

Abstract
In addition to being an amino acid that is incorporated into proteins, glutamate is the most abundant neurotransmitter in the mammalian CNS, the precursor for the inhibitory neurotransmitter γ-aminobutyric acid, and one metabolic step from the tricarboxylic acid cycle intermediate α-ketoglutarate. Extracellular glutamate is cleared by a family of Na+-dependent transporters. These transporters are variably expressed by all cell types in the nervous system, but the bulk of clearance is into astrocytes. GLT-1 and GLAST (also called EAAT2 and EAAT1) mediate this activity and are extremely abundant proteins with their expression enriched in fine astrocyte processes. In this review, we will focus on three topics related to these astrocytic glutamate transporters. First, these transporters co-transport three Na+ ions and a H+ with each molecule of glutamate and counter-transport one K+; they are also coupled to a Cl− conductance. The movement of Na+ is sufficient to cause profound astrocytic depolarization, and the movement of H+ is linked to astrocytic acidification. In addition, the movement of Na+ can trigger the activation of Na+ co-transporters (e.g. Na+ - Ca2+ exchangers). We will describe the ways in which these ionic movements have been linked as signals to brain function and/or metabolism. Second, these transporters co-compartmentalize with mitochondria, potentially providing a mechanism to supply glutamate to mitochondria as a source of fuel for the brain. We will provide an overview of the proteins involved, discuss the evidence that glutamate is oxidized, and then highlight some of the un-resolved issues related to glutamate oxidation. Finally, we will review evidence that ischemic insults (stroke or oxygen/glucose deprivation) cause changes in these astrocytic mitochondria and discuss the ways in which these changes have been linked to glutamate transport, glutamate transport-dependent signaling, and altered glutamate metabolism. We conclude with a broader summary of some of the unresolved issues.
Keywords: Glutamate transport, metabolism, mitochondria, astrocyte, sodium, calcium
Introduction
It is well-established that the amino acid glutamate is the predominant excitatory neurotransmitter in the mammalian CNS, while γ-aminobutyric acid (GABA) is the predominant inhibitory neurotransmitter in brain (1–3). There are several remarkable aspects of these neurotransmitters. First, the levels of glutamate and GABA are extraordinarily high. Glutamate levels are 5–10 mmol/Kg and the levels of GABA are approximately 1 mmol/Kg (4). If the brain were 100% water and glutamate were uniformly distributed, the levels of glutamate would be 10 mM. This is 1,000- to 10,000-fold higher than those of other important neurotransmitters, including all of the biogenic amines. It is worthwhile to note that the levels of glucose, the accepted predominant fuel for brain, are only ~1 mM in the brain (5). Second, glutamate is only one metabolic step from GABA. Third, both these neurotransmitters can be converted to fuel that can be oxidized in mitochondria. In fact, many of the enzymes that metabolize glutamate are found in mitochondria, and the enzyme that metabolizes GABA, GABA-transaminase, is a mitochondrial enzyme, converting GABA to succinyl semialdehyde while also converting α-ketoglutarate to glutamate. Finally, it is clear that an extracellular accumulation of glutamate is sufficient to cause neuronal cell death through a mechanism known as ‘excitotoxicity’ (6–10).
Although several studies have detected micromolar extracellular concentrations of glutamate using microdialysis probes (11, 12), analyses of baseline glutamate receptor activation put the number at closer to 25 nM (13–15). With total brain concentrations of 10 mmol/kg, this means intracellular concentrations of glutamate are ~250,000-fold higher than those in the extracellular space. This is an oversimplification because glutamate is packaged into vesicles by vesicular glutamate transporters and glutamate is likely quickly metabolized in astrocytes (16). There is still no evidence for extracellular metabolism of glutamate (2); therefore, all released glutamate needs to be cleared from the extracellular space by transporters. There are two families of these transporters, the first are Na+-symporters and the second exchange glutamate for cystine. These transporters have been the subject of several different reviews (17–27). We will focus on the family of Na+-dependent transporters. In brief, these transporters are called GLAST, GLT-1, EAAC1 (alternatively, these are called EAAT1, EAAT2, and EAAT3, respectively), EAAT4, and EAAT5. Some investigators use the two nomenclatures to selectively refer to rodent or human homologs, respectively, but this is not always the case. Both GLT-1 and GLAST are enriched in glial cells in the nervous system, but lower levels of GLT-1 are also found on neurons (28, 29). Interestingly, GLT-1 requires only 100-fold enrichment to obtain purified protein; this indicates that GLT-1 represents 1% of brain protein (30). The energetic cost of maintaining very low levels of extracellular glutamate is quite high, and this is accomplished by capturing the energy from moving three molecules of Na+ down its concentration gradient with each molecule of glutamate. In addition, the transport of glutamate is accompanied by the inward movement of one H+ and the counter-transport a K+ with each cycle (31).
Given the high level of transport activity, it should not be surprising that the transport of glutamate into astrocytes has significant implications for astrocytic ionic signaling and astrocytic metabolism (Fig. 1). These two topics will be first addressed below. This will be followed by a summary of relatively recent observations that suggest that glutamate transport triggers astrocytic mitochondrial fragmentation after hypoxia/ischemia.
Fig. 1.
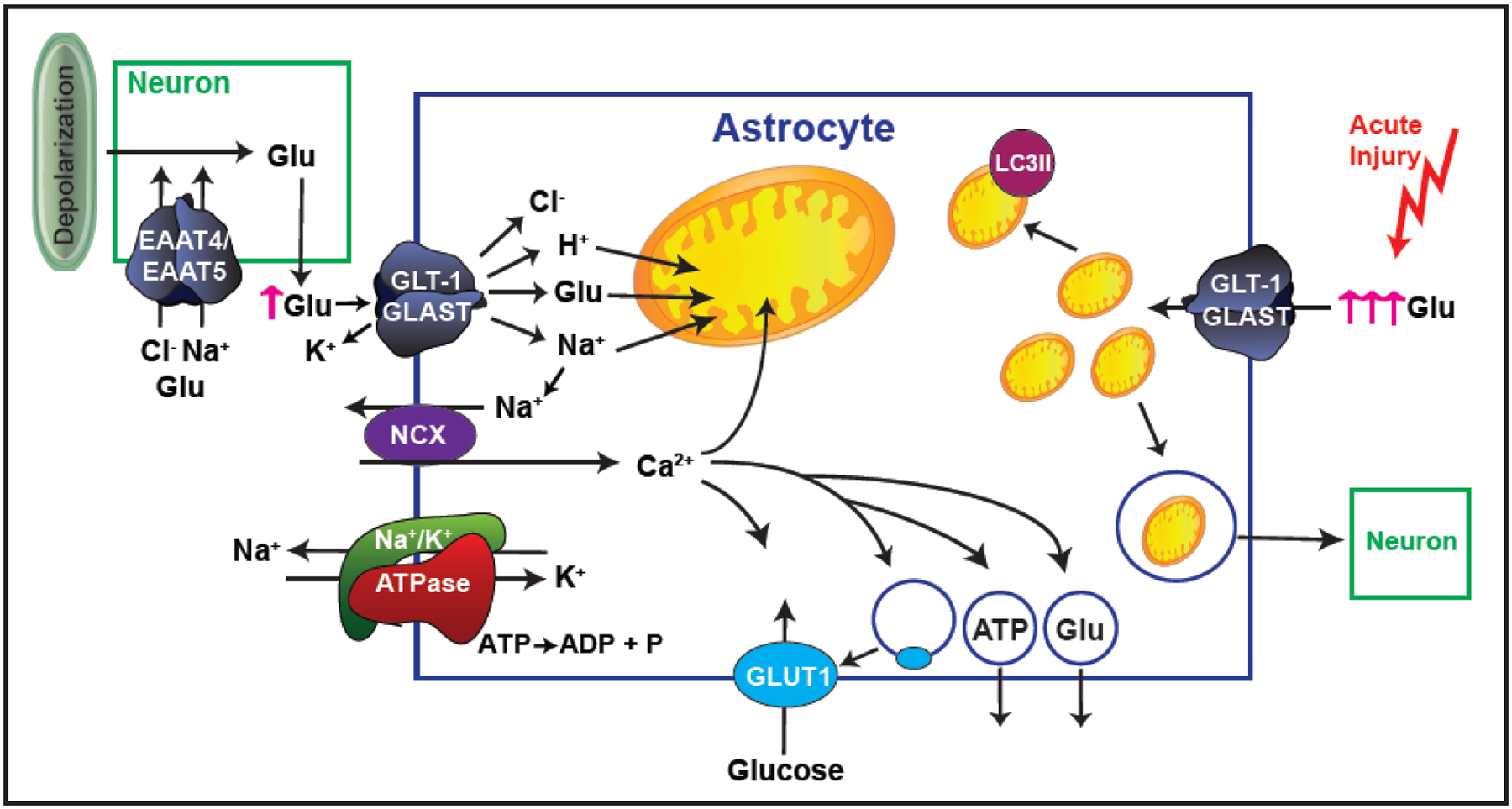
Schematic representation of the effects of glutamate uptake into astrocytes. Glutamate (Glu) uptake into astrocytes through Na+-dependent transporters is accompanied by an influx of 3 molecules of Na+ and one H+. The concentration gradient of Na+, the driving force behind Glu uptake, is established by the Na+/K+ ATPase. Increased intracellular Na+ causes reversed operation of the Na+/Ca2+ exchanger (NCX), resulting in increased intracellular Ca2+, which in turn can be taken up into mitochondria, increase glucose transport into the astrocyte, and/or cause vesicular release of ATP or gliotransmitters (including Glu) out of the astrocyte. These Glu transporters can also conduct Cl−, and this is especially relevant in the neuronal transporters where it can inhibit depolarization at the presynaptic nerve terminal. Upon acute injury such as ischemia, there is increased extracellular Glu and increased transport, which results in changes in astrocytic mitochondrial morphology and either degradation of the mitochondria through mitophagy or vesicular release for transfer to other cells, including neurons.
Glutamate transporters as signaling molecules
After release, glutamate diffuses out of the synapse and/or is cleared by the Na+-dependent transporters (for review, see 32). When one considers glutamate-mediated, inter-cellular signaling, one generally thinks of ligand-gated ion channels and G-protein coupled, so-called ‘metabotropic’ receptors (for reviews, see 33–39). In addition to limiting activation of these prototypic glutamate receptors, there is now substantial evidence that activation of transport, itself, causes downstream signaling. Transporter cycle times are relatively slow; even the fastest require between 10 and 75 msec to move a molecule of the glutamate across the plasma membrane (40, 41). However, the levels of transporters are high enough that the clearance of either exogenous or endogenous glutamate results in significant astrocytic depolarization (41–43). The membrane is repolarized by K+ channels and by the Na+/K+ ATPase (44), which has the stoichiometry of 3 Na+ (outward) and 2 K+ (inward) for each molecule of ATP hydrolyzed (45). This movement of Na+ and the depolarization both places a substantial energetic demand on the cell and contributes to signaling within and between astrocytes.
