

Abstract
Early mechanical reperfusion of the epicardial coronary artery by primary percutaneous coronary intervention (PCI) is the guideline-recommended treatment for ST-elevation myocardial infarction (STEMI). Successful restoration of epicardial coronary blood flow can be achieved in over 95% of PCI procedures. However, despite angiographically complete epicardial coronary artery patency, in about half of the patients perfusion to the distal coronary microvasculature is not fully restored, which is associated with increased morbidity and mortality. The exact pathophysiological mechanism of post-ischaemic coronary microvascular dysfunction (CMD) is still debated. Therefore, the current review discusses invasive and non-invasive techniques for the diagnosis and quantification of CMD in STEMI in the clinical setting as well as results from experimental in vitro and in vivo models focusing on ischaemic-, reperfusion-, and inflammatory damage to the coronary microvascular endothelial cells. Finally, we discuss future opportunities to prevent or treat CMD in STEMI patients.
Keywords: Coronary microvascular dysfunction, ST-elevation myocardial infarction, Microvascular reperfusion injury, Intramyocardial haemorrhage, Coronary microvascular endothelial cells
Graphical Abstract
Graphical Abstract.

This article is part of the Spotlight Issue on Coronary Microvascular Dysfunction.
1. Introduction
The guideline-recommended treatment for ST-elevation myocardial infarction (STEMI) is early mechanical reperfusion of the epicardial coronary artery by primary percutaneous coronary intervention (PCI). In over 95% of the PCI procedures, successful restoration of epicardial coronary blood flow is achieved, which has dramatically reduced mortality rates in STEMI patients.1 However, despite angiographic evidence of complete epicardial coronary artery patency, in about half of the patients, the perfusion of the distal coronary microvasculature is not fully restored, which is associated with increased morbidity and mortality.2 In experimental animal models, the extent of poorly or non-perfused regions of the coronary microvasculature evolves during reperfusion.3,4 This indicates that reperfusion itself paradoxically can have additional harmful effects. The phenomenon in which structural evidence of microvascular damage was linked to poorly or non-perfused regions of the intramural myocardium was first described in 1974.5 Since then, reperfusion injury has been extensively described in the literature and over the last several years, the coronary microcirculation has evolved from passive bystander to primary target for therapies aiming to diminish reperfusion injury. The exact pathophysiology is still debated,6 partly due to the fact that—despite multiple efforts—there are still no effective therapeutic options to prevent reperfusion injury in clinical practice.6–9 In this review, we present an overview of current knowledge on coronary microvascular dysfunction (CMD) in STEMI. We will summarize invasive and non-invasive techniques for the diagnosis and quantification of CMD in the clinical setting, as well as results from experimental in vitro and in vivo models. Furthermore, we will highlight future opportunities to prevent or treat CMD which could further improve clinical outcome and prognosis of STEMI patients.
1.1. Nomenclature and definitions
Throughout the literature, different terms have been used to describe the phenomenon of diminished myocardial perfusion after reperfused STEMI. As a guide to the reader, we first provide a short overview with nomenclature and definitions (Table 1).
Table 1.
Recommendations to use terminology of reperfusion injury
| Nomenclature | Definition |
|---|---|
| No-reflow |
|
| MVO |
|
| IMH |
|
| CMD | Umbrella term comprises no-reflow, MVO, IMH, and MVI |
| MVI | General term for microvascular damage after ischaemia–reperfusion |
| Reperfusion injury | General term for tissue damage after ischaemia–reperfusion (i.e. not specific for the coronary microvasculature) |
CMD, coronary microvascular dysfunction; CMR, cardiac magnetic resonance imaging; IMH, intramyocardial haemorrhage; MVI, microvascular injury; MVO, microvascular obstruction.
1.1.1 No-reflow
Already in 1966, Krug et al.10 observed disturbances of blood supply after removing a ligation of the coronary artery in the cat. However, this observation was not related to microvascular damage in that study. The term no-reflow was first mentioned in a rabbit model of cerebral ischaemia.11 A few years later, Kloner et al.5 reported coronary no-reflow in a canine model of acute myocardial infarction (AMI). They observed a persistently poor or even absent perfusion in large areas of the reperfused myocardium despite complete epicardial coronary artery patency and linked it to microvascular injury (MVI). Consequently, the term no-reflow has been used to describe the inability to reperfuse regions of previously ischaemic myocardium after re-opening the occluded coronary artery. The following years, this term was maintained mainly based on experimental animal studies using markers for flow, such as carbon black or microspheres, or staining for endothelial cells, such as Thioflavin S. It was assumed that lack of these markers in certain areas of the myocardium represented post-ischaemic no-reflow areas.12 In patients, coronary no-reflow was reported only years after the first mentioning in experimental studies. Schofer et al.13 provided scintigraphic evidence of no-reflow in a STEMI patient treated with thrombolysis. Shortly after, Bates et al.14 first reported angiographic no-reflow, as estimated coronary blood flow by angiographic contrast density. After the introduction of primary PCI, no-reflow was witnessed much more frequently by immediate angiographic visualization.15 Because patients with angiographic no-reflow had poor outcomes, absence of this angiographic sign served as a measure of procedural success for many years. However, the term no-reflow does not provide much information about its pathophysiology. In fact, no-reflow refers to multiple manifestations of which microvascular obstruction (MVO), MVI, intramyocardial haemorrhage (IMH), and CMD are the most prominent. Also, in contemporary practice, angiographic no-reflow is only encountered in <5% of patients,16 which is a clear underestimation when compared to the number of patients with myocardial perfusion deficits on cardiovascular magnetic resonance (CMR) post-STEMI.
1.1.2 Microvascular obstruction
When it became apparent that angiographic no-reflow was not sensitive enough to detect microvascular perfusion deficits, the use of CMR was introduced for this purpose. With CMR, a typical pattern with contrast-enhanced infarct area and contrast-void infarct core was observed and was coined MVO, since it was thought that this was the underlying mechanism preventing contrast to reach the infarct core. It was hypothesized that distal atherothrombotic embolization, plugging of circulating blood cells, de novo microvascular thrombus formation and extravascular compression attribute to MVO. However, none of the clinical trials targeting the aforementioned factors have led to positive results,6,17 indicating that true MVO might only play a limited role in reperfusion injury. Moreover, CMR-defined MVO is reversible in some patients.18 Furthermore, it has become clear that CMR-defined MVO often reflects MVI comprising complete microvascular destruction and IMH.19,20 Therefore, the term MVO should be reserved to describe the histologically proven obstruction of microvessels rather than the complete clinical entity of failed primary reperfusion.
1.1.3 Intramyocardial haemorrhage
IMH is an irreversible pathological consequence of severe MVI.21 Whilst MVO might resolve,18 e.g. recovery of perfusion with resorption of oedema, IMH represents capillary destruction which is irreversible. Experimentally, reperfusion causes IMH22,23 and is reflected by the loss of interendothelial cell junctions and extravasation of erythrocytes in the perivascular space.23 Furthermore, a large overlap was found in size and location of CMR-defined MVO and histologically proven IMH.19
1.1.4 Coronary microvascular dysfunction
The pathophysiology of reperfusion injury is of multifactorial origin and may include impaired vasomotor function, MVO, MVI, IMH, and inflammation.6 Therefore, the term CMD in STEMI better reflects the multifaceted pathophysiology of myocardial reperfusion deficits caused by a constellation of pathological mechanisms. We note that the term CMD is currently also used in the setting of ischaemia and no obstructive coronary artery disease. In the present review, we will use the term CMD (in STEMI) unless specific knowledge on the pathophysiological substrate is available.
1.2. Incidence and prognosis of CMD in STEMI patients
Occurrence of CMD after reperfused STEMI is associated with unfavourable clinical outcome and prognosis. As stated above, surrogates of CMD in STEMI can be measured with angiography or CMR. Using angiography, CMD is often denoted no-reflow. Angiographic no-reflow was reported in only 2.7% of STEMI patients. Patients with no-reflow showed higher in-hospital mortality and higher rates of reinfarction, cardiogenic shock, and heart failure compared to patients without no-reflow.16 Using CMR, which is nowadays the most used entity to assess myocardial damage, CMD is denoted MVO or IMH. MVO, which was defined by a contrast-devoid infarct core, was present in 54.9% of patients with angiographic optimal flow [defined as thrombolysis in myocardial infarction (TIMI) 3 flow]. The presence of MVO was found to be an independent predictor of major adverse cardiac events (MACE) at 2 years of follow-up. Furthermore, the presence of MVO has even incremental detrimental effects in addition to the extent of infarct size at 2 years of follow-up (Figure 1). In addition, MVO was an independent predictor of cardiac death.2 Not only the presence of MVO but also the extent of MVO is associated with poor clinical outcome. Both presence and extent of MVO assessed 15 min after contrast injection were identified as the strongest independent predictors of MACE, defined as a composite of death, myocardial reinfarction and congestive heart failure, and the individual outcome mortality.24 Furthermore, IMH, which was defined by a contrast-devoid infarct core on T2-weigthed imaging, was present in 34% of patients. IMH was an independent predictor of MACE.25
Figure 1.
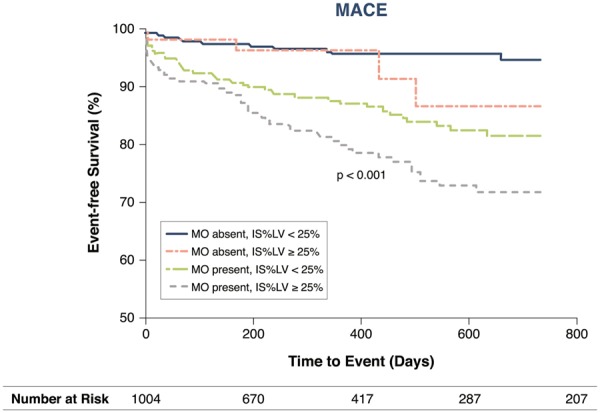
Relationship between CMR-defined microvascular obstruction, infarct size, and major adverse cardiac events. IS, infarct size; IS%LV, infarct size as percentage of left ventricle; LV, left ventricle; MACE, major adverse cardiac events; MO, CMR-defined microvascular obstruction. Values are Kaplan–Meier estimates in patients with IS%LV ≥25% vs. <25%, grouped by the presence or absence of MO, indicating the time to MACE during 2 years of follow-up. MACE is defined as a composite of cardiac death, myocardial reinfarction, and new congestive heart failure at 2 years of follow-up. Reprinted with permission from Van Kranenburg et al.2
2. Invasive and non-invasive techniques for diagnosis and quantification of CMD in STEMI
Different methods—both invasive and non-invasive—have been described to diagnose and/or quantify CMD in STEMI. We will summarize these methods and discuss their main strengths and limitations.
2.1. Invasive approaches
2.1.1 Angiographic methods
2.1.1.1 TIMI flow
TIMI flow is an angiographic visual scoring system grading epicardial antegrade flow. No antegrade flow is denoted TIMI 0 flow. Complete antegrade flow, including the distal bed, is denoted TIMI 3 flow. TIMI 1 and 2 flow are respectively denoted antegrade flow beyond the obstruction but without distal perfusion, and complete antegrade, but delayed flow.26 Post-procedural TIMI 3 flow is associated with fewer MACE and better survival compared to TIMI ≤2 flow.27,28 Although the angiographic TIMI flow assessment is fast and easy to perform, more than half of the patients with normal TIMI 3 flow show CMR-defined MVO.29 Thus, preserved TIMI flow does not necessarily guarantee adequate microvascular perfusion and is not sensitive enough to assess post-ischaemic CMD. Therefore, in current practice with fast mechanical reperfusion by primary PCI in the majority of patients and hence TIMI 3 flow, the predictive power of post-procedural TIMI flow is less evident. Because of a high interobserver variability and poor interobserver agreement in grading TIMI flow, the corrected TIMI frame count (CTFC) has been introduced, providing a quantitative index to assess coronary flow by counting the number of frames required for contrast to reach a standardized distal landmark. Advantages of CTFC are its high reproducibility, low intra-, and interobserver errors and it enables a quantitative estimation of flow.30–32 However, the prognostic value of the CTFC is not clear.33–36
2.1.1.2 Myocardial blush grade
Myocardial blush grade (MBG) is an angiographic visual scoring system, assessing myocardial perfusion by myocardial contrast blush after injection, grading from no myocardial contrast density to normal myocardial contrast density in the infarct-related coronary artery.37 MBG has been defined as 0, in case of no myocardial blush or contrast density or persisting myocardial blush; 1, with minimal myocardial blush or contrast density; 2, with moderate myocardial blush or contrast density, but less than that obtained during angiography of a contralateral or ipsilateral non-infarct-related coronary artery; and 3, with normal myocardial blush or contrast density, comparable with that obtained during angiography of a contralateral or ipsilateral non-infarct-related coronary artery. MBG provides additional diagnostic value to TIMI flow; of patients presenting with post-procedural TIMI 3 flow, two-third of patients had MBG 0–1. Furthermore, it has prognostic value; multivariate logistic regression showed that MBG is associated with long-term survival rates independent of TIMI flow.37 CMR-defined MVO was less frequently observed in patients with MBG ≥2 compared to MBG 0–1.38 However, similar to preserved TIMI flow, preserved MBG does not necessarily indicate adequate microvascular perfusion. In patients presenting with MBG 2–3, still, a substantial proportion of patients showed CMR-defined MVO.29,38
2.1.1.3 Computer-assisted myocardial blush quantification by quantitative blush evaluator
Vogelzang et al.39 described a novel assessment of myocardial perfusion through computer-assisted myocardial blush quantification by quantitative blush evaluator (QuBE). Although QuBE was an independent predictor of mortality in the work by Vogelzang et al.,39,40 these results have yet to be investigated by other groups. Furthermore, ∼20% of angiograms could not be assessed using QuBE due to overlapping vessels or panning movements.
