

Abstract
Recurrent and chronic Major Depressive Disorder (MDD) accounts for a substantial part of the disease burden because this course is most prevalent and typically requires long-term treatment. We associated blood DNA methylation profiles from 581 MDD patients at baseline with MDD status 6 years later. A resampling approach showed a highly significant association between methylation profiles in blood at baseline and future disease status (P=2.0×10−16). Top MWAS results were enriched specific pathways, overlapped with genes found in GWAS of MDD disease status, autoimmune disease and inflammation, and co-localized with eQTLS and (genic enhancers of) of transcription sites in brain and blood. Many of these findings remained significant after correction for multiple testing. The major themes emerging were cellular responses to stress and signaling mechanisms linked to immune cell migration and inflammation. This suggests that an immune signature of treatment-resistant depression is already present at baseline. We also created a methylation risk score (MRS) to predict MDD status 6 years later. The AUC of our MRS was 0.724 and higher than risk scores created using a set of five putative MDD biomarkers, genome-wide SNP data, and 27 clinical, demographic and lifestyle variables. Although further studies are needed to examine the generalizability to different patient populations, these results suggest that methylation profiles in blood may present a promising avenue to support clinical decision making by providing empirical information about the likelihood MDD is chronic or will recur in the future.
INTRODUCTION
MDD is a leading cause of disability worldwide1. Recurrent or chronic MDD accounts for a substantial part of this disease burden because this course is most prevalent and typically requires long-term treatment2; 3. Our overarching goal is to identify DNA methylation signatures in peripheral blood samples associated with future MDD status. Although methylation signatures in blood are unlikely to impact MDD directly, blood provides a biological environment for brain and can indirectly point to disease processes. For example, environmental insults that affect disease trajectories (e.g., stress) can modify methylation patterns in blood4-6. Methylation marks are also potential biomarkers as they are very stable in collected samples. Furthermore, blood is a suitable tissue because, for a biomarker to be useful in clinical settings, it is important that the biosample is easy to collect with a relatively small risk to the patient. The methylation signatures will therefore also be used to start generating prediction algorithms that could eventually be used to improve treatment7.
Specifically, we associated blood DNA methylation data from 581 patients at baseline with their MDD status 6 years later. To avoid missing sites of possible importance we used a sequencing-based methylation assay that provides nearly complete coverage of all 28 million CpGs in the human genome8; 9.
MATERIALS AND METHODS
The supplemental material gives a detailed description of study participants, methylation assay, and data analyses. Here we provide a brief summary.
Participants
Whole blood samples were obtained from 581 MDD patients from the Netherlands Study of Depression and Anxiety (NESDA)10 at baseline. MDD DSM-IV diagnoses (6-month recency) were obtained using the lifetime version of the Composite International Diagnostic Interview. All participants were of Dutch descent. The outcome variable was the presence/absence of a MDD diagnosis at the 6-year follow-up (MDDYear6). The 6-year follow up was chosen because it is the latest NESDA wave, which is important to assess long-term disease risk. Of the 581 patients diagnosed with MDD at baseline, 199 also received a MDD diagnosis at the 6-year follow-up. All participants provided written informed consent and the current study was approved by ethical committees in the Netherlands and USA.
Methylation Assay
To assay the methylome at baseline, we used an optimized protocol for methyl-CG binding domain sequencing (MBD-seq)11-14. The optimizations involved the choice of the the MBD protein15 and adaptations of the enrichment and sequencing protocol16; 17. MBD-seq is frequently confused with methylated DNA immunoprecipitation followed by sequencing (MeDIP-seq).18 While there are similarities in the workflow, MeDIP-seq suffers from lower performance and higher sequence bias than MBD-seq11; 12; 17; 19; 20. In the supplemental material, we provide a summary of comparisons our optimized MBD-seq protocol and “gold standard” whole genome bisulfite sequencing. Results show that optimized MBD-seq provides comparable information about the methylome as whole genome bisulfite assays8. Furthermore, MBD-seq has shown to detect previously reported robust associations21 as well as small effects that replicate using targeted bisulfite-sequencing22. Thus, it is a suitable assay for methylomewide association study (MWAS).
We obtained an average of 59.4 million (SD=11.2 million) reads per sample. This resulted in an average nonCpG-to-CpG score ratio23 of 0.010 (SD=0.005). The low ratio shows that the average CpG signal is high and the nonCpG background noise level is exceptionally low, allowing for detection of differently methylated regions.
Methylome-wide association study
Data quality control and analyses were performed with the Bioconductor package RaMWAS24. MWAS was performed using multiple regression analyses with four sets of covariates. First, we regressed out assay-related variables (i.e., potential technical artifacts) including the quantity of methylation-enriched DNA captured, peak sensitivity, percentage reads aligned, and reagent batch9. Second, we regressed out a battery of 27 clinical, demographic and lifestyle characteristics (see Table 1) including sex, symptom severity at baseline, smoking, and antidepressant use. To avoid inclusion of uncorrelated covariates and unnecessary loss of degrees of freedom, these variables were regressed out by creating a risk score (see next section). Third, to avoid confounding due to cell type heterogeneity, we regressed out blood cell type proportions as estimated by the methylation data25 using MBD-seq specific “reference methylomes”. We have previously shown that this effectively controlled for confounding due to cell type heterogeneity26. Fourth, principle components were regressed out to capture any remaining unmeasured sources of variation.
Table 1.
