

Abstract
Acute Respiratory Distress Syndrome (ARDS) is a syndrome of acute respiratory failure caused by non-cardiogenic pulmonary edema. Despite five decades of basic and clinical research, there is still no effective pharmacotherapy for this condition and the treatment remains primarily supportive. It is critical to study the molecular and physiologic mechanisms that cause ARDS in order to improve our understanding of this syndrome and reduce mortality. The goal of this review is to describe our current understanding of the pathogenesis and pathophysiology of ARDS. First, we will describe how pulmonary edema fluid accumulates in ARDS due to lung inflammation and increased alveolar endothelial and epithelial permeability. Next, we will review how pulmonary edema fluid is normally cleared in the uninjured lung, and describe how these pathways are disrupted in ARDS. Finally, we will explain how clinical trials and preclinical studies of novel therapeutic agents have further refined our understanding of this condition, highlighting in particular the study of mesenchymal stromal cells (MSCs) in the treatment of ARDS.
Keywords: Acute Respiratory Distress Syndrome (ARDS), alveolar fluid clearance (AFC), vectorial ion transport, pulmonary edema, mesenchymal stromal cells (MSCs)
I. Introduction
Acute Respiratory Distress Syndrome (ARDS) is a syndrome of acute respiratory failure caused by non-cardiogenic pulmonary edema. The most common clinical disorders associated with the development of ARDS are bacterial and viral pneumonia. ARDS is also commonly caused by sepsis due to non-pulmonary sources, severe trauma, and aspiration of gastric contents, and less commonly by pancreatitis and drug reactions1. Criteria for the diagnosis for ARDS have changed over time; the current definition includes acute onset of impaired oxygenation (arterial hypoxemia with PaO2/FiO2 ratio <300 mmHg) and bilateral infiltrates on chest imaging in the absence of left atrial hypertension as the dominant cause of pulmonary edema2. Based on the Berlin definition, ARDS is divided into three categories of severity depending on the degree of hypoxemia: mild (PaO2/FiO2 200–300mm Hg), moderate (PaO2/FiO2 100–200mm Hg), and severe (PaO2/FiO2 <100mm Hg)2. The prevalence of ARDS in the United States is 5–35 cases/100,000 individuals annually, depending on the definitions utilized and study methodology3. The mortality of ARDS is approximately 25–40% in most studies4,5. Despite five decades of basic and clinical research, there is still no effective pharmacotherapy for this syndrome and the treatment remains primarily supportive with lung protective ventilation and a conservative fluid management strategy. Therefore, it is critical to study the pathogenesis and pathophysiology of ARDS in order to identify novel targeted therapies for this condition.
ARDS is a complex clinical syndrome with a heterogeneous clinical phenotype, which has made it more challenging to study. Nonetheless, since the first description of ARDS in 19676, advances in laboratory and clinical studies have yielded valuable insights into the mechanisms responsible for the pathogenesis and pathophysiology of this condition. When the lung is injured by infection, trauma, or inflammatory conditions, inflammatory pathways are activated. The inflammatory response can aid in pathogen clearance, but excess inflammation can also contribute to alveolar damage – specifically greater endothelial and epithelial permeability – resulting in the accumulation of protein-rich alveolar edema fluid. Once pulmonary edema fluid accumulates in the interstitium and air spaces of the lungs, it causes increased work of breathing and impaired gas exchange resulting in hypoxemia, reduced carbon dioxide excretion, and ultimately acute respiratory failure. In uninjured lungs, active ion transport across the alveolar epithelium creates an osmotic gradient that drives alveolar fluid clearance (AFC)7. However, in ARDS, the osmotic gradient is disrupted and AFC is reduced, further compounding the decreased capacity to remove edema fluid from the distal airspaces of the lung.
In this review, we will first describe how increased inflammation causes endothelial and epithelial permeability, ultimately resulting in the accumulation of pulmonary edema fluid. Second, we will explain why AFC is reduced in this condition, highlighting key molecular pathways involved. Finally, we will summarize how clinical trials and the study of novel therapeutics offer further insight into ARDS pathophysiology.
II. Pathogenesis of ARDS: Excess Inflammation, Endothelial, and Epithelial Permeability
There are several pathophysiologic derangements that are central to the development of ARDS, including dysregulated inflammation and increased lung endothelial and epithelial permeability. Initially, acute lung injury is driven by dysregulated inflammation. Microbial products or cell-injury associated endogenous molecules (danger associated molecular patterns) bind to Toll-like receptors on the lung epithelium and alveolar macrophages and activate the innate immune system8. Mechanisms of innate immune defense, such as the formation of neutrophil extracellular traps and histone release, can be beneficial in capturing pathogens, but may worsen alveolar injury9. The immune system also generates reactive oxygen species, leukocyte proteases, chemokines, and cytokines that help neutralize pathogens, but can also result in worsening lung injury10. In essence, there is a delicate balance between effective immune activation to combat infection and excessive or dysregulated activation that contributes to alveolar injury.
In addition to excessive inflammation in ARDS, another central pathophysiologic derangement is the disruption of the lung microvascular barrier due to increased endothelial and epithelial permeability (Figure 1A and 1B). In healthy lungs, endothelial stabilization is mediated by vascular endothelial cadherin (VE-cadherin), which is an endothelial-specific adherens junction protein that is required for to maintain endothelial barrier integrity in lung microvessels11. During lung injury, increased concentrations of thrombin, tumor necrosis factor-α (TNF-α), vascular endothelial growth factor (VEGF), and leukocyte signals in the lungs destabilize the VE-cadherin bonds, resulting in increased endothelial permeability and the accumulation of alveolar fluid12. The importance of VE-cadherin bonds has been confirmed in mouse models. Specifically, alveolar fluid accumulates in a mouse model of lipopolysaccharide (LPS)-induced lung injury, but when the VE-cadherin bonds are stabilized by genetic alterations that prevent breakdown or by blocking VE phosphodiesterase, there is reduced edema formation13,14. In sum, the inflammatory-induced damage to lung endothelium results in increased capillary permeability, and thus leads to pulmonary edema formation.
Figure 1: Increased alveolar endothelial permeability in ARDS.
