

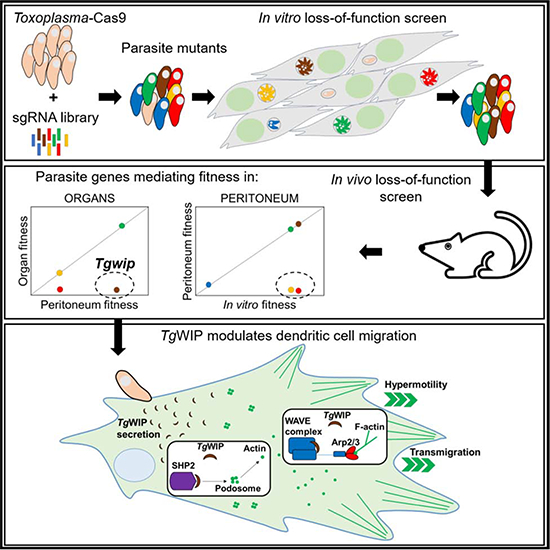
SUMMARY
Toxoplasma can reach distant organs, especially the brain, leading to a lifelong chronic phase. However, genes involved in related in vivo processes are currently unknown. Here, we use focused CRISPR libraries to identify Toxoplasma genes that affect in vivo fitness. We focus on TgWIP whose deletion affects Toxoplasma dissemination to distant organs. We show that TgWIP is secreted into the host cell upon invasion and interacts with the host WAVE regulatory complex and SHP2 phosphatase, both of which regulate actin dynamics. TgWIP affects the morphology of dendritic cells and mediates the dissolution of podosomes, which dendritic cells use to adhere to extracellular matrix. TgWIP enhances the motility and transmigration of parasitized dendritic cells, likely explaining its effect on in vivo fitness. Our results provide a framework for systemic identification of Toxoplasma genes with in vivo effects at the site of infection or on dissemination to distant organs, including the brain.
Keywords: Loss-of-function screen, Toxoplasma gondii, dendritic cell motility, migration, actin, WAVE-complex, dissemination
eTOC Blurb
• Sangaré et al. use a CRISPR screen to determine the in vivo fitness of Toxoplasma genes. They identify a secreted protein TgWIP, which determines dendritic cell motility and transmigration in the infected host. In mice, TgWIP determines T. gondii dissemination from the site of infection to distant organs.
Graphical Abstarct
INTRODUCTION
Toxoplasma can cause life-threating disease in the developing fetus and immunocompromised individuals, such as those suffering from AIDS, and blindness in otherwise healthy individuals (Desmonts and Couvreur, 1974; Holland, 1999; Munoz et al., 2011). The acute stage of infection is caused by rapidly dividing tachyzoites, which disseminate from the site of infection to distant organs. Eventually, the parasite converts to encysted bradyzoites, especially in the brain, that underlie the lifelong chronic phase of the infection. To survive and replicate in its host and reach the brain, Toxoplasma needs to evade the host immune response, acquire nutrients from its host, disseminate to distant organs from the site of infection, survive and replicate in those organs and convert to encysted bradyzoites. The complete set of Toxoplasma genes involved in these processes is currently unknown.
Toxoplasma replicates within a parasitophorous vacuole (PV), which is separated from the host cell cytoplasm by the PV membrane (PVM). The key to Toxoplasma’s successful co-option of host cells are proteins secreted from its rhoptry and dense granule organelles. Rhoptry bulb proteins (ROPs) are secreted into the host cell cytoplasm upon invasion while dense granule proteins (GRAs) are constitutively secreted once Toxoplasma has formed its PV. Some GRAs are secreted beyond the PVM into the host cell, most likely through a PVM-localized translocon consisting of the Toxoplasma proteins MYR1/2/3 (Franco et al., 2016; Marino et al., 2018). ROPs and GRAs are involved in modulating host cell signaling pathways, evasion of host immune responses, and Toxoplasma pathogenesis (reviewed in (Hakimi et al., 2017)).
Most Toxoplasma genes that determine virulence in mice have been identified using classical genetics: strains that differ in virulence were crossed in cats, F1 progeny were assessed for virulence in mice, and the parasite loci associated with virulence defects were identified using quantitative trait locus mapping. This led to the discovery of ROP18 (encoding a secreted kinase) (Saeij et al., 2006; Taylor et al., 2006) and ROP5 (encoding an expanded family of pseudokinases) (Behnke et al., 2011; Reese et al., 2011). Proteins encoded by these genes are highly polymorphic and determine strain differences in virulence in mice by cooperatively blocking, together with ROP17 and GRA7, the IFNγ-induced immunity-related GTPases (IRGs), which can bind and subsequently vesiculate the PVM eventually leading to the death of the parasite (Behnke et al., 2012; Fleckenstein et al., 2012; Khaminets et al., 2010; Martens et al., 2005; Niedelman et al., 2012; Zhao et al., 2009). However, genes involved in virulence and host cell modulation that are not different between Toxoplasma strains cannot be discovered with this method. Furthermore, virulence studies, which are mostly based on measuring host mortality, are unlikely to identify parasite genes that have more subtle effects but are still less fit than wild-type parasites. For example, parasite mutants that can no longer disseminate to the brain could still kill the host if replication is unaffected. Thus, although multiple ROPs, GRAs, and other genes have been individually investigated for their roles in virulence and formation of tissue cysts (Bai et al., 2018; Fox et al., 2016; Knoll, 2016; Rommereim et al., 2016), for many of these genes it is unknown what parasite function is defective and leading to the observed phenotype. Large scale screens are therefore needed to determine the role of individual Toxoplasma genes in immune evasion, dissemination, survival in distant organs and other processes that determine the parasite’s success in reaching the brain.
We recently described a CRISPR/Cas9 whole genome loss-of-function screen to identify Toxoplasma genes that determine fitness in cultured human foreskin fibroblasts (HFFs) (Sidik et al., 2016). Here we adapted this screen to show that focused loss-of-function screens can be used to identify Toxoplasma genes that determine in vivo fitness at the site of infection or at distant organs. Furthermore, we show that these screens can start to unravel the specific cellular functions modulated by individual Toxoplasma effectors. We show that one such effector encodes a yet unidentified rhoptry protein, TgWIP, that enhances the motility and transmigration of dendritic cells (DCs) likely explaining its effect on Toxoplasma in vivo fitness.
RESULTS
A Toxoplasma In Vivo Loss-of-Function Screen
To establish a chronic infection in its host, Toxoplasma needs to survive at the site of infection, disseminate, cross biological barriers, and evade immune responses. Our goal was to establish in vivo loss-of-function screens that could identify Toxoplasma genes involved in these processes. Large scale in vivo loss-of-function screens with bacterial pathogens have shown that such screens are often limited by in vivo bottlenecks (Cole et al., 2017; Stephens et al., 2015). If only a small fraction of the initial pool of mutants colonizes distant organs, the mutants that colonize the organs will be a random sample from the initial pool. It is currently unknown what the selective pressure (bottleneck) is after infection of mice with Toxoplasma. However, bottlenecks are likely much larger after oral infection compared to intraperitoneal (i.p.) infection, and much larger after infection with relatively avirulent parasite strains that can be rapidly eliminated by the host immune response. We therefore decided to first test a focused library of loss-of-function mutants generated in the virulent RH-Cas9 strain (Sidik et al., 2016) in i.p. infections. Our previously published whole-genome single guide RNA (sgRNA) library targeting all Toxoplasma genes contains built-in barcodes that can be used to amplify specific sub-libraries (see Table S1 for the identity of the 8 sub-libraries that can be amplified). We assembled the smallest sub-library consisting of 2,170 sgRNAs targeting 217 genes enriched in GRAs, but likely containing many other genes (Figure 1A). This sub-library was transfected into parasites constitutively expressing Cas9 and passaged three times in selective medium in HFFs. To determine which of the 217 targeted genes were important for in vitro fitness, sgRNAs amplified from parasite DNA isolated after the 3rd passage were sequenced and their relative abundance was compared to the input library. The average log2-fold change in abundance of sgRNAs in the input vs. the 3rd passage in HFFs is referred to as the in vitro fitness score. The in vitro fitness scores of the 217 genes was highly correlated (r = 0.85) with previously determined fitness scores in human foreskin fibroblasts (HFFs) (Sidik et al., 2016), highlighting the reproducibility of these screens (Figure 1B).
Figure 1. Toxoplasma gondii in vivo loss of function screen using a CRISPR/Cas9 sgRNA library.
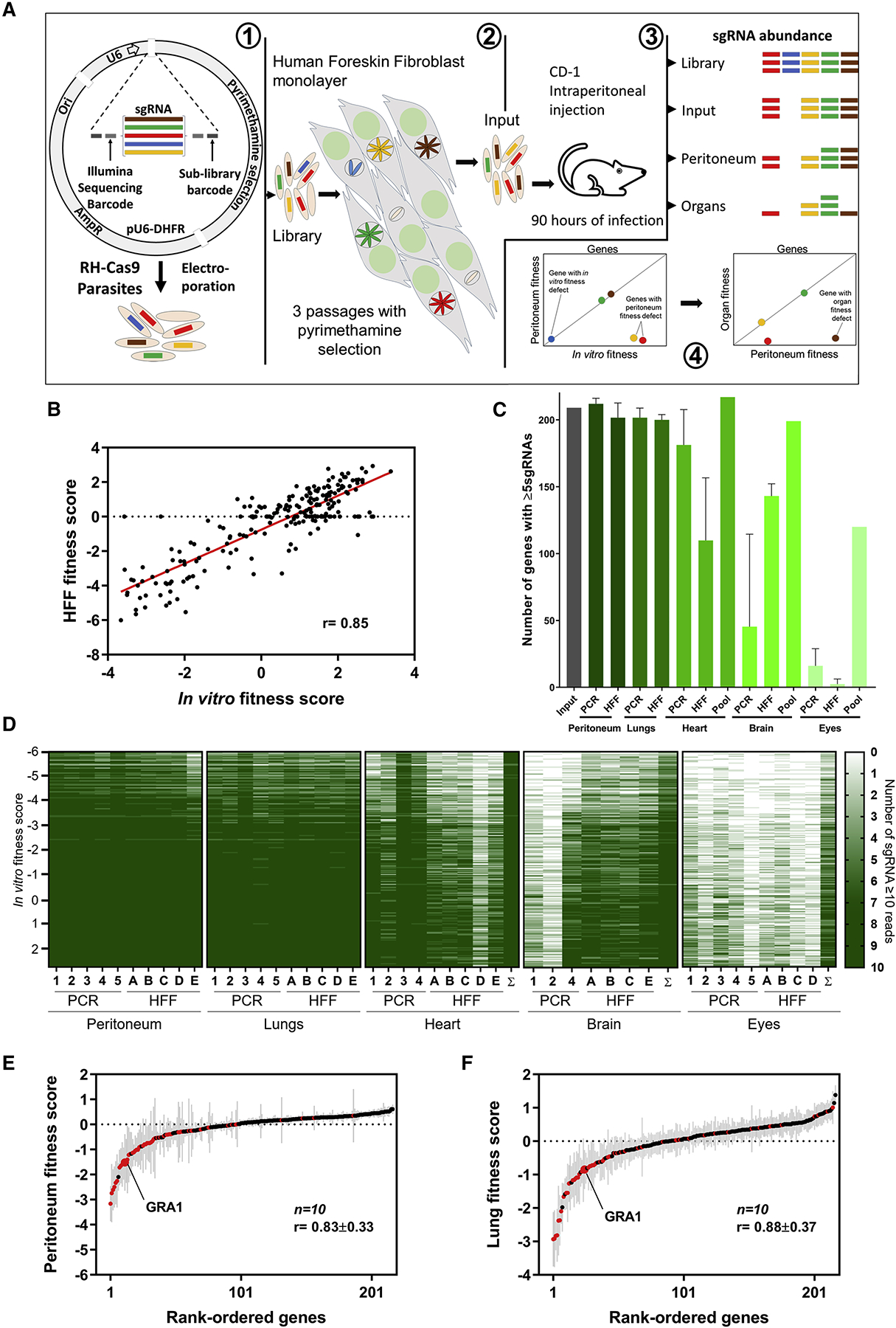
(A) 1) RH-Cas9 parasites are transfected with plasmids representing 50× coverage of a sub-library of sgRNAs. 2) Three in vitro passages in HFFs under pyrimethamine selection will remove parasites that integrated plasmids with sgRNAs targeting essential genes and non-transfected parasites. 3) CD-1 mice are i.p. injected with the mutant pool and 90 h later the mice are euthanized, organs and peritoneal lavage collected, genomic DNA extracted, and sgRNAs amplified and sequenced. 4) From the sgRNA in all the samples, the fitness score for in vitro, peritoneum and organs is calculated to identify in vivo fitness conferring genes. (B) Correlation between our in vitro fitness score vs. the published HFF fitness score (Sidik et al., 2016). (C) We obtained sgRNA abundance levels from 10 peritoneum and lung samples, 9 hearts, 7 brains, and 9 eye samples. The average number of genes (+SD) that have at least 5 sgRNAs with at least 10 reads for each sample when sgRNAs were directly amplified by PCR or from parasites amplified after HFF culture or from data pooled from all mice. (D) Genes with 7 or more sgRNAs amplified from the organ are colored dark green otherwise the gene is colored in a graded green scale based on how many sgRNAs were amplified. The genes are ranked according to the HFF fitness score with the genes on top having the lowest fitness score (Sidik et al., 2016). The sample sum (Σ) represents the pool of all reads from all mice of the same organ. (E) and (F) Rank-ordered genes according to the peritoneum or lung fitness scores. Genes that have fitness score of <−1.25 in HFF are indicated in red color. The grey lines indicate the standard error of averages (n=10).