Exogenous glutamate or the transportable glutamate transporter substrate D-aspartate cause increases in intracellular Na+ in primary astrocyte cultures (46), as well as in astrocytes in acute hippocampal slices (47). Neuronal activity also causes an increase in intracellular Na+ in hippocampal slices. This effect is almost completely blocked by the non-transportable blocker of glutamate transporters threo-β-benzyloxyaspartic acid (TBOA). Together, these results indicate that neuronal release of endogenous pools of glutamate activate Na+ signals in astrocytes. These signals rapidly propagate in processes at speeds of up to 120μm/sec (48 for review, see 49), and the “spread” of the Na+ signal is proportional to the intensity of the stimulation (47). Interestingly, Ziemens et al. recently showed that the increases in Na+ in cortical astrocytes were approximately twice the size of those observed in hippocampal astrocytes. Furthermore, they showed that inhibition of glutamate transport activity only partially blocked (~50%) the signal in cortical astrocytes; the remaining 50% was blocked by an NMDA receptor antagonist (50). This suggests that some subtypes of astrocytes differentially engage different sources of glutamate-evoked Na+ signaling. Finally, the short-term plasticity of these transport-dependent Na+ currents are sensitive to the intracellular Na+ concentrations, suggesting that other mechanisms of Na+ transport could modulate glutamate transport and the accompanying Na+ influx (51).
While it is well-established that this influx of Na+ serves to drive the transport of glutamate against its concentration gradient, an energetically-demanding process, there is also evidence that these Na+ currents serve additional signaling purposes within the astrocyte, particularly in metabolic signaling. Bernardinelli et al. showed that glutamate transport-dependent increases in astrocytic Na+ are associated with increased Ca2+ and increased glucose uptake (52). They suggest a model in which increases in intracellular Ca2+ cause the release of ATP and glutamate, followed by glutamate uptake (along with Na+) and ATP-signaling, thereby increasing intracellular Ca2+ and propagating the signal from astrocyte to astrocyte in a so-called ‘metabolic wave.’ Porras et al. also observed a glutamate- or D-aspartate-dependent increase in astrocytic glucose transporter (GLUT1) activity. This effect is blocked by an inhibitor of glutamate transport and is mimicked by increasing both Na+ and Ca2+ simultaneously in astrocytes (53). Several groups have documented a glutamate transport-dependent increase in glycolysis resulting in release of lactate, both in astrocyte cultures and brain slice preparations (54–56). This is the so-called ‘astrocyte-neuron lactate shuttle’. The fact that this is not consistently observed (57–61) suggests that an uncontrolled variable regulates the extent to which glutamate transport couples to glycolysis. Glutamate uptake also triggers an increase in NADH/NAD+ in astrocytes (62). This effect is dependent upon activation of the Na+-HCO3−-cotransporter NBCe1.
While glutamate transporters are not directly coupled to the movement of Ca2+, the glutamate transport-dependent influx of Na+ activates the reverse operation of the Na+/Ca2+ exchanger (NCX), whereby extracellular Ca2+ moves into the astrocyte and Na+ is extruded (63, 64 for reviews, see 65, 66). Ibáñez and colleagues observed that the glutamate transport-dependent increase in Ca2+ is only partially (~50%) blocked by an inhibitor of reversed operation of NCX in astrocytes; the remainder is dependent upon other sources (67). This implies that glutamate transport is also indirectly linked to activation of other pathways that increase astrocytic Ca2+; these are not well defined at this time. This group also showed that the inhibitor of reversed operation of NCX blocked activity-dependent internalization of GLT-1. In addition to regulation of glutamate transport, it is clear that Ca2+ plays several additional roles in astrocyte signaling. The glutamate transport-induced increase in Ca2+ through reversed operation of NCX also contributes to the maturation of oligodendrocytes (68). Ca2+ signals have been shown to trigger release of gliotransmitters, including glutamate, which can in turn cause an increase in Ca2+ levels in neurons (Fig. 1) (69, 70); these astrocytic Ca2+ signals have also been linked to modulation of synaptic activity (71). As previously mentioned, Ca2+ oscillations have been linked to release of ATP from astrocytes, leading to propagation of the Ca2+ waves by inducing Ca2+ release from the endoplasmic reticulum (72). Finally, astrocytes serve as the intermediary between neurons and blood vessels in neurovascular coupling, and astrocytic Ca2+ contributes to this process (73), for reviews, see 74), 75).
The ionic signals discussed above are not limited to the cytosol of astrocytes. In fact, there is evidence that these signals have effects on mitochondria. As will be discussed in the next section of this review, there is a close physical association between glutamate transporters, the Na+/K+ ATPase (which is responsible for establishing the Na+ gradient necessary for glutamate transport), and mitochondria (76, 77). Bernardinelli et al. found that Na+ signals propagate to mitochondria in a Ca2+-dependent manner (78). The same group later observed spontaneous Na+ spiking activity in mitochondria that was actually decreased by glutamate, unaffected by TBOA, and positively correlated with increases in ATP (79). We showed that glutamate transport and the reversed operation of NCX decrease mitochondrial mobility (80). These changes in ionic concentrations almost certainly also regulate mitochondrial metabolism, but this is still relatively unexplored.
Finally, several members of the EAAT family also function as glutamate-activated Cl− channels. In fact, the cycle times for one of the neuronal transporters is so slow that it seems unlikely that these transporters contribute much to the clearance of glutamate; rather, it is more likely that they function as ligand-gated anion channels to reduce excitability (81). EAAT4 is localized to Purkinje cells in the cerebellum and conducts Cl− in a manner that is activated by neuronal activity (82–85). Similarly, EAAT5 expression is essentially restricted to the presynaptic terminal of bipolar cells and photoreceptors in the retina (86). It has been observed that glutamate-generated Cl− currents in photoreceptors hyperpolarize the cells from which they originate (87), and that Cl− conductance through EAAT5 in bipolar cells serves to oppose depolarization and inhibit synaptic transmission (88, 89), indicating a possible inhibitory function of EAAT5 in these cells, apart from merely taking up glutamate. The neuronal EAATs are not the only members of the family that exhibit anion fluxes—all five of the EAATs display an anion conductance (90, 91). In fact, Untiet et al. demonstrated that the astrocytic transporters EAAT1/GLAST and EAAT2/GLT-1 are important for Cl− balance and resting membrane potential in developing Bergmann glia (92).
Glutamate transporters and mitochondria co-compartmentalize
The levels of GLT-1 and GLAST (EAAT2 and EAAT1, respectively) mRNA and protein are consistently higher in astrocytes than in other cells in the nervous system (for reviews, see 17, 18, 29, 93, 94). Furthermore, electron microscopic studies have documented that expression of both these transporters is enriched in fine processes of astrocytes (95, 96). Approximately a decade ago, our laboratory used an anti-GLT1 antibody to immunoprecipitate GLT-1 from rat cortex and identify proteins that coimmunoprecipitate with GLT-1 by mass spectrometry. We had hypothesized that GLT-1 might form complexes with signaling molecules previously implicated in regulation of GLT-1 activity (e.g. protein kinase C), proteins that formed scaffolds for GLT-1, and possibly proteins that support the ion gradients needed to fuel the activity of this transporter (e.g. the Na+/K+ ATPase). While some of the subunits of the Na+/K+ ATPase were identified, we unexpectedly identified many mitochondrial proteins and most of the enzymes in glycolysis (77). Around this same time, Erin Rose from Dave Hampson’s group performed a mass spectroscopy analysis of proteins that co-purified with the Na+/K+ ATPase from cerebellum and identified GLAST and many other proteins that overlapped with our analysis (76). Several things prompted us to doubt our results. First, our colleagues/collaborators had a hard time believing that mitochondria might form a complex with glutamate transporters. Although there is some precedent for mitochondrial interactions with a subtype of glucose transporter (97, 98), this was, and remains, a reasonable concern because no direct linkages have documented to date. Second, as described above, several observations had prompted the hypothesis that astrocytes might preferentially metabolize glucose to lactate and export the lactate for oxidative phosphorylation (99, 100). Finally, although mitochondria had been identified in fine astrocyte processes by electron microscopy (101–103), some investigators made the reasonable assumption that these very fine processes were too small to accommodate mitochondria (104). At about the time that we had confirmed many of the interactions with reverse immunoprecipitations, Dr. Joshua Jackson joined the laboratory. We spent considerable time trying different sets of experiments (trying to map domains of GLT-1 required for the various interactions, studying lateral mobility of GLT-1 in the plasma membrane, etc). Ultimately, Dr. Jackson prepared organotypic slice cultures and selectively transduced astrocytes with fluorescently tagged molecules. This led to several observations. First, ~45% of the typical astrocyte process is occupied by mitochondria. Second, ~42% of the typical process is occupied by fluorescently-tagged GLT-1. By definition, this means there will be considerable overlap of these two signals by chance. In fact, we found that GLT-1 puncta overlap mitochondria ~60% of the time, and using Monte Carlo simulations, we found that this overlap was significantly more than would occur by chance (77). Similarly, we found that mitochondria also overlap with exogenously expressed/fluorescently tagged GLAST (105). While this does suggest that there is significant co-compartmentalization of mitochondria with GLT-1 and GLAST under these circumstances, there are several issues that have not been addressed. First, these experiments were conducted with exogenously expressed GLT-1 or GLAST. Second, these organotypic slices have many advantages, but they are grown under conditions that are quite different from those observed in vivo (e.g. concentration of glucose, concentration of oxygen, lack of blood vessels, etc). Finally, it is still not clear if the co-immunoprecipitation is an artifact of solubilization followed by aggregation or if it reflects a true interaction.