Correlation of clinical and lifestyle characteristics, and depression biomarkers with Year 6 depression status
| Depressed | Remitted | Corr.a | P-value | |
|---|---|---|---|---|
| Clinical | ||||
| Symptom severity (IDS) | 36.4(11.2) | 31.3(10.1) | 0.225 | 4.43×10−08 |
| Co-morbid Anxiety | 79.40% | 70.60% | 0.094 | 0.024 |
| Any antidepressant | 50.70% | 41.10% | 0.092 | 0.026 |
| SSRI | 68.40% | 71.30% | 0.055 | 0.187 |
| SNRI | 21.70% | 23.50% | 0.022 | 0.605 |
| TCA | 9.90% | 8.90% | 0.032 | 0.435 |
| Other | 22.70% | 23.50% | 0.029 | 0.483 |
| Problematic benzodiazepine use | 7.30% | 7.32% | −0.001 | 0.988 |
| Psychotherapy | 87.40% | 86.10% | 0.072 | 0.082 |
| Family history of depression | 80.40% | 76.90% | 0.039 | 0.342 |
| NEO - Neuroticism | 44.0(5.8) | 41.4(6.1) | 0.175 | 2.22×10−05 |
| NEO - Extraversion | 31.2(6.3) | 33.8(6.5) | −0.195 | 2.29×10−06 |
| NEO - Openness | 38.4(6.4) | 38.5(6.2) | −0.011 | 0.793 |
| NEO - Agreeableness | 42.8(5.6) | 43.0(5.0) | −0.022 | 0.603 |
| Childhood trauma index score | 1.28(1.24) | 1.08(1.2) | 0.082 | 0.049 |
| Stressful life events (past year) | 0.99(1.23) | 0.95(1.1) | 0.015 | 0.718 |
| Lifestyle | ||||
| Smoker | −0.005 | 0.908 | ||
| Never | 33.60% | 29.50% | ||
| Former | 25.10% | 32.50% | ||
| Current | 41.20% | 37.90% | ||
| Alcohol use (AUDIT Score) | 6.19(5.03) | 5.88(5.0) | 0.029 | 0.491 |
| Body mass index (BMI) | 26.1(5.4) | 25.5(5.1) | 0.049 | 0.238 |
| Physical activity (MET-min/week) | 3479(3236) | 3708(3278) | −0.033 | 0.424 |
| Number of chronic diseases under treatment | 0.77(0.99) | 0.65(0.9) | 0.043 | 0.300 |
| Disability (WHO-DASII) | 37.7(14.7) | 30.9(14.5) | 0.216 | 1.52×10−07 |
| Biomarkers | ||||
| Brain derived neurotrophic factor | 9.12(3.59) | 9.03(3.48) | 0.013 | 0.759 |
| Interleukin-6 | 1.13(1.22) | 1.15(1.22) | −0.009 | 0.831 |
| Tumor necrosis factor- alpha | 1.19(1.81) | 1.09(1.37) | 0.035 | 0.406 |
| Vitamin D | 59.8(26.5) | 62.4(26.8) | −0.046 | 0.276 |
| Telomere length | 1.10(0.29) | 1.12(0.31) | −0.024 | 0.556 |
Corr. is the correlation between the characteristic and Year 6 depression status (depressed\ remitted).
Bioinformatics
Pathway analyses were performed using of the Reactome27 database. These analyses used circular permutations that properly control the Type I error in the presence of correlated sites (see28 and Figure S4). Furthermore, as the permutations are performed on a CpG level they account for gene size, as genes with more CpGs are more likely to be among the top results in the permutations. To correct for testing multiple pathways, we determined the threshold that resulted in one or more significant pathways in 5% of the 100,000 permutations (i.e., this controls the family-wise error rate at the 0.05 level). Furthermore, after removing the pathways that survived multiple testing, we used the permutations to examine whether genes from the other pathways collectively were still overrepresented in the top MWAS.
Circular permutation tests were also used to study whether MWAS findings were i) enriched for top findings from GWAS studies and ii) co-localized with potential regulatory sites in brain and blood tissue. If multiple thresholds were specified to define “top findings”, we corrected for this multiple testing by using the same thresholds in the permutations and selecting the most significant result to generate the empirical null distribution.
Methylation risk score
To predict MDDYear6 we used elastic nets that are suitable when there are many more variables than observations and effects are small and correlated29-31 (e.g., techniques such as lasso can select no more variables than there are samples and tend to only assign a non-zero coefficient to a single variable out of a set of correlated variables32). Elastic nets are akin to multiple regression but place a penalty on the size of the regression coefficients that is controlled by the alpha parameter. For the main analysis aimed at deriving the best possible prediction, we set alpha to zero, which results in all sites having non-zero regression coefficients and being retained in the model. However, for our exploratory analyses aimed at examining whether we can approximate the predictive power of this full site MRS with a smaller number of sites, we also fit elastic nets with alpha at 0.5.
We used k-fold cross-validation to avoid overfitting33. Specifically, we randomly partitioned the sample into k=10 equal sized subsamples. Of the k subsamples, k − 1 were used as a “training” set to fit the elastic net and obtain regression coefficients. This model is then used in “test” set to obtain predictions for the samples that were set aside. The entire cycle of CpG selection through MWAS followed by training the elastic net is repeated for each of the k folds. Because both the selection of CpGs and estimation of the prediction model is not affected by the participants in the test set, this yields an unbiased estimate of the predictive power.
RESULTS
Table S1 shows that MDD cases and controls at the six-year follow-up were generally similar in terms of demographic profiles.