The pool of mutants harvested from the 3rd passage in HFFs was used to infect mice and perform in vivo loss-of-function screens. A pilot experiment in C57BL/6J mice with ~2,500× coverage of each mutant in the library led to the death of mice after 60 h; however, at that time point few parasites were present in lungs, heart, and brain. Further, sequencing of the sgRNAs, PCR-amplified from DNA isolated from these organs, showed that the Toxoplasma mutants present in these organs were a random sample of the initial pool of mutants (not shown). C57BL/6 mice are extremely susceptible to Toxoplasma (Araujo et al., 1976; Hassan et al., 2019; Klesius and Hinds, 1979; Tuttle et al., 2018) and a high-dose infection with Toxoplasma is needed to prevent random disappearance of mutants because of a strong in vivo bottleneck. We therefore repeated the screen in the more resistant CD-1 mouse strain (Klesius and Hinds, 1979; Tuttle et al., 2018) in which the parasites would have more time to disseminate. We i.p. injected 20 CD-1 mice, each with 1×107 viable loss-of-function parasite mutants collected after the 3rd passage in HFFs and representing ~4,600× coverage of the 2,170 unique sgRNAs. Seven mice were used to examine pathology and parasite load in the different organs on different days post infection (p.i.). On day one the liver is the most infected organ, followed by the spleen, probably because these are intra-peritoneal organs that are in close proximity to the site of infection and because of their function in filtering blood. After three days the infection of the lungs becomes more prominent, but fewer parasites are detected in the heart and brain (Figure S1A). Parasites were mostly detected inside tissues, sometimes inside macrophages (Figure S1B), and rarely in blood vessels. When the first mouse died, we euthanized all other mice (~90 h p.i.). Because we were interested in parasite genes affecting survival at the site of infection or dissemination to distant organs, we decided to focus on peritoneum and non-peritoneal organs (lungs, heart, brain, and eyes).
For the amplification of sgRNAs from DNA we used two different methods: (i) we directly amplified sgRNAs from each organ from five mice (PCR, Figure 1C), or (ii) we expanded the parasites from various organs from another five mice, by innoculating HFFs with suspensions from each organ (HFF, Figure 1C). Upon lysis of the HFF monolayer parasite DNA was isolated and sgRNAs amplified and sequenced. Our data show that 95 ± 4% and 93 ± 2% (mean of 10 mice ±SD) of the 217 genes were represented by at least 5 sgRNAs in individual samples of the peritoneum and lungs, respectively, using either method indicating that there was no strong bottleneck (Figure 1C). The presence of mutants for a given gene in each samples is reflected in the relative abundance of sgRNAs. Coverage profiles for sgRNAs obtained from direct PCR from organs vs. parasite amplification via HFF culture followed by PCR were similar for peritoneum and lungs indicating that both methods are reliable (PCR vs. HFF, Figure 1D). However, fewer sgRNAs were recovered from the other organs, especially from the eyes (Figure 1C), indicating that at 90 h p.i. there is a stronger bottleneck to colonize heart, brain and eyes, compared to the lungs. For samples from the heart and eye direct PCR consistently amplified more unique sgRNAs while for samples from the brain amplifying the parasites first in HFFs recovered more unique sgRNAs. This difference is likely due to the difficulty in homogenizing heart and eye tissues.
Ranking the genes according to the in vitro fitness score (Sidik et al., 2016), it was evident that sgRNAs that were not amplified in the peritoneum and lungs target genes important for in vitro fitness (low in vitro fitness scores). However, for many samples from heart, brain, and eye even sgRNAs against genes dispensable in vitro fitness failed to amplify (Figure 1D), consistent with a severe bottleneck for parasites colonizing these organs within 90 h. To account for this bottleneck we pooled the sgRNA counts from all mice for samples from brain, heart and eye (Figure 1C, D). For each mouse we calculated the relative abundance of sgRNAs in the peritoneum vs. input (the parasite pool used to infect the mice) (peritoneal fitness) and lungs vs. input (lung fitness). Among the ten mice the fitness scores were highly reproducible (r = 0.83 ± 0.33 for the peritoneum and r = 0.88 ± 0.37 for the lungs (mean ± SD, n=10)) (Figure 1E, F). Parasite mutants that do not survive in the peritoneum will generally be absent from the lungs. Indeed, the correlation between peritoneal and lung fitness scores in the same mouse were high (r = 0.77 ± 0.1 (mean ± SD, n=10)). From the pooled data for heart, brain and eyes, we calculated the relative abundance of sgRNAs in each organ vs. input (heart fitness, brain fitness, and eye fitness). These fitness scores derived from the pooled data were also highly correlated (mean r = 0.69 ± 0.1) with the average peritoneal and lung fitness scores (Table S2). Overall, these data indicate that the in vivo fitness scores obtained are highly reproducible and that there is a high correlation between the fitness scoress in different organs.
Identification of Toxoplasma Genes that Determine Survival at the Site of Infection
A significant fraction of the 217 genes targeted by our sgRNA library are important for fitness in vitro in HFFs as they have negative in vitro fitness scores (Figure 1B and (Sidik et al., 2016)). We wanted to exclude genes with negative in vitro fitness scores from our analysis as the biological reason for their impact on parasite fitness can be studied in vitro. We used our published dataset that determined in vitro fitness scores for all Toxoplasma genes (Sidik et al., 2016) to determine an appropriate fitness score cut-off that identifies in vitro fitness-conferring genes. To do this we curated a list of 497 control genes (Table S3) that are not expressed in tachyzoites grown in HFFs or macrophages (ToxoDB, (Melo et al., 2013)), and mainly expressed in other life stages (e.g. in sexual stages or tissue cysts), since these genes are unlikely to have a fitness defect in in vitro screens. We used Chi-square tests to determine that a fitness score cut-off of −1.25 significantly excluded these 497 control genes from 3,300 fitness-conferring genes (genes with a fitness score less than −1.25). 59 of the 217 genes in our library had an in vitro fitness score less than −1.25 and were excluded from further analysis (Table S4).
We ranked the remaining 158 genes based on the peritoneum fitness score in each mouse and calculated the average rank for each gene. Genes with a rank of at least 2 standard deviations (SD) below the average rank (Z-score ≥ 2) were considered as affecting peritoneum fitness (Table 1). We calculated the lung fitness score and lung Z-scores for the 158 genes and for heart, brain and eyes, we ranked genes according to the fitness score derived from the pooled data of all mice in these organs. As expected, 86% of the peritoneum fitness genes also had negative fitness scores in lungs, heart, brain and eyes. Notable peritoneum fitness genes were: the BT1 gene (Tg266366) encoding a transporter-family member involved in the uptake of folates, an important metabolite in the eukaryotic cell biosynthesis (Massimine et al., 2005). TgDHHC8 (Tg255650) an ER/endosome-localized protein S-acyl transferase, responsible for protein palmitoylation (Frenal et al., 2013) and Tg205350 (GSN1/SUR4 family protein) a palmitoylated protein (Foe et al., 2015), likely involved in long-chain fatty acid elongation producing the 26-carbon precursors for ceramide and sphingolipid synthesis (Oh et al., 1997). Palmitoylation has been shown to be important for virulence and dissemination of Toxoplasma and other eukaryotic parasites (Brown et al., 2017). GRA22 (Tg215220) involved in regulating Toxoplasma egress in HFFs (Okada et al., 2013). Three members of a family of genes only present in Toxoplasma, Toxoplasma family C, were also present in our list of peritoneum fitness hits but the function of this gene family is unknown.
Table 1. Genes with a peritoneum fitness defect.
Upper panel: the top 15 (out of 22 genes) peritoneum fitness conferring genes with a peritoneum Z-score ≥2 after removing the genes with an HFF fitness score ≤−1.25 (Sidik et al., 2016). Lower panel: organ fitness conferring genes with a lung Z-score ≥2 or a fitness score of ≤−1 in heart, brain and eyes. The 22 peritoneum fitness conferring genes were excluded. The genes are ranked according to the lung Z-score. The peritoneum and lung fitness scores are averages (n=10). Heart, brain, and eye fitness scores were calculated from the pooled sequence data of 9, 7 and 9 mice, respectively. See Table S4 for the complete list.
| Gene ID | Peritoneum fitness score | Peritoneum Z-score | Lung fitness score | Lung Z-score | Heart fitness score | Brain fitness score | Eye fitness score | Product description |
|---|---|---|---|---|---|---|---|---|
| Genes with a peritoneum fitness defect | ||||||||
| TGGT1_203030 | −1.2 | 20.5 | −1.6 | 17.1 | −0.5 | −1.8 | −5.6 | N-methyl-D-aspartate receptor |
| TGGT1_266366 | −2.1 | 18.0 | −2.0 | 15.7 | −1.6 | −1.8 | −8.2 | BT1 family protein |
| TGGT1_205350 | −0.9 | 3.3 | −1.2 | 11.3 | −1.8 | −2.3 | −1.7 | GNS1/SUR4 family protein, |
| TGGT1_269950 | −0.4 | 3.3 | −0.8 | 6.6 | −1.7 | −1.4 | −8.0 | Hypothetical protein |
| TGGT1_215220 | −0.5 | 3.0 | −0.6 | 5.1 | −1.3 | −1.2 | −1.7 | GRA22 |
| TGGT1_200590 | −0.2 | 2.5 | −0.4 | 3.3 | −1.1 | −0.2 | −0.8 | Toxoplasma gondii family C protein |
| TGGT1_321170 | −0.2 | 3.9 | −0.2 | 3.0 | −1.1 | −1.1 | −0.9 | Toxoplasma gondii family C protein |
| TGGT1_259940 | −0.5 | 3.3 | −0.5 | 2.6 | −1.5 | −1.5 | −0.6 | Hypothetical protein |
| TGGT1_307260 | −0.5 | 4.6 | −0.6 | 2.5 | −0.8 | −0.3 | −4.5 | Toxoplasma gondii family C protein |
| TGGT1_273905 | −0.3 | 2.4 | −0.2 | 2.4 | −0.8 | −1.3 | −3.1 | Hypothetical protein |
| TGGT1_228770 | −0.2 | 2.4 | −0.2 | 2.3 | −0.6 | −1.3 | −6.3 | Hypothetical protein |
| TGGT1_269420 | −0.3 | 3.8 | −0.1 | 1.4 | −1.4 | −0.9 | −3.0 | Hypothetical protein |
| TGGT1_297100 | −0.2 | 2.3 | −0.5 | 0.9 | −0.7 | −1.9 | −5.2 | Hypothetical protein |
| TGGT1_231625 | −0.1 | 2.1 | −0.1 | 0.9 | −1.0 | −1.1 | −2.7 | Hypothetical protein |
| TGGT1_255650 | −0.3 | 3.2 | −0.1 | 0.9 | −1.3 | −1.9 | −3.4 | TgDHHC8 |
| Genes with an organ fitness defect | ||||||||
| TGGT1_288650 | −0.1 | 0.1 | −0.9 | 7.9 | −1.1 | −1.0 | −5.4 | GRA12 |
| TGGT1_290700 | −0.1 | 0.6 | −0.5 | 6.4 | −1.0 | −0.5 | −1.9 | GRA25 |
| TGGT1_280380 | 0.3 | −1.6 | −0.3 | 4.0 | −1.3 | −0.3 | −1.4 | Poly (ADP-ribose) glycohydrolase |
| TGGT1_247520 | 0.1 | −0.2 | −0.3 | 3.1 | −0.9 | −1.1 | −1.9 | Hypothetical protein-TgWIP |
| TGGT1_254470 | −0.1 | 1.3 | −0.3 | 3.0 | −1.1 | −1.3 | −1.7 | MYR1 |
| TGGT1_224090 | −0.3 | 1.4 | −0.3 | 2.8 | −1.0 | −0.6 | −3.6 | Enoyl-CoA hydratase/isomerase |
| TGGT1_270240 | 0.1 | 0.4 | −0.2 | 2.4 | −0.8 | −1.2 | −2.9 | MAG1 |
| TGGT1_312420 | −0.2 | 1.2 | −0.3 | 1.3 | −1.2 | −2.1 | −2.0 | GRA38 |
| TGGT1_261400 | 0.0 | 0.3 | −0.4 | 0.9 | −1.8 | −2.4 | −2.1 | Hypothetical protein-PfVFT1 orthologue |
MYR1, and the secreted effectors GRA16 and GRA18, which were all shown to be important for virulence of a type II strain (Bougdour et al., 2013; Franco et al., 2016; He et al., 2018) did not have a significant peritoneum fitness effect in our screen. This is consistent with the fact that mice infected with RHΔmyr1 only have a slight delay in time till death suggesting that GRAs secreted beyond the PVM are less important for RH virulence (Franco et al., 2016). Alternatively, the high infectious dose might have obscured the role of MYR1 and secreted effectors in cells infected with multiple mutants.
We selected two candidate genes Tg269950 and GRA22 that seemed to be involved in peritoneum fitness and generated individual knockouts in the RH strain, which we confirmed by PCR analysis (Figure S2B). To confirm both as peritoneum fitness genes, we infected 5 CD-1 mice i.p. with a 1:1 ratio of wild type (GFP−) and knockout parasites (Δgra22 or ΔTg269950 (GFP+)) and determined their ratio in peritoneum and lungs six days p.i. Both knockouts significantly decreased in abundance in the peritoneum and lungs compared to wild type (Figure 2A, B). Thus our in vivo screen successfully identified parasite genes that determine in vivo fitness.
Figure 2. Validation of candidate genes from in vivo loss-of-function screen.