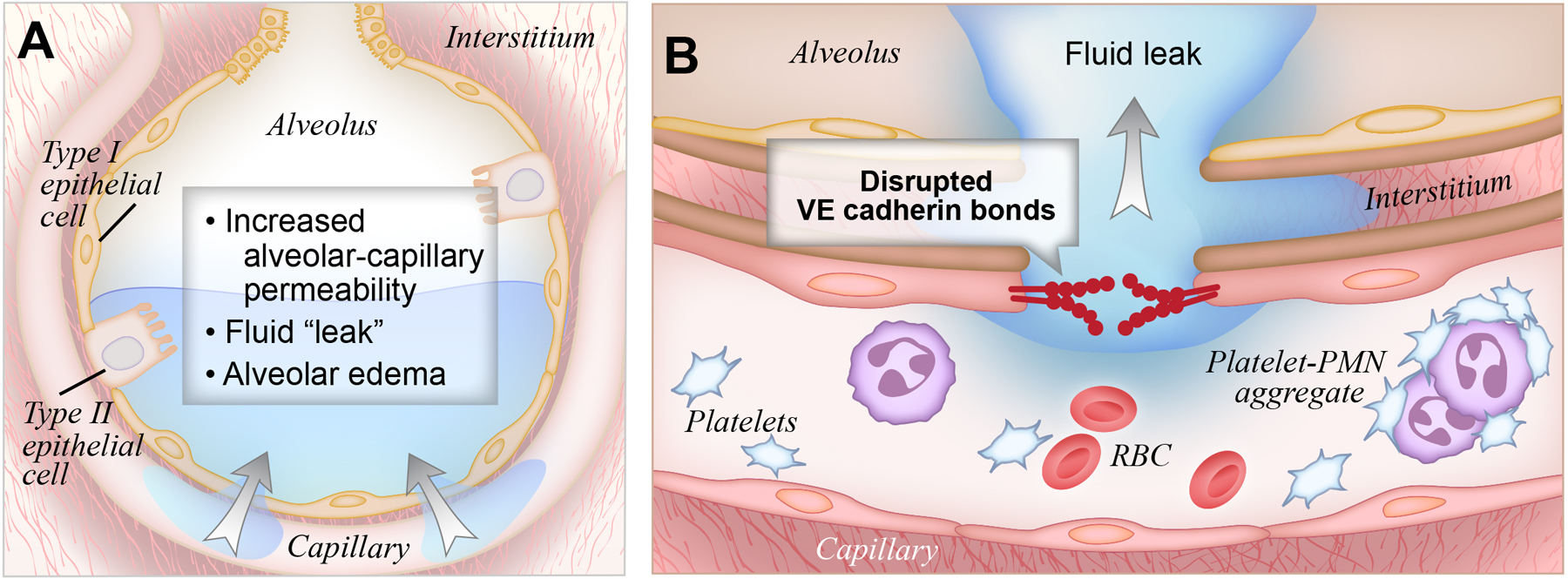
(A). In ARDS, inflammatory molecules disrupt alveolar barrier function, resulting in the accumulation of alveolar edema fluid.
(B). Specifically, disruption of VE-Cadherin bonds causes increased endothelial permeability, and subsequent leakage of water, solutes, leukocytes, platelets, and other inflammatory molecules into the alveolar space.
In addition to endothelial permeability, lung epithelial permeability is also an important factor in ARDS pathogenesis15,16. The alveolar epithelial barrier is similar to its endothelial counterpart, but has E-cadherin junctions instead of VE-cadherin junctions and it is substantially less permeable. Under pathologic conditions, neutrophil migration causes epithelial injury by disrupting intercellular junctions and causing apoptosis and denudation, ultimately resulting in increased epithelial permeability17. Restitution of epithelial integrity is critical for recovery and survival in acute lung injury. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling18, offering a potential therapeutic target to accelerate epithelial repair.
Finally, it is also important to note that environmental and genetic factors contribute to the susceptibility and severity of ARDS. Exposure to ambient air pollutants has been linked to risk of ARDS; this association is strongest in patients at risk for ARDS due to severe traumatic injuries19,20. Active and passive cigarette smoke exposure has been associated with the development of ARDS after blunt force trauma21, lung transplantation22 and non-pulmonary sepsis23. Chronic alcohol use increases the risk of acute lung injury24. Genetic variants have also been identified that confer increased risk of developing ARDS and are predictive of disease severity25. For example, genes involved in the inflammatory response and endothelial cell function, such as PPFIA1and ANGPT2, were identified as candidate genes for ARDS risk following major trauma26,27. Genetic variants in the FAS pathway, which regulates apoptosis and endothelial cell injury, were also associated with increased risk of ARDS28. In African Americans, a candidate gene study identified that a T-46C polymorphism in the promoter region of the Duff antigen/receptor for chemokines (DARC) gene was associated with higher mortality29. Additional studies are needed to better understand these environmental and genetic associations, which may further contribute to our understanding of the molecular pathways involved in ARDS pathogenesis.
III. Slow Resolution: Alveolar Fluid Clearance is Impaired in ARDS
Pulmonary edema can develop from increased pulmonary vascular pressure from left heart failure (cardiogenic pulmonary edema)30 or due to lung parenchymal damage from increased endothelial and epithelial permeability (non-cardiogenic pulmonary edema, as in ARDS, as described above)31. In both cases, the mechanism for the resolution of alveolar edema is the same: active ion transport across the alveolar epithelium creates an osmotic gradient that drives alveolar fluid clearance32.
Before discussing alveolar fluid clearance in ARDS, it is important to review how alveolar fluid is normally cleared in the uninjured lung. In the uninjured lung, vectorial ion transport across the intact alveolar epithelial layer creates an osmotic gradient that drives fluid from the alveoli into the lung interstitium (Figure 2A). After fluid is cleared into the interstitium, it can be drained by lymphatics or reabsorbed into the vasculature based on the balance of forces described in Starling equation. It was initially thought that only alveolar epithelial type II cells were involved in vectorial ion transport, but subsequent studies demonstrated an important role for alveolar epithelial type I cells as well33. The transport of sodium ions is the most important driver for the generation of the osmotic gradient: sodium is transported through the sodium channel (ENaC) on the apical surface, driven by the Na/K ATPase on the basolateral surface34,35. In animal models, this pathway is essential for survival; knockout of the alpha-subunit of ENaC in mice results in the inability to remove alveolar fluid at birth, causing respiratory failure and premature death36. In addition to ENaC, nonselective cation channels, cyclic nucleotide-gated channels, and the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels also help maintain the osmotic gradient37. Once the vectorial ion gradient is established, aquaporins facilitate the movement of water across the epithelial surface, but are not required for fluid transport38. This system of active ion-driven alveolar fluid reabsorption is the primary mechanism that removes alveolar edema fluid under both physiologic and pathological conditions39,40. However, in the setting of ARDS, the capacity to remove alveolar edema fluid is reduced, which is termed impaired alveolar fluid clearance (AFC). Patients with ARDS who have impaired AFC have decreased survival41,42.
Figure 2: Alveolar fluid clearance pathways in the uninjured lung vs. the lung affected by ARDS.