Methylome-wide association study
The Quantile-Quantile (QQ) plot (Figure 1A) for the MWAS shows that many P-values are above the upper 95% confidence interval. This implied many CpGs with small or modest effects. The Manhattan plot (Figure 1B) suggested that associated sites are distributed across the methylome.
Figure 1. MWAS of baseline methylation and MDD status six years later.

(A) Quantile-Quantile plot. The main diagonal of the plot indicates the P-values we would obtain assuming none of the CpGs predict MDD status six years later (MDDYear6) with the yellow curved lines denoting the 95% confidence intervals. The red points in the QQ plot are the observed MWAS P-values. (B) Manhattan plot. The plot shows the MWAS P-values by their chromosomal location. The dashed line indicates the threshold used for inclusion of CpGs in the MRS.
Studying the top MWAS findings (Table S2), the most significant CpG was in MAPKAPK5 (P < 5.76×10−8), a gene previously associated with anxiety34 that is highly co-morbid with MDD. The second top CpG mapped to LINC01192 (CT64) with prior evidence of shared association across MDD, bipolar disorder, and schizophrenia35. The fourth top finding was NOL4 (P < 8.51×10−8) that has been shown to predict antidepressant treatment response36. Other notable top results involves genes linked to neuronal development (MYO1037, RNF11138) and late-onset depression (SLC36A139). None of these individual top findings remained significant after corrections (false discovery rate or Bonferroni) for multiple testing.
Pathway analyses
The collective top findings (P < 5×10−5) involved 2,785 CpGs and 1,146 genes that were subjected to pathway analyses. Several pathways remained significant after correcting for multiple testing (Table S3). The mean odds ratio of the remaining pathways was 1.69 with P value 9.0×10−4. Thus, although they did not reach significance individually after correcting for multiple testing, collectively genes from these pathways were still significantly overrepresented among the top MWAS findings.
Pathways with P < 0.05 were clustered using the Louvain Method for community detection40 as implemented in igraph41 based on their overlapping member genes (Figure 2). Clusters that emerged included golgi-related processes, small RNA transcription, and cell-cell communication. The Golgi apparatus (red cluster) is a cellular hub of protein processing and is central in secretory and stimuli-sensing pathways42. Golgi fragmentation, that is commonly observed following cellular stress, e.g., oxidative stress or infection43, can induce the mitochondrial apoptotic pathway44. There is literature suggesting that neurons may be particularly sensitive to Golgi stress, especially under excitotoxic and inflammatory conditions44-46.
Figure 2. Summary of pathway analysis.
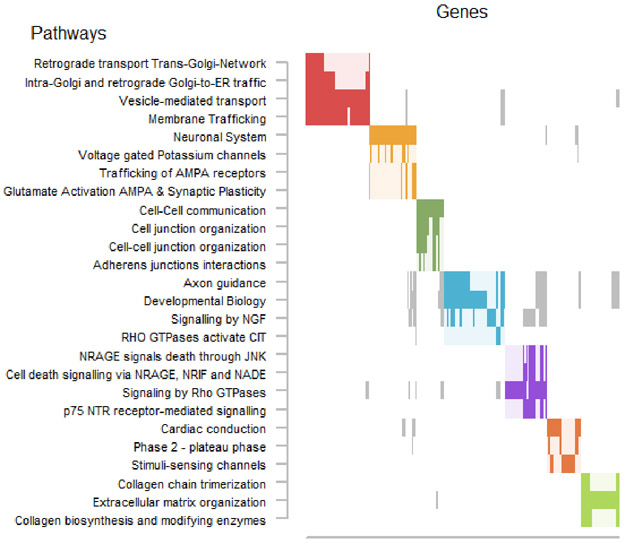
We only show pathways that overlapped with at least 4 top MWAS genes and yielded over-representation P-values < 0.05. Pathways often share genes. The raster plot visualizes the clustering of pathways (y-axis) as determined on the basis of their overlapping genes (x-axis). The solid rectangles indicate genes that were both among the top MWAS results and a member of the listed pathway.
Multiple pathway clusters were related to small non-coding RNA expression (orange, green, light blue clusters). Small nuclear RNA (snRNA) and PIWI-interacting RNA (piRNA) are primarily transcribed by RNA polymerase II and are implicated in splicing and transposon silencing, respectively47-50. While some evidence implicates other non-coding RNAs in MDD51, the potential roles for snRNA and piRNA in disease are less well characterized. One report52 demonstrated snRNA-dependent RNA editing of serotonin receptor subtype 2C mRNA in depression following interferon-α treatment. Interestingly, interferon and cytokine signaling were also implicated by our pathway analyses (brown cluster).
While cell-cell communication (blue cluster) is an absolute requirement of all cells in an organism, the enriched members of these pathways were particularly interesting. Many genes belonging to the claudin family were among those enriched. Social stress has been shown to lead to depression-like behavior via downregulation of claudin-5 and subsequent disruption of the blood-brain barrier (BBB)53. Collagens are major components of the extracellular matrix (ECM) and are key for immune cell attachment and infiltration54. These ECM proteins are recognized by leukocyte integrin receptors55 which were also among the enriched pathway members (pink cluster). Together, these results suggest changes in immune cell and BBB interactions are present in MDD.
Methylation risk score
We determined the MRS should contain 75,000 CpGs (Figure 3A). This MRS does not yield the best AUC but corresponds to the point where the predictive power reaches a stable plateau (Figure 3A, e.g., using 75 thousand or 300 thousand CpGs results in the same AUC). Thus, a substantial number of CpGs are needed to avoid excluding predictive sites with small effects. The ROC curve for the MRS (red line Figure 3B) corresponded to an AUC of 0.724 (P=2.0×10−16) suggesting a highly significant association between methylation profiles in blood and future disease status.