2.1.1.4 Fluoroscopy assisted scoring of myocardial hypoperfusion
A novel non-invasive estimate for coronary blood flow is the fluoroscopy assisted scoring of myocardial hypoperfusion (FLASH) represented by FLASH flow and FLASH ratio. FLASH flow is calculated by contrast passage velocity multiplied with the mean cross-sectional area. FLASH ratio is calculated as the relative difference between FLASH flows in the infarct-related coronary artery compared to that in a non-infarct-related coronary artery. In STEMI patients, FLASH flow is significantly lower in the infarct-related coronary artery as compared to non-infarct-related coronary arteries. FLASH ratio is an independent predictor of cardiac mortality (optimal cut-off value −49% for cardiac mortality at 6 months) and might be more sensitive than TIMI flow and MBG.41 However, FLASH has not been validated in an independent prospective cohort.
2.1.2 Intracoronary physiology
With intracoronary pressure and/or flow wires the function of the coronary microcirculation can directly be assessed. First, intracoronary nitroglycerin is infused to ensure complete epicardial vasodilation. Subsequently, the function and the capacity of the distal microvascular bed are assessed by infusion of vasodilators like adenosine that act specifically on the microcirculation. Measurements to assess the functionality of the coronary microcirculation include coronary flow reserve (CFR), index of microcirculatory resistance (IMR), hyperaemic microvascular resistance (HMR), resistive reserve ratio (RRR), instantaneous hyperaemic diastolic flow velocity–pressure slope (IHDVPS), and coronary zero-flow pressure (Pzf) (Figure 2).
Figure 2.

Intracoronary physiology: invasive measurements for coronary microvascular function after ST-elevation myocardial infarction. (A) Schematic overview of Pa, Pd, Tmn, APV, BR, CFR, IMR, HMR, RRR, IHDVPS, and Pzf. (B) With an intracoronary pressure and/or Doppler flow wire the following indices can be derived: Pa (red), Pd (green), BR, hyperaemic (dark blue lines) and rest (light blue lines) Tmn, hyperaemic (dark blue box) and rest (light blue box) Doppler APV, IHDVPS (pink dotted line) and Pzf (yellow dot). The right box shows the pressure–flow velocity loops and the calculation of the linear relationship between pressure and flow velocity over the mid-late phases of diastole (coloured in black). The higher the values of IHDVPS (the β term in the equation y = a + βx, where y = flow velocity and x = intracoronary pressure), the better the conductance of the microcirculation. Pzf is calculated from the intercept of the regression line with the pressure axis. (C) Formulas for CFR, IMR, HMR, RRR, IHDVPS, and Pzf. The arrow indicates higher or lower values for that formula when coronary microvascular dysfunction is present. Evidence on the ability of IHDVPS to reflect coronary microvascular dysfunction is controversial. See text for details. APV, average peak velocity; BR, basal resistance, CFR, coronary flow reserve; HMR, hyperaemic microvascular resistance; IHDVPS, instantaneous hyperaemic diastolic flow velocity–pressure slope; IMR, index of microcirculatory resistance; Pa, aortic pressure; Pd, distal pressure; Pzf, pressure at zero flow velocity; RRR, resistive reserve ratio; Tmn, mean transit time.
2.1.2.1 Coronary flow reserve
CFR provides information on the epicardial and microvascular compartments and can be derived using intracoronary wires with Doppler or thermodilution techniques.42 Using Doppler, CFR is defined as the ratio of hyperaemic blood flow divided by resting blood flow. Using thermodilution, CFR is defined as the ratio of mean transit time (Tmn) in rest divided by Tmn during hyperaemia. As CFR reflects both the epicardial- and the microvasculature, CFR can be used to assess the microcirculation in the absence of an epicardial stenosis. A value of <2.0 has a sensitivity of 79% and specificity of 34% for CMR-defined MVO.43 CFR corresponds with the extent of CMR-defined MVO.44 CFR measured in the infarct-related coronary artery is a prognostic marker for left ventricular function recovery after AMI and is associated with long-term mortality.45,46 A limitation of CFR is its reproducibility. Because resting flow is included in the calculation, changes in heart rate, blood pressure, and left ventricular contractility influence the CFR value.47
2.1.2.2 Index of microcirculatory resistance
While CFR reflects the combined epicardial and microvascular resistance, IMR and HMR specifically assess microvascular resistance. The distal coronary pressure multiplied with the hyperaemic Tmn provides IMR. The hyperaemic Tmn is the average of three separate transit time measurements after intracoronary injection of room temperature saline during adenosine-induced hyperaemia. Calculation of IMR is not affected by variations in hemodynamic conditions (because resting measurements are not required) or the severity of epicardial stenosis.48 A meta-analysis found that IMR is significantly increased in patients with CMR-defined MVO.49 IMR is more closely associated with MVO and clinical outcomes than TIMI myocardial perfusion grade or CFR.50 With regard to clinical outcome, a higher IMR is associated with worse outcome like adverse left ventricular remodelling at 6 months50 and even death.51 A limitation of IMR is the manual injection of saline, which can be a source of variability.
2.1.2.3 Hyperaemic microvascular resistance
HMR is calculated as the distal pressure divided by the mean Doppler flow velocity at peak hyperaemia simultaneously measured using a coronary guidewire with a combined pressure sensor and Doppler transducer.52 An HMR value >2.5 mmHg cm−1 s has been shown to be indicative of CMR-defined MVO with a sensitivity of 71% and a specificity of 63%. Furthermore, an HMR value >2.82 mmHg cm−1 s is a strong predictor of a composite of death and rehospitalization for heart failure.53 The role of HMR in STEMI has been studied less compared with IMR, and a limitation of HMR is the complexity of acquiring high-quality measurements with the Doppler wire.
2.1.2.4 Resistive reserve ratio
RRR is a measure of the ability to achieve maximal coronary hyperaemia. RRR is the ratio of basal resistance to IMR. Whereas IMR is a measure at peak hyperaemia and reflects structure, RRR quantifies the vasodilator response of the coronary microcirculation to a hyperaemic stimulus such as adenosine.54 In a prospective study of 50 patients with stable angina, 40 patients with STEMI and 50 patients with non-STEMI (NSTEMI), no difference was observed in RRR between non-culprit vessels in stable angina and either culprit vessels in stable angina or NSTEMI. However, RRR was significantly lower in STEMI patients.55
A novel measurement to assess microvascular function is thermodilution-based absolute flow and resistance measurement using continuous infusion of saline through a dedicated coronary catheter. This potentially reduces introduced variability by avoiding the manual injection of saline. Although larger studies in STEMI are awaited, early studies have shown the feasibility of this approach in both stable angina and STEMI patients and recently the technique was validated with positron emission tomography.56,57
2.1.2.5 IHDVPS and coronary Pzf
Alternative approaches to interrogate the coronary microcirculation are the measurement of the IHDVPS and Pzf. In this approach, coronary pressure–flow velocity loops are generated. As shown in Figure 2B, a linear relationship between pressure and flow values exists during the mid-late phases of diastole. The slope of the regression line obtained over this period expresses microcirculatory conductance. The described index, called IHDVPS, correlates well with structural changes in the coronary microcirculation.58 However, evidence on its ability to reflect reperfusion injury in patients with STEMI is controversial, with some studies suggesting a potential role59 and others failing to demonstrate its value as a predictor of CMD in the context of STEMI.52,60 A second index derived from the mid-late diastole linear relationship between pressure and flow values is Pzf. This index, which stems from the concept of vascular waterfalls,61 is calculated by extrapolation from the regression line relating pressure and flow values that have been described above for IHDVPS calculation. The value of Pzf in assessing reperfusion injury associated to STEMI is that, from a theoretical standpoint, it should reflect of the intraluminal pressure required to maintain patent compressible elements against extravascular compression. In the context of STEMI, extravascular compression may result from increased interstitial pressure secondary to myocardial haemorrhage and/or oedema. Pzf may also reflect extravascular compression due to raised intraventricular filling pressures resulting from acute myocardial injury.62 Available evidence is largely consistent on the predictive value of Pzf in the acute phase of STEMI in predicting subsequent myocardial injury and infarct size.52,60,63,64
2.2. Non-invasive approaches
2.2.1 CMR imaging
CMR plays a pivotal role in the characterization of post-infarction myocardial injury, most essentially with the use of a contrast agent. The CMR gadolinium-based contrast agent is a contrast medium with extracellular distribution, meaning that after injection it slowly diffuses in areas with increased extracellular or interstitial space, such as the acutely infarcted myocardium with ruptured cell membranes. While the contrast agent washes out from the viable myocardium with intact myocytes, the wash-out in infarcted myocardium is delayed, causing a hyperenhanced bright signal on the T1-weighted images, hence the name delayed contrast enhancement (LGE).65 In some patients, the hyperenhanced infarcted myocardium surrounds a subendocardial area of decreased signal in the core of the infarct, which slowly becomes hyperenhanced over time if repeated images are acquired.29 These patients with so-called MVO on CMR have worse functional recovery after STEMI, with the best predictive value if the images are taken 10–15 min after contrast injection.66 This predictive value is better than angiographic assessment or the electrocardiogram.29 Since the amount of MVO is dependent on timing of the image acquisition due to the pharmacokinetic dynamics of the contrast agent, it is clear that the area with MVO as visualized with CMR is not a well-defined area with a totally obstructed microvasculature, but reflecting severely injured myocardium hampering wash-in of the contrast agent. In a recently published recommendation paper for CMR endpoints in experimental and clinical trials, LGE has been defined as the most important primary endpoint, with MVO and left ventricular ejection fraction (LVEF) as main secondary endpoints since these were strongly associated with MACE and had consistent evidence in multiple studies.67
With the use of additional newer techniques, CMR provides further infarct characterization, with a surrounding oedematous border zone, an infarcted central zone with ruptured myocytes and in some patients a necrotic core with MVI and haemorrhage.68 The most used non-contrast technique is T2-weighted imaging, in which the oedematous myocardium causes prolongation of T2 decay times, and therefore, a relatively higher signal in acutely infarcted myocardium compared to remote myocardium. Additionally, this technique has been used for the assessment of IMH as well,69 since the paramagnetic effects of haemoglobin cause an attenuated low signal which correspond to regions of haemorrhaged infarcts on pathology.70 Newer mapping techniques allow a quantitative per pixel analysis of the myocardium without contrast, quantifying the degree of oedema (T2 mapping), myocyte loss (T1 mapping), and if present haemorrhage (T2* mapping). T1 relaxation and T2 decay times are prolonged in acutely infarcted tissue, whereas T1 and T2 are lower in the infarct core if IMH is present. T2* is merely reduced in the presence of IMH, and is not altered in remote or infarcted myocardium, and currently accepted as the preferred method to identify haemorrhage in the infarct core71 (Figure 3). For future trials investigating treatment strategies to prevent reperfusion injury, it is, therefore, important to include T2* mapping and T1/T2 mapping to study its effect on the different infarct components.67
Figure 3.

CMR images of a patient after acute anterior myocardial infarction. Typical example of a patient after acute anterior myocardial infarction with microvascular injury, demonstrating hyperintense oedematous myocardium on the T2-weighted image (A, V) compared to remote non-infarcted myocardium (O), with the corresponding delayed contrast-enhanced image showing the infarcted hyperenhanced infarcted myocardium (B, V) with a hypointense infarct core (asterisk) with microvascular injury. The area with microvascular injury has low T1 on the non-contrast T1 map (C, asterisk) and low T2*, whereas the area of infarction has increased T1 relaxation times and normal T2* decay times, not containing haemoglobin breakdown products. LV, left ventricle; RV, right ventricle.
3. Experimental models of CMD in STEMI
Experimental models show that microvascular endothelial cell dysfunction plays a prominent role in reperfusion injury. In the following paragraphs, subsequently ischaemic damage, reperfusion damage, vascular permeability, IMH, inflammatory damage, and the role of platelets and pericytes will be reviewed (Figure 4).
Figure 4.