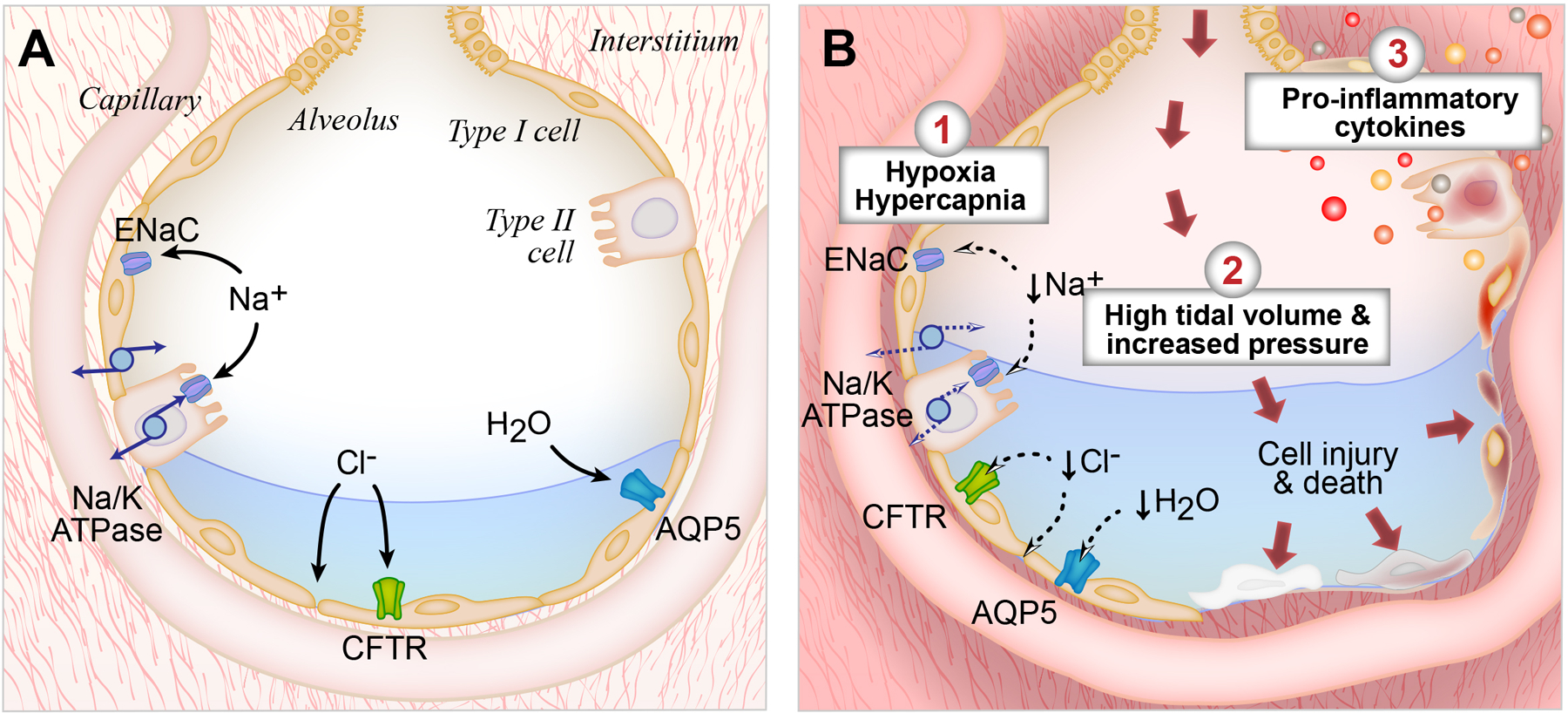
(A). In the uninjured lung, fluid is effectively cleared from the alveolar space by vectorial ion transport. Shown are the interstitial, capillary, and alveolar compartments of the lung, with pulmonary edema fluid in the alveolus. Both type I and type II alveolar cells are involved in transepithelial ion transport. Sodium (Na+) is transported across the apical side of the type I and type II cells through the epithelial sodium channel (ENaC), and then across the basolateral side via the sodium/potassium ATPase pump (Na/K-ATPase). Chloride (Cl-) is transported via the CFTR channel or by a paracellular route. Additional cation channels also transport ions across the alveolar epithelium (not shown). This vectorial ion transport creates an osmotic gradient that drives the clearance of fluid. Specifically, water (H20) moves down the osmotic gradient through aquaporin channels, such as aquaporin 5 (AQP5) or via an intracellular route (not shown). In the uninjured lung, this vectorial ion transport helps achieve effective alveolar fluid clearance.
(B). In lungs affected by ARDS, fluid is less effectively cleared from the lungs. First, hypoxia/hypercapnia result in downregulation of ENaC transcription and trafficking and less efficient function of the Na/K-ATPase. Second, high tidal volumes and elevated airway pressures injure the alveolar epithelium, inducing inflammation and cell death. Third, ARDS results in the formation of pro-inflammatory cytokines, which induce alveolar injury and cause reduced alveolar fluid clearance.
There are multiple physiologic and molecular mechanisms that cause a reduction in AFC in ARDS (Figure 2B). First, the primary physiologic respiratory impairments that characterize ARDS, hypoxia and hypercapnia, can directly impair AFC. ENaC transcription and trafficking are downregulated and the Na/K-ATPase functions less efficiently under states of low oxygen or high carbon dioxide, in part because reactive oxygen species trigger endocytosis and cell necrosis43,44,45. Therefore, supplemental oxygen and correction of hypercapnia can enhance the resolution of alveolar edema by helping to maintain active sodium transport across the lung epithelium.
Second, biomechanical stress in the lung can reduce AFC. High tidal volumes and elevated airway pressures injure the alveolar epithelium, inducing cell death and inflammation, which further reduces AFC46. If pulmonary hydrostatic pressures are elevated, the rate of net AFC is also reduced. These findings help explain the success of lung protective ventilation and conservative fluid strategies in reducing the morbidity and mortality of ARDS47,48.
Third, we now better understand the molecular mechanisms that contribute to the reduction in AFC in ARDS. Specifically, ARDS pulmonary edema fluid contains high levels of pro-inflammatory cytokines including interleukin (IL)-1β, IL-8, TNFα, and transforming growth factor-β1 (TGFβ1)49,50,51. When excessive levels of cytokines are present, they cause alveolar injury and reduced AFC52,53,54,55. This was elegantly demonstrated in an in vitro model of polarized human type II alveolar cells in 2006. Specifically, the authors showed that there are increased levels of cytokines and decreased levels of ion transport proteins in the presence of ARDS edema fluid compared to a plasma control56,57. Specifically, it is thought that the inflammatory edema fluid causes alveolar cell injury and necrosis, negating the tight epithelial barrier needed to establish an osmotic gradient and offsetting the effects of vectorial ion transport58,59. Cell necrosis and fluid accumulation in turn can trigger an even more pronounced inflammatory and immune response60. There are no current therapies that directly modify AFC, although lung protective ventilation itself reduces pro-inflammatory cytokines such as IL-6 and IL-861.
Ultimately, resolution of ARDS requires repair of the endothelial and epithelial barriers to allow for effective reabsorption of the alveolar edema fluid, as well as removal of inflammatory cells and cytokines from the airspaces and the lung interstitium. In order to repair the alveolar epithelial barrier, type II cells must proliferate and differentiate62. Progenitor cells are present in the bronchoalveolar junctions that aid in the regeneration of the endothelial and epithelial barrier63 and macrophages also contribute to tissue repair64. With repair of the endothelial and epithelial barrier, reabsorption of alveolar edema fluid can occur more efficiently via vectorial transport.
In addition to repairing the microvascular barrier, the resolution of ARDS requires clearance of neutrophils, monocytes, and anti-inflammatory molecules by macrophages65,66 and lymphocytes67. In a mouse model of influenza pneumonia, depletion of alveolar macrophages lead to an increased number of neutrophils and neutrophil extracellular traps, as well as slower recovery from lung injury68. Similarly, in a mouse model of endotoxin-induced lung injury, CD4+CD25+ regulatory T cells suppressed cytokine secretion and enhanced neutrophil apoptosis, aiding in faster resolution of lung injury67.
IV. Clinical Trials and Novel Therapeutics offer Further Insight About ARDS Pathogenesis
Since the first description of ARDS fifty years ago, there have been numerous clinical trials evaluating the efficacy of physiologic and pharmacologic interventions. Not only have these trials defined clinical practices, but they have enhanced our understanding of the pathophysiology of this condition.