Figure 3. Predictive power of the MRS.

(A) The number of CpG sites in the MRS (x-axis) is plotted against the AUC (y-axis). The asterix indicates the point where predictive power stabilizes. When many sites with no predictive power are added, it becomes more difficult for the elastic net to “find” the sites that predict. This explains the very minor gradual decrease when using more than 75,000 CpGs. (B) Receiver operating characteristic (ROC) curves for different predictor domains. Domain abbreviations are: MRS for genome-wide methylation data, CDL for the 27 Clinical, Demographic or Life style variables, SNPs for genome-wide SNP data, and Biomarkers for the five putative MDD biomarkers. (C) Receiver operating characteristic (ROC) curves for combined risk domains. MRS + CDL combines the predictive power of MRS and CDL. MRS + SNPs combines the predictive power of MRS and CDL.
When we repeated the analyses using 50-fold cross validation, the AUC remained 0.724. Furthermore, as can be seen from Fig S5, the AUC is not driven by outliers as folds consistently indicate a similar AUC. Thus, results were robust to any way the random subsamples are drawn. Excluding lab technical covariates reduced the AUC to 0.688, justifying our choice to include them. To examine whether the predictive power of the 75,000 site MRS could be approximated with a smaller number, we used a version of the elastic net that selected only the most important predictors. This resulted in 771 sites. However, the ROC curve of the reduced set MRS was well below the curve for the full set MRS (Figure S6). In addition, the use of only 771 sites decreased the AUC from 0.724 to 0.681.
We also attempted to predict MDD status at the intermediate waves, 2 and 4 years after baseline. Using 75,000 sites, the predictive power was more modest with AUC=0.571 for year 2 and 0.566 for year 4. To discriminate reliably between transient versus long-term MDD, it is critical that a distant time point is used. Thus, if only a short amount of time has passed between baseline and the time point where MDD is assessed, it will be impossible to distinguish cases with transient versus long-term MDD. This reduced ability to assess long-term disease status at the intermediate waves may have negatively affected predictive power.
Smith et al.56 observed an upward bias with k-fold cross validation, particularly with small sample sizes. However, they studied a scenario with only a very small number of predictors (3/5) that were likely correlated with the outcome of interest. In contrast, we have thousands of predictors, a substantial proportion of which may not be associated with the outcome. To test the risk of overfitting, we created 25 data sets where case-control status was randomly permuted. The mean correlation with MDD status at year 6 was −0.004 with an AUC of 0.513. Furthermore, whereas using all samples the AUC was 0.724, taking a random selection of 50% of the total number of samples resulted in an AUC of 0.647 and with 75% of all samples the AUC became 0.693. The finding that predictive power improves with larger sample sizes may be explained by the fact that it becomes easier to detect the CpGs with effects among all CpGs. Thus, there was no evidence the AUC was the result of overfitting and it may even have been higher if our sample size would have been bigger.
Comparing the MRS to other predictors
We used the same method to predict MDDYear6 from baseline data on (1) five other putative MDD biomarkers, (2) genome-wide SNP data, and (3) 27 Clinical, Demographic and Lifestyle characteristics (CDL). Table 1 lists all these variables and detailed descriptions are in the Supplemental Material. The biomarkers showed no significant correlations with MDDYear6. From the CDL domain, symptom severity, use of any antidepressant, co-morbid anxiety, neuroticism, openness and level of disability were significantly correlated with MDDYear6 (Table 1). To explore the SNP data, we performed a genome-wide association study, assuming an additive model for the SNP effects (see QQ-plot Figure S7).
The AUC of the CDLs and SNP risk score were 0.642 (P=3.3×10−9) and 0.549 (P=5.8×10−2), respectively (Figure 3A). The biomarkers AUC of 0.437 implied that these variables had no predictive value (Figure 3B). We investigated whether the predictive power of the MRS could be increased by including either the CDL or SNP risk score (Figure 3C). Only the inclusion of the CDL predictors showed a marginal prediction improvement (AUC = 0.742 for CDL and MRS combined versus 0.724 for the MRS only and 0.642 for CDL only). This increase was significant according to the DeLong test57 (P=0.01).
We studied which predictors from the CDL group were most critical. For example, smoking did not account for much of the predictive power. This is consistent with the lack of a significant association with MDDYear6 (Table 1). We did find a CpG 20,000 bp apart from the CpG in the AHRR gene that is a very reliable indicator of smoking(15, 16) among the 75,000 sites of the MRS. Anti-depressant use was associated with future disease status (Table 1, P=0.026) but effects were small and had no major effect on methylome (Figure S8). The best “clinical” predictor was baseline MDD symptom severity, which also showed the strongest correlation with MDDYear6 (Table 1). Thus, the AUC was 0.635 for MDD symptom severity only and 0.642 for the entire CDL group.
Overlap with GWAS and co-localization analyses
Table 2 reports results after testing overlap with GWAS and co-localization analyses. The overall P-value in the final column corrects for performing two tests for each data set (one for top MWAS findings the other for the 75,000 CpGs in the MRS). After further correcting this overall P-value for testing 8 data sets (Bonferroni corrected threshold 0.05/8=0.006), many tests remained significant.
Table 2.
