondialdehyde, CGRP and c-fos positive cells (Kraig et al., 1991).
Images of malondialdehyde and CGRP were analyzed in blinded pairs so that they could be converted once decoded to ratios of experimental/sham image integrated optical intensity, a sensitive metric that not only accounts for the area but also the pixel intensity of related image fluorescence. Immunostaining ratios of 1 indicated no difference between experimental and sham conditions, while a ratio of less than 1 indicates that experimental treatment reduced immunostaining compared to sham. These ratios were converted to natural logarithms whereby 0 corresponded to no difference between experimental and sham conditions, and a t-test (two-tailed) could be used to determine if differences of logarithms varied significantly from 0. In the text we report p-values and power of related log ratio statistical testing (Kraig et al., 1991). In addition, a more general listing of percent changes in intensity ratios is shown.
c-fos positive cell quantifications followed a similar pattern to that used for malondialdehyde and CGRP immunostaining. Specifically, c-fos positive cell counts were converted to a ratio (e.g., IGF-1/sham) and the natural logarithm of the results quantified statistically by comparison to “zero” (i.e., no difference ratio of 1.00 or Ln =0. Since maximal trigeminocervical complex changes in the superficial laminae are evident at −4.5 mm caudal to the obex, we report quantitative measurements at this level.
All image pairs (experimental and sham) were adjusted equally using Adobe Photoshop, which along with CorelDraw was used for final figure construction. Digital electronic files of spreading depression recordings were processed first in Origin (2019; Microcal, Northhampton, MA) and placed in final form using CorelDraw and Photoshop. Data were analyzed using SigmaPlot software (v. 12.5; Systat Software, Inc. San Jose, CA). All data passed normality testing (p-value to reject: 0.05) and equal variance testing (p-value to reject: 0.05) and power (1β: > 0.8). Animal groups consisted of ≥ 5 biological replicates.
Differences in blood glucose values from intranasal vehicle or IGF-1 at various post-treatment times versus zero-time were determined using one way ANOVA plus post hoc Holm-Sidak test for multiple comparisons.
Article Highlights.
Nasal IGF-1 stops spreading depression, a migraine model by reducing oxidative stress
Nasal IGF-1 similarly inhibits trigeminal system activation from spreading depression
Trigeminal ganglion oxidative stress and calcitonin gene-related peptide were reduced
Trigeminocervical c-fos activation was reduced
Thus, nasal IGF-1 warrants further development as a novel therapeutic for migraine
Acknowledgements
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NS-019108 and R43-NS108824–01); the Innovation Fund from the Polsky Center for Entrepreneurship and Innovation at the University of Chicago; by the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR000430); and a Sponsored Research Agreement from Seurat Therapeutics, Inc.
We thank Drs. Aya D. Pusic, Kae M. Pusic and Aaron Fox for reading and commenting on the manuscript.
Abbreviations:
- CGRP
calcitonin gene-related peptide
- IGF-1
insulin-like growth factor-1
- PBS
phosphate buffered saline
- PBS-T
phosphate buffered saline-triton
- IGF-1Rβ
insulin-like growth factor-1 receptor beta
Footnotes
Disclosures
Richard P. Kraig is a co-inventor (along with others) on issued and pending patent applications dealing with “Treatments for migraine and related disorders”.
Richard P. Kraig is also a co-founder and the Chief Scientific Officer of Seurat Therapeutics, Inc., a company formed to develop IGF-1 as a novel treatment for migraine.
Dr. Lisa Won has no conflict.
All aspects of this study passed review by the Provost’s Office of the University of Chicago which included assurances on an external oversight monitor for the work and blinding of all experiments.
Drs. Lisa Won and Richard Kraig participated equally in the design, execution and interpretation of all data as well as writing the final manuscript.