Multiple clinical trials have supported the use of lung protective ventilation, with lower tidal volumes and airway pressures, to reduce morality in ARDS47,69,70,71. Follow-up studies investigated why this strategy is effective. In a rat model of ARDS, resolution of alveolar edema was three-fold faster with a tidal volume of 6 mg/kg as opposed to 12 mg/kg, in part due to decreased lung epithelial injury45. Similarly in human studies, patients who were subjected to lung protective ventilation had reduced markers of lung epithelial injury72 and reduced pro-inflammatory molecules (neutrophils, IL6, IL8, soluble TNF receptor 1)61,73.
Another central concept in ARDS treatment is the utilization of a conservative fluid management strategy, which was first suggested to be effective in the late 1970s74 and was later confirmed by a large ARDS Network Trial75. The beneficial effect of a conservative fluid strategy is thought to be due to the fact that lowering vascular pressures reduces transvascular fluid filtration across the injured alveolar capillary barrier. There is also evidence that a conservative fluid strategy results in decreased plasma levels of angiopoietin-2, suggesting that this strategy also has a protective effect on the vascular endothelium76. Further studies are needed to better understand the molecular mechanisms underlying this process.
Given that ARDS is a pro-inflammatory state, there have also been numerous clinical trials evaluating anti-inflammatory agents as a potential treatment for ARDS. However, clinical trials of glucocorticoids77,78,79, granulocyte macrophage-colony stimulating factor (GM-CSF)80, and antioxidants81 have not shown clinical utility to date. Similarly, it was hypothesized that anticoagulant therapy may be effective in ARDS treatment given the known interplay between procoagulant and proinflammatory pathways. However, a trial testing activated protein C (APC) did not reduce mortality in patients with non-septic ARDS82. While none of these trials suggest clinical benefit in ARDS treatment, it is possible that these therapies only improve outcomes in certain subphenotypes of ARDS. There is significant clinical and biological heterogeneity in ARDS, and recent studies suggest that there are two distinct and consistent subphenotypes of this condition83,84. Approximately 30% of patients have a hyperinflammatory subphenotype, which is characterized by increased inflammatory markers, more severe acidosis and shock, and worse clinical outcomes. Future clinical trials should consider these subphenotypes, as they may help us better understand ARDS pathophysiology and also may respond differentially to therapeutic interventions. For example, a large randomized controlled clinical trial of simvastatin therapy in ARDS showed no mortality difference in the treatment vs. placebo groups85, but secondary analysis showed decreased mortality in the patients with the hyperinflammatory subphenotype of ARDS86, suggesting some role for anti-inflammatory treatments in this subphenotype.
Another therapeutic strategy that has been proposed is to target molecules that stabilize endothelial and epithelial cell-cell junctions, given the central role of alveolar endothelial and epithelial permeability in ARDS pathogenesis. Sphingosine 1-phosphate (S1P) is a lipid that is recognized by G protein-coupled receptors on endothelial cells (eg S1Pr1) and mediates endothelial barrier integrity87. In both in vitro and in vivo models, S1P enhances pulmonary and systemic endothelial integrity88, and small-molecule agonists of endothelial S1Pr1 decrease cytokine and leukocyte recruitment in mouse models of influenza infection89. Specifically, S1P binds to S1Pr1 which induces actin cytoskeleton reorganization and localization of catenin and VE-cadherin molecules to the endothelial surface90. The Robo4/Slit signaling system also stabilizes the endothelial barrier. Slit2N inhibits tyrosine phosphorylation of VE-cadherin, preventing the internalization of VE-cadherin and the resultant increased endothelial permeability triggered by TNF-alpha, IL1, or LPS91. Studying proteins that help stabilize the endothelial and epithelial barrier has therapeutic potential, and also may offer further insight into the mechanisms that underlie endothelial and epithelial permeability.
Aside from targeting specific proteins that have therapeutic potential, most recently, mesenchymal stromal cells (MSCs) have been recognized as a promising new cell-based therapy for ARDS, further informing our understanding of ARDS pathogenesis. MSCs are bone-marrow-derived cells that can differentiate in vitro into chondrocytes, osteoblasts, and adipocytes, although they do not have true stem cell properties in vivo92. The therapeutic potential of MSCs has been studied in a number of medical and surgical conditions including sepsis93,94, diabetes95, myocardial infarction96, hepatic failure97, acute renal failure98,99, chronic obstructive pulmonary disease100, neurologic injuries101, graft-versus-host disease102, and trauma103. Therefore, it was hypothesized that MSCs may also be beneficial in the treatment of ARDS. To test this hypothesis, several groups studied whether MSCs reduce the severity of lung injury in pre-clinical models. Treatment with MSCs improved survival and reduced pulmonary edema in Escherichia coli endotoxin-induced lung injury in mice104. Subsequent studies showed that MSCs attenuated lung injury caused by live bacteria in mouse, rat, and in ex vivo human lung models of lung injury105,106,107. In addition, MSCs enhanced bacterial clearance and improved survival in murine models of sepsis94,108,109.
Given the potential therapeutic benefit of MSCs in the treatment of ARDS, many groups have sought to understand their mechanism of action, and several possible mechanisms have been proposed to date (Figure 3). Initially, it was thought that MSCs engrafted at the site of tissue injury and provided direct structural benefit110. However, with more detailed cell identification methods, engraftment is now thought to be a rare event of unclear physiologic significance111. Instead, the beneficial effect of MSCs does not require direct cell contact and several paracrine mechanisms have been proposed. First, it has been suggested that MSCs secrete proteins that have anti-inflammatory properties, and several have been identified to date: interleukin-1 receptor antagonist (IL1-ra)112, tumor necrosis factor alpha stimulated gene six (TSG-6)113, insulin-like growth factor 1 (IGF-1)114, and lipoxin A4115. Of note, clinical trials studying systemic anti-inflammatory agents have not been beneficial as previously described, but MSC therapy may provide anti-inflammatory effects that are multimodal and responsive to the cellular microenvironment in the lung. Second, there is evidence to suggest that MSCs affect lung endothelial and alveolar epithelial permeability via a paracrine mechanism, and the proteins angiopoietin-1, IL1-ra, and prostaglandin (PG)E2 have been implicated in this process116,117. Third, MSCs may secrete paracrine factors that improve AFC, with evidence to suggest that fibroblast growth factor 7 (FGF7) may be particularly important in this process118,119. Fourth, apoptosis of both immune and structural cells occurs during ARDS, so it has been suggested that paracrine factors such as interleukin 6 (IL6)120 and keratinocyte growth factor121 may have beneficial anti-apoptotic effects. Finally, MSCs have the capacity to alter the polarization of alveolar macrophages to an M2-like pro-resolving phenotype122.
Figure 3. Potential Mechanisms for the Therapeutic Effects of Mesenchymal Stromal Cells (MSCs) in ARDS.