Coronary microvascular endothelial cell dysfunction in reperfused STEMI. (A) TEM image of a capillary in non-infarcted myocardium (healthy reference) of a rat23 with thin endothelium, preserved interendothelial cell junctions and numerous pinocytotic vesicles (small arrow), surrounded by a basement membrane; 17 500×. (B) Schematic overview of a healthy reference capillary with an attached pericyte. (C) TEM image of a capillary in permanently ischaemic myocardium of a rat23 with a preserved endothelial cell lining and some localized endothelial cell swelling, surrounded by a basement membrane; 24 500×. (D) Schematic overview of a capillary in permanently ischaemic myocardium with some diffuse endothelial cell swelling and some destabilization of interendothelial cell junctions. (E) TEM image of a completely ruptured capillary—beyond the phase of endothelial cell swelling with blebs—in reperfused myocardium of a rat23 with ruptured endothelial cell lining (small arrows) and intramyocardial haemorrhage; 5800×. (F) Schematic overview of completely ruptured capillary in reperfused myocardium with massive production of ROS, elevated levels of cytosolic calcium activating the contractile elements of endothelial cells, platelet adhesion and aggregation, destabilization of interendothelial cell junctions, intramyocardial haemorrhage, and numerous inflammatory cells. ▸, endothelial cell; B, basement membrane; CM, cardiomyocyte; E, erythrocyte; J, interendothelial cell junctions; M, mitochondrion; MMP, matrix metalloprotease; N, nucleus; P, pericyte; ROS, reactive oxygen species; V, pinocytotic vesicle.
3.1. Ischaemic damage to endothelial cells
Endothelial cells are relatively resistant to hypoxia. In vitro studies with human umbilical vein endothelial cells (HUVEC) showed that three-quarter of all cells survived 24 h of hypoxia72 and more than half of all cells survived 48 h of hypoxia,73 indicating that endothelial cells tolerate prolonged periods of hypoxia before destructive damage occurs. Besides some reduction in endothelial cell viability, hypoxia can have several reversible effects on endothelial cell function, including decreased release of vasoactive substances such as the vasodilator endothelial nitric oxide synthase (eNOS) and increased release of vasoconstrictors such as endothelin-1 (ET-1), which could hamper flow restoration.74 However, after permanent left anterior descending artery (LAD) occlusion, ex vivo acetylcholine-induced coronary vasodilation was preserved,75–77 suggesting sufficient coronary vasodilator reserve after ischaemia. With regard to endothelial cell injury, hypoxia up to 6 h resulted in reduced levels of malondialdehyde, which is a marker for oxidative stress, and increased levels of the antioxidant superoxide dismutase,72 suggesting even an initial protective effect of hypoxia on endothelial cells. Prolonged periods of hypoxia in vitro resulted in some destabilization of tight junctions78 and adherence junctions,79 but 90 min of LAD occlusion did not reduce the number of interendothelial cell junctions,23 indicating that longer hypoxic periods are necessary to induce vascular leakage. Ultrastructurally, the coronary endothelial cell layer remained intact after ischaemia,76,77 even after 6 h of ischaemia.76 Addition of in vivo reperfusion, however, resulted in prominent and immediate coronary endothelial cell injury with subendothelial bleb formation and disruption of interendothelial cell junctions.23,77 With regard to the coronary microvasculature, permanent occlusion of the circumflex branch showed mild endothelial cell swelling, some loss of pinocytotic vessels and caveolae, and nuclear chromatin condensation, but no completely occluded capillaries.80 Also, in a rat model of 30 min of ischaemia without in vivo reperfusion, no clear damage to the coronary microvascular endothelium was observed, whereas 30 min of ischaemia in combination with 60 min of in vivo reperfusion led to visible damage to the coronary microvasculature with prominent extravasation of erythrocytes in the myocardium.23
3.2. Reperfusion damage to endothelial cells
A 90 min proximal coronary artery occlusion followed by reperfusion was associated with perfusion defects detected by injecting the fluorescent dye, Thioflavin S, into the vasculature after a clamp was removed from the epicardial coronary artery. These perfusion defects were observed within seconds of initial reperfusion and were most prominent in the subendocardial layer of the left ventricular wall. A detailed ultrastructural analysis was performed to try to determine the cause of these microvascular perfusion defects. The most consistent finding was the presence of membrane bound ‘blebs’ that appeared to pinch off of the endothelial lining of capillaries and small vessels, and obstruct the lumen. These ‘blebs’ really represented localized areas of endothelial swelling. Other evidence of endothelial damage was present including more diffuse endothelial swelling, loss of pinocytotic vesicles within the endothelium, visible breaks of the endothelial lining with platelet, fibrin tactoids, and neutrophils in the same regions, extracellular erythrocytes, and rouleaux formation. Occasional obstructions to capillaries were present that appeared to be due to compression from adjacent swollen cardiac myocytes and may have also contributed to no-reflow.5 Subsequent studies showed that the ultrastructural abnormalities to the microvasculature appeared to occur after irreversible injury to the myocytes had already occurred.81 In addition, the no-reflow zones appeared to occur within areas of the heart that were already dead as assessed by triphenyl tetrazolium chloride (TTC) staining. However, the size of the no-reflow zone expands during the first few hours after epicardial coronary artery patency is established,3 suggesting that no-reflow is a true manifestation of reperfusion injury.
Thus, reperfusion is paramount to CMD in STEMI and prolonged periods of ischaemia can enhance the extent of reperfusion injury.82–85 Part of this reperfusion injury might be explained by the massive production of reactive oxygen species (ROS), in turn, leading to oxidative stress and additional vascular damage. In the vasculature, the main sources for ROS are xanthine oxidase, nicotinamide adenine dinucleotide phosphate oxidase, uncoupled eNOS and vascular adhesion protein-1.86 These ROS sources have been frequently proposed as targets for cardioprotection, but the effects of antioxidants on reperfusion injury are conflicting and results of experimental animal studies failed to be translated into the clinical setting. However, most of these studies focused on reperfusion injury to cardiomyocyte function rather than endothelial cells.87–89
3.3. Vascular permeability and oedema
Reperfusion is associated with myocardial oedema.8,90 Oedema already begins during ischaemia and occurs intracellularly (due to a loss of function of energy requiring ion pumps) as well as in the interstitium (due to an increase in osmolality as a result of metabolite accumulation).8,90 Upon reperfusion, the coronary reactive hyperaemia with normo-osmotic blood causes a further worsening of oedema, but within hours interstitial oedema begins to wane as washout of catabolites eliminates the osmotic gradient between the interstitium and the intravascular compartment.91 The early wave of oedema is subsequently followed by a second wave of oedema that appears to be principally the result of an increase in coronary microvascular permeability, possibly as part of influx of inflammatory cells and healing response of the infarct zone.92 Indeed, serial CMR imaging studies have demonstrated such bimodal pattern of myocardial oedema after reperfusion both in pigs82,93,94 and in humans,95,96 especially when IMH was present.18 The results of these studies showing highly variable levels of oedema early after reperfusion also raise concern regarding the validity of studies attempting to estimate the area at risk, using T2-weighted CMR imaging techniques.90 Myocardial oedema is not only a consequence of a severe ischaemia–reperfusion insult but, in turn, can contribute to the perturbations in coronary microvascular perfusion during reperfusion by increasing the extravascular compressive forces acting on the coronary microcirculation.90,97 Studies in swine suggest that this may be particularly prominent during the early wave of oedema, at a time (2 h of reperfusion) when IMH was not yet apparent.82 In contrast, at 24 h of reperfusion, haemorrhage was now very prominent and was a more likely contributor to the MVO at this time point, when oedema had dissipated.82
The endothelial disruption and the subsequent extravasation of cells upon reperfusion are likely facilitated by destabilization of the cellular junctions.8,90 Following reperfusion, the endothelium undergoes significant alterations in calcium-homeostasis, characterized by elevated levels of cytosolic calcium activating the contractile elements of endothelial cells.98 The resultant contraction promotes the formation of intercellular gaps, resulting in enhanced permeability to large molecules.98 In addition, cytokine release further impairs the stability of cell junctions and subsequently increases vascular permeability, via activation of Src and dissociation of the VEGFR2/VE-cadherin complex.99 Mice deficient for the vascular permeability modulator angiopoietin-like 4 (ANGPTL4) showed increased vascular leakage assessed by Evans blue dye and fluorescent microspheres, disruption of adherence junctions assessed by VE-cadherin staining, increased phosphorylation of Src kinase, and dissociation of the VEGFR2/VE-cadherin complex. Furthermore, these mice showed increase in myocardial infarct size, oedema, haemorrhage, and inflammation. Conversely, injection of recombinant ANGPTL4 reduced vascular permeability, infarct size, no-reflow, and haemorrhage.100 Interestingly, a recent study in mice undergoing ischaemia–reperfusion reported that losartan inhibited the phosphorylation of Src and VE-cadherin resulting in increased VEGFR2-Src-VE-cadherin complex formation, increased cell surface VE-cadherin, and inhibition of vascular hyperpermeability. These effects were accompanied by a decrease in myocardial infarct size as well as IMH, oedema, and inflammation.101 Besides destabilization of cellular junctions, reperfusion results in increased matrix metalloproteases (MMP)-2 and -9, which are able to degrade collagen in basement membranes, also promoting vascular permeability.102,103
3.4. Intramyocardial haemorrhage
The most severe form of MVI is IMH, which is a result of destruction of coronary capillaries and subsequent extravasation of erythrocytes into the myocardium.21,23 In a porcine model of ischaemia–reperfusion, histological analysis showed an infarct core with disruption of the microvasculature, extravasation of erythrocytes, necrosis, and cellular debris. The border zone contained besides necrosis and cellular debris, infiltration of leucocytes, granulation tissue, but more importantly a preserved microvasculature.19 Also, in primates, IMH was present in the core zone and decreased towards the border zone.104 CMR-defined IMH was first described in dogs with 4 h of coronary artery occlusion followed by 1 h of in vivo reperfusion. IMH, which was described by a hypointense core of a hyperintense area on T2-weighted imaging, was present in 80% of animals. Furthermore, IMH assessed by CMR was closely correlated with IMH assessed by histology.105 In addition, combined porcine and patient data show overlap in MVO and IMH, assessed by histology, LGE-imaging, and T2-weighted imaging on CMR.19 Pathophysiologically, IMH will result in destruction of haemoglobin with subsequent iron release in the myocardium. In a canine ischaemia–reperfusion model, histological assessment in the acute phase of AMI showed extravasation of erythrocytes with iron deposition in the infarct core zone, but not in the border zone. In addition, 2 months after AMI, histological assessment showed persistent iron deposition in the infarct core zone. Furthermore, newly recruited macrophages were colocalized with the iron depositions in the core zone, suggesting an ongoing inflammatory response.106 In the study of Carrick et al.107 all patients with IMH assessed by T2* imaging had CMR-defined MVO. Bulluck et al.108 showed that more than 80% of patients with CMR-defined MVO had IMH. IMH can occur already within 4–12 h after primary PCI107 and show a peak incidence in the first week in both patient and experimental animals.18,82,107,108
3.5. Inflammatory damage to endothelial cells
In one experimental study with a large non-atherosclerotic epicardial coronary artery, several changes were observed when a segment of that artery was deprived of blood flow by mechanical clamping both proximally and distally for 3 h followed by reperfusion.109 Light microscopic analysis of the reperfused segment revealed an influx of neutrophils in the intimal and medial layer of the epicardial coronary artery. Neutrophil influx was not observed in the control non-ischaemic coronary bed. Ultrastructural analysis revealed that neutrophils in the reperfused coronary arteries often appeared to be located between the endothelial cells of the luminal surface of the vessel and the elastic lamina. Neutrophils are also primary sources for MMP-9, in turn, promoting vascular leakage.103,110
Also, in other animal models of coronary occlusion, reperfusion was shown to promote rapid adhesion of leucocytes to the microvascular endothelial lining.111 Aggregates of these immune cells, with platelets, or in the form of neutrophil extracellular traps (NETs) can directly impede blood flow, and cause further endothelial damage by the release of pro-inflammatory cytokines.112,113 In addition to this histological evidence of leucocyte adhesion early in reperfusion, several observational studies in patients with AMI reported an association between the presence and extent of CMR-defined MVO and circulating inflammatory markers.6 In more detail, the extent of MVO is associated with the inflammatory circulating neutrophil-to-lymphocyte ratio on admission,114 as well as peak concentration of leucocytes115 and with high levels of classical monocytes.116 More recently, a transcriptome analysis of whole blood in STEMI patients revealed that the extent of late MVO was associated with a differential higher expression of inflammatory genes.117 In addition, the presence of MVO is associated with levels of inflammatory cytokines, including high-sensitive C-reactive protein,115 interleukin-8,118 and interleukin-6.119 In murine112,120 and porcine121,122 models of coronary occlusion, several anti-inflammatory interventions have proven effective in limiting MVO and/or reducing final infarct size, including depletion of neutrophil DNase to reduce NET formation, and inhibition of transcription factor nuclear factor kappa B (NF-κB). In contrast, however, all clinical trials that addressed these inflammatory components in patients with AMI did not show any benefit, which was recently reviewed by Rymer and Newby.123 For example, inhibitors of the CD11/CD18 integrin receptor in STEMI patients did not result in smaller infarct size, nor improved microvascular flow.124 In addition, studies targeting the complement system did not show beneficial effects.123 It is hypothesized that effective anti-inflammatory therapies in animal models failed to be translated in human, probably partly because of the method of coronary artery occlusion (coronary artery ligation vs. coronary artery occlusion by the process of atherothrombosis) and also partly because of the timing of anti-inflammatory drug administration. Some significant beneficial effects in animal models were observed when drugs were administered before the onset of ischaemia. However, in humans, this strategy is not possible in the setting of STEMI.123 Moreover, most inflammatory processes appear to be secondary in the cascade of reperfusion injury, such as macrophage recruitment to iron depositions and the aggregation of leucocytes and platelets.