Results GWAS enrichment analyses and co-localization analyses
| External data | Top MWAS | MRS | Overall | ||
|---|---|---|---|---|---|
| OR | P-val. | OR | P-val. | P-val. | |
| PGC MDD GWAS meta | 2.50 | <10−5 | 2.36 | <10−5 | <10−5 * |
| GWAS cat. Inflammation* | 1.03 | 0.44 | 1.26 | <10−5 | <10−5 * |
| GWAS cat. Infection* | 1.20 | 0.13 | 1.09 | 0.08 | 0.08 |
| GWAS cat. Autoimmune* | 1.05 | 0.35 | 1.29 | <10−5 | <10−5 * |
| GTEx eQTLs BA24 | 1.46 | <10−5 | 1.66 | <10−5 | <10−5 * |
| GTEx eQTLs BA9 | 1.52 | <10−5 | 1.66 | <10−5 | <10−5 * |
| GTEx eQTLs whole blood | 1.47 | <10−5 | 1.62 | <10−5 | <10−5 * |
| McClay et. al. meQTLs | 0.91 | 0.95 | 1.01 | 0.83 | 0.83 |
OR is odds ratio,
significant after correction for multiple testing. Number of SNPs in GWAS cat. inflammation, GWAS cat. infection, GWAS cat. Autoimmune was 919, 520, and 1843 respectively.
We first looked for overlap of MWAS findings with the top 10,000 variants from the recent PGC MDD GWAS meta-analysis58, and indeed found overlap (Overall P-value <10−5). Many of the above pathways have been linked to autoimmunity and inflammation. We therefore tested whether our top findings were significantly enriched for loci containing SNPs that were reported to be associated with these disorders according to the NHGRI-EBI GWAS Catalog59. Significant enrichment was observed for loci associated with autoimmune disease and inflammation (Overall P-value <10−5). Results were mainly driven by the MRS likely because the GWAS Catalog results for a specific disease involve a limited number of SNPs resulting in low power when the top MWAS also contain few sites.
Next, we studied whether MWAS findings co-localized with cis expression quantitative trait loci (cis-eQTLs) in two brain regions (Brodmann area 9 and 24) and whole blood using GTEx version 760 as well as methylation quantitative trait loci (cis-meQTLs) in blood using a genome-wide study in 697 normal subjects21. Significant enrichment was observed for cis-eQTLs in both brain regions as well as blood, indicating MWAS findings were overrepresented at true regulatory sites. The cis-meQTL analysis did not reach significance.
Finally, MWAS results were tested against Roadmap Epigenomics Project chromHMM Core 15-state model chromatin tracks61. Figure S8 shows a very consistent pattern across fetal and adult brain as well as monocytes and groups of T cells in blood. Thus, MWAS signals were consistently found at weak/strong transcription sites with many tests having P-value <10−5 surviving corrections for testing 15 states (0.05/15=3.3×10−3). The MWAS results also showed enrichment for genic enhancers in brain tissue with potential importance for transcriptional regulation.
DISCUSSION
A resampling approach showed a highly significant association between methylation profiles in blood at baseline and MDD status 6 years later (AUC=0.724, P=2.0×10−16). Top MWAS results clustered in pathways, overlapped with findings of external GWAS studies, and co-localized with eQTLS and (genic enhancers of) of transcription sites in brain and blood. These findings remained significant after correcting for multiple testing. The major themes emerging were cellular responses to stress and signaling mechanisms linked to immune cell migration and inflammation.
External validation of a baseline immune component was obtained through significant enrichment of SNPs previously associated with autoimmune disease and inflammation. The exact molecular mechanisms this risk remain to be elucidated. However, several findings pointed to stress-induced immune cell activation and systemic inflammation. Namely, cross-talk between the central nervous system and peripheral immune cells across the BBB appears to be a redundant theme. Our results also implicated inflammatory cytokine signaling and small-RNA transcription in recurrent MDD. It is interesting to note that snRNA complexes are often antigens for autoantibodies in autoimmune/autoinflammatory disorders62; 63.
The possibility of autoimmunity in MDD pathogenesis appears to be supported by previous studies establishing clear links between immune responses and mood64; 65. In patients, autoimmune disorders and severe infections greatly increase the risk for mood disorders66. The outcome of ongoing clinical trials of anti-inflammatory biologics (tocilizumab) as therapeutics for MDD will be of great interest67 and could further corroborate MDD as a systemic immune disorder.
It has been challenging to find strong predictors of MDD disease course68-72. The AUC of our methylation risk score (MRS) predicting MDD status 6 years later was 0.724. We also calculated the predictive power of a set of five putative MDD biomarkers, genome-wide SNP data, and 27 clinical, demographic or lifestyle variables. Our methylation predictor outperformed all these predictor sets and seemed to incorporate most of their predictive power, as the inclusion of any other set only marginally increased the AUC of the MRS.
The MBD-seq assay used in this article has several properties that makes it potentially useful for clinical applications. First, in comparison to other approaches such as targeted bisulfite sequencing or pyrosequencing, it is cost-effective. The MRS we used contained 75,000 CpGs but it is prohibitive to assay that many sites with these targeted approaches. However, even for the reduced site MRS containing 771 CpGs the MBD-seq costs of about $300/sample compare favorably to targeted bisulfite sequencing (about $450/sample) and pyrosequencing (about $1,450/sample). With sequencing costs dropping steadily, MBD-seq may become even more cost-effective in the future. Second, as all predictive sites can be used, it prevents loosing predictive power merely because relevant CpG sites are not assayed. Third, as it does not restrict assays to a specific set of sites there, it can simultaneously be used to calculate risk scores for other clinical features or risk of co-morbid disorders. Fourth, MBD-seq can be performed with as little as 5–20ng of DNA14 meaning that it suffices to use blood left over from routine clinical tests or collected through non-invasive procedures such as finger-pricks. Fifth, MBD-seq assays can be automated using standard robotics. This enables “high-throughput” testing by diagnostic laboratories.