In vivo growth competition assay between wild type and (A) Δgra22-GFP+ or (B) ΔTg269950-GFP+ in CD-1 mice. Each column represents the average percentage (± SD) of knockout (GFP+) parasites vs. wild-type parasites in the total population of input, peritoneal and lungs for 5 different mice (n=1) 6 days after infection as determined by plaque assays. Asterisks (*) indicates significant difference: one-way ANOVA, Dunnett’s multiple comparisons test. Respective P values for peritoneum in A and B =0.009; 0.04. Respective P values for lungs in A and B =0.002; 0.04 when compared to the input. (C) In vivo growth competition assay between wild type and ΔTg261400-GFP+. Each column represents the average percentage (± SD) of ΔTg261400-GFP+ vs. wild-type parasites in the total population of input, peritoneum, lungs, heart and brain for 8 mice (2–3 mice each experiment, n=3) 6 days after infection as determined by plaque assay counting. Asterisks (*) indicates significant difference: one-way ANOVA, Dunnett’s multiple comparisons test. P values for: lungs =0.009, Heart =0.01 brain <0.0001. D) In vivo growth competition assay between wild type and ΔTg247520-GFP+ or ΔTg247520+Tg247520-GFP+ in CD-1 mice. Each column represents the average percentage (± SD) of ΔTg247520-GFP+ or ΔTg247520+Tg247520-GFP+ parasites vs. wild-type parasites in the total population of input, peritoneum, lungs, heart and brain for 6 different mice each (n=3, 2 mice each experiment) 6 days after infection as determined by plaque assay counting (GFP+ vs. GFP− plaques). Asterisks (*) indicates significant difference: Two-way ANOVA, Bonferroni’s multiple comparisons test. The respective P values are: lungs <0.0001, heart =0.0006, brain <0.0001. (E) In vivo growth competition assay between ΔTg247520-GFP+ and ΔTg247520+Tg247520-GFP+ parasites. Each column represents the average percentage (± SD) of ΔTg247520-GFP+ vs. ΔTg247520+Tg247520-GFP+ parasites in the total population of input, peritoneum, lungs, heart and brain for 6 different mice each (n=3, 3 mice each experiment) 6 days after infection as determined by IFA counting of HA+ vs. HA− parasites. Asterisks (*) indicates significant difference: one-way ANOVA, Dunnett’s multiple comparisons test. P values for lungs =0.02, brain =0.04, when compared to the input. Immunofluorescence assays on HFFs infected with parasites containing endogenously HA-tagged: (F) Tg261400, GRA7 was used as a PVM membrane marker. (G) Tg247520 in colocalization with the ROP2/3/4 as a marker for rhoptry organelles. (H) Immunofluorescence assay on HFFs infected with RHΔTg247520+Tg247520-GFP+ parasites. The white arrow indicates a cell infected with multiple parasites and presenting a cytosolic HA signal. The white dashed arrow indicates non-infected cell without cytosolic HA signal. Scale bar represents 10 μm. (I) The number of parasites per vacuole was measured in naïve C57BL/6J mouse BMDMs or BMDMs stimulated with IFNγ or IFNγ/TNFα for 24 h and subsequently infected with wild type or ΔTg247520 parasites (MOI = 0.5) for 24 h. A total of 100 to 200 vacuoles were analyzed per experiment. Data are displayed as average (± SD) values (n=3), ns (no significant difference) two-way ANOVA multiple comparisons.
Identification of Genes Important for Reaching and/or Surviving in Distant Organs
We next wanted to identify genes that are important to colonize distant organs (lungs, heart, brain and eyes). To identify these genes, we first excluded genes that mediate fitness in the peritoneum (Table 1 and Table S4) and subsequently identified genes that were significantly depleted in the lungs (Z-score ≥ 2) or with at least a 2-fold fitness defect in heart, brain and eyes (Table 1). Notable among these genes were: GRA25, which was previously shown to modulate host macrophage CCL2 and CXCL1 secretion and mediate Toxoplasma virulence in mice (Shastri et al., 2014). MYR1, which did not have a significant defect in the peritoneum but appeared important for fitness in distant organs. GRA12, which was recently shown to determine Toxoplasma virulence (Fox et al., 2019). GRA38, and MAG1, which are GRAs with unknown function were also important for fitness in distant organs.
We selected two uncharacterized genes Tg261400 and Tg247520 that seemed to be involved in reaching or surviving in distant organs but did not have a fitness defect in the peritoneum. We generated individual knockouts, complemented the ΔTg247520 parasites, and confirmed these parasites by PCR analysis (Figure S2C, 3B, 3C). To confirm that ΔTg261400 has a systemic fitness defect we performed in vivo competition assays by i.p. infecting 8 CD-1 mice (2–3 mice each experiment, n=3) with a 1:1 mixture of knockout (GFP+) and wild-type (GFP−) parasites and determined the ratio of wild type and knockout in peritoneum and organs 6 days p.i. Compared to the input, the average percentage of ΔTg261400 in the peritoneum did not significantly change. However, in lungs, heart and brain we observed a significant decrease in the average percentage of the ΔTg261400 parasites (Figure 2C). For ΔTg247520 we performed similar experiments but besides competing the knockout (GFP+) with wild type (GFP−) (Figure 2D) we also competed the complemented strain with wild type (Figure 2D) and the knockout with complemented parasites (both GFP+, distinguished by the HA-expression of the complemented strain) (Figure 2E) (2–3 mice each experiment, n=3). When ΔTg247520 was competed against wild-type parasites we observed a significant increase of ΔTg247520 in the peritoneum but no difference in distant organs. By contrast, the ΔTg247520 complemented parasites outcompeted wild type in peritoneum and distant organs (Figure 2D). ΔTg247520 was outcompeted by the complemented parasites in lungs, heart and brain but not in the peritoneum (Figure 2E). Overall, these data indicate that the ΔTg247520 parasites have similar fitness as the complemented parasites in the peritoneum but decreased fitness in distant organs. Furthermore, the presence of GFP and/or HXGPRT (hpt) in the knockout and complemented might have increased their fitness compared to the wild-type parasites used, which were GFP−/Δhpt.
To determine whether Tg261400 and Tg247520 code for secretory proteins that could directly modulate the host cell, we determined their subcellular localization by C-terminally tagging the endogenous gene with an HA-tag. Tg261400 has an ER, apical and basal vesicle localization profile but we did not detect any HA signal outside the parasite (Figure 2F). These results indicate that Tg261400 is unlikely a parasite effector that can directly modulate the host cell, but it might be involved in trafficking and/or function of other parasite proteins involved in mediating its dissemination to distant organs. Tg247520 showed a rhoptry localization profile, which was confirmed by co-staining with antibodies that detect ROP2, ROP3, and ROP4 (Figure 2G). Tg247520 was readily detected in the host cytoplasm of infected cells 2 h after invasion (Figure 2H). We therefore decided to focus on determining how Tg247520 affects the parasite’s ability to colonize distant organs. However, as other secreted ROPs have been shown to play a role in the evasion of the host immune response, we wanted to examine whether Tg247520 plays a role in the evasion of IFNγ-mediated growth inhibition. The growth of ΔTg247520 parasites was similar to wild type in naïve murine bone marrow derived macrophages (BMDM) and IFNγ-stimulated or IFNγ+TNFα-stimulated BMDM indicating that Tg247520 does not determine parasite resistance to IFNγ (Figure 2I).
Tg247520 Encodes for a Host WAVE Complex Interacting Protein
Because Tg247520 gets secreted into the host cytoplasm upon invasion we hypothesized that it might interact with host proteins. To determine putative host interaction partners of Tg247520, we engineered a stable HEK293-derived (TREX-293) cell line conditionally expressing Tg247520 with a C-terminal HA-FLAG epitope tag under the control of the tetracycline operator (Bougdour et al., 2013). Upon induction of Tg247520 through tetracycline addition, the protein accumulated mainly in the cytoplasm (data not shown). We immunoprecipitated Tg247520 using antibodies against the HA-tag. As a control we used a TREX-293 cell line conditionally expressing Toxoplasma GRA15. To control for potential spurious interactions detected by mass spectrometry, we refined the list of candidate host proteins which could have direct or indirect interaction with Tg247520 to those identified only in the Tg247520 overexpression condition and excluded proteins detected in the GRA15 immunoprecipitate (Table S5). Four components of the pentameric WAVE regulatory complex (WRC): WASF1/2/3, NCK1/2, CYFIP1/2, and ABI1/2 and the tyrosine phosphatase PTN11 (SHP2) were specifically identified in the Tg247520 immunoprecipitate. To confirm these interactions in cells infected with parasites, we infected the murine DC cell line (DC2.4) with parasites expressing HA-tagged Tg247520 and we harvested whole cell/parasite lysate 3 h p.i. We fractionated the lysate and immunoprecipitated Tg247520 using antibodies against the HA-tag. As a control we used cells infected with parasites expressing HA-tagged GRA15. We confirmed the interaction with three components of the WRC: CYFIP1, NCK2, and ABI1; and with the tyrosine phosphatases SHP2 (Table S5). The WRC controls actin cytoskeletal dynamics throughout the cell by stimulating the actin-nucleating activity of the Arp2/3 complex at distinct membrane sites (Chen et al., 2014). The WCR possesses distinct classes of ligands, which can activate formation and recruitment of the complex to the plasma membrane. One class of ligands named WIRS (WRC interacting receptor sequence) have a specific motif Φ-x-T/S-F-X-X (Φ = bulky hydrophobic residues; X = any residue) with a highly conserved interaction surface of the WCR (Chen et al., 2014). We identified a conserved WIRS motif FGTFVK in the amino acid sequence of Tg247520 and two SH3 consensus motifs, which are known to interact with the proline-rich regions of ABI2 and WASF1, and likely facilitate membrane recruitment and clustering of the WRC (Figure S3A). This WIRS motif is conserved in the orthologues of Tg247520 in other apicomplexans like Neospora caninum and Hammondia hammondi (Figure S3B). Taken together, Tg247520 possesses WIRS/SH3 motifs and is secreted into the host cytosol upon invasion where interacts with host WRC in both human and murine cells. Thus, we named Tg247520, TgWIP, for Toxoplasma gondii WAVE complex interacting protein.
TgWIP Modulates Murine DC Actin Dynamics
Toxoplasma can infect DCs and monocytes, modulate their motility and adhesive properties (Ueno et al., 2014; Weidner et al., 2013) and use them as Trojan horses to migrate to distant organs and cross biological barriers, such as the blood-brain-barrier. TgWIP, via its WIRS/SH3 motifs, could interact with the WRC and modulate host actin nucleation. We previously showed that upon Toxoplasma infection, both human and murine DCs lose podosomes, which are F-actin rich structures used by DCs to adhere to the extracellular matrix (Weidner et al., 2013).
To determine if TgWIP can modulate DC podosome formation, we infected primary murine bone marrow-derived DCs (BMDCs) and the murine DC2.4 cell line with wild type or ΔTgwip parasites for 4 h and visualized podosomes by staining for F-actin. Consistent with what we previously published, ~75% of uninfected BMDCs contained podosomes while only ~20% of BMDCs infected with wild-type parasites contained podosomes. In contrast to wild type-infected BMDCs, ΔTgwip parasite infection was unable to dissolve BMDCs podosomes (Figure 3A, B). For the DC2.4 cells the most drastic change was in the morphology of the cells. Uninfected cells were round-shaped and contained abundant actin-rich plasma membrane extensions and many podosomes. In contrast, cells infected with wild-type parasites appeared to spread out resulting in larger irregularly shaped cells that contained abundant F-actin filaments resembling “stress fibers” in absence of podosome structures (Figure 3C, D and E). Similar to what was observed in uninfected cells, ΔTgwip infected DC2.4 cells are small, round-shaped with actin-rich plasma membrane extensions and many small podosomes (Figure 3C, D and E). Taken together, these results show that TgWIP impacts DC actin dynamics resulting in significant cytoskeletal rearrangements with gross morphological changes and dissolution of podosomes.
Figure 3. TgWIP modulates DC morphology and presence of podosomes.

(A) Murine primary DCs or (B) murine DC2.4 cells were infected with wild type or ΔTgwip parasites for 4 h. The cell actin structure and podosomes (indicated by white arrows) were visualized with 488 Alexa-Fluor Phalloidin, the parasite with an antibody against SAG1 and the nucleus with DAPI. Scale bars represents 10 μm. (C) Quantification of the percentage of primary DCs containing podosomes 4 h p.i with wild type or ΔTgwip parasites. Asterisks (*) indicates significant difference: repeated measures one-way ANOVA, Tukey’s post-hoc test (D) The average cell area (± SD) and (E) average cell roundness (± SD) of uninfected, wild type-infected and ΔTgwip infected DC2.4 cells (n=3, 100 cells to 300 cells per replicate). Asterisks (*) indicates significant difference: one-way ANOVA, Dunnett’s multiple comparisons test. The respective P values are for: cell area =0.0021, and cell roundness =0.0006, when wild type-infected DCs are compared with uninfected DCs.
TgWIP Induces Hypermotility of Toxoplasma-Infected Human and Murine DCs
We previously showed that Toxoplasma-infected DCs become hypermotile (Weidner et al., 2013), which is consistent with the disappearance of podosomes shortly after infection as podosomes are used by these cells to strongly adhere to extracellular matrix (Olafsson et al., 2018). To investigate the impact of TgWIP on the motility of primary human and murine DCs, we infected cells with wild type, ΔTgwip or ΔTgwip+Tgwip complemented parasites (Figure S4A). As previously established (Fuks et al., 2012), DCs infected with wild-type parasites migrated longer distances compared to uninfected DCs (Figure 4A, B). Importantly, ΔTgwip-infected DCs exhibited significantly reduced average migration distances, while DCs infected with the ΔTgwip+Tgwip complemented parasites migrated similar distances compared to wild type-infected DCs (Figure 4A, B, Figure S4B). TgWIP dramatically affected DC hypermotility (cells migrating >150 μm). A highly significant reduction in the frequency of hypermotile cells was observed when DCs were challenged with ΔTgwip parasites, which was restored upon challenge with ΔTgwip+Tgwip complemented parasites for both human and mouse DCs (Figure 4C). We also observed a significant decrease in the average velocities of human and murine DCs infected with ΔTgwip parasites compared to DCs infected with wild type or ΔTgwip+Tgwip complemented parasites (Figure 4D). The enhanced velocity of Toxoplasma-infected human DCs appeared to be only partially mediated by TgWIP while the enhanced velocity of infected murine DCs was almost entirely dependent on TgWIP. These data show that TgWIP promotes hypermotility in Toxoplasma-infected DCs.
Figure 4. Impact of TgWIP on migrated distances and velocities of primary human and murine DCs.

(A) DCs were infected with freshly-egressed GFP+ wild type (WT), ΔTgwip or ΔTgwip+Tgwip complemented parasites or left unchallenged, as detailed in Materials and Methods. Accumulated migrated distances of unchallenged or Toxoplasma-infected primary human and murine DCs was recorded. Histograms show accumulated distances of 180 cells per condition, from 3 independent experiments (n = 3). Dotted and continuous lines show the median accumulated distance of unchallenged and Toxoplasma-infected DCs, respectively. (B) Representative motility plots of unchallenged or Toxoplasma-infected primary human and murine DCs. Data are representative of 3 independent experiments. (C) Frequency analyses of DCs with accumulated migrated distances > 150 μm, performed as in A (n=3). (D) Cell velocity analyses of unchallenged or Toxoplasma-infected primary human and murine DCs. Bar graphs show average velocity (± SD) from 3 independent experiments (n=3). Asterisks (*) indicate significant difference, Chi-square test (C), repeated measures one-way ANOVA, Tukey’s post-hoc test (D).