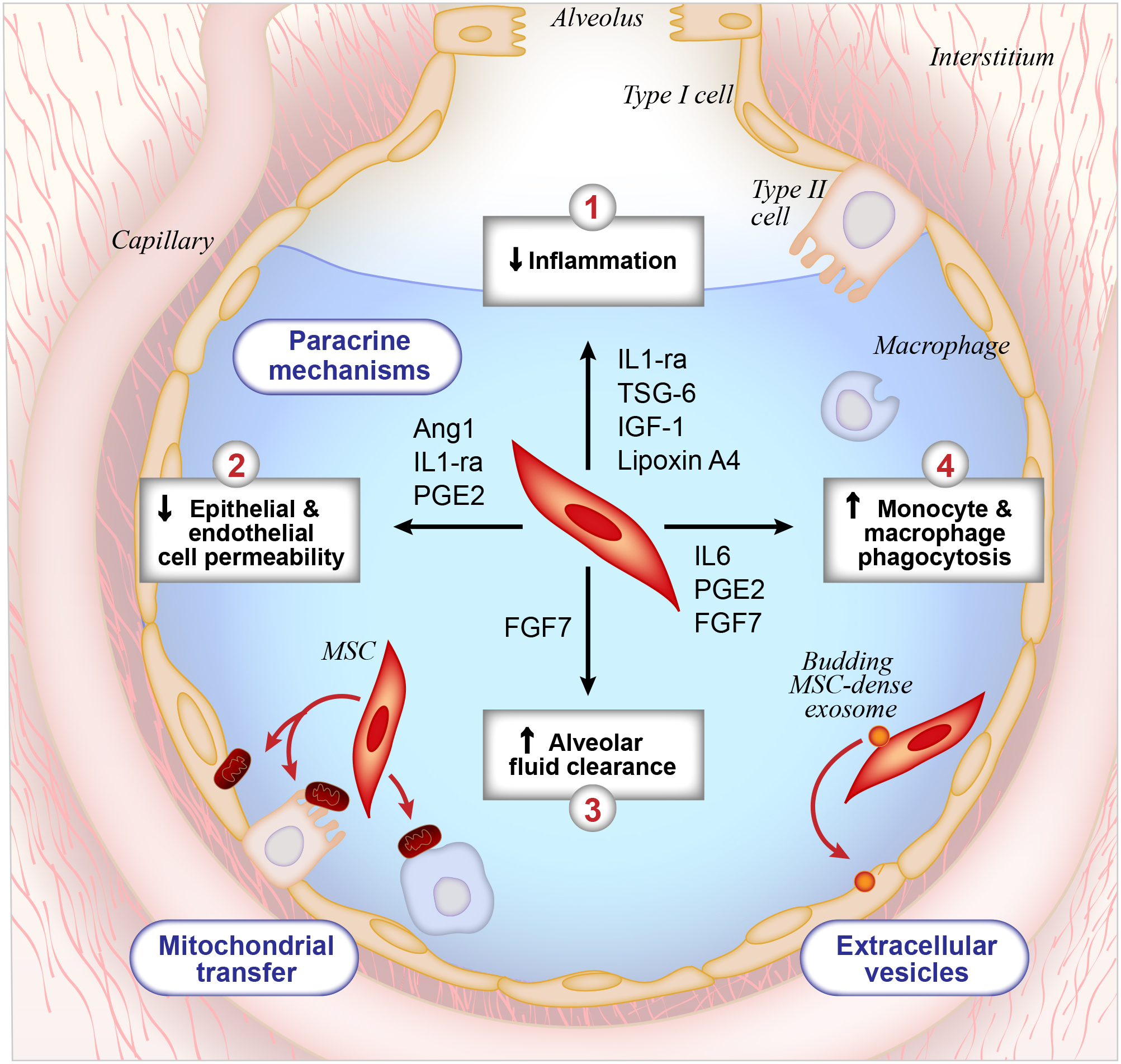
To date, multiple pre-clinical studies have demonstrated the therapeutic benefit of MSCs in the treatment of ARDS, and this diagram depicts our current mechanistic understanding of this therapeutic effect. First, MSCs secrete paracrine factors that modulate tissue repair through four mechanisms: (1) anti-inflammatory effects on host cells, (2) reduction of alveolar epithelial permeability in the lung, (3) increased rate of alveolar fluid clearance and (4) enhancement of host mononuclear cell phagocytic activity. Second, data suggests that MSCs directly transfer mitochondrial DNA to host cells, which also contributes to tissue repair and recovery. Third, MSCs secrete microvesicles that deliver micro RNA, RNA, proteins, and lipids to host cells.
While numerous studies suggest that paracrine factors are responsible for the beneficial effects of MSCs, more recent studies propose alternative mechanisms of action. For example, there is some evidence to suggest that MSCs mediate tissue repair through direct transfer of mitochondrial DNA to host cells123.124.125. Alternatively, it has been proposed that there are extracellular vesicles that bud off of MSCs and transfer biologically active material to host cells that have beneficial effects126,127. Thus, there may be several mechanisms by which MSCs mediate the resolution of lung injury, and further studies are needed to clarify key mechanisms of action.
Based on these pre-clinical data, phase 1 and 2 clinical trials are currently testing whether MSCs have therapeutic potential in humans128. Most recently, a prospective, double-blind, multicenter, Phase 2a randomized controlled trial demonstrated that it is safe to administer a single intravenous dose of MSCs in patients with moderate-severe ARDS129, so larger trials are needed to assess efficacy. Of note, the authors noted varying viability of the MSCs in this study, so it will be important to improve protocols to improve MSC viability in the future.
V. Conclusions
Over the past fifty years, there have been substantial advances in our understanding of ARDS pathogenesis. In vitro and in vivo studies have demonstrated that lung inflammation causes increased alveolar endothelial and epithelial permeability, resulting in the accumulation of pulmonary edema fluid. In ARDS, the mechanisms that typically remove pulmonary edema fluid are less effective. The study of novel therapeutic strategies, including cell-based MSC treatment, has further refined our understanding of ARDS pathophysiology, and may offer promising new treatment options for this condition. In addition, new therapies may be more effective if target to specific subphenotypes of ARDS defined by clinical and biological factors.
Acknowledgements
The authors thank Diana Lim for creating the figures for this article. Dr. Matthay was supported in part by NHLBI HL51856 and NHLBI HL108713. Dr. Ware was supported in part by NIH HL103836.