The logical next direction for this work has been to test for the presence of mitochondria in fine astrocyte processes in vivo. Using a couple of different strategies, several groups have confirmed that mitochondria are found throughout astrocyte processes in vitro and in vivo. A few of us have found that increasing neuronal activity recruits mitochondria to locations in astrocyte processes that are adjacent to synapses (80, 106–113). A recent study also demonstrated that deletion of neuronal pools of GLT-1 results in a doubling of the mitochondrial occupancy of astrocytic processes near excitatory synapses (114). It is not clear if this effect is due to increased neuronal activity or is dependent upon some other mechanism. To date, the extent of overlap of endogenous GLT-1 or GLAST with mitochondria has not been examined in vivo and given the high levels of GLT-1 and GLAST that are found in astrocytes, this may not be possible. Nevertheless, these studies provide strong evidence that astrocytic glutamate transporters will be overlapped by (co-compartmentalize with) mitochondria.
Implications of co-compartmentalization for glutamate metabolism
The glutamate-glutamine cycle was identified as the pathway for recycling of neurotransmitter pools of glutamate (115, 116). After clearance into astrocytes, glutamate is converted to glutamine by glutamine synthetase. The glutamine is then exported to neurons and re-converted to glutamate (117). This pathway is still the best explanation for the current data and likely represents a major, or the only, pathway for maintaining neurotransmitter pools of glutamate. Remarkably, there are still some issues with this pathway. First, it is relatively hard to demonstrate that excitatory, glutamate-mediated, transmission is dependent upon glutamine (118, 119). In addition, the glutamine transporters that were cloned in the early 2000’s are found on the cell body and are not enriched in presynaptic terminals; this makes it hard to believe that they contribute to recycling of neurotransmitter pools of glutamate (117, 120). A recent study from Jeff Erickson seems to be heading the field toward an activity that could mediate presynaptic glutamine uptake (121).
In addition to glutamine synthetase, there are several different enzymes and transporters involved in glutamate metabolism in astrocytes (Fig. 2). Glutamate can be converted to α-ketoglutarate in the cytosol by aspartate aminotransferase (also called glutamate-oxaloacetic acid transaminase); this is a pyridoxal phosphate (B6/thiamine)-dependent enzyme that simply transfers the amine to oxaloacetate to generate aspartate. These enzymes are readily reversible and the direction of flux is only driven by concentrations of substrates and products. The resulting cytosolic α-ketoglutarate can be imported by a mitochondrial transporter (mitochondrial 2-oxoglutarate/malate carrier protein, M2OM, or Slc25a11) (122). Glutamate can also be directly imported into mitochondria. There are two types of mitochondrial glutamate transporters. One type co-transports a H+ and glutamate into mitochondria (glutamate carrier 1, GC1, or Slc25a22). The second exchanges glutamate and aspartate (aralar or Slc25a12) across the mitochondrial membrane. There are several studies that suggest that aralar is not expressed by astrocytes, but there are also several studies that observe either protein or mRNA for aralar in astrocytes (94, 123–125). There are two mitochondrial enzymes that can convert glutamate to α-ketoglutarate (126). The first is a mitochondrial version of aspartate aminotransferase and the second is glutamate dehydrogenase. Unlike aspartate aminotransferase, the conversion of glutamate to α-ketoglutarate by glutamate dehydrogenase is associated with generation of NAD(P)H which can be converted to ATP or used as reducing power. The reader is referred to the following excellent discussions of the contributions of these enzymes to glutamate oxidation (126–128). It seems likely that both enzymes contribute to glutamate oxidation but under different, not well-defined conditions.
Fig. 2.
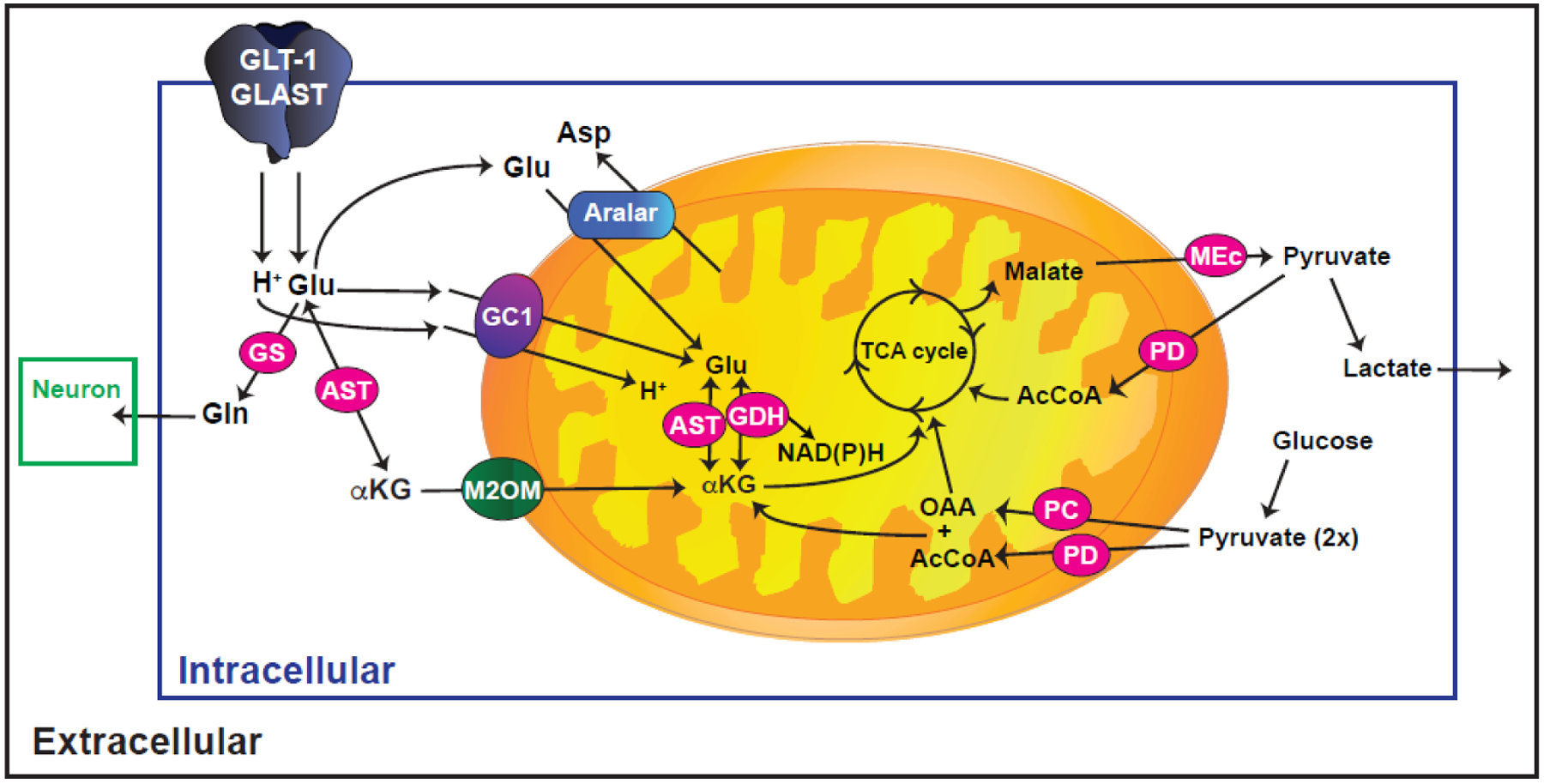
Schematic illustration of glutamate metabolism in astrocytes. In the cytosol, glutamate (Glu) can be transformed into glutamine (Gln) by glutamine synthetase (GS). This Gln is released into the extracellular space for subsequent transport into neurons and replenishment of neuronal Glu. Glutamate can also be transformed into α-ketoglutarate (αKG) by cytosolic aspartate aminotransferase (AST) which is transported into mitochondria via the mitochondrial 2-oxoglutarate/malate carrier protein (M2OM/Slc25a11). Glu can also be transported into mitochondria directly, either by glutamate carrier 1 (GC1) or by the glutamate/aspartate exchanger (aralar). In mitochondria, Glu is transformed into αKG by AST or by glutamate dehydrogenase (GDH). αKG is converted to malate which is exported into the cytosol and converted to pyruvate via malic enzyme (MEc) that can either be converted to lactate or to acetyl CoA by pyruvate dehydrogenase complex (PD) for complete oxidation. Glu (five carbon backbone) is replenished by converting pyruvate to oxaloacetate (OAA) by the astrocyte specific enzyme pyruvate carboxylase (PC) and the OAA is converted to αKG. The αKG can be converted to glutamate by either AST or GDH.
The α-ketoglutarate that is generated from glutamate is either partially oxidized to yield 5–7 molecules of ATP or completely oxidized to ultimately generate 20 molecules of ATP (129, 130). This occurs because tricarboxylic acid cycle intermediates cannot be depleted or accumulated. So after α-ketoglutarate is partially oxidized to malate, the malate needs to exit the mitochondria to the cytosol where it is converted to pyruvate (129). This pyruvate can either be converted to acetyl-CoA for re-entry into tricarboxylic acid cycle by pyruvate dehydrogenase (PD) or converted to lactate which could be exported into the extracellular space (131, 132).
The co-compartmentalization of glutamate transporters with mitochondria would seemingly provide an ideal mechanism for glutamate to be used as fuel to support its own uptake and provide ATP for glutamine synthesis. In fact, there are several different observations that demonstrate that some percentage of glutamate enters mitochondria and is oxidized. First, glutamate oxidation has been directly measured in astrocyte cultures either using glutamate that is radioactively labeled on the 1 carbon carboxyl group; one can capture and quantify the CO2 that is generated. Alternatively, non-radioactive, labeled glutamate can be used to follow glutamate oxidation using either NMR or mass spectrometry (133, 134 for reviews, see 104, 135). The percentage of glutamate that is oxidized is somewhat variable between laboratories ranging between 10% and 60% of the glutamate that enters an astrocyte (for review and primary references, see 75). Interestingly the percentage of glutamate oxidation increases with increasing extracellular glutamate, consistent with the notion that the amount of glutamate that is oxidized can be regulated (136, 137). Second, deletion of glutamate carrier 1 (Slc25a22) results in intracellular accumulation of glutamate in astrocytes, suggesting that this transporter is used to provide glutamate to mitochondria for oxidation (138). Finally, there has to be a net preservation of glutamate in the brain and it is generally thought that glutamate is not transported into or out of the brain, so one can estimate the amount of glutamate oxidation by following the incorporation of isotope from peripherally administered glucose into glutamate in vivo (139). The glucose supplies pyruvate to astrocytes where it can be converted to oxaloacetate by the astrocytic enzyme, pyruvate carboxylase, this is then conjugated with acetyl-CoA to generate α-ketoglutarate which can then be converted to glutamate (140, 141). This is the pathway that replenishes neuronal glutamate. Flux through this pathway has also been used to estimate that about 10 to 30% of brain glutamate is oxidized on a continuous basis (for reviews, see 115, 129, 130, 142).