3.6. Platelets
The role of platelets in reperfusion injury was recently reviewed by Ziegler et al.125 Several platelet-associated mechanisms of reperfusion injury have been proposed, including aggregation of platelets and formation of microthrombi in microvessels, release of vasoconstrictors like thromboxane A2, aggregation of platelets and leucocytes leading to additional pro-inflammatory leucocyte infiltration, release of extracellular vesicles (i.e. exosomes, microvesicles, and apoptotic vesicles) which can enhance inflammation, and stimulation of ischaemia-sensitive spinal afferent nerves. Aspirin and P2Y12 inhibitors have shown some cardioprotective effects but mainly because of their effects beyond platelet inhibition, such as their anti-inflammatory effects. The cardioprotective effects of glycoprotein IIb/IIIa (gpIIb/IIIa) inhibitors are unclear, but their use was accompanied by bleeding complications. Presently, the Platelet Inhibition to Target Reperfusion Injury trial (PITRI; NCT03102723) investigates whether the addition of the P2Y12 inhibitor cangrelor on top of conventional dual antiplatelet therapy is more effective in reducing infarct size and CMR-defined MVO than conventional dual antiplatelet therapy alone.
3.7. Pericytes
Pericytes are perivascular cells that are present along the endothelium lining the capillaries and venules.126 These types of cells are difficult to distinguish from other mural cells and as a consequence have been defined by a combination of morphology, anatomical localization, co-expression of markers, and functional characteristics.126,127 Pericytes are particularly recognized for their role in the diseased cerebral microvasculature, where they have been proposed to impede reperfusion by constricting capillaries upon ischaemia.128 Pericytes are also abundantly present in the heart126 and a role for pericytes in myocardial reperfusion injury was studied by O’Farrel and Attwell.128 LAD occlusion for 45 min followed by 15 min of reperfusion showed that 40% of the capillaries in the reperfused area were not perfused, which was accompanied by a 50% reduction in microvascular perfusion, despite complete epicardial coronary artery patency. Staining for leucocytes and erythrocytes failed to provide evidence for their presence at blocked capillaries. In contrast, unperfused capillaries were found in proximity of pericytes, identified as neuron-glial antigen 2 (NG2) positive cells. Furthermore, in close proximity of the soma of these pericytes, the capillary lumen diameter was reduced,128 leading to the hypothesis that cardiac pericytes actively constrict upon ischaemia, resulting in reduction of microvascular perfusion. Although the underlying molecular and cellular mechanisms by which pericytes contract are still incompletely understood,129 it is of interest that intravenous adenosine infusion started just prior to reperfusion attenuated constriction of capillaries at the site of pericytes, thereby reducing capillary blockage from 40% to 30%,123 while significantly improving microvascular perfusion.121,128
3.8. Reperfusion injury to endothelial cells in other organs
MVI is not limited to the heart but has also been described in the brain, kidney, and intestines. Although information on effects of ischaemia without reperfusion to endothelial cells is scarce, reperfusion itself has additional harmful effects on the cerebral, renal, and intestinal microvasculature and these effects are comparable to the coronary microvasculature. In the majority of studies however, the results are based on qualitative data rather than quantitative data.
Rats with a permanent middle cerebral artery occlusion showed in particular in the first 2 h of ischaemia endothelial nucleus swelling, moderate endothelial cell swelling, and an increase in endothelial mitochondria. After 12 h of ischaemia, endothelial cell necrosis became clearly apparent,130 indicating that ischaemia alone has limited effects on the cerebral endothelial cells. Similarities and differences in proposed mechanisms of cerebral and coronary reperfusion injury are recently reviewed by Kloner et al.131 After reperfusion, both organs show endothelial cell swelling causing blebs, inflammatory responses, and increased vascular permeability.
Mice subjected to renal ischaemia showed mild changes in the actin cytoskeleton of endothelial cells compared to controls. During ischaemia, interendothelial cell junctions were preserved. Reperfusion, however, resulted in disruption of the interendothelial cell junctions and subsequently increase in vascular permeability.132 Also rats subjected to renal ischaemia showed preserved vascular permeability.132 Renal reperfusion injury was reflected by endothelial- and interstitial cell swelling.133
Rats subjected to one hour of superior mesenteric ischaemia without reperfusion showed a decrease in NOS activity. Reperfusion was unable to restore this decreased NOS activity.134 In rats with 15 min of iliac artery ligation, intraluminal protrusions, but no endothelial gaps were described. Reperfusion resulted in severe endothelial damage and presence of leucocytes.135
4. Association between traditional cardiovascular risk factors and CMD in STEMI
Many patients presenting with AMI have one or more traditional cardiovascular risk factors such as diabetes mellitus, hypertension, hypercholesterolaemia, and smoking. These risk factors have also been associated with endothelial dysfunction, mainly explained by a decreased bioavailability of NO and an increased generation of ROS, as reviewed by Brunner et al.136 Potentially, pre-existing endothelial dysfunction increases the risk of reperfusion injury to the microcirculation (Table 2).
Table 2.
Clinical studies investigating the association between pre-existing cardiovascular risk factors and the presence and extent of coronary microvascular dysfunction after acute myocardial infarction
| Study | Variables | Measure of CMD | Main results | |
|---|---|---|---|---|
| Diabetes | Iwakura (2003)137 (n = 146) | Patient interview or medical record, abnormal oral glucose tolerance test, or HbA1c ≥6.5% | MCE | Categorized to MVO: no difference in diabetes and HbA1c |
| Eitel (2012)138 (n = 411) | Known diabetes | LGE-CMR | Categorized to diabetes: no difference in presence and extent of MVO | |
| Zia (2014)139 (n = 52) | Known diabetes or new diagnosis with HbA1c ≥6.5% or fasting BG ≥7.0 mmol/L | MVO by T1; IMH by T2* maps | Categorized to diabetes: no difference in presence and extent of MVO or IMH | |
| Ota (2015)140 (n = 93) | Known diabetes or abnormal oral glucose tolerance test | LGE-CMR | Categorized to MVO: no difference in diabetes | |
| Reinstadler (2017)141 (n = 792) | Known diabetes | LGE-CMR | Categorized to diabetes: no difference in presence and extent of MVO | |
| Blood glucose level (BG) | Iwakura (2003)137 (n = 146) | BG on admission; hyperglycaemia [BG ≥160 mg/dL (∼8.9 mmol/L)] | MCE | Categorized to MVO: BG was higher in MVO; BG and hyperglycaemia are independent predictors of MVO (OR 1.02 and RR 12.1) |
| Eitel (2012)138 (n = 411) | BG on admission | LGE-CMR | Graded relationship between BG on admission and MVO | |
| Ota (2015)140 (n = 93) | BG on admission | LGE-CMR | BG is an independent predictor of MVO (OR 1.014) | |
| Jensen (2011)142 (n = 107) | Hyperglycaemia (BG ≥7.8 mmol/L on admission). Diabetic patients excluded | LGE-CMR | Hyperglycaemia is an independent predictor of the presence and extent of MVO | |
| Hypertension | Reinstadler (2016)143 (n = 795) | Antihypertensive treatment or ≥3 SBP values >140 mmHg on at least two different days | LGE-CMR | Categorized to hypertension: no difference in presence and extent of MVO |
| Carrick (2018)144 (n = 324) | Antihypertensive treatment or ≥3 SBP values >140 mmHg on at least two different days | LGE-CMR, ST- resolution; IMH by T2* maps, intracoronary measurements | Pre-existing hypertension was dubious associated with IMH (OR 1.81 with CI 0.98–3.34). Blood pressure on admission was not associated with MVO | |
| Hyperchol-esteraemia | Iwakura (2006)145 (n = 293) | Previously diagnosed or total cholesterol >220 mg/dL (∼5.7 mmol/L) | MCE | Categorized to hypercholesterolaemia: no difference in incidence of no-reflow Statin pre-treatment was an independent predictor of no-reflow (OR 0.22) |
| Reindl (2017)146 (n = 235) | Blood samples on admission | LGE-CMR | Categorized to MVI: total cholesterol and LDL were higher in MVI Hypercholesterolaemia was associated with MVI (OR 1.02 with CI 1.01–1.02) No association with statin pre-treatment and MVI | |
| Smoking | Symons (2016)147 (n = 471) | Regularly smoking the past 12 months | LGE-CMR; IMH by T2 | Categorized to smoking: smokers had higher incidence of IMH (31% vs 20%) but comparable MVO; smoking was associated with IMH (OR 2.13–2.17) |
| Reinstadler (2017)148 (n = 727) | Patient interview | LGE-CMR; IMH is not described | Categorized to smoking: no difference in presence and extent of MVO or IMH | |
| Haig (2018)149 (n = 196) | ≥100 cigarettes in life and currently routinely smoking | LGE-CMR; IMH by T2* maps | Categorized to smoking: no difference in presence and extent of MVO or IMH Smoking was associated with IMH when corrected for IS (OR 2.76) |
BG, blood glucose; CI, confidence interval; IMH, intramyocardial haemorrhage; IS, infarct size; LGE-CMR, late gadolinium-enhanced cardiac magnetic resonance imaging; LV, left ventricle; MVI, microvascular injury; MVO, microvascular obstruction; n, number; OR, odds ratio; RR, relative risk ratio, SBP, systolic blood pressure.
4.1. Pre-existent diabetes and/or hyperglycaemia
Diabetes mellitus is a metabolic disease with macro- and microvascular complications. Patients with diabetes have a 2–4 times higher risk of cardiovascular complications and mortality.150 Systemic endothelial dysfunction136 and even CMD151,152 seem to play a key role in these cardiovascular complications. As endothelial dysfunction plays a prominent role in the cardiovascular complications of diabetes mellitus, an association between pre-existing diabetes, hyperglycaemia, and post-ischaemic CMD seems plausible. Insulin at physiological concentration increases coronary microvascular perfusion.153 The presence of absolute or relative insulin resistance in diabetes mellitus may, therefore, contribute to worsening microvascular function. However, in STEMI patients with pre-existing diabetes, diabetes per se is not associated with CMD.137–141 In contrast to pre-existing diabetes, blood glucose level and hyperglycaemia on admission, which is not uncommon during AMI, are both associated with CMR-defined MVO.137,138,140 A prospective trial in non-diabetic patients with first presentation of STEMI confirms that hyperglycaemia on admission was associated with more pronounced CMR-defined MVO.142 This suggests that hyperglycaemia, rather than diabetes mellitus, plays a pivotal role in CMD. Acute hyperglycaemia impairs endothelium-dependent vasodilatation in the brachial artery in healthy humans.154 In a hyperglycaemic canine model, endothelium-dependent coronary microvascular dilatation was impaired, whereas endothelium-independent coronary microvascular dilatation remained unaffected.155 Hyperglycaemia exerts its actions on endothelial cell function by perturbations in cell signalling, enhanced toxic metabolites, altered osmolarity, in turn, leading to oxidative stress and inflammatory cytokines.156 Therefore, it was hypothesized that lowering blood glucose level prior to or directly after primary PCI results in less CMD. In diabetic and non-diabetic rodent ischaemia–reperfusion models, treatment with metformin starting at the onset of reperfusion resulted in a reduction of myocardial infarct size.157–161 However, in a porcine ischaemia–reperfusion model, combined intravenous and intracoronary metformin started at the onset of reperfusion failed to reduce infarct size.162 Combined results of two large randomized controlled trials OASIS-6 and CREAT-ECLA show that glucose–insulin–potassium therapy did not improve survival after AMI and was even harmful in the first days post-AMI. However, the extent of CMD was not assessed.163 Whereas treatment with exenatide, a glucagon-like peptide-1 analogue, resulted in smaller infarct size in animal models,164–166 it failed to limit infarct size in STEMI patients.167–169
4.2. Pre-existent hypertension
Hypertension is strongly related to cardiovascular mortality.170 Also, STEMI patients with pre-existing hypertension have worse prognosis in terms of MACE and mortality.143,144 Essential hypertension is associated with impaired endothelium-dependent vasodilatation,171–174 mainly by reduced bioavailability of NO and increased production of ROS. However, assessment of reperfusion injury with invasive intracoronary parameters showed no association between pre-existing hypertension and TIMI flow, CFR, IMR, and RRR.144 Using LGE and T2-weighted imaging on CMR, there was no association between pre-existing hypertension and MVO143,144 or IMH.144
4.3. Hypercholesterolaemia
Hypercholesterolaemia is associated with reduced bioavailability of NO.174 Rabbits undergoing ischaemia–reperfusion after a cholesterol-enriched diet for 3 days showed increased infarct size as determined by TTC staining and increased MVO zone as determined by Thioflavin S.175 In patients, however, the relation between pre-existing hypercholesterolaemia and the development of post-ischaemic CMD is less clear. Reindl et al.146 showed that hypercholesterolaemia was associated with CMR-defined MVO, although the odds ratio was only 1.02. Iwakura et al.145 showed that the incidence of no-reflow determined by contrast echocardiography was comparable between patients with and without hypercholesterolaemia. Effect of chronic pre-treatment with statins on CMD is conflicting.145,146 However, early high-dose statin treatment with rosuvastatin or atorvastatin did not reduce myocardial injury in STEMI patients.176,177
4.4 Smoking
Smoking has a negative effect on vascular function, including microvascular function.178 Besides endothelial dysfunction,179 smoking produces a more thrombogenic state.180 How active smoking influences outcome with regard to post-ischaemic CMD is less clear and even conflicting.147–149 In a multicentre, prospective study with active smokers presenting with their first STEMI, the incidence of IMH on T2-weighted imaging on CMR was higher compared to non-smoking STEMI patients, whereas MVO was comparable between groups. In addition, smoking was independently associated with IMH when corrected for infarct size and MVO.147 In contrast, Reinstadler et al. and Haig et al.148,149 showed that presence and extent of MVO and IMH was comparable between smokers and non-smokers. However, the latter study did show that smoking was associated with IMH when corrected for infarct size.149
5. Treatment of CMD in STEMI in clinical practice: past, present, and future possibilities
In the practice guidelines of the European Society of Cardiology,181 a Class I recommendation is consistent with clear evidence and general agreement. Class IIa implies some incomplete or conflicting evidence, and the balance favours ‘should be considered’. A Class IIb recommendation indicates the efficacy is less well-established consistent with a ‘may be considered’ recommendation.