Footnotes
Conflict of Interest Statement
This work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. The Journal of clinical investigation 2012;122:2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R, Brochard L, Brower R, Esteban A, Gattinoni L, Rhodes A. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive care medicine 2012;38:1573–82. [DOI] [PubMed] [Google Scholar]
- 3.Villar J, Blanco J, Kacmarek RM. Current incidence and outcome of the acute respiratory distress syndrome. Current opinion in critical care 2016;22:1–6. [DOI] [PubMed] [Google Scholar]
- 4.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. New England Journal of Medicine 2005;353:1685–93. [DOI] [PubMed] [Google Scholar]
- 5.Máca J, Jor O, Holub M, Sklienka P, Burša F, Burda M, Janout V, Ševčík P. Past and present ARDS mortality rates: a systematic review. Respiratory care 2017;62:113–22. [DOI] [PubMed] [Google Scholar]
- 6.Ashbaugh D, Bigelow DB, Petty T, Levine B. Acute respiratory distress in adults. The Lancet 1967;290:319–23. [DOI] [PubMed] [Google Scholar]
- 7.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiological reviews 2002;82:569–600. [DOI] [PubMed] [Google Scholar]
- 8.Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. American journal of respiratory and critical care medicine 2010;181:1294–309. [DOI] [PubMed] [Google Scholar]
- 9.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nature Reviews Immunology 2011;11:519. [DOI] [PubMed] [Google Scholar]
- 10.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YC, Wang H, Liu H. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arteriosclerosis, thrombosis, and vascular biology 2008;28:223–32. [DOI] [PubMed] [Google Scholar]
- 12.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM. Vascular endothelial–cadherin is an important determinant of microvascular integrity in vivo. Proceedings of the National Academy of Sciences 1999;96:9815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D. Stabilizing the VE-cadherin–catenin complex blocks leukocyte extravasation and vascular permeability. The EMBO journal 2011;30:4157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, Vestweber D. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. Journal of Experimental Medicine 2011;208:2393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zemans RL, Matthay MA. Bench-to-bedside review: the role of the alveolar epithelium in the resolution of pulmonary edema in acute lung injury. Critical Care 2004;8:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. The Journal of clinical investigation 1991;88:864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginzberg HH, Shannon PT, Suzuki T, Hong O, Vachon E, Moraes T, Abreu MT, Cherepanov V, Wang X, Chow CW, Downey GP. Leukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and Akt. American Journal of Physiology-Gastrointestinal and Liver Physiology 2004;287:G286–98. [DOI] [PubMed] [Google Scholar]
- 18.Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, Yang IV, De Langhe S, Reynolds SD, Mason RJ, Kahn M. Neutrophil transmigration triggers repair of the lung epithelium via β-catenin signaling. Proceedings of the National Academy of Sciences 2011:201110144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ware LB, Zhao Z, Koyama T, May AK, Matthay MA, Lurmann FW, Balmes JR, Calfee CS. Long-term ozone exposure increases the risk of developing the acute respiratory distress syndrome. American journal of respiratory and critical care medicine 2016;193:1143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reilly JP, Zhao Z, Shashaty MG, Koyama T, Christie JD, Lanken PN, Wang C, Balmes JR, Matthay MA, Calfee CS, Ware LB. Low to moderate air pollutant exposure and acute respiratory distress syndrome after severe trauma. American journal of respiratory and critical care medicine 2019;199:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, Cohen MJ. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. American journal of respiratory and critical care medicine 2011;183:1660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diamond JM, Lee JC, Kawut SM, Shah RJ, Localio AR, Bellamy SL, Lederer DJ, Cantu E, Kohl BA, Lama VN, Bhorade SM. Clinical risk factors for primary graft dysfunction after lung transplantation. American journal of respiratory and critical care medicine 2013;187:527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calfee CS, Matthay MA, Kangelaris KN, Siew ED, Janz DR, Bernard GR, May AK, Jacob P, Havel C, Benowitz NL, Ware LB. Cigarette smoke exposure and the acute respiratory distress syndrome. Critical care medicine 2015;43:1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Critical care medicine 2003;31:S207–12. [DOI] [PubMed] [Google Scholar]
- 25.Gao L, Barnes KC. Recent advances in genetic predisposition to clinical acute lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology 2009;296:L713–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christie JD, Wurfel MM, Feng R, O’Keefe GE, Bradfield J, Ware LB, Christiani DC, Calfee CS, Cohen MJ, Matthay M, Meyer NJ. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS One 2012;7:e28268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer NJ, Li M, Feng R, Bradfield J, Gallop R, Bellamy S, Fuchs BD, Lanken PN, Albelda SM, Rushefski M, Aplenc R. ANGPT2 genetic variant is associated with trauma-associated acute lung injury and altered plasma angiopoietin-2 isoform ratio. American journal of respiratory and critical care medicine 2011;183:1344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glavan BJ, Holden TD, Goss CH, Black RA, Neff MJ, Nathens AB, Martin TR, Wurfel MM. Genetic variation in the FAS gene and associations with acute lung injury. American journal of respiratory and critical care medicine 2011;183:356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kangelaris KN, Sapru A, Calfee CS, Liu KD, Pawlikowska L, Witte JS, Vittinghoff E, Zhuo H, Auerbach AD, Ziv E, Matthay MA. The association between a Darc gene polymorphism and clinical outcomes in African American patients with acute lung injury. Chest 2012;141:1160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staub NC. The pathogenesis of pulmonary edema. Progress in cardiovascular diseases 1980;23:53–80. [DOI] [PubMed] [Google Scholar]
- 31.Staub NC. Plumonary Edema due to Increased Microvascular Permeability. Annual review of medicine 1981;32:291–312. [DOI] [PubMed] [Google Scholar]
- 32.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiological reviews 2002;82:569–600. [DOI] [PubMed] [Google Scholar]
- 33.Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proceedings of the National Academy of Sciences 2002;99:1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994;367:463. [DOI] [PubMed] [Google Scholar]
- 35.Matalon S, O’Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annual review of physiology 1999;61:627–61. [DOI] [PubMed] [Google Scholar]
- 36.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nature genetics 1996;12:325. [DOI] [PubMed] [Google Scholar]
- 37.Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. The Journal of general physiology 2002;119:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verkman AS, Matthay MA, Song Y. Aquaporin water channels and lung physiology. American Journal of Physiology-Lung Cellular and Molecular Physiology 2000;278:L867–79. [DOI] [PubMed] [Google Scholar]
- 39.Eaton DC, Chen J, Ramosevac S, Matalon S, Jain L. Regulation of Na+ channels in lung alveolar type II epithelial cells. Proceedings of the American Thoracic Society 2004;1:10–6. [DOI] [PubMed] [Google Scholar]
- 40.Mutlu GM, Sznajder JI. Mechanisms of pulmonary edema clearance. American Journal of Physiology-Lung Cellular and Molecular Physiology 2005;289:L685–95. [DOI] [PubMed] [Google Scholar]
- 41.Matthay MA, WIENER-KRONISH P. Intact Epithelial Barrier Function Is Critical for the Resolution of Alveolar Edema in Humans1–3. Am Rev Respir Dis 1990;142:1250–7. [DOI] [PubMed] [Google Scholar]
- 42.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. American journal of respiratory and critical care medicine 2001;163:1376–83. [DOI] [PubMed] [Google Scholar]
- 43.Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by β-adrenergic agonist treatment. American journal of respiratory cell and molecular biology 2001;25:554–61. [DOI] [PubMed] [Google Scholar]