Credit author statement
Drs. Lisa Won and Richard Kraig participated equally in the design, execution and interpretation of all data as well as writing the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aytac B, et al. , 2014. Decreased antioxidant status in migraine patients with brain white matter hyperintensities. Neurol. Sci. 35,1925–1929. [DOI] [PubMed] [Google Scholar]
- Bitar MS, et al. , 1996. Antinociceptive action of intrathecally administered IGF-I and the expression of its receptor in rat spinal cord. Brain Res. 737, 292–4. [DOI] [PubMed] [Google Scholar]
- Borkum JM, 2016. Migraine triggers and oxidative stress: A narrative review and synthesis. Headache 56, 12–35. [DOI] [PubMed] [Google Scholar]
- Chen SP et al. , 2017. Inhibition of the P2X7-PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain 140, 1643–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciucci F, et al. , 2007. Insulin-like growth factor 1 (IGF-1) mediates the effects of enriched environment (EE) on visual cortical development. PloS one 2, e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Logu F, et al. , 2019. Migraine-provoking substances evoke periorbital allodynia in mice. J. Headache Pain 20, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterle A, et al. , 2011. Increase in CGRP- and nNOS-immunoreactive neurons in the rat trigeminal ganglion after infusion of an NO donor. Cephalalgia 31, 31–42. [DOI] [PubMed] [Google Scholar]
- Dodick DW, 2018. Migraine. Lancet 39, 1315–1330. [DOI] [PubMed] [Google Scholar]
- Djupesland PG, et al. , 2014. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther. Deliv. 5, 709–33. [DOI] [PubMed] [Google Scholar]
- Durham PL, 2006. Calcitonin gene-related peptide (CGRP) and migraine. Headache 46 (Suppl 1), S3–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, et al. , 2018. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neurol. 14, 338–350. [DOI] [PubMed] [Google Scholar]
- Epaud R, et al. , 2012. Knockout of insulin-like growth factor-1 receptor impairs distal lung morphogenesis. PloS one 7, e48071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiz A, et al. , 2019. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. Cephalalgia 39, 354–365. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, et al. , 2017. Pathophysiology of migraine: A disorder of sensory processing. Physiol. Rev. 97, 553–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco MC, et al. , 2016. Lacosamide inhibits calcitonin gene-related peptide production and release at trigeminal level in the rat. Eur. J. Pain 20, 959–66. [DOI] [PubMed] [Google Scholar]
- Grinberg YY, et al. , 2012. Insulin-like growth factor-1 lowers spreading depression susceptibility and reduces oxidative stress. J. Neurochem. 122, 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg YY, et al. , 2013. Insulin-like growth factor-1 abrogates microglial oxidative stress and TNF-alpha responses to spreading depression. J. Neurochem. 126, 662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg YY, et al. , 2017. Intranasally administered IGF-1 inhibits spreading depression in vivo. Brain Res. 1677, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, et al. , 2011. Calcitonin gene-related peptide does not cause migraine attacks in patients with familial hemiplegic migraine. Headache 51, 544–53. [DOI] [PubMed] [Google Scholar]
- Ipsen Biopharmaceuticals I, 2016. INCRELEX. http://www.increlex.com/pdf/patient-fullprescribing-information.pdf.
- Irby MB, et al. , 2016. Aerobic exercise for reducing migraine burden: mechanisms, markers, and models of change processes. Headache 56, 357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar S, et al. , 2019. CGRP and the Trigeminal System in Migraine. Headache 59, 659–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, et al. , 2019. ROS/TRPA1/CGRP signaling mediates cortical spreading depression. J. Headache Pain 20, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn DF, Clifford CB, 2002. Biology and diseases of rats: in, Fox JG, Anderson LC, Lowe FM, Quimby F (Eds.), Laboratory Animal Medicine. Academic Press, New York, pp. 121–167. [Google Scholar]
- Kraig RP, et al. , 1991. Spreading depression increases immunohistochemical staining of glial fibrillary acidic protein. J. Neurosci. 11, 2187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraig RP, et al. , 2010. TNF-alpha and microglial hormetic involvement in neurological health & migraine. Dose-response 8, 389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, Kraig RP, 2003. Hippocampal spreading depression bilaterally activates the caudal trigeminal nucleus in rodents. Hippocampus 13, 835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, et al. , 2005. Optical current source density analysis in hippocampal organotypic culture shows that spreading depression occurs with uniquely reversing currents. J. Neurosci. 25, 3952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen LH, et al. , 2002. CGRP may play a causative role in migraine. Cephalalgia 22, 54–61. [DOI] [PubMed] [Google Scholar]
- Lemmens J, et al. , 2019. The effect of aerobic exercise on the number of migraine days, duration and pain intensity in migraine: a systematic literature review and meta-analysis. J. Headache Pain 20, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. , 2016. Valproate ameliorates nitroglycerin-induced migraine in trigeminal nucleus caudalis in rats through inhibition of NF-κB. J. Headache Pain. 16,17:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, et al. , 2015. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Cereb. Blood Flow Metab. 35, 371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, et al. , 2009. Neuronal mitochondrial toxicity of malondialdehyde: inhibitory effects on respiratory function and enzyme activities in rat brain mitochondria. Neurochem. Res. 34, 786–94. [DOI] [PubMed] [Google Scholar]