While there are differences in both the results and conclusions with the studies of glutamate metabolism, it seems likely that the differences are related to differences in the conditions under which the results were obtained. For example, many of the critical studies were performed in astrocyte-enriched cultures. From a metabolic perspective, this is the only system that allows specific interrogation of ‘astrocyte’ metabolism in the absence of contributions from other cells. While it seems clear that this will remain an important model system into the future, individuals who use this system will need to be aware of several issues. First, from a mRNA expression perspective, cultured astrocytes are quite different from those observed in vivo (143). We and several other groups have demonstrated that both neurons and endothelia affect expression of various proteins in astrocytes (144–146). In fact, co-culturing astrocytes with neurons increases astrocytic expression of several glycolytic enzymes (147). Second, the conditions under which these experiments are conducted can have consequences for the results. For example, the levels of O2 are known to change astrocyte Ca2+ signaling (148) and Ca2+ can activate several of the mitochondrial proteins involved in glutamate metabolism, including aralar and several of the mitochondrial dehydrogenases (149–154 for recent discussion, see 113). Although changing glucose concentrations does not affect the extent of glutamate oxidation in astrocyte cultures (155), it seems likely that glucose levels could easily affect measures of energy production. Finally, the buffers and ionic composition of the medium can also affect the results as illustrated by Rimmele and her colleagues (156). One suspects that as genetically-encoded indicators of brain metabolites are optimized, it will be possible to determine exactly how endogenous pools of glutamate affect astrocytic oxidative phosphorylation, to monitor lactate production from endogenous glutamate as opposed to endogenous glucose, and to test for possible linkages between neuronal activation and the relative disposition of glutamate.
While it is still not clear that co-compartmentalization of glutamate transporters with mitochondria necessarily favors import of glutamate into mitochondria and subsequent oxidation, there is evidence that glutamate transport is coupled to mitochondrial function. We found that several different inhibitors of glutamate dehydrogenase non-competitively block glutamate or D-aspartate uptake, consistent with the notion that endogenous pools of glutamate are oxidized to provide fuel for glutamate uptake, at least under some conditions (157). Conversely, glutamate transport also changes mitochondrial function. Originally, glutamate transport activation was linked to mitochondrial acidification, decreased oxygen consumption, and decreased ATP production (158). The mitochondrial import of H+ is associated with decreased oxidative phosphorylation by a mechanism that involves uncoupling protein 4 (159). These investigators also showed that D-aspartate, a substrate for the plasma membrane transporters but not for the mitochondrial transporters, also causes mitochondrial acidification. These studies were performed in a HEPES-based buffer. More recently some of these same investigators examined the effects of glutamate uptake on mitochondrial function in bicarbonate-based buffer. Under these conditions, glutamate triggers increased mitochondrial function as assessed by measuring the oxygen consumption rate (156). A similar glutamate transport-dependent increase in mitochondrial function (oxygen consumption) was recently observed by Juaristi and colleagues (132). They also showed that D-aspartate triggers an increase in oxygen consumption, consistent with the notion that this increase is not dependent upon glutamate per se but is instead triggered by the energetic demands imposed by the increase in Na+/K+ ATPase activity required to maintain Na+ gradients in the cell (for review, see 49).
While one might intuitively assume that the coupling of glutamate transport with mitochondrial function and the potential to oxidize glutamate (see above) would result in glutamate transport-dependent increases in ATP, this is not the case. In fact, glutamate transport activation consistently causes a decrease in astrocytic ATP, regardless of whether D-aspartate or L-glutamate is used as a transporter substrate (47, 132, 160, 161). In addition, neuronal activity and the Na+/K+ ATPase both increase extracellular K+. Increased extracellular K+ rapidly stimulates glycolytic activity and increases astrocytic ATP (54, for reviews see 162 163, 164, 165); it seems likely that this glycolytic activity is directly fueling transport. Together, these data suggest that the amount of glutamate that enters via the transporter and is oxidized does not generate excess ATP beyond that required to support re-generation of the Na+ gradient through the Na+/K+ ATPase and fuel the conversion of glutamate to glutamine. All of these studies were conducted either in cultured astrocytes or acute brain slices; it is possible that the balance of ATP generated changes in an in vivo setting, but this has not been tested to date. The effects of glutamate and/or K+ on ATP levels may follow different kinetics. In non-astrocyte systems, Magi and colleagues have demonstrated the presence of EAAC1 in purified mitochondria, and have documented a glutamate transport- and NCX-dependent increase in mitochondrial Ca2+ and ATP. They also show that EAAC1 co-immunoprecipitates with NCX1 (166–168). Together, these findings suggest that glutamate transport is physically and functionally coupled to Ca2+ influx via NCX1, and that activation of this system can trigger an increase in ATP in some cells.
Effects of Hypoxia/ischemia on astrocytic glutamate transport and mitochondria
The brain is highly dependent upon energy. In fact, it consumes ~20% of the basal metabolic rate, and it is generally thought that it is almost exclusively dependent upon glucose for energy production. (169–172). Remarkably, the brain contains relatively small reserve pools of glycogen, and these pools are essentially restricted to astrocytes (173, 174). Therefore, it should not be surprising that the brain is particularly sensitive to periods of hypoxia and/or decreased blood flow (ischemia). Over 30 years ago, several groups simultaneously demonstrated that glutamate receptor antagonists protect neurons from global and/or focal ischemia in in vitro and in vivo model systems (for reviews, see 6, 7, 175, 176). While this and several other lines of evidence are consistent with the notion that these insults cause a rise in extracellular concentrations of glutamate and excessive activation of glutamate receptors followed by excitotoxic cell death, excitatory amino acid receptor antagonists have not made it into clinical practice for reasons that are still unclear (177, 178).
Compared to neurons, astrocytes are relatively resistant to periods of hypoxia/ischemia. This relative sparing has been observed in vitro and in vivo (179, for reviews see 180, 181, 182). Depending on their location relative to a focal ischemic infarct, astrocytes undergo a progressive change in morphology with loss of highly branched processes, hypertrophy, and increased expression of glial fibrillary acidic protein (GFAP) (183 for review, see 184). These are termed ‘reactive astrocytes’. Astrocytes also proliferate and can generate new neurons (185–187 for review, see 188). Several different groups have observed decreases in the levels of GLT-1 and/or GLAST mRNA and/or protein in several different models of hypoxia/ischemia (189, 190 191 for reviews, see 22, 192). It is interesting to note that decreases in protein frequently occur within 24 hours after an insult. Although not necessarily translatable to in vivo, the half-lives of GLT-1 and GLAST proteins are longer than 24 h when measured in astrocyte cultures (193); this suggests that these insults may accelerate degradation of these transporters possibly by ubiquitination in addition to the decreased translation that presumably accompanies the decreases in mRNA for these transporters (194–196).
The effects of hypoxia and ischemia on astrocytic mitochondria have also been examined in vitro and in vivo. In primary cultures, where astrocytes have essentially no processes, oxygen-glucose deprivation (OGD) causes mitochondria to undergo a change from a highly inter-connected network to a fragmented, punctate morphology (197). In an organotypic culture in which astrocyte morphology is highly branched and more reminiscent of that observed in vivo with mitochondria found throughout the processes (for discussion see 198), transient OGD causes relatively rapid fragmentation of mitochondria in the processes followed by gradual decrease in the occupancy of the process by mitochondria (199). Similar effects have been observed in an in vivo model of global ischemia (183, 200). In preliminary studies, our laboratory has observed similar effects as early as 1 hour after photothrombosis-induced occlusion of the middle cerebral artery in mice (Shih et al., unpublished observations, Fig. 1).
The loss of astrocytic mitochondria has been linked to two mechanisms. First, damaged mitochondria (those that have been subjected to increased reactive oxygen species) undergo a specialized form of degradation called mitophagy (for reviews, see 201, 202). Quintana and colleagues showed that hypoxia and post-hypoxia reoxygenation of rat primary astrocytes results in a fragmentation of mitochondria followed by increased co-localization of mitochondria with a marker of mitophagy, microtubule-associated proteins 1A/1B light chain 3B (LC3-II) (197). This effect was associated with dephosphorylation of dynamin-related protein-1 (Drp-1) at Ser637, which results in activation of Drp1 and increased fission of mitochondria (197). Similarly, we observed an increase in co-localization of mitochondria with LC3-II after transient OGD in organotypic slices (199, Fig. 1). Second, Hayakawa and colleagues provide evidence for transfer of mitochondria from astrocytes to neurons (203). They showed that cultured astrocytes release functional mitochondria by a calcium-dependent mechanism involving CD38 and cyclic ADP ribose signaling and that mitochondria are transferred from astrocytes to neurons in culture. Furthermore, suppressing CD38 signaling with siRNA reduced extracellular mitochondria in culture, exacerbated OGD-induced neuronal damage in culture and worsened neurological damage after transient focal ischemia in vivo. These studies are consistent with the hypothesis that the loss of mitochondria from astrocytes can be attributed to both increased degradation and transfer to neighboring cells (for review, see 204). This is still a controversial observation (205), and the mechanisms that contribute to intercellular transfer of mitochondria have not been clarified (206, for review see 207).
We found that a glutamate transport inhibitor, 3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate (TFB-TBOA), completely blocks the decrease in mitochondrial size and the loss of mitochondria observed after transient OGD in an organotypic slice (199). We also showed that glutamate, in the presence of receptor antagonists, was sufficient to cause fragmentation and loss of astrocytic mitochondria. Although one is always concerned that the effects of an inhibitor might be caused by interactions with other targets, TFB-TBOA was first synthesized in 2004, has affinities in the low nM range for blocking glutamate uptake and has no effects on glutamate receptors at 100 μM concentrations (208). Furthermore, no off-target effects have been identified to date. This suggests that either prolonged glutamate metabolism or the ionic imbalances observed with glutamate uptake contributes to the loss of mitochondria after stroke. As discussed above, activation of glutamate transport accompanied by the influx of Na+ activates reversed operation of the Na+/Ca2+ exchanger and increases intracellular Ca2+. While blocking reversed Na+/Ca2+ exchange completely attenuates OGD-induced loss of neurons, it only partially attenuates the fragmentation and loss of astrocytic mitochondria (199).