TgWIP is Implicated in the Transmigration of Toxoplasma-Infected Human and Murine DCs
One additional feature of the hypermigratory phenotype (Weidner and Barragan, 2014) is the enhanced transmigration of parasitized DCs across endothelial cell monolayers (Lambert et al., 2006). To investigate the role of TgWIP in this process we used flow cytometry to measure the transmigration frequencies of Toxoplasma-infected DCs in a trans-well assay, taking advantage of the fact that all our parasites express GFP (Figure S4C). The infection ratio of DCs was similar between the different Toxoplasma strains suggesting that TgWIP does not affect parasite invasion (Figure 5A). The transmigration frequency of Toxoplasma-challenged DCs was partially but significantly impeded upon deletion of TgWIP and rescued upon reconstitution of TgWIP expression (Figure 5B). Transmigration frequency analyses of the total DC population (GFP− and GFP+, Figure 5B) vs. Toxoplasma infected-DCs (GFP+, Figure 5C) yielded similar results consistent with the observation that non-infected bystander DCs (GFP−) did not transmigrate (Figure 5D). Together, these results are consistent with a role for TgWIP in enhancing human and murine DC transmigration.
Figure 5. Effects of TgWIP on the transmigration of primary human and murine DCs.

DCs were infected with freshly egressed GFP+ wild type (WT), ΔTgwip or ΔTgwip+Tgwip complemented parasites, incubated on transwell filters and transmigrated cells were assessed by flow cytometry as detailed in Materials and Methods. (A) Bar graphs show the average infection frequencies (± SD) of each parasite line. Data are from 3 independent experiments performed in duplicate (n=3). (B – D) Transmigration frequencies of unchallenged and Toxoplasma-challenged primary human and murine DCs related to total added cell numbers. For the different conditions, bar graphs show the average percentage (%, ± SD) of (B) total transmigrated DCs (GFP+ and GFP− cells), (C) Toxoplasma-infected DCs (GFP+ cells) and (D) non-infected bystander DCs (GFP− cells) from 3 independent experiments performed in duplicate (n=3). Asterisks (*) indicate significant difference, ns: non-significant difference: repeated measures one-way ANOVA, Tukey’s post-hoc test (A, B, C, D).
TgWIP is Important for Type II Parasite Dissemination and Virulence.
Given that we observed that TgWIP has a significant effect on DC hypermotility and transmigration the relatively small in vivo effect on dissemination was somewhat surprising. However, we previously reported that type I parasites (such as RH) rely more on extracellular dissemination compared to type II parasites (e.g. ME49), which rely more on dissemination via immune cells. Type II-infected DCs are also more motile compared to type I-infected DCs (Lambert et al., 2009). To determine the effect of TgWIP on dissemination of a type II strain we created an ME49ΔTgwip strain (Figure S5A). ME49ΔTgwip parasites were significantly less virulent compared to wild-type parasites (Figure 6A). Serology confirmed that all mice were infected (Figure S5B) but none of the 10 mice surviving infection with ME49ΔTgwip contained visible cysts in the brain while the brains of wild-type ME49 infected mice contained on average ~1,500 cysts (Figure 6B). However, PCR amplification of the multi-copy B1 parasite gene in five of the ten brains from mice infected with ME49ΔTgwip indicated that a small number of parasites reached the brains of these mice (Figure S5C).
Figure 6. TgWIP is important for type II parasite dissemination and virulence.

(A) Survival curve of CD-1 mice i.p. infected with 1,000 wild-type ME49 or ME49ΔTgwip parasites (n=2, 5 mice in each group for a total of 10 mice/group). Asterisks (*) indicates significant difference for the survival experiments using the log rank Mantel-Cox test. P value =0.001. (B) Cyst counts from brains of surviving mice in A. The graph represents the average (± SD) number of cysts detected by FITC-DBA staining in the brain of each mouse 27 days p.i. Asterisks (*) indicates significant difference, P value =0.001, non-parametric unpaired t-test. (C) The number of parasites per vacuole was measured in naive BMDMs or BMDMs stimulated with IFNγ or IFNγ/TNFα for 24 h and subsequently infected with wild type or ME49ΔTgwip parasites for 24 h. A total of 100 to 200 vacuoles were analyzed per experiment. Data are displayed as average (± SD) values (n=3), ns (no significant difference) two-way ANOVA multiple comparisons. (D) In vivo growth competition assay between wild type ME49 and ME49ΔTgwip parasites in CD-1 mice. Each column represents the average percentage (± SD) of parasites that are ME49ΔTgwip in the total population of input, peritoneum, lungs, and heart for 6 individual mice (n=2, 3 mice in each experiment for a total of 6 mice) 7 days after infection as determined by plaque assay counting (GFP+ vs. GFP− plaques). Asterisks (*) indicates significant difference: one-way ANOVA, Dunnett’s multiple comparisons test. P values for peritoneum =0.01, lungs =0.03, and heart =0.005, when compared to the input.
To confirm that TgWIP does not play a role in mediating type II strain survival in naïve or activated macrophages we compared parasite growth of wild type and ME49ΔTgwip parasites in naïve, IFNγ-stimulated, or IFNγ+TNFα-stimulated BMDM but observed no difference between parasites (Figure 6C). To further confirm that ME49ΔTgwip parasites do not have a defect surviving the immune response we performed an in vivo competition assay by injecting an equal ratio of wild-type ME49 and ME49ΔTgwip parasites i.p. in CD-1 mice. Seven days after infection significantly fewer ΔTgwip parasites were present in the lungs while no ΔTgwip parasites were detected in the heart (Figure 6D). Interestingly, more ΔTgwip parasites were detected in the peritoneum compared to wild-type parasites and in 4 out of 6 mice only ΔTgwip parasites were detected in the peritoneum (Figure 6D). These data further confirm that ΔTgwip parasites are not more susceptible to the host immune response but have a strong defect in dissemination out of the peritoneum.
DISCUSSION
We recently published the first CRISPR/Cas9 whole genome loss-of-function screen to identify Toxoplasma genes that determine fitness in vitro in HFFs (Sidik et al., 2016). Here, we adapted this screen to identify Toxoplasma genes that determine in vivo fitness in the mouse.
We confirmed GRA22 and Tg269950 to be fitness-conferring genes in the peritoneum. Deletion of GRA22 was shown to induce earlier spontaneous egress in HFFs and faster calcium ionophore-induced egress (Okada et al., 2013). In vivo, it has been shown that macrophages in the peritoneal exudate can elicit early Toxoplasma egress called externally triggered egress. Newly egressed parasites are preferentially restricted in vivo, probably because they have to re-infect stimulated immune cells that are better able to restrict parasite growth (Tomita et al., 2009). Thus, the reduced fitness of Δgra22 parasites in the peritoneum is possibly due to early egress thereby forcing the parasite to invade cells that by then are activated by IFNγ. Tg269950 contains a thioredoxin-like domain and has homology to sulfhydryl oxidases such as the human QSOX1 protein family. IFNγ-stimulated murine peritoneal macrophages increase the production of reactive oxygen species (ROS) in response to Toxoplasma infection (Arsenijevic et al., 2001). Possibly, Tg269950, similar to what has been described for human QSOX1 (Ostrowski and Kistler, 1980; Ostrowski et al., 1979) F, is important for protection against oxidative stress (Caillard et al., 2018; Morel et al., 2007).
Toxoplasma’s capacity to use host immune cells as Trojan horses helps its dissemination to distant sites and enhances its ability to cross biological barriers (Lambert and Barragan, 2010). Immediately after Toxoplasma infection, DCs and monocytes lose actin-rich structures called podosomes (Weidner et al., 2013) and exhibit amoeboid high-velocity locomotion, termed hypermotility (Olafsson et al., 2019; Olafsson et al., 2018), which also increase their capacity to migrate in 3-dimensional matrix confinements and along a chemokine gradient (Kanatani et al., 2015). Transportation of Toxoplasma by parasitized DCs and monocytic cells has been associated with enhanced systemic dissemination and elevated parasite loads in the brain during murine toxoplasmosis (Courret et al., 2006; Fuks et al., 2012; Lambert et al., 2006). We previously demonstrated that the Toxoplasma-derived 14-3-3 protein, which is encoded by Tg263090, is sufficient to induce hypermotility in DCs (Weidner et al., 2016). Tg263090 was not present in our small library and our previously published data show it is important for fitness in HFFs, which makes it difficult to investigate its effect on in vivo parasite fitness (Sidik et al., 2016). However, the hypermigratory phenotype of DCs is determined by multiple pathways, most of which lack cognate Toxoplasma effectors (Bhandage et al., 2019). Our in vivo loss-of-function screen identified that Tg261400 and TgWIP are not important for peritoneum fitness but seemed to be involved in colonizing distant organs. Tg261400 is related to the Plasmodium falciparum protein 3D7_0606800, which contains a Venus Flytrap (VFT) domain involved in substrate binding (Parker et al., 2017). It remains to be determined what substrate the VFT domain of Tg261400 binds and how it is involved in dissemination and/or survival in distant organs. TgWIP is secreted into the host cell cytosol upon invasion where it can specifically bind to components of the WRC and a tyrosine phosphatase (SHP2). The WRC is well known to regulate actin polymerization by activating the actin-nucleating activity of the Arp2/3 complex (Chen et al., 2014). SHP2 phosphatase activity is important in the dissolution of podosomes (Pan et al., 2013) and the formation of invadopodia, lamellipodia persistence and cell migration(Hartman et al., 2013; Sztacho et al., 2016; Tsai et al., 2015). Contrary to Toxofilin, a secretory effector that can modulate host actin at the invasion stage (Delorme-Walker et al., 2012), TgWIP deletion did not appear to impair host cell invasion. We demonstrated that TgWIP plays an important role in Toxoplasma-mediated dissolution of podosomes, modulation of host cell morphology, induction of hypermotility in human and murine DCs and enhancing the transmigration of Toxoplasma-infected primary human and murine DCs. Jointly, these data likely explain the dissemination defect of TgWIP-deficient parasites in vivo in mice and situate TgWIP as a major contributor to the DC hypermigratory phenotype. If TgWIP also affects activation of the GABAergic pathway (Kanatani et al., 2017), the induction of CCR7 chemotaxis (Fuks et al., 2012) or TIMP-1-dependent inhibition of extracellular matrix proteolysis in Toxoplasma-infected DCs remains to be determined (Olafsson et al., 2019; Olafsson et al., 2018).
If TgWIP is also responsible for cytoskeletal changes in other immune cells known to interact with Toxoplasma, such as monocytes/macrophages, and the contribution of TgWIP to transmigration across endothelial barriers, such as the blood-brain-barrier, and through tissues is currently unknown. Toxoplasma-infected macrophages lose their adhesiveness to fibronectin, laminin and collagen IV and downregulate L-selectin and integrins (Da Gama et al., 2004). Under shear stress conditions or in the blood stream, Toxoplasma alters the adhesion dynamics of human monocytes (Ueno et al., 2014). Infected monocytes roll faster and farther on vascular endothelium compared to non-infected monocytes. Thus, Toxoplasma alters cytoskeletal functions of monocytes, DCs, macrophages and cortical microglia (Bhandage et al., 2019); however, these cell types differ in actin dynamics and cell migration (Varol et al., 2009). It is important to keep in mind that we previously observed that while infected DCs transmigrated readily upon infection (Lambert et al., 2006), macrophages exhibited significantly lower transmigration frequencies and infected monocytes exhibited non-significant differences compared to uninfected monocytes or even a slight reduction in transmigration (Lambert et al., 2011). Furthermore, adoptive transfer of parasitized DCs significantly potentiated Toxoplasma dissemination (Fuks et al., 2012; Lambert et al., 2006) while adoptive transfer of parasitized macrophages/monocytes did not confer a measurable dissemination advantage (Lambert et al., 2011). These observations are in line with recently published data (Drewry et al., 2019) demonstrating that infected monocytes have enhanced migration through tissues but reduced transmigration across endothelial cells. These differences need to be further explored to understand the relative contribution of TgWIP to Toxoplasma dissemination in different leukocytes.
Previously, we demonstrated that Neospora parasites induced hypermotility and transmigration of human DCs and bovine monocyte-derived macrophages in vitro and that adoptive transfer in of Neospora-infected DCs into mice increased vertical transmission of the parasite to the fetus and enhanced parasite brain-blood-barrier crossing to reach the central nervous system (Collantes-Fernandez et al., 2012; Garcia-Sanchez et al., 2019). TgWIP is well conserved in Neospora and therefore likely also plays a role in Neospora dissemination. In conclusion, TgWIP could be an excellent tool to better understand the molecular mechanisms by which Toxoplasma and other apicomplexan parasites use host cells to disseminate from the site of infection to distant organs. The methods described here should provide a framework for the design of other in vivo loss-of-function screens to identify Toxoplasma gene function in vivo. For example, some of the other sub-libraries, e.g. targeting ROPs or MICs, could be tested or screens could be performed using different parasite or host genetic backgrounds.
STAR* METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to the lead contact, Jeroen P.J. Saeij (jsaeij@ucdavis.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Parasite culture
All the Toxoplasma gondii parasite strains were routinely passaged in vitro in monolayers of HFFs at 37°C in 5% CO2 as previously described (Rosowski et al., 2011).
Culture of cell lines
The TREX-293 cell lines and DC2.4 were cultured in DMEM, 10% fetal bovine serum (FBS), 2mM L-glutamine, 10mM HEPES, 1xnon essential amino acids, 1mM sodium pyruvate, 100U/mL penicillin/streptomycin, 10μg/ml gentamicin.