5.1. Established clinical evidence
CMR-defined MVO is related to the size of infarction. From a pragmatic perspective, clinical measures to limit the severity of AMI, notably timely presentation to hospital to limit the duration of ischaemia, will intuitively be helpful to limit infarct size and prevent CMD on a case by case basis.182
In a randomized, controlled trial of intra-coronary vasodilator therapy in patients with acute coronary syndromes, Vijayalakshmi et al.183 compared intracoronary administration of heparinized saline, intracoronary verapamil (0.5 mg in 10 mL of heparinized saline) or intracoronary adenosine (30 µg in 10 mL of heparinized saline). They observed similar improvements in coronary blood flow as reflected by the TIMI Frame Count (TFC) acutely and left ventricular wall motion at 1 and 30 days after PCI in the verapamil and adenosine groups. However, a systematic review by Su et al.184 concluded there is insufficient evidence.
More recently, in the REFLOW-STEMI trial185 compared with standard primary PCI, high-dose intracoronary adenosine (2–3 mg), and intracoronary sodium nitroprusside (0.5 mg in total) during primary PCI did not reduce CMR-defined infarct size or MVO. In a per-protocol analysis, high-dose intracoronary adenosine was associated with an increase in infarct size, a reduction in LVEF and an increase in MACE at 6 months. In patients with no- or slow reflow, these findings point to a ‘do not treat’ recommendation for higher doses of intracoronary adenosine.
The practice guidelines indicate that gpIIb/IIIa inhibitor therapy should be considered for bailout therapy for patients with slow- or no-reflow.181 This is a Class IIa recommendation supported by a Level C recommendation since there are no clinical trials. Options for gpIIb/IIIa inhibitor therapy include abciximab, tirofiban, and epitifibitide. These drugs are given by either intravenous or intracoronary routes, initially with a bolus followed by a maintenance infusion for 12–24 h.
5.2. Evidence that does not support clinical use
There are four other trials of intracoronary reduced dose lytic therapy, including T-TIME (NCT02257294), STRIVE (NCT03335839), and the smaller OPTIMAL trial (NCT02894138) using alteplase and RESTORE-MI (ACTRN12618000778280) using tenecteplase. The recently published T-TIME study is a Phase 2 clinical trial of low-dose adjunctive intracoronary fibrinolysis with alteplase in reperfused STEMI.17 By targeting thrombus within the infarct-related artery and microcirculation with fibrinolytic therapy the aim was to restore microvascular blood flow at the earliest point after coronary reperfusion. CMR-defined MVO, as reflected by the amount (percentage of left ventricular mass) and percentage of affected patients, was not reduced by alteplase.17 The results do not support the use of low-dose alteplase either routinely in patients at risk of CMD or in those patients with slow- or no-reflow. The potential for intracoronary lytic therapy to prevent or treat CMD remains to be determined.
Routine deferral of stent implantation is also not associated with favourable results186 and is not recommended in practice guidelines.181
5.3. Future possibilities
In the DEFER-STEMI pilot trial,187 STEMI patients with successful reperfusion by initial thrombectomy and/or balloon angioplasty were randomized to deferred stenting with an intention-to-stent 4–16 h later or conventional treatment with immediate stenting. Fewer patients in the deferred stenting group had no/slow-reflow or intra-procedural thrombotic events. TIMI coronary flow grades at the end of PCI and myocardial salvage index at 6 months were higher in the deferred stenting group. CMR-defined MVO was numerically reduced, but the difference was not statistically significant. The paradigm of a selective approach to deferred stenting is being prospectively assessed in the PRIMACY study (NCT01542385). The investigators derived the Bayesian prior probability of efficacy with patient-level data from the DEFER-STEMI (UK),187 MIMI (France),188 DANAMI-3 (Denmark),187 and INNOVATION (South Korea)189 trials, which all tested the delayed stenting hypothesis. The investigators aim to determine whether a selective strategy of deferred stenting increases the probability of early urgent target vessel revascularization in the short-term and whether or not there may be meaningful protective effects against heart failure in the longer-term.
In smaller scale studies, intracoronary nitrate and intracoronary nicorandil190 have been associated with favourable improvements in TFC. These treatments are routinely used by clinicians to treat patients with no-reflow. However, larger trials will be needed to substantiate practice guideline recommendations.
The Early-Beta blocker Administration before reperfusion primary PCI in patients with ST-elevation Myocardial Infarction (EARLY-BAMI) trial involved a randomized, double-blind, matched-placebo-controlled clinical trial of intravenous metoprolol (intravenous bolus of metoprolol, 5 mg) in STEMI patients.191 There were no between-group differences in infarct size at 30 days post-MI or MACE. The effect on CMD was not reported.191 The Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial randomized patients with anterior STEMI who were undergoing primary PCI within 6 h of symptom onset to receive intravenous metoprolol (15 mg) or not (control). The first dose of oral metoprolol was scheduled within 24 h of admission. Infarct size was reduced and LVEF at 6 months was increased compared to control treatment.192 The incidence of MACE at 2 years was comparable between the early metoprolol and control group.193 Metoprolol treatment was associated with a 40% reduction in the extent of CMR-defined MVO and the mechanism might involve inhibition of neutrophil activation.194 These trials present seemingly conflicting results. The differences in the results might be explained by patient selection, design and power. Overall, the results support the case for a substantive, randomized, placebo-controlled, double-blind, clinical trial of intravenous metoprolol before primary PCI.
Ischaemic conditioning, which is defined by either repeated brief episodes of mechanical ischaemia/reperfusion or by pharmacological prevention of the opening of the mitochondrial permeability transition pore before primary PCI or at the onset of reperfusion, has been championed as a novel cardioprotective therapy.195,196 However, recent trials of intravenous cyclosporine,197 TRO40303,198 and post-conditioning199 have not supported the case. CONDI-2 (NCT02342522) and ERIC-PPCI (NCT02342522) are multicentre, multinational clinical trials that should provide conclusive results to clarify whether ischaemic conditioning has a role for cardioprotection.
Remote ischaemic preconditioning (RIC), which is defined as repeated brief episodes of ischaemia/reperfusion at a remote site before prolonged ischaemia/reperfusion in a target organ occurs, is proposed as a novel cardioprotective strategy to attenuate reperfusion injury. Cardioprotection by RIC may be mediated by microvesicles. Microvesicles (0.1–1 µm in diameter) are a subset of extracellular vesicles that can be secreted by activated endothelial cells, platelets, erythrocytes and immune cells.200,201 Currently, microvesicles are studied in the context of cardiovascular biomarkers,202 in relation to endothelial dysfunction after AMI203 and in relation to microvascular dysfunction after AMI.204 However, there is also some evidence that microvesicles, when obtained during RIC, have cardioprotective effects by reducing myocardial infarct size.205–207 Therefore, the use of RIC-derived microvesicles may be a promising cardioprotective strategy.
CMD is a common complication of AMI, presenting an unmet therapeutic need. Moving forward, we advocate targeted approaches to identify patients who might respond to effective therapy.208 Use of stratified medicine, coupling novel diagnostics to stratify at-risk patients for targeted therapy, holds future promise.208 Therapeutic hypothermia during primary PCI may be an option. The EUROpean Intracoronary Cooling Evaluation in Patients With ST-elevation Myocardial Infarction (EURO-ICE) is currently investigating the feasibility, safety and efficacy of localized cooling of the ischaemic area-at-risk by infusion of chilled saline into the infarct-related LAD in patients with anterior STEMI (NCT03447834).
6. Conclusion
In conclusion, reperfusion after STEMI can lead to incremental detrimental effects to the coronary microvasculature. This phenomenon has been described by multiple terms, including no-reflow, MVO, MVI, reperfusion injury, and IMH. All aforementioned pathophysiological mechanisms taken together, we recommend to use the term post-ischaemic CMD as an umbrella term unless specific knowledge on the pathophysiological substrate is available. Post-ischaemic CMD can be predicted using invasive intracoronary measurements such as IMR, HMR, RRR, IHDVPS, Pzf, and visualized using LGE imaging and T2* maps on CMR. Traditional cardiovascular risk factors do not seem to contribute to the development of post-ischaemic CMD. Although multiple clinical trials aimed to target post-ischaemic CMD, none of them were consistently able to reduce the extent of CMD. Targeting coronary microvascular endothelial cells, thereby aiming to prevent vascular leakage and IMH may be a new promising cardioprotective strategy. However, at present, CMD still constitutes an unmet therapeutic need.
Acknowledgements
The authors wish to thank Regina Konst for her help with designing the figures.
Conflict of interest: none declared.
Funding
N.P. Riksen is supported by a CVON grant from the Netherlands Heart Foundation (CVON2018-27), a grant of the ERA-CVD Joint Transnational Call 2018, which is supported by the Dutch Heart Foundation (JTC2018, project MEMORY; 2018T093), and by funding from the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 667837). D.J. Duncker is supported by a CVON grant from the Netherlands heart Foundation (CVON2014-11; RECONNECT). C. Berry is supported by funding from the British Heart Foundation (RE/18/6134217; PG/11/2/2874).