The functional consequences of this loss of mitochondria have not been systematically examined. When mitochondria undergo a change in shape from long tubular structures to small rounded ball-like structures, this increases the ratio of mitochondrial surface area to matrix, presumably increasing mitochondrial efficiency (For review, see 209). Fragmentation is also a prerequisite for degradation of mitochondria (202). In addition, fragmentation of mitochondria may be required to enable the transfer of mitochondria from astrocytes to other cells (Fig. 1, see above). Although the technology to selectively tag and isolate astrocytic mitochondria has recently been developed (210), nobody has yet used this approach to perform an analysis of astrocytic mitochondrial function after an in vivo stroke. However, several groups have taken advantage of the fact that the mitochondrial enzyme pyruvate carboxylase is selectively expressed in astrocytes in the mammalian brain (for reviews, see 211, 212, 213) to determine if stroke has a functional consequence for astrocytic mitochondria. In several different models and in both adult and neonatal animals, stroke or hypoxia/ischemia causes a decrease in flux through the pyruvate carboxylase pathway in vivo (214–216). It does appear that increasing glutamate oxidation can be beneficial within the context of stroke. For example, mice that over-express glutamate dehydrogenase have smaller lesions and reduced edema following ischemia-reperfusion (217). Similarly, a GDH activator increases oxidative glutamate metabolism and enhances neuronal survival in an in vivo model of stroke (218). Finally, mitochondria are known to provide a sink for cytosolic Ca2+, essentially shaping Ca2+ responses by decreasing amplitude of the responses and/or accelerating the decay of these signals (for review, see 219). In astrocytes, mitochondria also generate small transient Ca2+ elevations (111). In organotypic slices, we found that the lateral spread of spontaneous Ca2+ signals was bounded by mitochondria and that interfering with mitochondria or their docking increased the amplitude and spread of these Ca2+ signals (66, 199). We also found that transient OGD resulted in a dramatic increase in the size and lateral spread of spontaneous Ca2+ signals (199). Similar increases in Ca2+ signals in astrocyte cell bodies have been observed in an in vivo model of stroke (for review, see 220).
Conclusions/summarize future directions
Over the last forty years, glutamate has gained acceptance as the predominant excitatory neurotransmitter in the mammalian nervous system. It seems remarkably efficient that the predominant inhibitory neurotransmitter GABA is only one metabolic step from glutamate. It is clear that glutamate transporters contribute to signaling within astrocytes, between astrocytes, and in other cell types. Na+ influx contributes to metabolic signaling and could indirectly change the concentrations of other ions such as Ca2+. Ca2+ signaling in turn plays various signaling roles in astrocytes, including metabolism, neurovascular coupling, morphological signaling, and release of gliotransmitters. Finally, the chloride conductance that has been observed in these transporters is also used for signaling. While this has mostly been studied in the presynaptic EAATs, there is some evidence for a developmental role of these Cl− currents in glial cells. Most of these findings have been in cultured cells in vitro or in acute slices, and the contributions of glutamate transport-dependent signals to in vivo astrocyte biology have not been defined.
It is also clear that the astrocytic glutamate transporters co-compartmentalize with mitochondria and that a variable amount of glutamate is oxidized in mitochondria. It seems like there are redundant (or complementary) enzymes involved in the oxidation of glutamate. However, there are essentially no selective pharmacological agents that can be used to explore the functional consequences of acute inhibition of any of these transporters/enzymes, and genetic deletion of many of the key enzymes is likely to trigger homeostatic compensation that will make it difficult to define functional roles. As indicated above, there is some evidence that the higher levels of glutamate results in greater fractional oxidation of glutamate in astrocyte cultures. One wonders if this occurs during normal synaptic transmission or if pathologic elevations of extracellular glutamate are required. The ability to generate genetically-encoded optical sensors for virtually any molecule is likely to dramatically impact our understanding of how these various biochemical pathways operate in vivo.
Finally, stroke or stroke-like insults cause rapid changes in the morphology of astrocytic mitochondria, and there is evidence that this is associated with a deficiency in anaplerotic re-generation of the carbon backbone required for synthesis of glutamate as well as exaggerated astrocytic Ca2+ signaling. Whether this process contributes to the pathologic progression of stroke-induced damage or is a protective response to the insult is un-resolved.
Acknowledgements:
The authors regret that we were not able to cite all of the numerous studies that have been published in this area. The authors are partially supported by a grant from the National Institutes of Neurologic Disorders and Stroke of the National Institutes of Health (R01 NS106693). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Robinson MB, Coyle JT (1987) Glutamate and related acidic excitatory neurotransmitters: from basic science to clinical application. Faseb J 1:446–455 [DOI] [PubMed] [Google Scholar]
- 2.Schousboe A (1981) Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol 22:1–45 [DOI] [PubMed] [Google Scholar]
- 3.Bowery NG, Smart TG (2006) GABA and glycine as neurotransmitters: a brief history. Br J Pharmacol 147 Suppl 1:S109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellison DW, Beal MF, Martin JB (1987) Amino acid neurotransmitters in postmortem human brain analyzed by high performance liquid chromatography with electrochemical detection. J Neurosci Methods 19:305–315 [DOI] [PubMed] [Google Scholar]
- 5.Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, Shulman RG, Tamborlane WV (1992) Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci U S A 89:1109–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi DW (1992) Excitotoxic cell death. J Neurobiol 23:1261–1276 [DOI] [PubMed] [Google Scholar]
- 7.Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695 [DOI] [PubMed] [Google Scholar]
- 8.Greene JG, Greenamyre JT (1996) Bioenergetics and glutamate excitotoxicity. Prog Neurobiol 48:613–634. [DOI] [PubMed] [Google Scholar]
- 9.Doble A (1999) The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacology and Therapeutics 81:163–221. [DOI] [PubMed] [Google Scholar]
- 10.Olney J (2003) Excitotoxicity, apoptosis and neuropsychaitric disorders. Curr Opin Pharm 3:101–109 [PubMed] [Google Scholar]
- 11.Timmerman W, Westerink BHC (1997) Brain microdialysis of GABA amd glutamate: What does it signify? Synapse 27:242–261 [DOI] [PubMed] [Google Scholar]
- 12.Miele M, Berners M, Boutelle MG, Kusakabe H, Fillenz M (1996) The determination of the extracellular concentration of brain glutamate using quantitative microdialysis. Brain Res 707:131–133 [DOI] [PubMed] [Google Scholar]
- 13.Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman MA, Nahir B, Jahr CE (2011) Distribution of extracellular glutamate in the neuropil of hippocampus. PLoS One 6:e26501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu DN, Jahr CE (2017) Extracellular Glutamate in the Nucleus Accumbens Is Nanomolar in Both Synaptic and Non-synaptic Compartments. Cell Rep 18:2576–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attwell D, Barbour B, Szatkowski M (1993) Nonvesicular release of neurotransmitter. Neuron 11:401–407 [DOI] [PubMed] [Google Scholar]
- 17.Sims KD, Robinson MB (1999) Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol 13:169–197 [DOI] [PubMed] [Google Scholar]
- 18.Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105 [DOI] [PubMed] [Google Scholar]
- 19.Slotboom DJ, Konings WN, Lolkema JS (2001) Glutamate transporters combine transporter- and channel-like features. Trends Biochem Sci 26:534–539 [DOI] [PubMed] [Google Scholar]
- 20.Huang YH, Bergles DE (2004) Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol 14:346–352. [DOI] [PubMed] [Google Scholar]
- 21.Beart PM, O’Shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150:5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo M, Wang YZ, Gout PW (2008) The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. Journal of cellular physiology 215:593–602 [DOI] [PubMed] [Google Scholar]
- 24.Tzingounis AV, Wadiche JI (2007) Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8:935–947 [DOI] [PubMed] [Google Scholar]
- 25.Vandenberg RJ, Ryan RM (2013) Mechanisms of glutamate transport. Physiol Rev 93:1621–1657 [DOI] [PubMed] [Google Scholar]
- 26.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P (2013) The cystine/glutamate antiporter system x(c)(−) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & redox signaling 18:522–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy-Royal C, Dupuis J, Groc L, Oliet SHR (2017) Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res 95:2140–2151 [DOI] [PubMed] [Google Scholar]
- 28.Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA (2002) Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35:5187–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danbolt NC, Pines G, Kanner BI (1990) Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry 29:6734–6740 [DOI] [PubMed] [Google Scholar]
- 31.Zerangue N, Kavanaugh MP (1996) Flux coupling in a neuronal glutamate transporter. Nature 383:634–637 [DOI] [PubMed] [Google Scholar]
- 32.Conti F, Weinberg RJ (1999) Shaping excitation at glutamatergic synapses. Trends Neurosci 22:451–458. [DOI] [PubMed] [Google Scholar]
- 33.Conn PJ, Patel J (1994) The metabotropic glutamate receptors. Humana Press, Totowa, NJ [Google Scholar]
- 34.Ferraguti F, Shigemoto R (2006) Metabotropic glutamate receptors. Cell Tissue Res 326:483–504 [DOI] [PubMed] [Google Scholar]
- 35.Hollman M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108 [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi S (1992) Molecular diversity of glutamate receptors and implications for brain function. Science 258:597–603 [DOI] [PubMed] [Google Scholar]
- 37.Nakanishi S (1994) Metabotropic glutamate receptors: Synaptic transmission, modulation, and plasticity. Neuron 13:1031–1037 [DOI] [PubMed] [Google Scholar]
- 38.Nicolletti F, Bruno V, Capani A, Casabona G, Knöpfel T (1996) Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci 19:267–271 [DOI] [PubMed] [Google Scholar]
- 39.Pin J-P, Duvoisin R (1995) The metabotropic glutamate receptors: Structure and functions. Neuropharmacology 14:1–26 [DOI] [PubMed] [Google Scholar]