Primary host cell culture
Bone marrow-derived macrophages (BMDM) were isolated from 5 to 8 weeks old female C57BL/6J mice (The Jackson laboratory) as previously described (Jensen et al., 2013). BMDMs were obtained by culturing murine bone marrow cells in DMEM, 10% fetal bovine serum (FBS), 2mM L-glutamine, 10mM HEPES, 1xnon essential amino acids, 1mM sodium pyruvate, 100U/mL penicillin/streptomycin, 10μg/ml gentamicin and 20% L929 conditioned medium for 7 days. Primary murine bone marrow-derived DCs (DCs) were generated as previously described (Fuks et al., 2012). Briefly, cells from bone marrow of 6 to 10 weeks old female C57BL/6 mice (The Jackson Laboratory (Stock No: 000664) or Charles River Laboratories (strain code: 027)) were cultivated in RPMI 1640 with 10% FBS, gentamicin (20 g/ml), L-glutamine (2 mM), HEPES (0.01 M) and 1× non-essential amino acids, referred to as complete medium (CM), and supplemented with recombinant mouse GM-CSF (10 ng/ml, Peprotech). Loosely adherent cells were harvested after 6 or 10 days of maturation. To generate primary human monocyte-derived DCs, buffy coats from healthy blood donors were incubated with monocyte enrichment cocktail (RosetteSep™, StemCell Technologies), followed by centrifugation on density gradient medium (Lymphoprep™, StemCell Technologies). The in-between layer containing monocytes was transferred and washed twice with PBS. The residual red blood cells were removed by using red blood cell lysis buffer (15 mM NH4 Cl, 1.4 mM NaHCO3, 0.03 mM EDTA, pH7.3). The cell population obtained was composed mainly of CD14+ (DakoCytomation) with < 1% CD3+/CD19+ cells (BD), as evaluated by flow cytometry (FACS-Calibur, BD). Human DCs were generated by culturing the purified population in CM supplemented with 75 ng/ml human GM-CSF (Peprotech) and 30 ng/ml human IL-4 (Peprotech) for 6 days. The medium was changed after 3 days in culture.
Mice
CD-1 female mice were purchased from Charles River Laboratories (strain code:022) and were used for all infection experiments. C57BL/6 mice were purchased from The Jackson Laboratory (Stock No: 000664) or Charles River Laboratories (strain code: 027) and were used to generate BMDM (all from Jackson Laboratory mice) or BMDC (from Jackson Laboratory or Charles River Laboratories). At UC Davis, mice were housed in ventilated cages on corn bedding and provided water and mouse chow ad libitum. Cages were all on one rack at a housing density of five mice per cage and mice were allowed to acclimatize in our vivarium for at least a week undisturbed. The animal room was on a 12 light/12 dark cycle, and temperature was maintained at 22–25 °C. Mice were monitored twice daily by veterinarians, body weights were monitored daily, and cage bedding changed every two weeks. Mice were housed under specific pathogen-free conditions at University of California, Davis animal facility. The pathogen-free conditions are regularly tested by placing sentinel animals (Nu/+ mice and Nu/Nu) in the facility. These mice are tested for Mouse Hepatitis Virus, Sendai virus, Pneumonia Virus of Mice, Mouse Parvo Virus, Minute Virus of Mice, Mycobacterium pulmonis and arthriditis, Theiler’s Murine Encephalomyelitis Virus part of GDVII strain, Ectromelia Virus, Epidemic Diarrhea of Infant Mice virus, Mouse Adeno DNA Virus 1 and 2, Lymphocytic ChorioMeningitis Virus, Reovirus 3, Mouse Noro Virus, and fur mite. All animal experiments were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the Animal Welfare Act, approved by the Institutional Animal Care and Use Committee at the University of California, Davis (UC Davis) (assurance number A-3433–01). 5–9 weeks old mice at the start of the experiment and littermates were randomly assigned to experimental groups (the exact number of mice per group is indicated in the figure legends). Mice were challenged by intraperitoneal (i.p.) injection with 1000 or 1×107 Toxoplasma gondii tachyzoites.
At Stockholm University, C57BL/6NCrl mice were purchased from Charles River Laboratories (strain code: 027). Mice were housed in ventilated cages on sterilized bedding and provided water and mouse chow ad libitum. Housing density was five mice per cage and mice were allowed to acclimatize for at least a week undisturbed. The animal room was on a 12 light/12 dark cycle, and temperature was maintained at 22–25 °C. Mice were monitored daily and cage bedding changed every week. Mice were housed under specific pathogen-free conditions at Stockholm University and in strict accordance with the regulations of the Swedish Board of Agriculture, SJVSF 2017:40, saknr L150 (permit number 5.2.18–2966/17, facility approval 5.2.18–18145/17) and following proceedings described in EUlegislation (Council Directive 2010/63/EU). The experimental procedures and protocols involving the extraction of cells from mice were approved by the Regional Animal Research Ethical Board, Stockholm, Sweden (approval number 9707–2018).
Human blood donors
The Regional Ethics Committee, Stockholm, Sweden, approved protocols involving human cells (2006/116–31). All donors were healthy with a serum tested negative for Toxoplasma infection and received written and oral information upon a donation of blood. Written consent was obtained for the utilization of white blood cells for research purposes.
Method Details
Plasmid construction
The vector pcDNA-LIC-HF was a gift from M.A. Hakimi and A. Bougdour (Braun et al., 2013) and used as vector to making TREX-293 cells expressing Toxoplasma proteins. Primers were designed to amplify the gene of interest from after the predicted signal peptide to the predicted stop codon. Forward primers to amplify TGME49_24752 together with reverse primers contained LIC sequences (Table S6) were used to amplify TGME49_247520 from PRU (type II) genomic DNA. Forward primers to amplify GRA15 (TGME49_275470) and reverse primers contained Ligation independent cloning (LIC) sequences and were used to amplify GRA15 (TGME49_275470) from PRU genomic DNA. PCR products were treated with T4 DNA polymerase (using only TTP at 100mM). The pcDNA-LIC-HF vector was digested with SmaI and treated with T4 DNA polymerase (using only ATP at 100mM) to generate long overhangs. The PCR fragment and vector were then annealed for 15 minutes at room temperature, generating expression vectors with Toxoplasma genes C-terminally tagged with HA-FLAG. sgRNA targeting candidate genes were cloned into the pU6-Universal vector (Sidik et al., 2014). For C-terminal HA epitope tagging, a region of the TgWIP and Tg261400 genes, upstream of the stop codon, was amplified by PCR with primers (Table S6) and inserted into pLIC-HA-DHFR by ligation-independent cloning (Huynh and Carruthers, 2009). For the construction of the complementation plasmid, the pUPRT::DHFR-D (Addgene: plasmid #58528) plasmid backbone was PCR-amplified to remove the DHFR cassette. Subsequently the 5’UTR region of TgWIP containing the promoter (1000 bp upstream to the start codon) and the coding region was amplified and flanked with the HA tag sequence before the stop codon. The 3’UTR region (1000 bp) was also amplified. The open plasmid and the two inserts were assembled using the Gibson assembly kit. To generate the pTKO2-HXGPRT-ClickLuc plasmid for insertion of Click Beetle luciferase into the HXGPRT locus, the 5’UTR (~3.4kb) and 3’UTR (~3.3kb) of the HXGPRT gene were amplified from genomic DNA of RH parasites by using primers in Table S6. The Click Beetle luciferase coding sequence flanked by DHFR 5’UTR and 3’UTR were amplified from pDHFR-ClickLuc by using corresponding primers (Table S6). These PCR products were cloned into pDONRs plasmid via BP reaction of Gateway cloning. Three-way gateway cloning was performed to integrate the three fragments into the destination vector pTKO2 (Rosowski et al., 2011) by LR recombination (Invitrogen).
Inducible TREX-293 cell line construction
The TREX-293 cell line was a gift from J. Niles. TREX-293 cells were seeded at 75% confluency, and cotransfected with expression vectors pcDNA-LIC-TGME49_247520-HF or TGME49_275470-HF and a puromycin resistance vector (ratio of 10:1), using XtremeGENE 9 DNA transfection reagent (Roche). Cells were split 2 days post-transfection and subjected to puromycin selection at 1μg/ml. Foci were picked and expanded at least one-week post selection, and positive foci were identified by HA expression using immunofluorescence and immunoblotting.
Construction of parasite strains
Toxoplasma parasites were cultured on human foreskin fibroblasts (HFFs) as previously described (Rosowski et al., 2011). Individual knockouts of candidate genes were generated using clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9. Sequences targeting candidate genes were cloned into the pU6-Universal vector (Sidik et al., 2014). The sequences are available in Table S6. To generate the luciferase-expressing RH-Luc+/Δhpt parasites the RH strain was transfected with NotI (New England Biolabs)-linearized pTKO2-HXGPRT-ClickLuc, which contains HXGPRT homology regions surrounding the Click Beetle luciferase coding sequence driven by the DHFR promoter. Selection by growth in 6-thioxanthine (177 μg/ml) was used to obtain stably resistant clones that were subsequently screened by PCR for correct integration of the luciferase coding sequence into the HXGPRT locus. The expression of luciferase in PCR-positive clones was further confirmed by luciferase assay.
To generate the knockout strains for the candidate hits from the in vivo screen, plasmids containing sgRNAs were co-transfected with NotI (New England Biolabs)-linearized pTKOatt, which contains the HXGPRT selection cassette and GFP (Rosowski et al., 2011), into RH-Luc+/Δhpt or RHΔhpt (parasites at a 5:1 ratio of sgRNA to linearized pTKOatt plasmid). 24 h post-transfection, populations were selected with mycophenolic acid (50 μg/ml) and xanthine (50 μg/ml) and cloned by limiting dilution. Individual knockout clones were confirmed by PCR (Figure S2B, S2C). Endogenously tagged parasites were made in the RHΔku80 strain (Huynh and Carruthers, 2009) by transfection with plasmid pLIC-TgWIP-HA-DHFR or pLIC-Tg261400-HA-DHFR. 24 h post-transfection, populations were selected with 1 μM of pyrimethamine and cloned by limiting dilution. The presence of the tagged gene was determined by immunofluorescence assay (IFA). For generating complemented strains, the RHΔTgwip strain was co-transfected with plasmids containing sgRNA specific for the UPRT locus and NarI (New England Biolabs)-linearized pUPRT::TgWIP-HA plasmid at a ratio 1:5 (sgRNA/linearized plasmid). After the first complete lysis cycle, populations were selected with 10 μM of 5-fluoro-2-deoxyuridine (FUDR) for 2 complete lysis cycles and individual clones isolated by limiting dilution. The presence of the tagged gene was determined by immunofluorescence assay (IFA) and by PCR to confirm the integration into the UPRT locus (Figure S2C).
Sub-library construction
Putative ROP, MIC, and IMC-encoding genes were identified based on their cyclical expression profiles. In short, the expression data from Behnke et al. (Behnke et al., 2010) was normalized and clustered as previously described (Camejo et al., 2014) and clusters enriched in ROP, MIC or IMC-encoding genes identified. The GRA sub-library was based on most known GRAs identified before 2013 (when this library was designed) and putative novel GRA-encoding genes were identified based on genes with high non-cyclical expression (most GRA genes have a constant expression level during the cell cycle unlike ROP, MIC, and IMC genes) (Behnke et al., 2010), and presence of a signal peptide. Although, it is likely that this library also contains many non-GRA-encoding genes ~55 of the genes in this library encode for proteins that have been identified in GRA BioID studies (Chen et al., 2015; Nadipuram et al., 2016) indicating that it is indeed enriched for GRA-encoding genes. The other sublibraries are putative CDPK-targets, non-ROP kinases and two large sublibraries containg all other genes subdivided in hypothetical vs. non-hypothetical proteins (Table S1).
RH-Cas9 parasite transfection with the GRA sgRNA sub-library
1×107 RH-Cas9 parasites were transfected with 100 μg of pU6-DHFR plasmids (Sidik et al., 2016) containing the 2,170 sgRNA designed against 217 genes enriched in GRA-encoding genes. The transfected parasites were used to infect (MOI: 0.5) two T175 flasks containing a confluent monolayer of HFFs grown in DMEM with 1% FBS, 2mM L-glutamine, 100U/mL penicillin/streptomycin, 10μg/ml gentamicin and 40 μM Chloramphenicol. The next day the medium in each flask was removed and replaced with a selective medium containing DMEM, 10% FBS, 2mM L-Glutamine, 10mM HEPES, 1xNon Essential Amino Acids, 1mM Sodium Pyruvate, 100U/mL penicillin/streptomycin, 10μg/ml gentamicin, 40μM Chloramphenicol, 1μM Pyrimethamine, and 10μg/ml DNase I for 3 lytic cycles.
Infection of mice with the pool of Toxoplasma mutants
Toxoplasma mutants isolated after the 3rd lytic cycle in HFF (input) were intraperitoneally injected (with 1×107 viable parasites representing ~4,600× coverage of the 2,170 unique sgRNA) into 20 CD-1 mice. After 90 h of infection, the mice were euthanized. For 5 mice the peritoneal lavage, lungs, heart, brain, and eyes were collected and weighed and the peritoneal lavage was centrifuged and the cell pellet collected. According to the weight, the whole organ was digested using DNeasy Blood and Tissue Kits (Qiagen), the genomic DNA was extracted, and the concentration of the DNA was measured. The whole organ genomic DNA was PCR amplified using Q5 polymerase (NEB) (1μg of genomic DNA per PCR, the whole extracted gDNA was used for each sample) with specific primers to amplify the sgRNAs and to introduce a specific barcode for each sample from each mouse (Table S6) to distinguish them during the Illumina sequencing. For 5 other mice, the peritoneal lavage and organs were collected and were passed through a 70 μm cell strainer to make single cell suspensions, which were added to two tissue culture dishes containing HFFs (2× T175/organ). When the parasites completely lysed out from the HFF culture, they were counted and adjusted to 1×107 parasites per pellet after centrifugation. The sgRNAs were amplified from isolated parasite DNA and barcoded with three PCRs (1μg of genomic DNA per PCR) per sample with Q5 polymerase (NEB). sgRNAs from the plasmid were PCR also amplified from parasite DNA collected after the first, and the third lytic cycle. The resulting libraries were sequenced at the University of California Davis Genomic Center on a NEXT Seq (Illumina) with single-end reads using primers P150 and P151 (Table S6).