Several limitations should be noted. Our study involved DNA obtained from whole blood. Because buccal epithelial and brain cells are derived from the same ectodermal layer during development, DNA methylation in buccal epithelia cells (e.g., as white blood are the main source of DNA in spit/saliva this can be collected through buccal swabs that scrape loose these cells) may potentially be more consistent with methylation patterns in brain73. Although our methylation predictor shows promise (e.g., AUC in the NESDA study is comparable to that of the Framingham Risk Score 74, one of the most widely used prediction algorithms in medicine), replication in external samples is needed for establishing the veracity of the findings and generalizability to other patient populations. Thus, to estimate predictive power, we used k-fold cross validation. This ensures that in independent samples with the same properties and outcome measures, our findings would ”replicate” with an expected AUC of 0.724. However, it does not imply that the MRS will yield the same predictive power in patient populations that have different characteristics or with other outcome measures. As all our participants were diagnosed with MDD at the time of the methylation assessment, our analyses essentially control for disease status. However, peripheral methylation changes may still be a consequence of disease. Clearly, it would be better to use causal variables for predictions but identifying all these variable and establishing their etiological role will be a challenging and long process. In the meantime, being able to predict future disease status may have clinical utility (even if the all changes we observed at baseline would be the results of the disease) as it would enable the identification of patients at risk for recurrent MDD.
In summary, our results indicate that an immune signature of treatment-resistant depression is already present at baseline in NESDA and may confer long-term MDD risk. Although further studies are needed to study the generalizability to different patient populations, results suggest that methylation profiles in blood may present a promising avenue to support clinical decision making by providing empirical information about the likelihood MDD is chronic or will recur in the future.
Supplementary Material
ACKNOWLEDGEMENTS
The NESDA study is supported by the Geestkracht program of the Netherlands Organization for Health Research and Development (Zon-Mw, grant number 10–000-1002) and the participating institutions (VU University Medical Center, Leiden University Medical Center, University Medical Center Groningen. The current methylation project was supported by grant R01MH099110 from the National Institute of Mental Health. The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
CONFLICT OF INTEREST
Brenda Penninx has obtained research funding – not related to current study – from Jansen Research and Boehringer Ingelheim. Other co-authors have no conflicts to declare.
REFERENCES
- 1.(2017). Depression and Other Common Mental Disorders: Global Health Estimates. In. (Geneva, World Health Organization. [Google Scholar]
- 2.Hardeveld F, Spijker J, De Graaf R, Nolen WA, and Beekman AT (2009). Prevalence and predictors of recurrence of major depressive disorder in the adult population. Acta PsychiatrScand. [DOI] [PubMed] [Google Scholar]
- 3.Mueller TI, Leon AC, Keller MB, Solomon DA, Endicott J, Coryell W, Warshaw M, and Maser JD (1999). Recurrence after recovery from major depressive disorder during 15 years of observational follow-up. The American journal of psychiatry 156, 1000–1006. [DOI] [PubMed] [Google Scholar]
- 4.Bonasio R, Tu S, and Reinberg D (2010). Molecular signals of epigenetic states. Science 330, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vialou V, Feng J, Robison AJ, and Nestler EJ (2012). Epigenetic Mechanisms of Depression and Antidepressant Action. Annu Rev Pharmacol Toxicol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, and Spengler D (2009). Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 12, 1559–1566. [DOI] [PubMed] [Google Scholar]
- 7.Volkow ND, Koob G, and Baler R (2015). Biomarkers in substance use disorders. ACS Chem Neurosci 6, 522–525. [DOI] [PubMed] [Google Scholar]
- 8.Chan RF, Shabalin AA, Xie LY, Adkins DE, Zhao M, Turecki G, Clark SL, Aberg KA, and Van den Oord EJCG (2017). Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res epub 25 February 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aberg KA, Chan RF, Shabalin AA, Zhao M, Turecki G, Heine Staunstrup N, Starnawska A, Mors O, Xie LY, and van den Oord E (2017). A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penninx B, Beekman A, and Smit J (2008). The Netherlands Study of Depression and Anxiety (NESDA): Rationales, Objectives and Methods. International Journal of Methods in Psychiatric Research 17, 121–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreland B, Oman K, Curfman J, Yan P, and Bundschuh R (2016). Methyl-CpG/MBD2 Interaction Requires Minimum Separation and Exhibits Minimal Sequence Specificity. Biophys J 111, 2551–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MD, and Clark SJ (2011). Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 6, 34–44. [DOI] [PubMed] [Google Scholar]
- 13.Aberg KA, McClay JL, Nerella S, Xie LY, Clark SL, Hudson AD, Bukszar J, Adkins D, Swedish Schizophrenia C, Hultman CM, et al. (2012). MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case--control samples. Epigenomics 4, 605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aberg KA, Xie LY, Nerella S, Copeland WE, Costello EJ, and van den Oord EJ (2013). High quality methylome-wide investigations through next-generation sequencing of DNA from a single archived dry blood spot. Epigenetics 8, 542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aberg KA, Xie L, Chan RF, Zhao M, Pandey AK, Kumar G, Clark SL, and van den Oord EJ (2015). Evaluation of Methyl-Binding Domain Based Enrichment Approaches Revisited. PLoS One 10, e0132205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aberg KA, Chan RF, Shabalin AA, Zhao M, Turecki G, Staunstrup NH, Starnawska A, Mors O, Xie LY, and van den Oord EJ (2017). A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics 12, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan RF, Shabalin AA, Xie LY, Adkins DE, Zhao M, Turecki G, Clark SL, Aberg KA, and van den Oord E (2017). Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res 45, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, and Schubeler D (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37, 853–862. [DOI] [PubMed] [Google Scholar]