References
- 1. Cohen M, Boiangiu C, Abidi M.. Therapy for ST-segment elevation myocardial infarction patients who present late or are ineligible for reperfusion therapy. J Am Coll Cardiol 2010;55:1895–1906. [DOI] [PubMed] [Google Scholar]
- 2. Van Kranenburg MV, Magro M, Thiele H, DeWaha S, Eitel I, Cochet A, Cottin Y, Atar D, Buser P, Wu E, Lee D, Bodi V, Klug G, Metzler B, Delewi R, Bernhardt P, Rottbauer W, Boersma E, Zijlstra F, Geuns RJ.. Prognostic value of microvascular obstruction and infarct size, as measured by CMR in STEMI patients. JACC Cardiovasc Imaging 2014;7:930–939. [DOI] [PubMed] [Google Scholar]
- 3. Reffelmann T, Kloner RA.. Microvascular reperfusion injury: rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am J Physiol Circ Physiol 2002;283:H1099–H1107. [DOI] [PubMed] [Google Scholar]
- 4. Lima JAC, Wu KC, Becker LC, Melin JA, Gerber BL, Rochitte CE, Bluemke DA, McVeigh ER.. Microvascular obstruction and left ventricular remodeling early after acute myocardial infarction. Circulation 2012;101:2734–2741. [DOI] [PubMed] [Google Scholar]
- 5. Kloner RA, Ganote CE, Jennings RB.. The ‘no reflow’ phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974;54:1496–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sezer M, van Royen N, Umman B, Bugra Z, Bulluck H, Hausenloy DJ, Umman S.. Coronary microvascular injury in reperfused acute myocardial infarction: a view from an integrative perspective. J Am Heart Assoc 2018;7:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heusch G. Critical issues for the translation of cardioprotection. Circ Res 2017;120:1477–1486. [DOI] [PubMed] [Google Scholar]
- 8. Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, van RN, Schulz R, Heusch G.. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res 2019;115:1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bulluck H, Yellon DM, Hausenloy DJ.. Reducing myocardial infarct size: challenges and future opportunities. Heart 2016;102:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krug A, Mesnil de Rochemont D, Korb G.. Blood supply of the myocardium after temporary coronary occlusion. Circ Res 1966;19:57–62. [DOI] [PubMed] [Google Scholar]
- 11. Ames A, Wright RL, Kowada M, Thurston JM, Majno G.. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol 1968;52:437–453. [PMC free article] [PubMed] [Google Scholar]
- 12. Kloner RA. No-reflow phenomenon: maintaining vascular integrity. J Cardiovasc Pharmacol Ther 2011;16:244–250. [DOI] [PubMed] [Google Scholar]
- 13. Schofer J, Montz R, Mathey DG.. Scintigraphic evidence of the “No reflow” phenomenon in human beings after coronary thrombolysis. J Am Coll Cardiol 1985;5:593–598. [DOI] [PubMed] [Google Scholar]
- 14. Bates ER, Krell MJ, Dean EN, O'Neill WW, Vogel RA.. Demonstration of the “no-reflow” phenomenon by digital coronary arteriography. Am J Cardiol 1986;57:177–178. [DOI] [PubMed] [Google Scholar]
- 15. Feld H, Lichstein E, Schachter J, Shani J.. Early and late angiographic findings of the ‘no-reflow’ phenomenon following direct angioplasty as primary treatment for acute myocardial infarction. Am Heart J 1992;123:782–784. [DOI] [PubMed] [Google Scholar]
- 16. Harrison RW, Aggarwal A, Ou FS, Klein LW, Rumsfeld JS, Roe MT, Wang TY.. Incidence and outcomes of no-reflow phenomenon during percutaneous coronary intervention among patients with acute myocardial infarction. Am J Cardiol 2013;111:178–184. [DOI] [PubMed] [Google Scholar]
- 17. McCartney PJ, Eteiba H, Maznyczka AM, McEntegart M, Greenwood JP, Muir DF, Chowdhary S, Gershlick AH, Appleby C, Cotton JM, Wragg A, Curzen N, Oldroyd KG, Lindsay M, Rocchiccioli JP, Shaukat A, Good R, Watkins S, Robertson K, Malkin C, Martin L, Gillespie L, Ford TJ, Petrie MC, Macfarlane PW, Tait RC, Welsh P, Sattar N, Weir RA, Fox KA, Ford I, McConnachie A, Berry C.. Effect of low-dose intracoronary alteplase during primary percutaneous coronary intervention on microvascular obstruction in patients with acute myocardial infarction: a randomized clinical trial. JAMA 2019;321:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrick D, Haig C, Ahmed N, Rauhalammi S, Clerfond G, Carberry J, Mordi I, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay M, Mahrous A, Welsh P, Sattar N, Ford I, Oldroyd KG, Radjenovic A, Berry C.. Temporal evolution of myocardial hemorrhage and edema in patients after acute ST-segment elevation myocardial infarction: pathophysiological insights and clinical implications. J Am Heart Assoc 2016;5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Robbers L, Eerenberg ES, Teunissen PF, Jansen MF, Hollander MR, Horrevoets AJG, Knaapen P, Nijveldt R, Heymans MW, Levi MM, Rossum AC, Van Niessen HWM, Marcu CB, Beek AM, Royen N.. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur Heart J 2013;34:2346–2353. [DOI] [PubMed] [Google Scholar]
- 20. Van Den Bos BE, Baks T, Moelker AD, Kerver W, Geuns RV, Giessen WJ, Van Der Duncker DJ, Wielopolski PA.. Magnetic resonance imaging of haemorrhage within reperfused myocardial infarcts: possible interference with iron oxide-labelled cell tracking? Eur Heart J 2006;27:1620–1626. [DOI] [PubMed] [Google Scholar]
- 21. Betgem RP, Waard GD, Nijveldt R, Beek AM, Escaned J, Van Royen N.. Intramyocardial haemorrhage after acute myocardial infarction. Nat Rev Cardiol 2015;12:156–167. [DOI] [PubMed] [Google Scholar]
- 22. Roberts CS, Schoen FJ, Kloner RA.. Effect of coronary reperfusion on myocardial hemorrhage and infarct healing. Am J Cardiol 1983;52:610–614. [DOI] [PubMed] [Google Scholar]
- 23. Hollander MR, Waard G. D, Konijnenberg LSF, Meijer-van Putten RME, Brom C. V D, Paauw N, Vries H. D, Ven P. V D, Aman J, Nieuw-Amerongen GV, Hordijk PL, Niessen HWM, Horrevoets AJG, Royen NV.. Dissecting the effects of ischemia and reperfusion on the coronary microcirculation in a rat model of acute myocardial infarction. PLoS One 2016;11:e0157233.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waha SD, Desch S, Eitel I, Fuernau G, Zachrau J, Leuschner A, Gutberlet M, Schuler G, Thiele H.. Impact of early vs. late microvascular obstruction assessed by magnetic resonance imaging on long-term outcome after ST-elevation myocardial infarction: a comparison with traditional prognostic markers. Eur Heart J 2010;31:2660–2668. [DOI] [PubMed] [Google Scholar]
- 25. Husser O, Monmeneu JV, Sanchis J, Nunez J, Lopez-Lereu MP, Bonanad C, Chaustre F, Gomez C, Bosch MJ, Hinarejos R, Chorro FJ, Riegger GAJ, Llacer A, Bodi V.. Cardiovascular magnetic resonance-derived intramyocardial hemorrhage after STEMI: influence on long-term prognosis, adverse left ventricular remodeling and relationship with microvascular obstruction. Int J Cardiol 2013;167:2047–2054. [DOI] [PubMed] [Google Scholar]
- 26.TIMI Study Group. The thrombolysis in myocardial infarction (TIMI) trial. Phase I findings. N Engel J Med 1985;312:932–936. [DOI] [PubMed] [Google Scholar]
- 27. Mehta RH, Harjai KJ, Cox D, Stone GW, Brodie B, Boura J, O'Neill W, Grines CL.. Clinical and angiographic correlates and outcomes of suboptimal coronary flow in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention. J Am Coll Cardiol 2003;42:1739–1746. [DOI] [PubMed] [Google Scholar]
- 28. Caixeta A, Lansky AJ, Mehran R, Brener SJ, Claessen B, Genereux P, Palmerini T, Witzenbichler B, Guagliumi G, Brodie BR, Dudek D, Fahy M, Dangas GD, Stone GW.. Predictors of suboptimal TIMI flow after primary angioplasty for acute myocardial infarction: results from the HORIZONS-AMI trial. Eurointervention 2013;9:220–227. [DOI] [PubMed] [Google Scholar]
- 29. Nijveldt R, Beek AM, Hirsch A, Stoel MG, Hofman MBM, Umans V, Algra PR, Twisk JWR, van Rossum AC.. Functional recovery after acute myocardial infarction: comparison between angiography, electrocardiography, and cardiovascular magnetic resonance measures of microvascular injury. J Am Coll Cardiol 2008;52:181–189. [DOI] [PubMed] [Google Scholar]
- 30. Kunadian V, Harrigan C, Zorkun C, Palmer AM, Ogando KJ, Biller LH, Lord EE, Williams SP, Lew ME, Ciaglo LN, Buros JL, Marble SJ, Gibson WJ, Gibson CM.. Use of the TIMI frame count in the assessment of coronary artery blood flow and microvascular function over the past 15 years. J Thromb Thrombolysis 2009;27:316–328. [DOI] [PubMed] [Google Scholar]
- 31. Abaci A, Oguzhan A, Eryol NK, Ergin A.. Effect of potential confounding factors on the thrombolysis in myocardial infarction (TIMI) trial frame count and its reproducibility. Circulation 1999;100:2219–2223. [DOI] [PubMed] [Google Scholar]
- 32. French JK, Ellis CJ, Webber BJ, Williams BF, Amos DJ, Ramanathan K, Whitlock RM, White HD.. Abnormal coronary flow in infarct arteries 1 year after myocardial infarction is predicted at 4 weeks by corrected thrombolysis in myocardial infarction (TIMI) frame count and stenosis severity. Am J Cardiol 1998;81:665–671. [DOI] [PubMed] [Google Scholar]
- 33. Gibson CM, Murphy SA, Rizzo MJ, Ryan K. A, Marble SJ, McCabe CH, Cannon CP, Van de Werf F, Braunwald E.. Relationship between TIMI frame count and clinical outcomes after thrombolytic administration. Thrombolysis In Myocardial Infarction (TIMI) Study Group. Circulation 1999;99:1945–1950. [DOI] [PubMed] [Google Scholar]
- 34. Gibson CM, Cannon CP, Murphy SA, Marble SJ, Barron HV, Braunwald E.. Relationship of the TIMI myocardial perfusion grades, flow grades, frame count, and percutaneous coronary intervention to long-term outcomes after thrombolytic administration in acute myocardial infarction. Circulation 2002;105:1909–1913. [DOI] [PubMed] [Google Scholar]
- 35. Bhatt DL, Ellis SG, Ivanc TB, Crowe T, Balazs E, Debowey D, Pangerl A, Chew PH.. Corrected TIMI frame count does not predict 30-day adverse outcomes after reperfusion therapy for acute myocardial infarction. Am Heart J 1999;138:785–790. [DOI] [PubMed] [Google Scholar]
- 36. Haager PK, Christott P, Heussen N, Lepper W, Hanrath P, Hoffmann R.. Prediction of clinical outcome after mechanical revascularization in acute myocardial infarction by markers of myocardial reperfusion. J Am Coll Cardiol 2003;41:532–538. [DOI] [PubMed] [Google Scholar]
- 37. van’T Hof AW, Liem A, Suryapranata H, Hoorntje JCA, de Boer M-J, Zijlstra F.. Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction—myocardial blush grade. Circulation 1998;97:2302–2306. [DOI] [PubMed] [Google Scholar]
- 38. Marra MP, Corbetti F, Cacciavillani L, Tarantini G, Ramondo AB, Napodano M, Basso C, Lacognata C, Marzari A, Maddalena F, Iliceto S.. Relationship between myocardial blush grades, staining, and severe microvascular damage after primary percutaneous coronary intervention: a study performed with contrast-enhanced magnetic resonance in a large consecutive series of patients. Am Heart J 2010;159:1124–1132. [DOI] [PubMed] [Google Scholar]
- 39. Vogelzang M, Vlaar PJ, Svilaas T, Amo D, Nijsten MWN, Zijlstra F.. Computer-assisted myocardial blush quantification after percutaneous coronary angioplasty for acute myocardial infarction: a substudy from the TAPAS trial. Eur Heart J 2009;30:594–599. [DOI] [PubMed] [Google Scholar]
- 40. Haeck JDE, Gu YL, Vogelzang M, Bilodeau L, Krucoff MW, Tijssen JGP, Winter RD, Zijlstra F, Koch KT.. Feasibility and applicability of computer-assisted myocardial blush quantification after primary percutaneous coronary intervention for ST-segment elevation myocardial infarction. Catheter Cardiovasc Interv 2010;75:701–706. [DOI] [PubMed] [Google Scholar]
- 41. Biesbroek PS, Roos ST, Hout MV, Gragt JVD, Teunissen PFA, Waard GD, Knaapen P, Kamp O, Royen NV.. Fluoroscopy assisted scoring of myocardial hypoperfusion (FLASH) ratio as a novel predictor of mortality after primary PCI in STEMI patients. Int J Cardiol 2016;202:639–645. [DOI] [PubMed] [Google Scholar]
- 42. Everaars H, de Waard GA, Driessen RS, Danad I, Van de Ven PM, Raijmakers PG, Lammertsma AA, van Rossum AC, Knaapen P, van Royen N.. Doppler flow velocity and thermodilution to assess coronary flow reserve: a head-to-head comparison with [15O]H2O PET. JACC Cardiovasc Interv 2018;11:2044–2054. [DOI] [PubMed] [Google Scholar]