Immunofluorescence assay (IFA)
To check the localization of TgWIP and Tg261400, HFFs grown on glass coverslips were infected with parasites (endogenously tagged strains) for 16 h and were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 20 minutes, permeabilized/blocked with PBS with 3% (w/v) BSA, 5% (v/v) goat serum and 0.1% Triton-X100. To check for TgWIP secretion into the host cell cytosol, HFFs grown on glass coverslips were infected with parasites RHΔTgwip+Tgwip-HA with MOI: 3 to 5 for 2 h of infection then processed as described below. The coverslips were incubated with antibodies against the HA epitope tag, ROP2,3,4, GRA7 or SAG1 at room temperature for 1 h, washed and incubated with the respective fluorescent secondary antibodies and DAPI to stain the nucleus for 30 minutes. The coverslips were mounted with Vecta-Shield mounting oil and the microscopy was performed with NIS-Elements software (Nikon) and a digital camera (CoolSNAP EZ; Roper Scientific) connected to an inverted fluorescence microscope (eclipse Ti-S; Nikon) and either phase contrast or DIC imaging.
In vivo growth competition in CD-1 mice
CD-1 mice were i.p. infected with a 1:1 ratio of GFP-negative wild type (RH-Luc+/Δhpt, RH/hpt+, RHΔhpt or ME49-RFP+/hpt+) and GFP-positive knockout parasites (RH-Luc+/Δgra22, RH-Luc+/ΔTg269950, RH-Luc+/ΔTg261400 or RHΔTgwip, RHΔTgwip+Tgwip-HA and ME49 ΔTgwip/RFP+). Or HA-negative RHΔTgwip, and HA-positive RHΔTgwip+Tgwip-HA parasite. The real ratio of viable parasites was determined with a plaque assay by counting the ratio of GFP positive vs. GFP negative plaques formed five days after addition of 100 parasites of the mix to a monolayer of HFFs in 24-well plates. Six days post infection the mice were euthanized, and the peritoneal lavage and organs were collected. The parasites were counted from the peritoneal lavage, and a serial dilution of 100, 50, 25, 10 parasites per ml of media were made and plated in triplicate onto 24-well plates with HFFs. Or the parasites from the peritoneum were stained with HA antibody to determine the ratio HA-positive vs. HA negative parasites by Immunofluorescence assay (IFA) counting. Lungs, and heart were passed through a 70 μm cell strainer to make single cell suspensions in 10 ml of media. Subsequently, serial dilutions of 1/2, 1/4, 1/8, 1/16 were made and plated in triplicate on 24-well plates with HFFs. Five days later the ratio of GFP positive (knockout) vs. GFP negative (wild type) plaques was determined by microscopy. The brain was passed through a 70 μm cell strainer to make single cell suspensions in 10 ml of media and added to two T175 HFF dishes until the parasites completely lysed out. The parasites were counted and diluted to 100 parasites per ml of media and plated in triplicate onto 24-well plates with HFFs for 5 days. The ratio of GFP positive vs. GFP negative plaques was determined by microscopy. Or brain, lungs and heart were passed through 70 μm cell strainer to make single cell suspensions and added to HFF dishes until the parasites completely lysed out. The parasites from these cultures were stained with HA antibody to determine the ratio HA-positive vs. HA negative parasite by IFA counting.
Parasite growth in murine BMDM
BMDM on coverslips were stimulated or not for 24 h with 5ng/ml of IFNγ or 5ng/ml+10ng/ml of IFNγ + TNFα. BMDM were then infected with freshly lysed out parasites (MOI: 0.5 for type I parasite or 2 for type II parasite) for 24 h. The cells were washed with PBS and fixed with 4% PFA for 20 minutes, permeabilized/blocked with PBS with 3% (w/v) BSA, 5% (v/v) goat serum and 0.1% Triton-X100. GRA7 and SAG1 were used to determine the presence of the parasitophorous vacuole and the number of parasites per vacuole, respectively. Mounted coverslips were evaluated by microscopy. 100 vacuoles were analyzed to determine the average number of parasites per vacuole for each condition.
Co-immunoprecipitation
TREX-293 cells expressing HA-FLAG-tagged Tg247520 or GRA15II from a tetracycline-inducible promoter were grown in a T175 flask until 100% confluency after which expression was induced with tetracycline (1μg/mL) for 24 h. DC2.4 cells were grown in 3× T175 flask until 100% confluency and infected (MOI: 3 to 5) for 3 h with RHΔTgwip+Tgwip-HA parasites or RH parasite expressing GRA15II-HA. Cells were then scraped in ice-cold PBS, centrifuged and resuspended in 1 or 3 ml of lysis buffer (HEPES 10mM pH7.9, MgCl2 1.5mM, KCl 10mM, EDTA 0.1mM, dithiothreitol (DTT), 0.5mM, NP40 0.65%, cocktail of protease inhibitor (Roche), phenylmethylsulfonyl fluoride (PMSF) 0.5mM.) for 30 minutes at 4°C. The lysate was centr ifuged for 30 minutes at 18,000 × g, 4°C. Each sample was incubated with 25 or 90 μl of magnetic beads coupled with HA antibodies (Thermo scientific) and placed on a rotator overnight at 4°C. The beads were washed three times with Tris-HCl 10mM pH7.5, NaCl 150mM, Triton-100× 0.2%, PMSF 0.5mM, a cocktail of protease inhibitors (Roche), once more with Tris-HCl 62.5mM pH6.8 and beads were resuspended in 100 μl of this buffer.
Mass spectrometry-based proteomics and analysis.
Magnetic beads coupled with antibodies against the HA tag were sent to the Proteomic Core Facility of the University of California, Davis or Mass spectrometry facility of Massachusetts Institute of Technology (MIT) for mass spectrometry analysis. Briefly, the proteins were digested using Promega modified trypsin overnight at room temperature on a gently shaking device. Resulting peptides were analyzed by online LC-MS/MS Q-Exactive. All MS/MS samples were analyzed using X! Tandem (The GPM, thegpm.org; version X! Tandem Alanine (2017.2.1.4)). X! Tandem was set up to search the uniprotHSTG_crap database assuming the digestion enzyme trypsin. X! Tandem was searched with a fragment ion mass tolerance of 20 PPM and a parent ion tolerance of 20 PPM. Glu->pyro-Glu of the n-terminus, ammonia-loss of the n-terminus, gln->pyro-Glu of the n-terminus, deamidated of asparagine and glutamine, oxidation of methionine and tryptophan, dioxidation of methionine and tryptophan and dicarbamidomethyl of lysine were specified in X! Tandem as variable modifications. Scaffold (version Scaffold_4.8.6, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 50.0% probability by the Scaffold Local FDR algorithm. Peptide identifications were required to exceed specific database search engine thresholds and X! Tandem identifications were also required. Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve an FDR less than 5.0% and contained at least 1 identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. MoDPepInt online software (see Key Resources Table) was used to identify SH3 motifs in the Tg247520 protein sequence. PRALINE multiple sequence alignment online software (see Key Resources Table) was used to align the protein sequences of Toxoplasma gondii type I (TGGT1_247520), Neospora caninum (BN1204_065230) and the Tg247520 homolog in Hammondia hammondi.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ROP2,3,4 (mouse mAb; T3 4A7) | (Sadak et al., 1988) | N/A |
| SAG1 (mouse mAb; DG52) | (Nagel and Boothroyd, 1988) | N/A |
| GRA7 (rabbit antibody) | (Dunn et al., 2008) | N/A |
| Alexa Fluor™ 488 Phalloidin | Life Technologies | Cat#A12379 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies | Cat#A11037 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Fisher Scientific | Cat#A11029 |
| Goat anti Rat (IgG H+L) Alexa Fluor 594 | Invitrogen | Cat#A-11007 |
| Goat Anti-Mouse Alexa Fluor 594 (IgG H+L) | Invitrogen | Cat#A11032 |
| Anti-HA High Affinity | Sigma-Aldrich | Cat#11867431001 |
| Bacterial and Virus Strains | ||
| 5-ALPHA F’ COMPETENT CELLS | NEB | Cat#C2992H |
| E. cloni 10G SUPREME Electrocompetent Cells | Lucigen (VWR) | Cat#60080-2 |
| Biological Samples | ||
| Primary Human Monocyte-Derived DCs from blood donors | This study | N/A |
| Monocyte-Derived DCs from C57BL/6 mice | This study | The Jackson Laboratory Stock No:000664 or Charles River Laboratories (Strain Code: 027) |
| Bone Marrow Derived Macrophage from C57BL/6J mice | This study | The Jackson Laboratory Stock No: 000664 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Penicillin/Streptomycin | Life Technologies | Cat#15140-122 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378-5 |
| Puromycin | Fisher | Cat#540411 |
| Mycophenolic acid | Millipore | Cat#89287 |
| Pyrimethamine | Sigma-Aldrich | Cat#46706 |
| xanthine | Millipore | Cat#X3627 |
| Gentamicin | Life Technologies | Cat#15710072 |
| 6-Thioxanthine | Toronto Research | Cat#T385800 |
| 5-fluoro-2-deoxyuridine (FUDR) | Sigma-Aldrich | Cat#F0503 |
| Tetracycline | Sigma-Aldrich | Cat#87128 |
| DMEM Medium | Life Technologies | Cat#11965-118 |
| L-Glutamine | Life Technologies | Cat#25030-081 |
| HEPES | Life Technologies | Cat#15630-080 |
| Non-Essential Amino Acids | Thermo-Fisher | Cat#11140-050 |
| Sodium Pyruvate | Life Technologies | Cat#11360-070 |
| Fetal Bovine Serum (FBS) | Thermo Scientific (Gibco) | Cat#26400036 |
| Recombinant murine GM-CSF | PeproT ech | Cat#315-03 |
| Recombinant Human GM-CSF | PeproT ech | Cat#300-03 |
| IL-4 | PeproT ech | Cat#200-04 |
| Phosphate Saline Buffer (PBS) | Life Technologies | Cat#10010-049 |
| Bovine collagen | Life Technologies | Cat#A1064401 |
| Bovine Serum Albumin (BSA) | Fisher scientific | Cat#NC9227912 |
| Red blood cell lysis buffer | Sigma-Aldrich | Cat#R7757 |
| Fluorescein labeled Dolichos Biflorus Agglutinin (DBA) | Vector Laboratories | Cat#FL-1031-5 |
| VECTA SHIELD MOUNTING MEDIUM | Vector Laboratories | Cat#H-1000 |
| Critical Commercial Assays | ||
| X-tremeGENE 9 DNA transfection reagent | Sigma-Aldrich | Cat#XTG9-RO |
| DNeasy Blood and Tissue Kits | Qiagen | Cat#69504 |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | Cat#M0494S |
| T4 DNA polymerase | Thermo Scientific | Cat#EP0061 |
| Halt Protease and phosphatase inhibitor cocktail | Thermo-Fisher | Cat#78444 |
| Gibson assembly kit | NEB | Cat#E2611L |
| Anti-HA Magnetic Beads | Pierce | Cat#88836 |
| RosetteSep™ | StemCell Technologies | Cat#15068 |
| Lymphoprep™ | StemCell Technologies | Cat#07801 |
| Transwell® filters | Corning | Cat#3428 |
| 70 μm cell strainer | Sigma-Aldrich | Cat#CLS431751 |
| Experimental Models: Cell Lines | ||
| Human foreskin fibroblasts | Gift from John Boothroyd | N/A |
| Mouse DC2.4 | Millipore-Sigma | Cat#SCC142 |
| TREX-293 cells | gift from Jacquin Niles | N/A |
| Experimental Models: Organisms/Parasite Strains | ||
| CD-1 mice | Charles River Laboratories | Strain Code: 022 |
| RH-Δku80 | (Huynh & Carruthers, 2009) | N/A |
| RH88 (Background of RH-Luc+/Δhpt) | This study | N/A |
| RH-Luc+/Δhpt (Background of RHΔgra22, RHΔTg269950 and RHΔTg261400) | This study | N/A |
| RHΔhpt (Background of RHATgwip) | This study | N/A |
| ME49-RFP+/hpt+ (background of ME49-RFP+/hpt−) | gift from Michael Grigg | N/A |
| ME49-RFP+/hpt− (Background of ME49ΔTgwip) | gift from Michael Grigg | N/A |
| Oligonucleotides | ||
| Primers for PCR listed in Table S1 and Table S6 | ||
| Recombinant DNA | ||
| pTKOatt | (Rosowski et al., 2011) | N/A |
| pU6-DHFR | (Sidik et al., 2016) | N/A |
| pcDNA-LIC-HF | gift from Mohamed-Ali Hakimi and Alexandre Bougdour | N/A |
| pLIC-HA-DHFR | (Huynh and Carruthers, 2009) | N/A |
| pUPRT::DHFR-D | Addgene | Plasmid #58528 |
| pU6-Universal vector | (Sidik et al., 2014) | N/A |
| Software and Algorithms | ||
| ImageJ | bundled with 64-bit Java 1.8.0_112 (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Prism | Graphpad | https://www.graphpad.com |
| R | N/A | www.R-project.org |
| Microsoft Excel | N/A | Office.com |
| Scaffold V4.9 | v4.9 (Searle, 2010) | http://www.proteomesoftware.com |
| Chemotaxis and migration tool | N/A | https://ibidi.com/chemotaxis-analysis/171-chemotaxis-and-migration-tool.html |
| FlowJo | N/A | https://www.flowjo.com |
| MoDPepInt (Modular Domain Peptide Interaction) | (Kundu et al., 2014) | http://modpepint.informatik.uni-freiburg.de/ |
| PRALINE multiple sequence alignment | (Simossis and Heringa, 2005) | http://www.ibi.vu.nl/programs/pralinewww/ |
Podosome assay and morphology analysis
DCs (primary cells or DC2.4) were seeded on gelatin or poly-L-lysine coated coverslips 2 h before infection after which freshly egressed parasites were added at the desired MOI, and the cells were processed for immunofluorescence assays 4 h p.i. F-actin was visualized by incubating fixed (4% PFA for 20 minutes) and permeabilized cells (5 minutes with 0.1% Triton-X100) with Alexa Fluor 488 Phalloidin for 20 minutes. Coverslips were mounted with Vecta-Shield mounting medium and visualized by microscopy with NIS-Elements software (Nikon) and a digital camera (CoolSNAP EZ; Roper Scientific) connected to an inverted fluorescence microscope (eclipse Ti-S; Nikon), DIC imaging. For primary mouse DCs, podosomes were identified and quantified as described in (Weidner et al., 2013). For DC2.4 cell area and roundness measurements, pictures of 100 to 300 cells were analyzed for each experiment (n=3). Using the software ImageJ, the threshold of the green channel (Alexa Fluor 488 Phalloidin) was adjusted to distinguish single cells. The tool “Analyze particles” was used to count cells and measure the area (50 to 1000 μm2) and the roundness (0 to 1) of the cells.