- 19.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, and Meissner A (2010). Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28, 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lentini A, Lagerwall C, Vikingsson S, Mjoseng HK, Douvlataniotis K, Vogt H, Green H, Meehan RR, Benson M, and Nestor CE (2018). A reassessment of DNA-immunoprecipitation-based genomic profiling. Nat Methods 15, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClay JL, Shabalin AA, Dozmorov MG, Adkins DE, Kumar G, Nerella S, Clark SL, Bergen SE, Swedish Schizophrenia C, Hultman CM, et al. (2015). High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol 16, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aberg KA, McClay JL, Nerella S, Clark S, Kumar G, Chen W, Khachane AN, Xie L, Hudson A, Gao G, et al. (2014). Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 71, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shabalin AA, Hattab MW, Clark SL, Chan RF, Kumar G, Aberg KA, van den Oord E, and Birol I (2018). RaMWAS: Fast Methylome-Wide Association Study Pipeline for Enrichment Platforms. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shabalin AA, Clark S, Hattab MW, Aberg KA, and Van den Oord EJCG RaMWAS: Fast Methylome-Wide Association Study Pipeline for Enrichment Platforms. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, and Kelsey KT (2012). DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattab MW, Shabalin AA, Clark SL, Zhao M, Kumar G, Chan RF, Xie LY, Jansen R, Han LK, Magnusson PK, et al. (2017). Correcting for cell-type effects in DNA methylation studies: reference-based method outperforms latent variable approaches in empirical studies. Genome Biol 18, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, et al. (2016). The Reactome pathway Knowledgebase. Nucleic Acids Res 44, D481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabrera CP, Navarro P, Huffman JE, Wright AF, Hayward C, Campbell H, Wilson JF, Rudan I, Hastie ND, Vitart V, et al. (2012). Uncovering networks from genome-wide association studies via circular genomic permutation. G3 (Bethesda) 2, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J, and Tibshirani RJ (2012). Strong rules for discarding predictors in lasso-type problems. J R Stat Soc Series B Stat Methodol 74, 245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon N, Friedman J, Hastie T, and Tibshirani R (2011). Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J Stat Softw 39, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman J, Hastie T, and Tibshirani R (2010). Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 32.Zou H, and Hastie T (2005). Regularization and variable selection via the elastic net. J Roy Stat Soc B 67, 301–320. [Google Scholar]
- 33.Hastie T, Tibshirani R, and Friedman J (2001). The Elements of Statistical Learning: Data Mining, Inference, and Prediction.(New York: Springer Verlag; ). [Google Scholar]
- 34.Gerits N, Van Belle W, and Moens U (2007). Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Behav Brain Funct 3, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Long F, Cai B, Chen X, and Chen G (2018). A novel relationship for schizophrenia, bipolar and major depressive disorder Part 3: Evidence from chromosome 3 high density association screen. Journal of Comparative Neurology 526, 59–79. [DOI] [PubMed] [Google Scholar]
- 36.Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, Reinalda MS, Slager SL, McGrath PJ, and Hamilton SP (2010). A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry 67, 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ju XD, Guo Y, Wang NN, Huang Y, Lai MM, Zhai YH, Guo YG, Zhang JH, Cao RJ, Yu HL, et al. (2014). Both Myosin-10 isoforms are required for radial neuronal migration in the developing cerebral cortex. Cereb Cortex 24, 1259–1268. [DOI] [PubMed] [Google Scholar]
- 38.Tonazzini I, Meucci S, Van Woerden GM, Elgersma Y, and Cecchini M (2016). Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv Healthc Mater 5, 850–862. [DOI] [PubMed] [Google Scholar]
- 39.Miyata S, Kurachi M, Okano Y, Sakurai N, Kobayashi A, Harada K, Yamagata H, Matsuo K, Takahashi K, Narita K, et al. (2016). Blood Transcriptomic Markers in Patients with Late-Onset Major Depressive Disorder. PLoS One 11, e0150262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincent DB, Jean-Loup G, Renaud L, and Etienne L (2008). Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008, P10008. [Google Scholar]
- 41.Csardi G, and Nepusz T (2006). The igraph software package for complex network research. InterJournal Complex Systems 1695. [Google Scholar]
- 42.Cancino J, and Luini A (2013). Signaling circuits on the Golgi complex. Traffic 14, 121–134. [DOI] [PubMed] [Google Scholar]