- 43. Carrick D, Haig C, Carberry J, May VTY, McCartney P, Welsh P, Ahmed N, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Mahrous A, Rauhalammi SMO, Mordi I, Ford I, Radjenovic A, Sattar N, Oldroyd KG, Berry C.. Microvascular resistance of the culprit coronary artery in acute ST-elevation myocardial infarction. JCI Insight 2016;1:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hirsch A, Nijveldt R, Haeck JDE, Beek AM, Koch KT, Henriques JPS, van der Schaaf RJ, Vis MM, Baan J, de Winter R, Tijssen JGP, van Rossum A, Piek JJ.. Relation between the assessment of microvascular injury by cardiovascular magnetic resonance and coronary Doppler flow velocity measurements in patients with acute anterior wall myocardial infarction. J Am Coll Cardiol 2008;51:2230–2238. [DOI] [PubMed] [Google Scholar]
- 45. Bulluck H, Foin N, Tan JW, Low AF, Sezer M, Hausenloy DJ.. Invasive assessment of the coronary microcirculation in reperfused ST-segment-elevation myocardial infarction patients: where do we stand? Circ Cardiovasc Interv 2017;10:1–11. [DOI] [PubMed] [Google Scholar]
- 46. Hoef TVD, Bax M, Meuwissen M, Damman P, Delewi R, Winter RD, Koch KT, Schotborgh C, Henriques JPS, Tijssen JGP, Piek JJ.. Impact of coronary microvascular function on long-term cardiac mortality in patients with acute ST-segment-elevation myocardial infarction. Circ Cardiovasc Interv 2013;6:207–215. [DOI] [PubMed] [Google Scholar]
- 47. Fearon WF. Invasive coronary physiology for assessing intermediate lesions. Circ Cardiovasc Interv 2015;8:1–8. [DOI] [PubMed] [Google Scholar]
- 48. Aarnoudse W, Fearon WF, Manoharan G, Geven M, Vosse FVD, Rutten M, Bruyne BD, Pijls N.. Epicardial stenosis severity does not affect minimal microcirculatory resistance. Circulation 2004;110:2137–2142. [DOI] [PubMed] [Google Scholar]
- 49. Bulluck H, Foin N, Cabrera-Fuentes HA, Yeo KK, Wong AS, Fam JM, Wong PE, Tan JW, Low AF, Hausenloy DJ.. Index of microvascular resistance and microvascular obstruction in patients with acute myocardial infarction. JACC Cardiovasc Interv 2016;9:2172–2174. [DOI] [PubMed] [Google Scholar]
- 50. Carrick D, Haig C, Ahmed N, Carberry J, Yue May VT, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Davie A, Mahrous A, Mordi I, Ford I, Radjenovic A, Oldroyd KG, Berry C.. Comparative prognostic utility of indexes of microvascular function alone or in combination in patients with an acute ST-segment-elevation myocardial infarction. Circulation 2016;134:1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fearon WF, Low AF, Yong AS, McGeoch R, Berry C, Shah MG, Ho MY, Kim HS, Loh JP, Oldroyd KG.. Prognostic value of the index of microcirculatory resistance measured after primary percutaneous coronary intervention William. Circulation 2013;127:2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Teunissen PFA, Waard GD, Hollander MR, Robbers L, Danad I, Biesbroek PS, Amier RP, Echavarría-Pinto M, Quirós A, Broyd C, Heymans MW, Nijveldt R, Lammertsma AA, Raijmakers PG, Allaart CP, Lemkes JS, Appelman YE, Marques KM, Bronzwaer JGF, Horrevoets AJG, Rossum AV, Escaned J, Beek AM, Knaapen P, Royen NV.. Doppler-derived intracoronary physiology indices predict the occurrence of microvascular injury and microvascular perfusion deficits after angiographically successful primary percutaneous coronary intervention. Circ Cardiovasc Interv 2015;8:1–11. [DOI] [PubMed] [Google Scholar]
- 53. Jin XJ, Yoon MH, Seo KW, Tahk SJ, Lim HS, Yang HM, Choi BJ, Choi SY, Hwang GS, Shin JH, Park JS.. Usefulness of hyperemic microvascular resistance index as a predictor of clinical outcomes in patients with ST-segment elevation myocardial infarction. Korean Circ J 2015;45:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hennigan B, Layland J, Fearon WF, Oldroyd KG.. Fractional flow reserve and the index of microvascular resistance in patients with acute coronary syndromes. Eurointervention 2014;10(Suppl. T):T55–63. [DOI] [PubMed] [Google Scholar]
- 55. Layland J, Carrick D, McEntegart M, Ahmed N, Payne A, McClure J, Sood A, McGeoch R, MacIsaac A, Whitbourn R, Wilson A, Oldroyd K, Berry C.. Vasodilatory capacity of the coronary microcirculation is preserved in selected patients with non-ST-segment-elevation myocardial infarction. Circ Cardiovasc Interv 2013;6:231–236. [DOI] [PubMed] [Google Scholar]
- 56. Xaplanteris P, Fournier S, Keulards DCJ, Adjedj J, Ciccarelli G, Milkas A, Pellicano M, van’t Veer M, Barbato E, Pijls NHJ, Bruyne BD.. Catheter-based measurements of absolute coronary blood flow and microvascular resistance. Circ Cardiovasc Interv 2018;11:1–8. [DOI] [PubMed] [Google Scholar]
- 57. Wijnbergen I, van't Veer M, Lammers J, Ubachs J, Pijls NHJ.. Absolute coronary blood flow measurement and microvascular resistance in ST-elevation myocardial infarction in the acute and subacute phase. Cardiovasc Revasc Med 2016;17:81–87. [DOI] [PubMed] [Google Scholar]
- 58. Escaned J, Flores A, García-Pavía P, Segovia J, Jimenez J, Aragoncillo P, Salas C, Alfonso F, Hernández R, Angiolillo DJ, Jiménez-Quevedo P, Bañuelos C, Alonso-Pulpón L, Macaya C.. Assessment of microcirculatory remodeling with intracoronary flow velocity and pressure measurements. Circulation 2009;120:1561–1568. [DOI] [PubMed] [Google Scholar]
- 59. Shibata T, Watanabe H, Tsurusaki T, Minai K, Ogawa T, Iwano K, Tamura T, Yoshida S, Mutou M, Imai K, Horie T, Mochizuki S.. Do indices of coronary conductance after reperfusion reflect the extent of salvaged myocardium? Jpn Heart J 2004;45:387–396. [DOI] [PubMed] [Google Scholar]
- 60. Shimada K, Sakanoue Y, Kobayashi Y, Ehara S, Hirose M, Nakamura Y, Fukuda D, Yamagishi H, Yoshiyama M, Takeuchi K, Yoshikawa J.. Assessment of myocardial viability: using coronary pressure and flow after acute myocardial infarction. Heart 2003;89:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bellamy RF. Diastolic coronary artery pressure-flow relations in the dog. Circ Res 1978;43:92–101. [DOI] [PubMed] [Google Scholar]
- 62. Herck PV, Carlier SG, Claeys MJ, Haine SE, Gorissen P, Miljoen H, Bosnians JM, Vrints CJ.. Coronary microvascular dysfunction after myocardial infarction: increased coronary zero flow pressure both in the infarcted and in the remote myocardium is mainly related to left ventricular filling pressure. Heart 2007;93:1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kitabata H, Imanishi T, Kubo T, Takarada S, Kashiwagi M, Matsumoto H, Tsujioka H, Ikejima H, Arita Y, Okochi K, Kuroi A, Ueno S, Kataiwa H, Tanimoto T, Yamano T, Hirata K, Nakamura N, Tanaka A, Mizukoshi M, Akasaka T.. Coronary microvascular resistance index immediately after primary percutaneous coronary intervention as a predictor of the transmural extent of infarction in patients with ST-segment elevation anterior acute myocardial infarction. JACC Cardiovasc Imaging 2009;2:263–272. [DOI] [PubMed] [Google Scholar]
- 64. Patel N, Petraco R, Dall’Armellina E, Kassimis G, Maria GD, Dawkins S, Lee R, Prendergast BD, Choudhury RP, Forfar JC, Channon KM, Davies J, Banning AP, Kharbanda RK.. Zero-flow pressure measured immediately after primary percutaneous coronary intervention for ST-segment elevation myocardial infarction provides the best invasive index for predicting the extent of myocardial infarction at 6 months: an OxAMI study (Oxford Acute Myocardial Infarction). JACC Cardiovasc Interv 2015;8:1410–1421. [DOI] [PubMed] [Google Scholar]
- 65. Kim RJ, Fieno DS, Parrish TB, Harris K, Chen E, Simonetti O, Bundy J, Finn JP, Klocke FJ, Judd RM.. Irreversible injury, infarct age, and contractile function. Circulation 1999;100:1992–2002. [DOI] [PubMed] [Google Scholar]
- 66. Nijveldt R, Hofman MBM, Hirsch A, Beek AM, Umans V, Algra PR, Piek JJ, van Rossum AC.. Assessment of microvascular obstruction and prediction of short-term remodeling after acute myocardial infarction: cardiac MR imaging study. Radiology 2009;250:363–370. [DOI] [PubMed] [Google Scholar]
- 67. Ibanez B, Aletras AH, Arai AE, Arheden H, Bax J, Berry C, Bucciarelli-Ducci C, Croisille P, Dall'Armellina E, Dharmakumar R, Eitel I, Fernández-Jiménez R, Friedrich MG, García-Dorado D, Hausenloy DJ, Kim RJ, Kozerke S, Kramer CM, Salerno M, Sánchez-González J, Sanz J, Fuster V.. Cardiac MRI endpoints in myocardial infarction experimental and clinical trials: JACC scientific expert panel. J Am Coll Cardiol 2019;74:238–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Demirkiran A, Everaars H, Amier RP, Beijnink C, Bom MJ, Götte MJW, van Loon R, Selder JL, van Rossum A, Nijveldt R.. Cardiovascular magnetic resonance techniques for tissue characterization after acute myocardial injury. Eur Heart J Cardiovasc Imaging 2019;20:723–734. [DOI] [PubMed] [Google Scholar]
- 69. Beek AM, Nijveldt R, Rossum AV.. Intramyocardial hemorrhage and microvascular obstruction after primary percutaneous coronary intervention. Int J Cardiovasc Imaging 2010;26:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Basso C, Corbetti F, Silva C, Abudureheman A, Lacognata C, Cacciavillani L, Tarantini G, Marra MP, Ramondo A, Thiene G, Iliceto S.. Morphologic validation of reperfused hemorrhagic myocardial infarction by cardiovascular magnetic resonance. Am J Cardiol 2007;100:1322–1327. [DOI] [PubMed] [Google Scholar]
- 71. Kumar A, Green JD, Sykes JM, Ephrat P, Carson JJL, Mitchell AJ, Wisenberg G, Friedrich MG.. Detection and quantification of myocardial reperfusion hemorrhage using T2 (*)-weighted CMR. JACC Cardiovasc Imaging 2011;4:1274–1283. [DOI] [PubMed] [Google Scholar]
- 72. Baldea I, Teacoe I, Olteanu DE, Vaida-Voievod C, Clichici A, Sirbu A, Filip GA, Clichici S.. Effects of different hypoxia degrees on endothelial cell cultures—time course study. Mech Ageing Dev 2018;172:45–50. [DOI] [PubMed] [Google Scholar]
- 73. Stempien-Otero A, Karsan A, Cornejo CJ, Xiang H, Eunson T, Morrison RS, Kay M, Winn R, Harlan J.. Mechanisms of hypoxia-induced endothelial cell death. J Biol Chem 1999;274:8039–8045. [DOI] [PubMed] [Google Scholar]
- 74. Faller DV. Endothelial cell responses to hypoxic stress. Experimental biology 1998 symposium on molecular approaches to understanding cellular responses to stress. Clin Exp Pharmacol Physiol 1999;26:74–84. [DOI] [PubMed] [Google Scholar]
- 75. Tsao PS, Aoki N, Lefer DJ, Johnson G, Lefer AM.. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 1990;82:1402–1412. [DOI] [PubMed] [Google Scholar]
- 76. Viehman GE, Ma XL, Lefer DJ, Lefer AM.. Time course of endothelial dysfunction and myocardial injury during coronary arterial occlusion. Am J Physiol Circ Physiol 1991;261:H874–H881. [DOI] [PubMed] [Google Scholar]
- 77. VanBenthuysen KM, McMurtry IF, Horwitz LD.. Reperfusion after acute coronary occlusion in dogs impairs endothelium-dependent relaxation to acetylcholine and augments contractile reactivity in vitro. J Clin Invest 1987;79:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma X, Zhang H, Pan Q, Zhao Y, Chen J, Zhao B, Chen Y.. Hypoxia/aglycemia-induced endothelial barrier dysfunction and tight junction protein downregulation can be ameliorated by citicoline. PLoS One 2013;8:e82604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Funamoto K, Yoshino D, Matsubara K, Zervantonakis IK, Funamoto K, Nakayama M, Masamune J, Kimura Y, Kamm RD.. Endothelial monolayer permeability under controlled oxygen tension. Integr Biol 2017;9:529–538. [DOI] [PubMed] [Google Scholar]
- 80. Gavin JB, Seelye RN, Nevalainen TJ, Armiger LC.. The effect of ischaemia on the function and fine structure of the microvasculature of myocardium. Pathology 1978;10:103–111. [DOI] [PubMed] [Google Scholar]
- 81. Kloner RA, Rude RE, Carlson N, Maroko PR, DeBoer LW, Braunwald E.. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: which comes first? Circulation 1980;62:945–952. [DOI] [PubMed] [Google Scholar]