Motility assay
Motility assays were performed as previously described (Olafsson et al., 2018; Weidner et al., 2013). Briefly, DCs were cultured in 96-well plates with CM ± freshly egressed Toxoplasma tachyzoites (MOI 3) for 4 h. Bovine collagen I (0,75 mg/ml, Life Technologies) was then added and live cell imaging was performed for 1 h, 1 frame/min at 10× magnification (Z1 Observer with Zen 2 Blue v. 4.0.3, Zeiss). Time-lapse images were consolidated into stacks and motility data was obtained from 60 cells/condition (Manual Tracking, ImageJ) yielding average velocities and accumulated distance (Chemotaxis and migration tool, v. 2.0, Ibidi). Infected cells were defined by GFP-cell co-localization.
Transmigration assay
Transmigration assays were performed as previously described (Lambert et al., 2006; Olafsson et al., 2018). Briefly, human and murine DCs were cultured in CM ± freshly egressed Toxoplasma tachyzoites (MOI 3) for 6 h. DCs were subsequently transferred to Transwell® filters (8 μm pore size; Corning) or collected, fixed (PFA 2%) and used to determine infection rate and the number of DCs per well. Transmigrated DCs were collected from the lower chamber after 18 h of transmigration. Cell numbers were quantified by flow cytometry (easyCyte™ 8HT, Millipore) in accordance with the manufacturer’s guidelines. Samples were gated on FSC, SSC and GFP. Data was analyzed in FlowJo (Tree Star Inc, OR). Percentages are from cells added at time 0 relative to cells in the lower chamber after 18 h for each indicated population.
CD-1 mice single infection, cyst counting, diagnostic PCR and serological detection
20 CD-1 mice were i.p. infected with 1,000 type II ME49-RFP/hpt+ parasites or ME49ΔTgwip-RFP+/GFP+/hpt+ parasites (n=2 experiments) and monitored for 27 days p.i. Every day the number of dead mice per group was observed and the brains and blood of the surviving mice were harvested. Following homogenization of brains by passage through 21- and 25-gauge needles, cysts were stained with FITC-DBA. To detect the presence of parasites in the brains of infected mice, genomic DNA from homogenized brains was isolated using Qiagen DNeasy Blood & Tissue Kits. Diagnostic PCR targeting the B1 gene was performed as previously described (Wang et al., 2019). To make sure the mice were successfully infected with the parasites, serum anti-Toxoplasma IgG of infected mice was measured using an enzyme-linked immunosorbent assay (ELISA) following established protocols(Wang et al., 2019). Briefly, the plates were coated with 0.25 μg of whole parasite lysate and blocked with 2% BSA in PBS-0.05% Tween-20. Subsequently, 1/100 dilutions of serum were added and incubated at room temperature for at least 2 h, followed by incubation with 1/2000 diluted HRP-conjugated goat anti-mouse IgG at room temperature for 2 h. Finally, 100 μl of substrate solution (ABTS solution from Sigma) was added to the wells and the optical density was measured at 405 nm after 20 minutes of ABTS incubation.
Quantification and Statistical Analysis
Bioinformatic analysis of the loss-of-function screens
sgRNA selection and screen analysis were performed using custom software as described in (Sidik et al., 2016; Sidik et al., 2018) that will be provided upon request. Statistical analyses were performed in R (www.R-project.org) and Excel (Microsoft Office). Illumina sequencing reads were matched against the sequences of the sgRNA sub-library. The number of exact matches were counted and considered as raw read numbers. For heart, brain and eyes raw read data was pooled from all mice into one unique sample. The abundance of each sgRNA was calculated and normalized to the total number of reads. sgRNAs that had zero reads were assigned a pseudo-count corresponding to 90% of the lowest value in that sample. Only sgRNA whose abundance was above the 5th percentile in the preparation of the sgRNA library were considered for the HFF in vitro screen. Only sgRNA whose abundance was above the 5th percentile in the input were considered for the in vivo screen. The “fitness” score for each gene was calculated as the average log2 fold change for the top five scoring sgRNAs, which minimized the effect of stochastic losses. The Pearson correlation between each sample from the same organ was calculated. In each sample the genes were ranked according to the fitness score (lowest fitness score to the highest), and for peritoneum and lungs samples (10 samples each) the average rank and SD were also calculated for each gene in each organ. To identify genes with consistent top rankings Z-scores for each gene were calculated by subtracting the average rank from that gene’s rank and dividing the result by the standard deviation (SD) of all the ranks for each gene. Genes with a Z-score≥2) in the peritoneum, were considered as peritoneum fitness confering genes (Table 1 and Table S4). Genes (excluding peritoneum fitness confering genes) with a Z-score≥2) in the lungs and/or with a fitness score of −1 (2 fold decrease in abundance of sgRNAs) in at least three other organs (heart, brain and eyes) were considered as organ fitness confering genes (Table 1 and Table S4).
Statistical tests
All statistical analyses were performed using Prism (Graph Pad) version 8.1.1 All data are presented as average ± standard deviation (SD), and the exact n values are mentioned in each of the figure legends. For all the calculations p<0.05 are considered as significant and represented with the Asterix. For one variable test with two groups, the two-way ANOVA was used followed with Bonferroni’s multiple comparisons test. For more than three groups with one variable, One-way ANOVA followed with Dunnett’s multiple comparisons test or repeated measures one-way ANOVA with Tukey’s post-hoc test was used. For comparison between two fractions/percentages with different standard deviations the unpaired t-test with Welch’s correction was used. Survival experiments were analyzed using the log rank Mantel-Cox test. Chi square test was used to compare the frequency of cells with accumulated migration distances above and below 150 μm.
Data and Code Availability
All the sub libraries sgRNA and primers sequences are available in Table S1 and will be provided upon request. All the data from the in vivo screen is available in Table S4. All the data from the TgWIP IP mass spectrometry analysis is available in Table S5.
Supplementary Material
Table S1. Related to STAR Methods. sgRNA sequences for the eight sub-libraries available.
Table S2. Related to Figure 1. Pearson correlation on in vivo sample fitness scores.
Table S3. Related to Figure 1. List of control genes that are not expressed.
Table S4. Related to Table 1 and 2. Illumina sequencing raw reads, Fitness scores, and Z-scores.
Table S5. Identification of host interaction partners of Tg247520, Related to Figure 3. Immunoprecipitation (IP) with HA antibody on stable HEK293-derived (TREX-293) cells expressing Tg247520 or GRA15 with a C-terminal HA-FLAG epitope tag under the control of the tetracycline operator (first sheet, upper panel) or IP with HA antibody on immortalized murine DC cell line (DC2.4) infected for 3 h with parasites expressing HA-tagged Tg247520 or GRA15 (first sheet, lower panel). The table represents the putative host interaction partners identified through mass spectrometry analysis specific to Tg247520 IP. The total number of spectra of peptides for each protein and MW= Molecular weight are indicated (raw data in second and third sheet).
Table S6. Related to STAR Methods. sgRNAs for generation of individual gene knockout and PCR primers used in this study.
Highlights.
CRISPR screen reveals Toxoplasma gondii genes that confer in vivo fitness
The screen identifies genes important for colonization of distant organs
TgWIP, a novel secreted rhoptry protein, induces dendritic cell hypermotility
TgWIP determines parasite dissemination from the site of infection to the brain
ACKNOWLEDGEMENTS
Swedish Research Council (Vetenskapsrådet, 2018-02411) to AB, NIH R01AI080621 to JS, NIH Director’s Early Independence Award (1DP5OD017892) and a Mathers Foundation grant to SL. UC Davis and Whitehead Institute (Eric Spooner) Proteomic cores for assistance.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Araujo FG, Williams DM, Grumet FC, and Remington JS (1976). Strain-dependent differences in murine susceptibility to toxoplasma. Infect Immun 13, 1528–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Bilbao FD, Giannakopoulos P, Girardier L, Samec S, and Richard D (2001). A role for interferon-gamma in the hypermetabolic response to murine toxoplasmosis. Eur Cytokine Netw 12, 518–527. [PubMed] [Google Scholar]
- Bai MJ, Wang JL, Elsheikha HM, Liang QL, Chen K, Nie LB, and Zhu XQ (2018). Functional Characterization of Dense Granule Proteins in Toxoplasma gondii RH Strain Using CRISPR-Cas9 System. Front Cell Infect Microbiol 8, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke MS, Fentress SJ, Mashayekhi M, Li LX, Taylor GA, and Sibley LD (2012). The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Pathog 8, e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke MS, Khan A, Wootton JC, Dubey JP, Tang K, and Sibley LD (2011). Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc Natl Acad Sci U S A 108, 9631–9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke MS, Wootton JC, Lehmann MM, Radke JB, Lucas O, Nawas J, Sibley LD, and White MW (2010). Coordinated progression through two subtranscriptomes underlies the tachyzoite cycle of Toxoplasma gondii. PLoS One 5, e12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandage AK, Kanatani S, and Barragan A (2019). Toxoplasma-Induced Hypermigration of Primary Cortical Microglia Implicates GABAergic Signaling. Front Cell Infect Microbiol 9, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougdour A, Durandau E, Brenier-Pinchart MP, Ortet P, Barakat M, Kieffer S, Curt-Varesano A, Curt-Bertini RL, Bastien O, Coute Y, et al. (2013). Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13, 489–500. [DOI] [PubMed] [Google Scholar]
- Braun L, Brenier-Pinchart MP, Yogavel M, Curt-Varesano A, Curt-Bertini RL, Hussain T, Kieffer-Jaquinod S, Coute Y, Pelloux H, Tardieux I, et al. (2013). A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J Exp Med 210, 2071–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RW, Sharma AI, and Engman DM (2017). Dynamic protein S-palmitoylation mediates parasite life cycle progression and diverse mechanisms of virulence. Crit Rev Biochem Mol Biol 52, 145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard A, Sadoune M, Cescau A, Meddour M, Gandon M, Polidano E, Delcayre C, Da Silva K, Manivet P, Gomez AM, et al. (2018). QSOX1, a novel actor of cardiac protection upon acute stress in mice. J Mol Cell Cardiol 119, 75–86. [DOI] [PubMed] [Google Scholar]
- Camejo A, Gold DA, Lu D, McFetridge K, Julien L, Yang N, Jensen KD, and Saeij JP (2014). Identification of three novel Toxoplasma gondii rhoptry proteins. Int J Parasitol 44, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AL, Kim EW, Toh JY, Vashisht AA, Rashoff AQ, Van C, Huang AS, Moon AS, Bell HN, Bentolila LA, et al. (2015). Novel components of the Toxoplasma inner membrane complex revealed by BioID. MBio 6, e02357–02314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, and Rosen MK (2014). The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole BJ, Feltcher ME, Waters RJ, Wetmore KM, Mucyn TS, Ryan EM, Wang G, Ul-Hasan S, McDonald M, Yoshikuni Y, et al. (2017). Genome-wide identification of bacterial plant colonization genes. PLoS Biol 15, e2002860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collantes-Fernandez E, Arrighi RB, Alvarez-Garcia G, Weidner JM, Regidor-Cerrillo J, Boothroyd JC, Ortega-Mora LM, and Barragan A (2012). Infected dendritic cells facilitate systemic dissemination and transplacental passage of the obligate intracellular parasite Neospora caninum in mice. PLoS One 7, e32123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, and Tardieux I (2006). CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Gama LM, Ribeiro-Gomes FL, Guimaraes U Jr., and Arnholdt AC (2004). Reduction in adhesiveness to extracellular matrix components, modulation of adhesion molecules and in vivo migration of murine macrophages infected with Toxoplasma gondii. Microbes Infect 6, 1287–1296. [DOI] [PubMed] [Google Scholar]
- Delorme-Walker V, Abrivard M, Lagal V, Anderson K, Perazzi A, Gonzalez V, Page C, Chauvet J, Ochoa W, Volkmann N, et al. (2012). Toxofilin upregulates the host cortical actin cytoskeleton dynamics, facilitating Toxoplasma invasion. J Cell Sci 125, 4333–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmonts G, and Couvreur J (1974). Congenital toxoplasmosis. A prospective study of 378 pregnancies. N Engl J Med 290, 1110–1116. [DOI] [PubMed] [Google Scholar]
- Drewry LL, Jones NG, Wang Q, Onken MD, Miller MJ, and Sibley LD (2019). The secreted kinase ROP17 promotes Toxoplasma gondii dissemination by hijacking monocyte tissue migration. Nat Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JD, Ravindran S, Kim SK, and Boothroyd JC (2008). The Toxoplasma gondii dense granule protein GRA7 is phosphorylated upon invasion and forms an unexpected association with the rhoptry proteins ROP2 and ROP4. Infect Immun 76, 5853–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein MC, Reese ML, Konen-Waisman S, Boothroyd JC, Howard JC, and Steinfeldt T (2012). A Toxoplasma gondii pseudokinase inhibits host IRG resistance proteins. PLoS Biol 10, e1001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe IT, Child MA, Majmudar JD, Krishnamurthy S, van der Linden WA, Ward GE, Martin BR, and Bogyo M (2015). Global Analysis of Palmitoylated Proteins in Toxoplasma gondii. Cell Host Microbe 18, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox BA, Guevara RB, Rommereim LM, Falla A, Bellini V, Petre G, Rak C, Cantillana V, Dubremetz JF, Cesbron-Delauw MF, et al. (2019). Toxoplasma gondii Parasitophorous Vacuole Membrane-Associated Dense Granule Proteins Orchestrate Chronic Infection and GRA12 Underpins Resistance to Host Gamma Interferon. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox BA, Rommereim LM, Guevara RB, Falla A, Hortua Triana MA, Sun Y, and Bzik DJ (2016). The Toxoplasma gondii Rhoptry Kinome Is Essential for Chronic Infection. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco M, Panas MW, Marino ND, Lee MC, Buchholz KR, Kelly FD, Bednarski JJ, Sleckman BP, Pourmand N, and Boothroyd JC (2016). A Novel Secreted Protein, MYR1, Is Central to Toxoplasma’s Manipulation of Host Cells. MBio 7, e02231–02215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenal K, Tay CL, Mueller C, Bushell ES, Jia Y, Graindorge A, Billker O, Rayner JC, and Soldati-Favre D (2013). Global analysis of apicomplexan protein S-acyl transferases reveals an enzyme essential for invasion. Traffic 14, 895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks JM, Arrighi RB, Weidner JM, Kumar Mendu S, Jin Z, Wallin RP, Rethi B, Birnir B, and Barragan A (2012). GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog 8, e1003051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sanchez M, Jimenez-Pelayo L, Horcajo P, Regidor-Cerrillo J, Olafsson EB, Bhandage AK, Barragan A, Werling D, Ortega-Mora LM, and Collantes-Fernandez E (2019). Differential Responses of Bovine Monocyte-Derived Macrophages to Infection by Neospora caninum Isolates of High and Low Virulence. Front Immunol 10, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Olias P, and Sibley LD (2017). Toxoplasma Effectors Targeting Host Signaling and Transcription. Clin Microbiol Rev 30, 615–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ZR, Schaller MD, and Agazie YM (2013). The tyrosine phosphatase SHP2 regulates focal adhesion kinase to promote EGF-induced lamellipodia persistence and cell migration. Mol Cancer Res 11, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MA, Olijnik AA, Frickel EM, and Saeij JP (2019). Clonal and atypical Toxoplasma strain differences in virulence vary with mouse sub-species. Int J Parasitol 49, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Brenier-Pinchart MP, Braun L, Kraut A, Touquet B, Coute Y, Tardieux I, Hakimi MA, and Bougdour A (2018). Characterization of a Toxoplasma effector uncovers an alternative GSK3/beta-catenin-regulatory pathway of inflammation. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland GN (1999). Reconsidering the pathogenesis of ocular toxoplasmosis. Am J Ophthalmol 128, 502–505. [DOI] [PubMed] [Google Scholar]