- Mason BN, et al. , 2017. Induction of migraine-like photophobic behavior in mice by both peripheral and central CGRP mechanisms. J. Neurosci. 37, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Carrillo A, et al. , 2017. Selective inhibition of trigeminovascular neurons by fremanezumab: A humanized monoclonal anti-CGRP antibody. J. Neurosci. 37, 7149–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, et al. , 2012. CGRP and NO in the trigeminal system: mechanisms and role in headache generation. Headache 52, 1411–27. [DOI] [PubMed] [Google Scholar]
- Mitsikostas DD, Reuter U, 2017. Calcitonin gene-related peptide monoclonal antibodies for migraine prevention: comparisons across randomized controlled studies. Curr. Opin. Neurol. 30, 272–280. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, et al. , 1993. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J. Neurosci. 13, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C 2001. Diffusion and related transport mechanisms in brain tissue. Rep. Prog. Phys. 64, 815–884. [Google Scholar]
- Nishijima T, et al. , 2010. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 67, 834–46. [DOI] [PubMed] [Google Scholar]
- Petersen KA, et al. , 2005. BIBN4096BS antagonizes human alpha-calcitonin gene related peptide-induced headache and extracerebral artery dilatation. Clin. Pharmacol. Ther. 77, 202–13. [DOI] [PubMed] [Google Scholar]
- Pusic KM, et al. , 2014. Spreading depression requires microglia and is decreased by their M2a polarization from environmental enrichment. Glia 62, 1176–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddant AC, Russo AF, 2014. Reactive oxygen species induce procalcitonin expression in trigeminal ganglia glia. Headache. 54, 472–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, et al. , 2014. Nitric oxide synthase, calcitonin gene-related peptide and NK-1 receptor mechanisms are involved in GTN-induced neuronal activation. Cephalalgia 34, 136–47. [DOI] [PubMed] [Google Scholar]
- Schwartz JH, 2000. Neurotransmitters: in Kandel RR, Schwartz JHG, Jessell TM (Eds.), Principles of Neural Science. McGraw Hill, New York, pp. 280–297. [Google Scholar]
- Shatillo A, et al. , 2013. Cortical spreading depression induces oxidative stress in the trigeminal nociceptive system. Neuroscience 253, 341–9. [DOI] [PubMed] [Google Scholar]
- Sixt ML, et al. , 2009. Calcitonin gene-related peptide receptor antagonist olcegepant acts in the spinal trigeminal nucleus. Brain 132, 3134–41. [DOI] [PubMed] [Google Scholar]
- Skerratt S, 2017. Recent progress in the discovery and development of TRPA1 modulators. Prog. Med. Chem. 56, 81–115. [DOI] [PubMed] [Google Scholar]
- Thorne RG, et al. , 2004. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127, 481–96. [DOI] [PubMed] [Google Scholar]
- Tremoleda JL, et al. ,. 2012. Anaesthesia and physiological monitoring during in vivo imaging of laboratory rodents: considerations on experimental outcomes and animal welfare. EJNMMI Res. 2, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncel D, et al. , 2008. Oxidative stress in migraine with and without aura. Biol. Trace Elem. Res. 126, 92–7. [DOI] [PubMed] [Google Scholar]
- Vigano A, et al. , 2019. Treating chronic migraine with neuromodulation: The role of neurophysiological abnormalitiesand maladaptive plasticity. Front. Pharmacol. doi: 10.3389/fpha.2019.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. , 2014. Insulin-like growth factor-1 receptor-mediated inhibition of A-type K(+) current induces sensory neuronal hyperexcitability through the phosphatidylinositol 3kinase and extracellular signal-regulated kinase 1/2 pathways, independently of Akt. Endocrinology 155, 168–79. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. , 2016. Induction of calcitonin gene-related peptide expression in rats by cortical spreading depression. Cephalalgia. 39, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won L, Kraig RP, 2019a. Development of nasal insulin-like growth factor-1 as a novel treatment for migraine. Am. Headache Soc. 59 (Supplement 1), P221LB. [Google Scholar]
- Won L, Kraig RP, 2019b. Nose-to-brain delivery of IGF-1 abrogates trigeminal system activation including oxidative stress and CGRP from recurrent spreading depression. Soc. Neurosci. 45, Abstract # 053.02. [Google Scholar]
- Yigit M et al. , 2018. Oxidative/antioxidative status, lymphocyte DNA damage, and urotensin-2 receptor level in patients with migraine attacks. Neuropsychiatr. Dis. Treat. 14, 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, et al. , 2007. Increased nitrosative and oxidative stress in platelets of migraine patients. Tohoku J Exp Med 211(1), 23–30 [DOI] [PubMed] [Google Scholar]
- Zhang X, et al. , 2011. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 69, 855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuurbier CJ, et al. , 2008. Anesthesia’s effects on plasma glucose and insulin and cardiac hexokinase at similar hemodynamics and without major surgical stress in fed rats. Anesth. Analg. 106, 135–42. [DOI] [PubMed] [Google Scholar]