- 40.Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP (1995) Kinetics of a human glutamate transporter. Neuron 14:1019–1027 [DOI] [PubMed] [Google Scholar]
- 41.Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19:1297–1308. [DOI] [PubMed] [Google Scholar]
- 42.Bowman CL, Kimelberg HK (1984) Excitatory amino acids directly depolarize rat brain astrocytes in primary culture. Nature 311:656–659 [DOI] [PubMed] [Google Scholar]
- 43.Mennerick S, Zorumski CF (1994) Glial contribution to excitatory neurotransmission in cultured hippocampal cells. Nature 368:59–62 [DOI] [PubMed] [Google Scholar]
- 44.Bellot-Saez A, Kekesi O, Morley JW, Buskila Y (2017) Astrocytic modulation of neuronal excitability through K(+) spatial buffering. Neurosci Biobehav Rev 77:87–97 [DOI] [PubMed] [Google Scholar]
- 45.Kaplan JH (2002) Biochemistry of Na,K-ATPase. Annu Rev Biochem 71:511–535 [DOI] [PubMed] [Google Scholar]
- 46.Rose CR, Ransom BR (1996) Mechanisms of H+ and Na+ changes induced by glutamate, kainate, and D-aspartate in rat hippocampal astrocytes. J Neurosci 16:5393–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langer J, Rose CR (2009) Synaptically induced sodium signals in hippocampal astrocytes in situ. J Physiol 587:5859–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langer J, Gerkau NJ, Derouiche A, Kleinhans C, Moshrefi-Ravasdjani B, Fredrich M, Kafitz KW, Seifert G, Steinhauser C, Rose CR (2017) Rapid sodium signaling couples glutamate uptake to breakdown of ATP in perivascular astrocyte endfeet. Glia 65:293–308 [DOI] [PubMed] [Google Scholar]
- 49.Rose CR, Felix L, Zeug A, Dietrich D, Reiner A, Henneberger C (2017) Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front Mol Neurosci 10:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziemens D, Oschmann F, Gerkau NJ, Rose CR (2019) Heterogeneity of Activity-Induced Sodium Transients between Astrocytes of the Mouse Hippocampus and Neocortex: Mechanisms and Consequences. J Neurosci 39:2620–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Unichenko P, Myakhar O, Kirischuk S (2012) Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 60:605–614 [DOI] [PubMed] [Google Scholar]
- 52.Bernardinelli Y, Magistretti PJ, Chatton JY (2004) Astrocytes generate Na+-mediated metabolic waves. Proc Natl Acad Sci U S A 101:14937–14942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porras OH, Ruminot I, Loaiza A, Barros LF (2008) Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 56:59–68 [DOI] [PubMed] [Google Scholar]
- 54.Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San Martin A, Gutierrez R, Zambrano M, Barros LF (2011) Fast and reversible stimulation of astrocytic glycolysis by k+ and a delayed and persistent effect of glutamate. J Neurosci 31:4709–4713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I, Baeza-Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, Weber B (2016) In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab 23:94–102 [DOI] [PubMed] [Google Scholar]
- 56.Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91:10625–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab 26:361–374 e364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gramsbergen JB, Leegsma-Vogt G, Venema K, Noraberg J, Korf J (2003) Quantitative online monitoring of hippocampus glucose and lactate metabolism in organotypic cultures using biosensor technology. J Neurochem 85:399–408 [DOI] [PubMed] [Google Scholar]
- 59.Hertz L, Swanson RA, Newman GC, Marrif H, Juurlink BH, Peng L (1998) Can experimental conditions explain the discrepancy over glutamate stimulation of aerobic glycolysis? Dev Neurosci 20:339–347 [DOI] [PubMed] [Google Scholar]
- 60.Peng L, Swanson RA, Hertz L (2001) Effects of L-glutamate, D-aspartate, and monensin on glycolytic and oxidative glucose metabolism in mouse astrocyte cultures: further evidence that glutamate uptake is metabolically driven by oxidative metabolism. Neurochem Int 38:437–443 [DOI] [PubMed] [Google Scholar]
- 61.Swanson RA, Yu AC, Chan PH, Sharp FR (1990) Glutamate increases glycogen content and reduces glucose utilization in primary astrocyte culture. J Neurochem 54:490–496 [DOI] [PubMed] [Google Scholar]
- 62.Kohler S, Winkler U, Sicker M, Hirrlinger J (2018) NBCe1 mediates the regulation of the NADH/NAD(+) redox state in cortical astrocytes by neuronal signals. Glia 66:2233–2245 [DOI] [PubMed] [Google Scholar]
- 63.Rojas H, Colina C, Ramos M, Benaim G, Jaffe EH, Caputo C, DiPolo R (2007) Na+ entry via glutamate transporter activates the reverse Na+/Ca2+ exchange and triggers Ca(i)2+-induced Ca2+ release in rat cerebellar Type-1 astrocytes. J Neurochem 100:1188–1202 [DOI] [PubMed] [Google Scholar]
- 64.Rojas H, Colina C, Ramos M, Benaim G, Jaffe E, Caputo C, Di Polo R (2013) Sodium-calcium exchanger modulates the L-glutamate Ca(i) (2+) signalling in type-1 cerebellar astrocytes. Adv Exp Med Biol 961:267–274 [DOI] [PubMed] [Google Scholar]
- 65.Parpura V, Sekler I, Fern R (2016) Plasmalemmal and mitochondrial Na(+) -Ca(2+) exchange in neuroglia. Glia 64:1646–1654 [DOI] [PubMed] [Google Scholar]
- 66.Jackson JG, Robinson MB (2015) Reciprocal Regulation of Mitochondrial Dynamics and Calcium Signaling in Astrocyte Processes. J Neurosci 35:15199–15213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ibanez I, Bartolome-Martin D, Piniella D, Gimenez C, Zafra F (2019) Activity dependent internalization of the glutamate transporter GLT-1 requires calcium entry through the NCX sodium/calcium exchanger. Neurochem Int 123:125–132 [DOI] [PubMed] [Google Scholar]
- 68.Martinez-Lozada Z, Waggener CT, Kim K, Zou S, Knapp PE, Hayashi Y, Ortega A, Fuss B (2014) Activation of sodium-dependent glutamate transporters regulates the morphological aspects of oligodendrocyte maturation via signaling through calcium/calmodulin-dependent kinase IIbeta’s actin-binding/-stabilizing domain. Glia 62:1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG (1994) Glutamate-mediated astrocyte-neuron signalling. Nature 369:744–747 [DOI] [PubMed] [Google Scholar]
- 70.Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A (2014) Gliotransmitters travel in time and space. Neuron 81:728–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marchaland J, Cali C, Voglmaier SM, Li H, Regazzi R, Edwards RH, Bezzi P (2008) Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J Neurosci 28:9122–9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB (1999) ATP released from astrocytes mediates glial calcium waves. J Neurosci 19:520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006) Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267 [DOI] [PubMed] [Google Scholar]
- 74.Petzold GC, Murthy VN (2011) Role of astrocytes in neurovascular coupling. Neuron 71:782–797 [DOI] [PubMed] [Google Scholar]
- 75.Robinson MB, Jackson JG (2016) Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem Int 98:56–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR (2009) Glutamate transporter coupling to Na,K-ATPase. J Neurosci 29:8143–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernardinelli Y, Azarias G, Chatton JY (2006) In situ fluorescence imaging of glutamate-evoked mitochondrial Na+ responses in astrocytes. Glia 54:460–470 [DOI] [PubMed] [Google Scholar]
- 79.Azarias G, Van de Ville D, Unser M, Chatton JY (2008) Spontaneous NA+ transients in individual mitochondria of intact astrocytes. Glia 56:342–353 [DOI] [PubMed] [Google Scholar]
- 80.Jackson JG, O’Donnell JC, Takano H, Coulter DA, Robinson MB (2014) Neuronal activity and glutamate uptake decrease mitochondrial mobility in astrocytes and position mitochondria near glutamate transporters. J Neurosci 34:1613–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mim C, Balani P, Rauen T, Grewer C (2005) The glutamate transporter subtypes EAAT4 and EAATs 1–3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J Gen Physiol 126:571–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino acid transporter with properties of a ligand-gated chloride channel. Nature (Lond) 375:599–603 [DOI] [PubMed] [Google Scholar]
- 83.Kataoka Y, Morii H, Watanabe Y, Ohmori H (1997) A postsynaptic excitatory amino acid chloride conductance functionally regulated by neuronal activity in cerebellar purkinje cells. J Neurosci 17:7017–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Otis TS, Jahr CE (1998) Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci 18:7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proceedings of the National Academy of Sciences USA 94:4155–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Picaud S, Larsson HP, Wellis DP, Lecar H, Werblin F (1995) Cone photoreceptors respond to their own glutamate release in the tiger salamander. Proc Natl Acad Sci U S A 92:9417–9421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Veruki ML, Morkve SH, Hartveit E (2006) Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat Neurosci 9:1388–1396 [DOI] [PubMed] [Google Scholar]
- 89.Wersinger E, Schwab Y, Sahel JA, Rendon A, Pow DV, Picaud S, Roux MJ (2006) The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J Physiol 577:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wadiche JI, Amara SG, Kavanaugh MP (1995) Ion fluxes associated with excitatory amino acid transport. Neuron 15:721–728 [DOI] [PubMed] [Google Scholar]
- 91.Divito CB, Borowski JE, Glasgow NG, Gonzalez-Suarez AD, Torres-Salazar D, Johnson JW, Amara SG (2017) Glial and Neuronal Glutamate Transporters Differ in the Na(+) Requirements for Activation of the Substrate-Independent Anion Conductance. Front Mol Neurosci 10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Untiet V, Kovermann P, Gerkau NJ, Gensch T, Rose CR, Fahlke C (2017) Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 65:388–400 [DOI] [PubMed] [Google Scholar]
- 93.Robinson MB (1999) The family of sodium-dependent glutamate transporters: A focus on the GLT-1/EAAT2 subtype. Neurochem Int 33:479–491 [DOI] [PubMed] [Google Scholar]
- 94.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720 [DOI] [PubMed] [Google Scholar]
- 96.Minelli A, Barbaresi P, Reimer RJ, Edwards RH, Conti F (2001) The glial glutamate transporter GLT-1 is localized both in the vicinity of and at distance from axon terminals in the rat cerebral cortex. Neuroscience 108:51–59 [DOI] [PubMed] [Google Scholar]
- 97.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V (2008) Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem 283:13482–13490 [DOI] [PubMed] [Google Scholar]
- 98.Zaid H, Talior-Volodarsky I, Antonescu C, Liu Z, Klip A (2009) GAPDH binds GLUT4 reciprocally to hexokinase-II and regulates glucose transport activity. Biochem J 419:475–484 [DOI] [PubMed] [Google Scholar]
- 99.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55:1251–1262 [DOI] [PubMed] [Google Scholar]
- 100.Allaman I, Belanger M, Magistretti PJ (2011) Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 34:76–87 [DOI] [PubMed] [Google Scholar]