- Huynh MH, and Carruthers VB (2009). Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell 8, 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KD, Hu K, Whitmarsh RJ, Hassan MA, Julien L, Lu D, Chen L, Hunter CA, and Saeij JP (2013). Toxoplasma gondii rhoptry 16 kinase promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect Immun 81, 2156–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatani S, Fuks JM, Olafsson EB, Westermark L, Chambers B, Varas-Godoy M, Uhlen P, and Barragan A (2017). Voltage-dependent calcium channel signaling mediates GABAA receptor-induced migratory activation of dendritic cells infected by Toxoplasma gondii. PLoS Pathog 13, e1006739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatani S, Uhlen P, and Barragan A (2015). Infection by Toxoplasma gondii Induces Amoeboid-Like Migration of Dendritic Cells in a Three-Dimensional Collagen Matrix. PLoS One 10, e0139104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Hunn JP, Konen-Waisman S, Zhao YO, Preukschat D, Coers J, Boyle JP, Ong YC, Boothroyd JC, Reichmann G, et al. (2010). Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol 12, 939–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesius PH, and Hinds SE (1979). Strain-dependent differences in murine susceptibility to coccidia. Infect Immun 26, 1111–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll LJ (2016). Functional Analysis of the Rhoptry Kinome during Chronic Toxoplasma gondii Infection. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu K, Mann M, Costa F, and Backofen R (2014). MoDPepInt: an interactive web server for prediction of modular domain-peptide interactions. Bioinformatics 30, 2668–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert H, and Barragan A (2010). Modelling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol 12, 292–300. [DOI] [PubMed] [Google Scholar]
- Lambert H, Dellacasa-Lindberg I, and Barragan A (2011). Migratory responses of leukocytes infected with Toxoplasma gondii. Microbes Infect 13, 96–102. [DOI] [PubMed] [Google Scholar]
- Lambert H, Hitziger N, Dellacasa I, Svensson M, and Barragan A (2006). Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol 8, 1611–1623. [DOI] [PubMed] [Google Scholar]
- Lambert H, Vutova PP, Adams WC, Lore K, and Barragan A (2009). The Toxoplasma gondii-shuttling function of dendritic cells is linked to the parasite genotype. Infect Immun 77, 1679–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ND, Panas MW, Franco M, Theisen TC, Naor A, Rastogi S, Buchholz KR, Lorenzi HA, and Boothroyd JC (2018). Identification of a novel protein complex essential for effector translocation across the parasitophorous vacuole membrane of Toxoplasma gondii. PLoS Pathog 14, e1006828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, and Howard JC (2005). Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 1, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimine KM, Doan LT, Atreya CA, Stedman TT, Anderson KS, Joiner KA, and Coppens I (2005). Toxoplasma gondii is capable of exogenous folate transport. A likely expansion of the BT1 family of transmembrane proteins. Mol Biochem Parasitol 144, 44–54. [DOI] [PubMed] [Google Scholar]
- Melo MB, Nguyen QP, Cordeiro C, Hassan MA, Yang N, McKell R, Rosowski EE, Julien L, Butty V, Darde ML, et al. (2013). Transcriptional analysis of murine macrophages infected with different Toxoplasma strains identifies novel regulation of host signaling pathways. PLoS Pathog 9, e1003779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel C, Adami P, Musard JF, Duval D, Radom J, and Jouvenot M (2007). Involvement of sulfhydryl oxidase QSOX1 in the protection of cells against oxidative stress-induced apoptosis. Exp Cell Res 313, 3971–3982. [DOI] [PubMed] [Google Scholar]
- Munoz M, Liesenfeld O, and Heimesaat MM (2011). Immunology of Toxoplasma gondii. Immunol Rev 240, 269–285. [DOI] [PubMed] [Google Scholar]
- Nadipuram SM, Kim EW, Vashisht AA, Lin AH, Bell HN, Coppens I, Wohlschlegel JA, and Bradley PJ (2016). In Vivo Biotinylation of the Toxoplasma Parasitophorous Vacuole Reveals Novel Dense Granule Proteins Important for Parasite Growth and Pathogenesis. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel SD, and Boothroyd JC (1988). The alpha- and beta-tubulins of Toxoplasma gondii are encoded by single copy genes containing multiple introns. Mol Biochem Parasitol 29, 261–273. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Niedelman W, Gold DA, Rosowski EE, Sprokholt JK, Lim D, Farid Arenas A, Melo MB, Spooner E, Yaffe MB, and Saeij JP (2012). The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog 8, e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh CS, Toke DA, Mandala S, and Martin CE (1997). ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem 272, 17376–17384. [DOI] [PubMed] [Google Scholar]
- Okada T, Marmansari D, Li ZM, Adilbish A, Canko S, Ueno A, Shono H, Furuoka H, and Igarashi M (2013). A novel dense granule protein, GRA22, is involved in regulating parasite egress in Toxoplasma gondii. Mol Biochem Parasitol 189, 5–13. [DOI] [PubMed] [Google Scholar]
- Olafsson EB, Ross EC, Varas-Godoy M, and Barragan A (2019). TIMP-1 promotes hypermigration of Toxoplasma-infected primary dendritic cells via CD63-ITGB1-FAK signaling. J Cell Sci 132. [DOI] [PubMed] [Google Scholar]
- Olafsson EB, Varas-Godoy M, and Barragan A (2018). Toxoplasma gondii infection shifts dendritic cells into an amoeboid rapid migration mode encompassing podosome dissolution, secretion of TIMP-1, and reduced proteolysis of extracellular matrix. Cell Microbiol 20. [DOI] [PubMed] [Google Scholar]
- Ostrowski MC, and Kistler WS (1980). Properties of a flavoprotein sulfhydryl oxidase from rat seminal vesicle secretion. Biochemistry 19, 2639–2645. [DOI] [PubMed] [Google Scholar]
- Ostrowski MC, Kistler WS, and Williams-Ashman HG (1979). A flavoprotein responsible for the intense sulfhydryl oxidase activity of rat seminal vesicle secretion. Biochem Biophys Res Commun 87, 171–176. [DOI] [PubMed] [Google Scholar]
- Pan YR, Cho KH, Lee HH, Chang ZF, and Chen HC (2013). Protein tyrosine phosphatase SHP2 suppresses podosome rosette formation in Src-transformed fibroblasts. J Cell Sci 126, 657–666. [DOI] [PubMed] [Google Scholar]
- Parker ML, Ramaswamy R, van Gordon K, Powell CJ, Bosch J, and Boulanger MJ (2017). The structure of Plasmodium falciparum 3D7_0606800 reveals a bi-lobed architecture that supports re-annotation as a Venus Flytrap protein. Protein Sci 26, 1878–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing.
- Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, and Boyle JP (2011). Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci U S A 108, 9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommereim LM, Bellini V, Fox BA, Petre G, Rak C, Touquet B, Aldebert D, Dubremetz JF, Cesbron-Delauw MF, Mercier C, et al. (2016). Phenotypes Associated with Knockouts of Eight Dense Granule Gene Loci (GRA2–9) in Virulent Toxoplasma gondii. PLoS One 11, e0159306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, and Saeij JP (2011). Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med 208, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadak A, Taghy Z, Fortier B, and Dubremetz JF (1988). Characterization of a family of rhoptry proteins of Toxoplasma gondii. Mol Biochem Parasitol 29, 203–211. [DOI] [PubMed] [Google Scholar]
- Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, and Boothroyd JC (2006). Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314, 1780–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle BC (2010). Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10, 1265–1269. [DOI] [PubMed] [Google Scholar]
- Shastri AJ, Marino ND, Franco M, Lodoen MB, and Boothroyd JC (2014). GRA25 is a novel virulence factor of Toxoplasma gondii and influences the host immune response. Infect Immun 82, 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik SM, Hackett CG, Tran F, Westwood NJ, and Lourido S (2014). Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9, e100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC, et al. (2016). A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435 e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik SM, Huet D, and Lourido S (2018). CRISPR-Cas9-based genome-wide screening of Toxoplasma gondii. Nat Protoc 13, 307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simossis VA, and Heringa J (2005). PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res 33, W289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens WZ, Wiles TJ, Martinez ES, Jemielita M, Burns AR, Parthasarathy R, Bohannan BJ, and Guillemin K (2015). Identification of Population Bottlenecks and Colonization Factors during Assembly of Bacterial Communities within the Zebrafish Intestine. MBio 6, e01163–01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztacho M, Segeletz S, Sanchez-Fernandez MA, Czupalla C, Niehage C, and Hoflack B (2016). BAR Proteins PSTPIP1/2 Regulate Podosome Dynamics and the Resorption Activity of Osteoclasts. PLoS One 11, e0164829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, et al. (2006). A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780. [DOI] [PubMed] [Google Scholar]
- Tomita T, Yamada T, Weiss LM, and Orlofsky A (2009). Externally triggered egress is the major fate of Toxoplasma gondii during acute infection. J Immunol 183, 6667–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Chen CL, and Chen HC (2015). Protein tyrosine phosphatase SHP2 promotes invadopodia formation through suppression of Rho signaling. Oncotarget 6, 23845–23856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle AH, Philip VM, Chesler EJ, and Mogil JS (2018). Comparing phenotypic variation between inbred and outbred mice. Nat Methods 15, 994–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno N, Harker KS, Clarke EV, McWhorter FY, Liu WF, Tenner AJ, and Lodoen MB (2014). Real-time imaging of Toxoplasma-infected human monocytes under fluidic shear stress reveals rapid translocation of intracellular parasites across endothelial barriers. Cell Microbiol 16, 580–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Yona S, and Jung S (2009). Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol 87, 30–38. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cirelli KM, Barros PDC, Sangare LO, Butty V, Hassan MA, Pesavento P, Mete A, and Saeij JPJ (2019). Three Toxoplasma gondii Dense Granule Proteins Are Required for Induction of Lewis Rat Macrophage Pyroptosis. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner JM, and Barragan A (2014). Tightly regulated migratory subversion of immune cells promotes the dissemination of Toxoplasma gondii. Int J Parasitol 44, 85–90. [DOI] [PubMed] [Google Scholar]
- Weidner JM, Kanatani S, Hernandez-Castaneda MA, Fuks JM, Rethi B, Wallin RP, and Barragan A (2013). Rapid cytoskeleton remodelling in dendritic cells following invasion by Toxoplasma gondii coincides with the onset of a hypermigratory phenotype. Cell Microbiol 15, 1735–1752. [DOI] [PubMed] [Google Scholar]
- Weidner JM, Kanatani S, Uchtenhagen H, Varas-Godoy M, Schulte T, Engelberg K, Gubbels MJ, Sun HS, Harrison RE, Achour A, et al. (2016). Migratory activation of parasitized dendritic cells by the protozoan Toxoplasma gondii 14-3-3 protein. Cell Microbiol 18, 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YO, Khaminets A, Hunn JP, and Howard JC (2009). Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 5, e1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to STAR Methods. sgRNA sequences for the eight sub-libraries available.
Table S2. Related to Figure 1. Pearson correlation on in vivo sample fitness scores.
Table S3. Related to Figure 1. List of control genes that are not expressed.
Table S4. Related to Table 1 and 2. Illumina sequencing raw reads, Fitness scores, and Z-scores.
Table S5. Identification of host interaction partners of Tg247520, Related to Figure 3. Immunoprecipitation (IP) with HA antibody on stable HEK293-derived (TREX-293) cells expressing Tg247520 or GRA15 with a C-terminal HA-FLAG epitope tag under the control of the tetracycline operator (first sheet, upper panel) or IP with HA antibody on immortalized murine DC cell line (DC2.4) infected for 3 h with parasites expressing HA-tagged Tg247520 or GRA15 (first sheet, lower panel). The table represents the putative host interaction partners identified through mass spectrometry analysis specific to Tg247520 IP. The total number of spectra of peptides for each protein and MW= Molecular weight are indicated (raw data in second and third sheet).
Table S6. Related to STAR Methods. sgRNAs for generation of individual gene knockout and PCR primers used in this study.
Data Availability Statement
All the sub libraries sgRNA and primers sequences are available in Table S1 and will be provided upon request. All the data from the in vivo screen is available in Table S4. All the data from the TgWIP IP mass spectrometry analysis is available in Table S5.