- 43.Hansen MD, Johnsen IB, Stiberg KA, Sherstova T, Wakita T, Richard GM, Kandasamy RK, Meurs EF, and Anthonsen MW (2017). Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc Natl Acad Sci U S A 114, E3462–E3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machamer CE (2015). The Golgi complex in stress and death. Frontiers in Neuroscience 9, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Machamer CE (2015). The Golgi complex in stress and death. Front Neurosci 9, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez-Miranda EA, Sinnl M, and Farhan H (2015). Alteration of Golgi Structure by Stress: A Link to Neurodegeneration? Frontiers in Neuroscience 9, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jawdekar GW, and Henry RW (2008). Transcriptional regulation of human small nuclear RNA genes. Biochimica et biophysica acta 1779, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andersen PR, Tirian L, Vunjak M, and Brennecke J (2017). A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karijolich J, and Yu Y-T (2010). Spliceosomal snRNA modifications and their function. RNA Biology 7, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, and Hannon GJ (2008). A piRNA Pathway Primed by Individual Transposons Is Linked to De Novo DNA Methylation in Mice. Molecular Cell 31, 785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin R, and Turecki G (2017). Noncoding RNAs in Depression In Neuroepigenomics in Aging and Disease, Delgado-Morales R, ed. (Cham, Springer International Publishing; ), pp 197–210. [Google Scholar]
- 52.Yang W, Wang Q, Kanes SJ, Murray JM, and Nishikura K (2004). Altered RNA editing of serotonin 5-HT2C receptor induced by interferon: implications for depression associated with cytokine therapy. Molecular Brain Research 124, 70–78. [DOI] [PubMed] [Google Scholar]
- 53.Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, Takahashi A, Flanigan ME, Aleyasin H, LeClair KB, et al. (2017). Social stress induces neurovascular pathology promoting depression. Nature Neuroscience 20, 1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luster AD, Alon R, and von Andrian UH (2005). Immune cell migration in inflammation: present and future therapeutic targets. Nature Immunology 6, 1182. [DOI] [PubMed] [Google Scholar]
- 55.Barreiro O, De La Fuente H, Mittelbrunn M, and Sánchez-Madrid F (2007). Functional insights on the polarized redistribution of leukocyte integrins and their ligands during leukocyte migration and immune interactions. Immunological Reviews 218, 147–164. [DOI] [PubMed] [Google Scholar]
- 56.Smith GC, Seaman SR, Wood AM, Royston P, and White IR (2014). Correcting for optimistic prediction in small data sets. Am J Epidemiol 180, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeLong ER, DeLong DM, and Clarke-Pearson DL (1988). Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845. [PubMed] [Google Scholar]
- 58.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, et al. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics 50, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Research 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gamazon ER, Segre AV, van de Bunt M, Wen X, Xi HS, Hormozdiari F, Ongen H, Konkashbaev A, Derks EM, Aguet F, et al. (2018). Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 50, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coppo P, Clauvel JP, Bengoufa D, Oksenhendler E, Lacroix C, and Lassoued K (2002). Inflammatory myositis associated with anti‐U1‐small nuclear ribonucleoprotein antibodies: a subset of myositis associated with a favourable outcome. Rheumatology 41, 1040–1046. [DOI] [PubMed] [Google Scholar]
- 63.Kattah NH, Kattah MG, and Utz PJ (2010). The U1-snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunological Reviews 233, 126–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wohleb ES, Franklin T, Iwata M, and Duman RS (2016). Integrating neuroimmune systems in the neurobiology of depression. Nature Reviews Neuroscience 17, 497. [DOI] [PubMed] [Google Scholar]
- 65.Crawford B, Craig Z, Mansell G, White I, Smith A, Spaull S, Imm J, Hannon E, Wood A, Yaghootkar H, et al. (2018). DNA methylation and inflammation marker profiles associated with a history of depression. Human molecular genetics 27, 2840–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benros ME, Waltoft BL, Nordentoft M, and et al. (2013). Autoimmune diseases and severe infections as risk factors for mood disorders: A nationwide study. JAMA Psychiatry 70, 812–820. [DOI] [PubMed] [Google Scholar]
- 67.Kappelmann N, Lewis G, Dantzer R, Jones PB, and Khandaker GM (2016). Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Molecular Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Loo HM, Aggen SH, Gardner CO, and Kendler KS (2015). Multiple risk factors predict recurrence of major depressive disorder in women. J Affect Disord 180, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wardenaar KJ, van Loo HM, Cai T, Fava M, Gruber MJ, Li J, de Jonge P, Nierenberg AA, Petukhova MV, Rose S, et al. (2014). The effects of co-morbidity in defining major depression subtypes associated with long-term course and severity. Psychol Med 44, 3289–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Loo HM, Cai T, Gruber MJ, Li J, de Jonge P, Petukhova M, Rose S, Sampson NA, Schoevers RA, Wardenaar KJ, et al. (2014). Major depressive disorder subtypes to predict long-term course. Depress Anxiety 31, 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson JC, Zhang Q, Deberdt W, Marangell LB, Karamustafalioglu O, and Lipkovich IA (2012). Predictors of remission with placebo using an integrated study database from patients with major depressive disorder. Curr Med Res Opin 28, 325–334. [DOI] [PubMed] [Google Scholar]
- 72.Riedel M, Moller HJ, Obermeier M, Adli M, Bauer M, Kronmuller K, Brieger P, Laux G, Bender W, Heuser I, et al. (2011). Clinical predictors of response and remission in inpatients with depressive syndromes. J Affect Disord 133, 137–149. [DOI] [PubMed] [Google Scholar]
- 73.Langie SAS, Moisse M, Declerck K, Koppen G, Godderis L, Vanden Berghe W, Drury S, and De Boever P (2017). Salivary DNA Methylation Profiling: Aspects to Consider for Biomarker Identification. Basic Clin Pharmacol Toxicol 121 Suppl 3, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tzoulaki I, Liberopoulos G, and Ioannidis JP (2009). Assessment of claims of improved prediction beyond the Framingham risk score. JAMA 302, 2345–2352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.