- 101.Mugnaini E (1964) Helical Filaments in Astrocytic Mitochondria of the Corpus Striatum in the Rat. The Journal of cell biology 23:173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pysh JJ, Khan T (1972) Variations in mitochondrial structure and content of neurons and neuroglia in rat brain: an electron microscopic study. Brain Res 36:1–18 [DOI] [PubMed] [Google Scholar]
- 103.Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27:12255–12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27:219–249 [DOI] [PubMed] [Google Scholar]
- 105.Bauer DE, Jackson JG, Genda EN, Montoya MM, Yudkoff M, Robinson MB (2012) The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem Int 61:566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stephen TL, Gupta-Agarwal S, Kittler JT (2014) Mitochondrial dynamics in astrocytes. Biochem Soc Trans 42:1302–1310 [DOI] [PubMed] [Google Scholar]
- 107.Ugbode CI, Hirst WD, Rattray M (2014) Neuronal influences are necessary to produce mitochondrial co-localization with glutamate transporters in astrocytes. J Neurochem 130:668–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Benjamin Kacerovsky J, Murai KK (2016) Stargazing: Monitoring subcellular dynamics of brain astrocytes. Neuroscience 323:84–95 [DOI] [PubMed] [Google Scholar]
- 109.Derouiche A, Haseleu J, Korf HW (2015) Fine Astrocyte Processes Contain Very Small Mitochondria: Glial Oxidative Capability May Fuel Transmitter Metabolism. Neurochem Res 40:2402–2413 [DOI] [PubMed] [Google Scholar]
- 110.Stephen TL, Higgs NF, Sheehan DF, Al Awabdh S, Lopez-Domenech G, Arancibia-Carcamo IL, Kittler JT (2015) Miro1 Regulates Activity-Driven Positioning of Mitochondria within Astrocytic Processes Apposed to Synapses to Regulate Intracellular Calcium Signaling. J Neurosci 35:15996–16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, Wirtz D, Bergles DE (2017) Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 93:587–605 e587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gobel J, Motori E, Bergami M (2018) Spatiotemporal control of mitochondrial network dynamics in astroglial cells. Biochem Biophys Res Commun 500:17–25 [DOI] [PubMed] [Google Scholar]
- 113.Jackson JG, Robinson MB (2018) Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 66:1213–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McNair LF, Andersen JV, Aldana BI, Hohnholt MC, Nissen JD, Sun Y, Fischer KD, Sonnewald U, Nyberg N, Webster SC, Kapur K, Rimmele TS, Barone I, Hawks-Mayer H, Lipton JO, Hodgson NW, Hensch TK, Aoki CJ, Rosenberg PA, Waagepetersen HS (2019) Deletion of Neuronal GLT-1 in Mice Reveals Its Role in Synaptic Glutamate Homeostasis and Mitochondrial Function. J Neurosci 39:4847–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hertz L (2013) The Glutamate-Glutamine (GABA) Cycle: Importance of Late Postnatal Development and Potential Reciprocal Interactions between Biosynthesis and Degradation. Frontiers in endocrinology 4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hertz L, Chen Y (2017) Integration between Glycolysis and Glutamate-Glutamine Cycle Flux May Explain Preferential Glycolytic Increase during Brain Activation, Requiring Glutamate. Front Integr Neurosci 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chaudhry FA, Reimer RJ, Edwards RH (2002) The glutamine commute: take the N line and transfer to the A. J Cell Biol 157:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192–9200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ (2014) A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 81:888–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Varoqui H, Zhu H, Yao D, Ming H, Erickson JD (2000) Cloning and functional identification of a neuronal glutamine transporter. J Biol Chem 275:4049–4054 [DOI] [PubMed] [Google Scholar]
- 121.Erickson JD (2017) Functional identification of activity-regulated, high-affinity glutamine transport in hippocampal neurons inhibited by riluzole. J Neurochem 142:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Runswick MJ, Walker JE, Bisaccia F, Iacobazzi V, Palmieri F (1990) Sequence of the bovine 2-oxoglutarate/malate carrier protein: structural relationship to other mitochondrial transport proteins. Biochemistry 29:11033–11040 [DOI] [PubMed] [Google Scholar]
- 123.Pardo B, Rodrigues TB, Contreras L, Garzon M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrustegui J (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab 31:90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hertz L (2011) Brain glutamine synthesis requires neuronal aspartate: a commentary. J Cereb Blood Flow Metab 31:384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li B, Hertz L, Peng L (2012) Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem Int 61:1325–1332 [DOI] [PubMed] [Google Scholar]
- 126.McKenna MC, Stridh MH, McNair LF, Sonnewald U, Waagepetersen HS, Schousboe A (2016) Glutamate oxidation in astrocytes: Roles of glutamate dehydrogenase and aminotransferases. J Neurosci Res 94:1561–1571 [DOI] [PubMed] [Google Scholar]
- 127.Hertz L, Rothman DL (2017) Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology (Basel) 6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Karaca M, Frigerio F, Migrenne S, Martin-Levilain J, Skytt DM, Pajecka K, Martin-Del-Rio R, Gruetter R, Tamarit-Rodriguez J, Waagepetersen HS, Magnan C, Maechler P (2015) GDH-Dependent Glutamate Oxidation in the Brain Dictates Peripheral Energy Substrate Distribution. Cell Rep 13:365–375 [DOI] [PubMed] [Google Scholar]
- 129.Dienel GA (2013) Astrocytic energetics during excitatory neurotransmission: What are contributions of glutamate oxidation and glycolysis? Neurochem Int 63:244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McKenna MC (2013) Glutamate pays its own way in astrocytes. Frontiers in endocrinology 4:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dienel GA, McKenna MC (2014) A dogma-breaking concept: glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J Neurochem 131:395–398 [DOI] [PubMed] [Google Scholar]
- 132.Juaristi I, Llorente-Folch I, Satrustegui J, Del Arco A (2019) Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 67:759–774 [DOI] [PubMed] [Google Scholar]
- 133.Tiwari V, Ambadipudi S, Patel AB (2013) Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J Cereb Blood Flow Metab 33:1523–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lanz B, Xin L, Millet P, Gruetter R (2014) In vivo quantification of neuro-glial metabolism and glial glutamate concentration using 1H-[13C] MRS at 14.1T. J Neurochem 128:125–139 [DOI] [PubMed] [Google Scholar]
- 135.Sonnay S, Gruetter R, Duarte JMN (2017) How Energy Metabolism Supports Cerebral Function: Insights from (13)C Magnetic Resonance Studies In vivo. Frontiers in neuroscience 11:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66:386–393 [DOI] [PubMed] [Google Scholar]
- 137.Torres FV, Hansen F, Locks-Coelho LD (2013) Increase of extracellular glutamate concentration increases its oxidation and diminishes glucose oxidation in isolated mouse hippocampus: reversible by TFB-TBOA. J Neurosci Res 91:1059–1065 [DOI] [PubMed] [Google Scholar]
- 138.Goubert E, Mircheva Y, Lasorsa FM, Melon C, Profilo E, Sutera J, Becq H, Palmieri F, Palmieri L, Aniksztejn L, Molinari F (2017) Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front Cell Neurosci 11:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL (2011) 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed 24:943–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu AC, Drejer J, Hertz L, Schousboe A (1983) Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem 41:1484–1487 [DOI] [PubMed] [Google Scholar]
- 141.Shank RP, Bennett GS, Freytag SO, Campbell GL (1985) Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res 329:364–367 [DOI] [PubMed] [Google Scholar]
- 142.Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation - where do all the carbons go? J Neurochem 131:399–406 [DOI] [PubMed] [Google Scholar]
- 143.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC (1997) Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci 17:932–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB (1998) Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol 53:355–369 [DOI] [PubMed] [Google Scholar]
- 146.Lee ML, Martinez-Lozada Z, Krizman EN, Robinson MB (2017) Brain endothelial cells induce astrocytic expression of the glutamate transporter GLT-1 by a Notch-dependent mechanism. J Neurochem 143:489–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, Markus NM, McQueen J, Hampton DW, Torvell M, Tiwari SS, McKay S, Eraso-Pichot A, Zorzano A, Masgrau R, Galea E, Chandran S, Wyllie DJA, Simpson TI, Hardingham GE (2017) Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun 8:15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N, Korsak A, Zwicker J, Teschemacher AG, Ackland GL, Funk GD, Kasparov S, Abramov AY, Gourine AV (2015) Functional Oxygen Sensitivity of Astrocytes. J Neurosci 35:10460–10473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316 [DOI] [PubMed] [Google Scholar]
- 150.Denton RM, McCormack JG (1980) The role of calcium in the regulation of mitochondrial metabolism. Biochem Soc Trans 8:266–268 [DOI] [PubMed] [Google Scholar]
- 151.Denton RM, McCormack JG (1985) Physiological role of Ca2+ transport by mitochondria. Nature 315:635. [DOI] [PubMed] [Google Scholar]
- 152.Denton RM, McCormack JG, Edgell NJ (1980) Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J 190:107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Denton RM, Randle PJ, Martin BR (1972) Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128:161–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Denton RM, Richards DA, Chin JG (1978) Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J 176:899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McKenna MC (2012) Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem Res 37:2613–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rimmele TS, de Castro Abrantes H, Wellbourne-Wood J, Lengacher S, Chatton JY (2018) Extracellular Potassium and Glutamate Interact To Modulate Mitochondria in Astrocytes. ACS chemical neuroscience 9:2009–2015 [DOI] [PubMed] [Google Scholar]
- 157.Whitelaw BS, Robinson MB (2013) Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Frontiers in endocrinology 4:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Azarias G, Perreten H, Lengacher S, Poburko D, Demaurex N, Magistretti PJ, Chatton JY (2011) Glutamate Transport Decreases Mitochondrial pH and Modulates Oxidative Metabolism in Astrocytes. J Neurosci 31:3550–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Perreten Lambert H, Zenger M, Azarias G, Chatton JY, Magistretti PJ, Lengacher S (2014) Control of mitochondrial pH by uncoupling protein 4 in astrocytes promotes neuronal survival. J Biol Chem 289:31014–31028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shen Y, Tian Y, Shi X, Yang J, Ouyang L, Gao J, Lu J (2014) Exposure to high glutamate concentration activates aerobic glycolysis but inhibits ATP-linked respiration in cultured cortical astrocytes. Cell Biochem Funct 32:530–537 [DOI] [PubMed] [Google Scholar]