- 44.Vadász I, Raviv S, Sznajder JI. Alveolar epithelium and Na, K-ATPase in acute lung injury. Intensive care medicine 2007;33:1243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Briva A, Vadász I, Lecuona E, Welch LC, Chen J, Dada LA, Trejo HE, Dumasius V, Azzam ZS, Myrianthefs PM, Batlle D. High CO2 levels impair alveolar epithelial function independently of pH. PloS one 2007;2:e1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs. American journal of respiratory and critical care medicine 2002;165:242–9. [DOI] [PubMed] [Google Scholar]
- 47.Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New England Journal of Medicine 2000;342:1301–8. [DOI] [PubMed] [Google Scholar]
- 48.Schuller D, Schuster DP. Fluid-management strategies in acute lung injury. The New England journal of medicine 2006;355:1175. [DOI] [PubMed] [Google Scholar]
- 49.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Critical care medicine 1999;27:304–12. [DOI] [PubMed] [Google Scholar]
- 50.Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, Zhu S, Pugin J, Matthay MA. Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1β-induced IL-6 expression. The Journal of Immunology 2004;172:2668–77. [DOI] [PubMed] [Google Scholar]
- 51.Ware LB, Matthay MA. The acute respiratory distress syndrome. New England Journal of Medicine 2000;342:1334–49. [DOI] [PubMed] [Google Scholar]
- 52.Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Mechanisms of TNF-α stimulation of amiloride-sensitive sodium transport across alveolar epithelium. American Journal of Physiology-Lung Cellular and Molecular Physiology 2001;280:L1258–65. [DOI] [PubMed] [Google Scholar]
- 53.Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, ndler MA, Totsch M, De Baetselier P, Fransen L, Fukuda N, Morel DR. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-α. American journal of respiratory and critical care medicine. 2003;168:1043–50. [DOI] [PubMed] [Google Scholar]
- 54.Dagenais A, Fréchette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Massé C, Berthiaume Y. Downregulation of ENaC activity and expression by TNF-α in alveolar epithelial cells. American Journal of Physiology-Lung Cellular and Molecular Physiology 2004;286:L301–11. [DOI] [PubMed] [Google Scholar]
- 55.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. Interleukin-1β decreases expression of the epithelial sodium channel α-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. Journal of Biological Chemistry 2005;280:18579–89. [DOI] [PubMed] [Google Scholar]
- 56.Fang X, Song Y, Hirsch J, Galietta LJ, Pedemonte N, Zemans RL, Dolganov G, Verkman AS, Matthay MA. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. American Journal of Physiology-Lung Cellular and Molecular Physiology 2006;290:L242–9. [DOI] [PubMed] [Google Scholar]
- 57.Lee JW, Fang X, Dolganov G, Fremont RD, Bastarache JA, Ware LB, Matthay MA. Acute lung injury edema fluid decreases net fluid transport across human alveolar epithelial type II cells. Journal of Biological Chemistry 2007;282:24109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. American journal of respiratory cell and molecular biology 2009;40:519–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Calfee CS, Matthay MA. Clinical immunology: Culprits with evolutionary ties. Nature 2010;464:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hung CF, Mittelsteadt KL, Brauer R, McKinney BL, Hallstrand TS, Parks WC, Chen P, Schnapp LM, Liles WC, Duffield JS, Altemeier WA. Lung pericyte-like cells are functional interstitial immune sentinel cells. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017;312:L556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Critical care medicine 2005;33:1–6. [DOI] [PubMed] [Google Scholar]
- 62.Adamson IY. The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab Invest 1974;30:35–42. [PubMed] [Google Scholar]
- 63.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005;121:823–35. [DOI] [PubMed] [Google Scholar]
- 64.Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. American Journal of Physiology-Lung Cellular and Molecular Physiology 2014;306:L709–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. The FASEB journal 2007;21:325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bratton DL, Henson PM. Neutrophil clearance: when the party is over, clean-up begins. Trends in immunology 2011;32:350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.D’alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS. CD4+ CD25+ Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. The Journal of clinical investigation 2009;119:2898–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. The American journal of pathology 2011;179:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. New England Journal of Medicine 1998;338:347–54. [DOI] [PubMed] [Google Scholar]
- 70.Eichacker PQ, Gerstenberger EP, Banks SM, Cui X, Natanson C. Meta-analysis of acute lung injury and acute respiratory distress syndrome trials testing low tidal volumes. American journal of respiratory and critical care medicine 2002;166:1510–4. [DOI] [PubMed] [Google Scholar]
- 71.Villar J, Kacmarek RM, Pérez-Méndez L, Aguirre-Jaime A. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Critical care medicine 2006;34:1311–8. [DOI] [PubMed] [Google Scholar]
- 72.Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, Matthay MA, NHLBI ARDS Network. Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax 2008;63:1083–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. Jama 1999;282:54–61. [DOI] [PubMed] [Google Scholar]
- 74.Staub NC. Pulmonary edema: physiologic approaches to management. Chest 1978;74:559–64. [DOI] [PubMed] [Google Scholar]
- 75.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. New England Journal of Medicine 2006;354:2564–75. [DOI] [PubMed] [Google Scholar]
- 76.Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA, NHLBI ARDS Network. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Critical care medicine 2012;40:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, Kariman K, Higgins S, Bradley R, Metz CA, Harris TR. High-dose corticosteroids in patients with the adult respiratory distress syndrome. New England Journal of Medicine 1987;317:1565–70. [DOI] [PubMed] [Google Scholar]
- 78.Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, Tolley EA. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. Jama 1998;280:159–65. [DOI] [PubMed] [Google Scholar]
- 79.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. New England Journal of Medicine 2006;354:1671–84. [DOI] [PubMed] [Google Scholar]
- 80.Nemunaitis J, Rabinowe SN, Singer JW, Bierman PJ, Vose JM, Freedman AS, Onetto N, Gillis S, Oette D, Gold M, Buckner CD. Recombinant granulocyte-macrophage colony-stimulating factor after autologous bone marrow transplantation for lymphoid cancer. New England Journal of Medicine 1991;324:1773–8. [DOI] [PubMed] [Google Scholar]
- 81.Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, Wright PE, Antioxidant in ARDS Study Group. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. Chest 1997;112:164–72. [DOI] [PubMed] [Google Scholar]
- 82.Liu KD, Levitt J, Zhuo H, Kallet RH, Brady S, Steingrub J, Tidswell M, Siegel MD, Soto G, Peterson MW, Chesnutt MS. Randomized clinical trial of activated protein C for the treatment of acute lung injury. American journal of respiratory and critical care medicine 2008;178:618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, NHLBI ARDS Network. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. The Lancet Respiratory Medicine 2014;2:611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, Calfee CS. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. American journal of respiratory and critical care medicine 2017;195:331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McAuley DF, Laffey JG, O’kane CM, Perkins GD, Mullan B, Trinder TJ, Johnston P, Hopkins PA, Johnston AJ, McDowell C, McNally C. Simvastatin in the acute respiratory distress syndrome. New England Journal of Medicine 2014;371:1695–703. [DOI] [PubMed] [Google Scholar]
- 86.Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, McDowell C, Laffey JG, O’Kane CM, McAuley DF, Johnston AJ. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. The Lancet Respiratory Medicine 2018;6:691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Ramadan I, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999;99:301–12. [DOI] [PubMed] [Google Scholar]