- 161.Winkler U, Seim P, Enzbrenner Y, Kohler S, Sicker M, Hirrlinger J (2017) Activity-dependent modulation of intracellular ATP in cultured cortical astrocytes. J Neurosci Res 95:2172–2181 [DOI] [PubMed] [Google Scholar]
- 162.Magistretti PJ, Allaman I (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron 86:883–901 [DOI] [PubMed] [Google Scholar]
- 163.Barros LF, Brown A, Swanson RA (2018) Glia in brain energy metabolism: A perspective. Glia 66:1134–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Juaristi I, Contreras L, Gonzalez-Sanchez P, Perez-Liebana I, Gonzalez-Moreno L, Pardo B, Del Arco A, Satrustegui J (2019) The Response to Stimulation in Neurons and Astrocytes. Neurochem Res 44:2385–2391 [DOI] [PubMed] [Google Scholar]
- 165.Deitmer JW, Theparambil SM, Ruminot I, Noor SI, Becker HM (2019) Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Frontiers in neuroscience 13:1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Magi S, Lariccia V, Castaldo P, Arcangeli S, Nasti AA, Giordano A, Amoroso S (2012) Physical and functional interaction of NCX1 and EAAC1 transporters leading to glutamate-enhanced ATP production in brain mitochondria. PLoS One 7:e34015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Magi S, Arcangeli S, Castaldo P, Nasti AA, Berrino L, Piegari E, Bernardini R, Amoroso S, Lariccia V (2013) Glutamate-induced ATP synthesis: relationship between plasma membrane Na+/Ca2+ exchanger and excitatory amino acid transporters in brain and heart cell models. Mol Pharmacol 84:603–614 [DOI] [PubMed] [Google Scholar]
- 168.Magi S, Piccirillo S, Amoroso S (2019) The dual face of glutamate: from a neurotoxin to a potential survival factor-metabolic implications in health and disease. Cellular and molecular life sciences : CMLS 76:1473–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145 [DOI] [PubMed] [Google Scholar]
- 170.Stobart JL, Anderson CM (2013) Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front Cell Neurosci 7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Weber B, Barros LF (2015) The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harbor perspectives in biology 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, Espindola de Freitas A, Braga de Freitas G, Goncalves RA, Gupta D, Gupta R, Ha SR, Hemming IA, Jaggar M, Jakobsen E, Kumari P, Lakkappa N, Marsh APL, Mitlohner J, Ogawa Y, Kumar PR, Ribeiro FC, Salamian A, Saleem S, Sharma S, Silva JM, Singh S, Sulakhiya K, Tefera TW, Vafadari B, Yadav A, Yamazaki R, Seidenbecher CI (2019) The energetic brain - A review from students to students. J Neurochem 151:139–165 [DOI] [PubMed] [Google Scholar]
- 173.Alberini CM, Cruz E, Descalzi G, Bessieres B, Gao V (2018) Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 66:1244–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bak LK, Walls AB, Schousboe A, Waagepetersen HS (2018) Astrocytic glycogen metabolism in the healthy and diseased brain. J Biol Chem 293:7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Albers GW, Goldberg MP, Choi DW (1992) Do NMDA antagonists prevent neuronal injury? Yes. Arch Neurol 49:418–420 [DOI] [PubMed] [Google Scholar]
- 176.McDonald JW, Johnston MV (1990) Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41–70 [DOI] [PubMed] [Google Scholar]
- 177.Ikonomidou C, Turski L (2002) Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic injury?? Lancet Neurology 1:383–386. [DOI] [PubMed] [Google Scholar]
- 178.Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188 [DOI] [PubMed] [Google Scholar]
- 179.Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM (2002) Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem 81:207–217 [DOI] [PubMed] [Google Scholar]
- 180.Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149 [DOI] [PubMed] [Google Scholar]
- 181.Bambrick L, Kristian T, Fiskum G (2004) Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res 29:601–608 [DOI] [PubMed] [Google Scholar]
- 182.Shih EK, Robinson MB (2018) Role of Astrocytic Mitochondria in Limiting Ischemic Brain Injury? Physiology (Bethesda) 33:99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ito U, Hakamata Y, Kawakami E, Oyanagi K (2009) Degeneration of astrocytic processes and their mitochondria in cerebral cortical regions peripheral to the cortical infarction: heterogeneity of their disintegration is closely associated with disseminated selective neuronal necrosis and maturation of injury. Stroke 40:2173–2181 [DOI] [PubMed] [Google Scholar]
- 184.Pekny M, Pekna M (2016) Reactive gliosis in the pathogenesis of CNS diseases. Biochim Biophys Acta 1862:483–491 [DOI] [PubMed] [Google Scholar]
- 185.Shimada IS, LeComte MD, Granger JC, Quinlan NJ, Spees JL (2012) Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci 32:7926–7940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I, Dimou L, Gotz M (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16:580–586 [DOI] [PubMed] [Google Scholar]
- 187.Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O, Kokaia Z, Frisen J (2014) A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science 346:237–241 [DOI] [PubMed] [Google Scholar]
- 188.Becerra-Calixto A, Cardona-Gomez GP (2017) The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci 10:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP (1995) Reduced postischemic expression of a glial glutamate transporter, GLT-1, in the rat hippocampus. Exp Brain Res 103:51–58 [DOI] [PubMed] [Google Scholar]
- 190.Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS (2005) Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology 49:703–714 [DOI] [PubMed] [Google Scholar]
- 191.Yeh TH, Hwang HM, Chen JJ, Wu T, Li AH, Wang HL (2005) Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis 18:476–483 [DOI] [PubMed] [Google Scholar]
- 192.Krzyzanowska W, Pomierny B, Filip M, Pera J (2014) Glutamate transporters in brain ischemia: to modulate or not? Acta Pharmacol Sin 35:444–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zelenaia OA, Robinson MB (2000) Degradation of glial glutamate transporter mRNAs is selectively blocked by inhibition of cellular transcription. J Neurochem 75:2252–2258. [DOI] [PubMed] [Google Scholar]
- 194.Gonzalez-Gonzalez IM, Garcia-Tardon N, Gimenez C, Zafra F (2008) PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia 56:963–974 [DOI] [PubMed] [Google Scholar]
- 195.Sheldon AL, Gonzalez MI, Krizman-Genda EN, Susarla BT, Robinson MB (2008) Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int 53:296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ibanez I, Diez-Guerra FJ, Gimenez C, Zafra F (2016) Activity dependent internalization of the glutamate transporter GLT-1 mediated by beta-arrestin 1 and ubiquitination. Neuropharmacology 107:376–386 [DOI] [PubMed] [Google Scholar]
- 197.Quintana DD, Garcia JA, Sarkar SN, Jun S, Engler-Chiurazzi EB, Russell AE, Cavendish JZ, Simpkins JW (2019) Hypoxia-reoxygenation of primary astrocytes results in a redistribution of mitochondrial size and mitophagy. Mitochondrion 47:244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Benediktsson AM, Schachtele SJ, Green SH, Dailey ME (2005) Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods 141:41–53 [DOI] [PubMed] [Google Scholar]
- 199.O’Donnell JC, Jackson JG, Robinson MB (2016) Transient Oxygen/Glucose Deprivation Causes a Delayed Loss of Mitochondria and Increases Spontaneous Calcium Signaling in Astrocytic Processes. J Neurosci 36:7109–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Owens K, Park JH, Gourley S, Jones H, Kristian T (2015) Mitochondrial dynamics: cell-type and hippocampal region specific changes following global cerebral ischemia. J Bioenerg Biomembr 47:13–31 [DOI] [PubMed] [Google Scholar]
- 201.Kubli DA, Gustafsson AB (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111:1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Russo E, Napoli E, Borlongan CV (2018) Healthy mitochondria for stroke cells. Brain Circ 4:95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Berridge MV, Schneider RT, McConnell MJ (2016) Mitochondrial Transfer from Astrocytes to Neurons following Ischemic Insult: Guilt by Association? Cell Metab 24:376–378 [DOI] [PubMed] [Google Scholar]
- 206.Berridge MV, Herst PM, Rowe MR, Schneider R, McConnell MJ (2018) Mitochondrial transfer between cells: Methodological constraints in cell culture and animal models. Anal Biochem 552:75–80 [DOI] [PubMed] [Google Scholar]
- 207.Torralba D, Baixauli F, Sanchez-Madrid F (2016) Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y (2004) Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 65:1008–1015 [DOI] [PubMed] [Google Scholar]
- 209.Kaasik A, Safiulina D, Zharkovsky A, Veksler V (2007) Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 292:C157–163 [DOI] [PubMed] [Google Scholar]
- 210.Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kuhn R, Hartmann J, Karl RM, Konnerth A, Korn T, Wurst W, Merkler D, Lichtenthaler SF, Perocchi F, Misgeld T (2019) Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci 22:1731–1742 [DOI] [PubMed] [Google Scholar]
- 211.Sonnewald U, Rae C (2010) Pyruvate carboxylation in different model systems studied by (13)C MRS. Neurochem Res 35:1916–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC (2014) Glutamate metabolism in the brain focusing on astrocytes. Advances in neurobiology 11:13–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schousboe A, Waagepetersen HS, Sonnewald U (2019) Astrocytic pyruvate carboxylation: Status after 35 years. J Neurosci Res 97:890–896 [DOI] [PubMed] [Google Scholar]
- 214.Haberg A, Qu H, Saether O, Unsgard G, Haraldseth O, Sonnewald U (2001) Differences in neurotransmitter synthesis and intermediary metabolism between glutamatergic and GABAergic neurons during 4 hours of middle cerebral artery occlusion in the rat: the role of astrocytes in neuronal survival. J Cereb Blood Flow Metab 21:1451–1463 [DOI] [PubMed] [Google Scholar]
- 215.Brekke EM, Morken TS, Wideroe M, Haberg AK, Brubakk AM, Sonnewald U (2014) The pentose phosphate pathway and pyruvate carboxylation after neonatal hypoxic-ischemic brain injury. J Cereb Blood Flow Metab 34:724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Brekke E, Berger HR, Wideroe M, Sonnewald U, Morken TS (2017) Glucose and Intermediary Metabolism and Astrocyte-Neuron Interactions Following Neonatal Hypoxia-Ischemia in Rat. Neurochem Res 42:115–132 [DOI] [PubMed] [Google Scholar]
- 217.Badawi Y, Pal R, Hui D, Michaelis EK, Shi H (2015) Ischemic tolerance in an in vivo model of glutamate preconditioning. J Neurosci Res 93:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim AY, Jeong KH, Lee JH, Kang Y, Lee SH, Baik EJ (2017) Glutamate dehydrogenase as a neuroprotective target against brain ischemia and reperfusion. Neuroscience 340:487–500 [DOI] [PubMed] [Google Scholar]
- 219.Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578 [DOI] [PubMed] [Google Scholar]
- 220.Ding S (2014) Ca(2+) signaling in astrocytes and its role in ischemic stroke. Advances in neurobiology 11:189–211 [DOI] [PMC free article] [PubMed] [Google Scholar]