- 88.Xiong Y, Hla T. S1P control of endothelial integrity In Sphingosine-1-Phosphate Signaling in Immunology and Infectious Diseases 2014. (pp. 85–105). Springer, Cham. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011;146:980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Obinata H, Hla T. Sphingosine 1-phosphate in coagulation and inflammation InSeminars in immunopathology 2012. January 1 (Vol. 34, No. 1, pp. 73–91). Springer-Verlag. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Science translational medicine 2010;2:23ra19-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic transplants of bone marrow. Transplantation 1968;6:230–47. [PubMed] [Google Scholar]
- 93.Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X. Bone marrow stromal cells attenuate sepsis via prostaglandin E 2–dependent reprogramming of host macrophages to increase their interleukin-10 production. Nature medicine 2009;15:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, Stewart DJ. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. American journal of respiratory and critical care medicine 2010;182:1047–57. [DOI] [PubMed] [Google Scholar]
- 95.Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proceedings of the National Academy of Sciences 2006;103:17438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li TS, Hayashi M, Ito H, Furutani A, Murata T, Matsuzaki M, Hamano K. Regeneration of Infarcted Myocardium by Intramyocardial Implantation of Ex Vivo Transforming Growth Factor-β–Preprogrammed Bone Marrow Stem Cells. Circulation 2005;111:2438–45. [DOI] [PubMed] [Google Scholar]
- 97.Parekkadan B, Van Poll D, Suganuma K, Carter EA, Berthiaume F, Tilles AW, Yarmush ML. Mesenchymal stem cell-derived molecules reverse fulminant hepatic failure. PloS one 2007;2:e941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. American Journal of Physiology-Renal Physiology 2005;289:F31–42. [DOI] [PubMed] [Google Scholar]
- 99.Ullah I, Subbarao RB, Rho GJ. Human mesenchymal stem cells-current trends and future prospective. Bioscience reports 2015;35:e00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weiss DJ, Cruz FF. A Placebo-Controlled, Randomized Trial of Mesenchymal Stromal Cells Combined with One-Way Endobronchial Valve Therapy in Severe COPD. Cytotherapy 2016;18:S17. [Google Scholar]
- 101.Hu SL, Luo HS, Li JT, Xia YZ, Li L, Zhang LJ, Meng H, Cui GY, Chen Z, Wu N, Lin JK. Functional recovery in acute traumatic spinal cord injury after transplantation of human umbilical cord mesenchymal stem cells. Critical care medicine 2010;38:2181–9. [DOI] [PubMed] [Google Scholar]
- 102.Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M, Dini G. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. The Lancet 2008;371:1579–86. [DOI] [PubMed] [Google Scholar]
- 103.Matthay MA, Pati S, Lee JW. Concise review: mesenchymal stem (stromal) cells: biology and preclinical evidence for therapeutic potential for organ dysfunction following trauma or sepsis. Stem Cells 2017;35:316–24. [DOI] [PubMed] [Google Scholar]
- 104.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. The Journal of Immunology 2007;179:1855–63. [DOI] [PubMed] [Google Scholar]
- 105.Gupta N, Krasnodembskaya A, Kapetanaki M, Mouded M, Tan X, Serikov V, Matthay MA. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax 2012:thoraxjnl-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Devaney J, Horie S, Masterson C, Elliman S, Barry F, O’brien T, Curley GF, O’toole D, Laffey JG. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax. 2015. May 16:thoraxjnl-2015. [DOI] [PubMed] [Google Scholar]
- 107.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. American journal of respiratory and critical care medicine 2013;187:751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, Stewart DJ. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. American journal of respiratory and critical care medicine 2010;182:1047–57. [DOI] [PubMed] [Google Scholar]
- 109.Walter J, Ware LB, Matthay MA. Mesenchymal stem cells: mechanisms of potential therapeutic benefit in ARDS and sepsis. The Lancet Respiratory Medicine 2014;2:1016–26. [DOI] [PubMed] [Google Scholar]
- 110.Liechty KW, MacKenzie TC, Shaaban AF, Radu A, Moseley AB, Deans R, Marshak DR, Flake AW. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nature medicine 2000;6:1282. [DOI] [PubMed] [Google Scholar]
- 111.Wong AP, Dutly AE, Sacher A, Lee H, Hwang DM, Liu M, Keshavjee S, Hu J, Waddell TK. Targeted cell replacement with bone marrow cells for airway epithelial regeneration. American journal of physiology-lung cellular and molecular physiology 2007;293:L740–52. [DOI] [PubMed] [Google Scholar]
- 112.Ortiz LA, DuTreil M, Fattman C, Pandey AC, Torres G, Go K, Phinney DG. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proceedings of the National Academy of Sciences 2007;104:11002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Danchuk S, Ylostalo JH, Hossain F, Sorge R, Ramsey A, Bonvillain RW, Lasky JA, Bunnell BA, Welsh DA, Prockop DJ, Sullivan DE. Human multipotent stromal cells attenuate lipopolysaccharide-induced acute lung injury in mice via secretion of tumor necrosis factor-α-induced protein 6. Stem cell research & therapy 2011;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ionescu L, Byrne RN, van Haaften T, Vadivel A, Alphonse RS, Rey-Parra GJ, Weissmann G, Hall A, Eaton F, Thébaud B. Stem cell conditioned medium improves acute lung injury in mice: in vivo evidence for stem cell paracrine action. American Journal of Physiology-Lung Cellular and Molecular Physiology 2012;303:L967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fang X, Abbott J, Cheng L, Colby JK, Lee JW, Levy BD, Matthay MA. Human mesenchymal stem (stromal) cells promote the resolution of acute lung injury in part through lipoxin A4. The Journal of Immunology 2015:1500244. [DOI] [PubMed] [Google Scholar]
- 116.Fang X, Neyrinck AP, Matthay MA, Lee JW. Allogeneic human mesenchymal stem cells restore epithelial protein permeability in cultured human alveolar type II cells by secretion of angiopoietin-1. Journal of Biological Chemistry 2010:jbc-M110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goolaerts A, Pellan-Randrianarison N, Larghero J, Vanneaux V, Uzunhan Y, Gille T, Dard N, Planès C, Matthay MA, Clerici C. Conditioned media from mesenchymal stromal cells restore sodium transport and preserve epithelial permeability in an in vitro model of acute alveolar injury. American Journal of Physiology-Lung Cellular and Molecular Physiology 2014;306:L975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proceedings of the National Academy of Sciences 2009:pnas-0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McAuley DF, Curley GF, Hamid UI, Laffey JG, Abbott J, McKenna DH, Fang X, Matthay MA, Lee JW. Clinical grade allogeneic human mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. American Journal of Physiology-Heart and Circulatory Physiology. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, Ottonello L, Pistoia V. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem cells 2008;26:151–62. [DOI] [PubMed] [Google Scholar]
- 121.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. American journal of respiratory and critical care medicine 2013;187:751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Laffey JG, Matthay MA. Fifty years of research in ARDS. Cell-based therapy for acute respiratory distress syndrome. Biology and potential therapeutic value. American journal of respiratory and critical care medicine 2017;196:266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nature medicine 2012;18:759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, Stolz DB, Watkins SC, Di YP, Leikauf GD, Kolls J. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nature communications 2015;6:8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, O’Kane CM, Krasnodembskaya AD. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem cells 2016;34:2210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu YG, Feng XM, Abbott J, Fang XH, Hao Q, Monsel A, Qu JM, Matthay MA, Lee JW. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem cells 2014;32:116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, Krasnodembskaya AD. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. American journal of respiratory and critical care medicine 2017;196:1275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, Cosgrove K, Vojnik R, Calfee CS, Lee JW, Rogers AJ. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. The Lancet Respiratory Medicine 2015;3:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, Rogers AJ, Gotts JE, Wiener-Kronish JP, Bajwa EK, Donahoe MP. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. The Lancet Respiratory Medicine 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]