- 82. Fernández-Jiménez R, Galán-Arriola C, Sánchez-González J, Agüero J, López-Martín GJ, Gomez-Talavera S, Garcia-Prieto J, Benn A, Molina-Iracheta A, Barreiro-Pérez M, Martin-García A, García-Lunar I, Pizarro G, Sanz J, Sánchez PL, Fuster V, Ibanez B.. Effect of ischemia duration and protective interventions on the temporal dynamics of tissue composition after myocardial infarction. Circ Res 2017;121:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ghugre NR, Pop M, Barry J, Connelly KA, Wright GA.. Quantitative magnetic resonance imaging can distinguish remodeling mechanisms after acute myocardial infarction based on the severity of ischemic insult. Magn Reson Med 2013;70:1095–1105. [DOI] [PubMed] [Google Scholar]
- 84. Nevalainen T, Armiger LC, Gavin JB.. Effects of ischaemia on vasculature. J Mol Cell Cardiol 1986;18(Suppl. 4):7–10. [DOI] [PubMed] [Google Scholar]
- 85. Reffelmann T, Hale SL, Li G, Kloner RA.. Relationship between no reflow and infarct size as influenced by the duration of ischemia and reperfusion. Am J Physiol Circ Physiol 2002;282:H766–H772. [DOI] [PubMed] [Google Scholar]
- 86. Yu H, Kalogeris T, Korthuis RJ.. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radic Biol Med 2019;135:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rodrigo R, Libuy M, Feliú F, Hasson D.. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. Biomed Res Int 2013;2013:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qin C, Yap S, Woodman OL. Antioxidants in the prevention of myocardial ischemia/reperfusion injury. Expert Rev Clin Pharmacol 2009;2:673–695. [DOI] [PubMed] [Google Scholar]
- 89. Elmoselhi AB, Dhalla NS, Makino N, Hata T.. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc Res 2000;47:446–456. [DOI] [PubMed] [Google Scholar]
- 90. Heusch G. The coronary circulation as a target of cardioprotection. Circ Res 2016;118:1643–1658. [DOI] [PubMed] [Google Scholar]
- 91. Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I.. Myocardial edema: a translational view. J Mol Cell Cardiol 2012;52:931–939. [DOI] [PubMed] [Google Scholar]
- 92. Fernández-Jiménez R, García-Prieto J, Sánchez-González J, Agüero J, López-Martín GJ, Galán-Arriola C, Molina-Iracheta A, Doohan R, Fuster V, Ibáñez B.. Pathophysiology underlying the bimodal edema phenomenon after myocardial ischemia/reperfusion. J Am Coll Cardiol 2015;66:816–828. [DOI] [PubMed] [Google Scholar]
- 93. Fernández-Jiménez R, Sánchez-González J, Agüero J, García-Prieto J, López-Martín GJ, García-Ruiz JM, Molina-Iracheta A, Rosselló X, Fernández-Friera L, Pizarro G, García-Álvarez A, Dall'Armellina E, Macaya C, Choudhury RP, Fuster V, Ibáñez B.. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J Am Coll Cardiol 2015;65:315–323. [DOI] [PubMed] [Google Scholar]
- 94. Foltz WD, Yang Y, Graham JJ, Detsky JS, Wright GA, Dick AJ.. MRI relaxation fluctuations in acute reperfused hemorrhagic infarction. Magn Reson Med 2006;56:1311–1319. [DOI] [PubMed] [Google Scholar]
- 95. Alkhalil M, Borlotti A, Maria GL, De Gaughran L, Langrish J, Lucking A, Ferreira V, Kharbanda RK, Banning AP, Channon KM, Armellina ED, Choudhury RP.. Dynamic changes in injured myocardium, very early after acute myocardial infarction, quantified using T1 mapping cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2018;20:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fernández-Jiménez R, Barreiro-Pérez M, Martin-García A, Sánchez-González J, Agüero J, Galán-Arriola C, García-Prieto J, Díaz-Pelaez E, Vara P, Martinez I, Zamarro I, Garde B, Sanz J, Fuster V, Sánchez PL, Ibanez B.. Dynamic edematous response of the human heart to myocardial infarction. Circulation 2017;136:1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Manciet LH, Poole DC, McDonagh PF, Copeland JG, Mathieu-Costello O.. Microvascular compression during myocardial ischemia: mechanistic basis for no-reflow phenomenon. Am J Physiol Circ Physiol 1994;266:H1541–H1550. [DOI] [PubMed] [Google Scholar]
- 98. Kasseckert SA, Schäfer C, Kluger A, Gligorievski D, Tillmann J, Schlüter KD, Noll T, Sauer H, Piper HM, Abdallah Y.. Stimulation of cGMP signalling protects coronary endothelium against reperfusion-induced intercellular gap formation. Cardiovasc Res 2009;83:381–387. [DOI] [PubMed] [Google Scholar]
- 99. Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, Mcsharry H, Iwakura A, Yoon Y-S, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheres D.. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest 2004;113:885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Galaup A, Gomez E, Souktani R, Durand M, Cazes A, Monnot C, Teillon J, Jan S, Le Bouleti C, Briois G, Philippe J, Pons S, Martin V, Assaly R, Bonnin P, Ratajczak P, Janin A, Thurston G, Valenzuela DM, Murphy AJ, Yancopoulos GD, Tissier R, Berdeaux A, Ghaleh B, Germain S.. Protection against myocardial infarction and no-reflow through preservation of vascular integrity by angiopoietin-like 4. Circulation 2012;125:140–149. [DOI] [PubMed] [Google Scholar]
- 101. Li Y, Yao Y, Li J, Chen Q, Zhang L, Wang QK.. Losartan protects against myocardial ischemia and reperfusion injury via vascular integrity preservation. FASEB J 2019;33:8555–8564. [DOI] [PubMed] [Google Scholar]
- 102. Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R.. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 2000;101:1833–1839. [DOI] [PubMed] [Google Scholar]
- 103. Romanic AM, Harrison SM, Bao W, Burns-Kurtis CL, Pickering S, Gu J, Grau E, Mao J, Sathe GM, Ohlstein EH, Yue TL.. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardiovasc Res 2002;54:549–558. [DOI] [PubMed] [Google Scholar]
- 104. McNamara JJ, Lacro RV, Yee M, Smith GT.. Hemorrhagic infarction and coronary reperfusion. J Thorac Cardiovasc Surg 1981;81:498–501. [PubMed] [Google Scholar]
- 105. Lotan CS, Pohost GM, Bishop SP, Bouchard A, Cranney GB.. Assessment of postreperfusion myocardial hemorrhage using proton NMR imaging at 1.5 T. Circulation 1992;86:1018–1025. [DOI] [PubMed] [Google Scholar]
- 106. Kali A, Kumar A, Cokic I, Tang RLQ, Tsaftaris SA, Friedrich MG, Dharmakumar R.. Chronic manifestation of postreperfusion intramyocardial hemorrhage as regional iron deposition a cardiovascular magnetic resonance study with ex vivo validation. Circ Cardiovasc Imaging 2013;6:218–228. [DOI] [PubMed] [Google Scholar]
- 107. Carrick D, Haig C, Ahmed N, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay MM, Davie A, Mahrous A, Mordi I, Rauhalammi S, Sattar N, Welsh P, Radjenovic A, Ford I, Oldroyd KG, Berry C.. Myocardial hemorrhage after acute reperfused ST-segment-elevation myocardial infarction: relation to microvascular obstruction and prognostic significance. Circ Cardiovasc Imaging 2016;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bulluck H, Rosmini S, Abdel-Gadir A, White SK, Bhuva AN, Treibel TA, Fontana M, Ramlall M, Hamarneh A, Sirker A, Herrey AS, Manisty C, Yellon DM, Kellman P, Moon JC, Hausenloy DJ.. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging 2016;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kloner RA, Giacomelli F, Alker KJ, Hale SL, Matthews R, Bellows S.. Influx of neutrophils into the walls of large epicardial coronary arteries in response to ischemia/reperfusion. Circulation 1991;84:1758–1772. [DOI] [PubMed] [Google Scholar]
- 110. Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M.. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001;103:2181–2187. [DOI] [PubMed] [Google Scholar]
- 111. Sheridan FM, Cole PG, Ramage D.. Leukocyte adhesion to the coronary microvasculature during ischemia and reperfusion in an in vivo canine model. Circulation 1996;93:1784–1787. [DOI] [PubMed] [Google Scholar]
- 112. Ge L, Zhou X, Ji W-J, Lu R-Y, Zhang Y, Zhang Y-D, Ma Y-Q, Zhao J-H, Li Y-M.. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Circ Physiol 2015;308:H500–H509. [DOI] [PubMed] [Google Scholar]
- 113. Götz AK, Zahler S, Stumpf P, Welsch U, Becker BF.. Intracoronary formation and retention of micro aggregates of leukocytes and platelets contribute to postischemic myocardial dysfunction. Basic Res Cardiol 2005;100:413–421. [DOI] [PubMed] [Google Scholar]
- 114. Lee MJ, Park SD, Kwon SW, Woo SI, Lee MD, Shin SH, Kim DH, Kwan J, Park KS.. Relation between neutrophil-to-lymphocyte ratio and index of microcirculatory resistance in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol 2016;118:1323–1328. [DOI] [PubMed] [Google Scholar]
- 115. Reindl M, Reinstadler SJ, Feistritzer HJ, Klug G, Tiller C, Mair J, Mayr A, Jaschke W, Metzler B.. Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care 2017;6:640–649. [DOI] [PubMed] [Google Scholar]
- 116. Laan AM, Van Der Hirsch A, Robbers LF, Nijveldt R, Lommerse I, Delewi R, Vleuten PA, Van Der Biemond BJ, Zwaginga JJ, Giessen WJ, Van Der Zijlstra F, Rossum AC, Van Voermans C, Schoot CE, Van Der Shoot CE, Piek JJ.. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: monocytes and myocardial infarction. Am Heart J 2012;163:57–65.e2. [DOI] [PubMed] [Google Scholar]
- 117. Teren A, Kirsten H, Beutner F, Scholz M, Holdt LM, Teupser D, Gutberlet M, Thiery J, Schuler G, Eitel I.. Alteration of multiple leukocyte gene expression networks is linked with magnetic resonance markers of prognosis after acute ST-elevation myocardial infarction. Sci Rep 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shetelig C, Limalanathan S, Hoffmann P, Seljeflot I, Gran JM, Eritsland J, Andersen G.. Association of IL-8 with infarct size and clinical outcomes in patients with STEMI. J Am Coll Cardiol 2018;72:187–198. [DOI] [PubMed] [Google Scholar]
- 119. Guo F, Dong M, Ren F, Zhang C, Li J, Tao Z, Yang J, Li G.. Association between local interleukin-6 levels and slow flow/microvascular dysfunction. J Thromb Thrombolysis 2014;37:475–482. [DOI] [PubMed] [Google Scholar]
- 120. Zeng M, Yan H, Chen Y, Jun ZH, Lv Y, Liu C, Zhou P, Zhao B.. Suppression of NF-κB reduces myocardial no-reflow. PLoS One 2012;7:e47306.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yetgin T, Uitterdijk A, Lintel Hekkert M, Merkus D, Te Krabbendam-Peters I, Beusekom HMM, Falotico R, van Serruys PW, Manintveld OC, Geuns R-J, van Zijlstra F, Duncker DJ.. Limitation of infarct size and no-reflow by intracoronary adenosine depends critically on dose and duration. JACC Cardiovasc Interv 2015;8:1990–1999. [DOI] [PubMed] [Google Scholar]
- 122. Uitterdijk A, Yetgin T, Lintel Hekkert M, Te Sneep S, Krabbendam-Peters I, Beusekom HMM, van Fischer TM, Cornelussen RN, Manintveld OC, Merkus D, Duncker DJ.. Vagal nerve stimulation started just prior to reperfusion limits infarct size and no-reflow. Basic Res Cardiol 2015;110:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rymer JA, Newby LK.. Failure to launch: targeting inflammation in acute coronary syndromes. JACC Basic Transl Sci 2017;2:484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F.. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol 2002;40:1199–1204. [DOI] [PubMed] [Google Scholar]
- 125. Ziegler M, Wang X, Peter K.. Platelets in cardiac ischaemia/reperfusion injury : a promising therapeutic target. Cardiovasc Res 2019;115:1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dijk CGM, Van Nieuweboer FE, Pei JY, Xu YJ, Burgisser P, Mulligen E, Van Azzouzi H, El Duncker DJ, Verhaar MC, Cheng C.. The complex mural cell: pericyte function in health and disease. Int J Cardiol 2015;190:75–89. [DOI] [PubMed] [Google Scholar]
- 127. Avolio E, Madeddu P.. Discovering cardiac pericyte biology: from physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascul Pharmacol 2016;86:53–63. [DOI] [PubMed] [Google Scholar]
- 128. O’Farrell FM, Attwell D.. A role for pericytes in coronary no-reflow. Nat Rev Cardiol 2014;11:427–432. [DOI] [PubMed] [Google Scholar]
- 129. Costa MA, Paiva AE, Andreotti JP, Cardoso MV, Cardoso CD, Mintz A, Birbrair A.. Pericytes constrict blood vessels after myocardial ischemia. J Mol Cell Cardiol 2018;116:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Garcia JH, Liu KF, Yoshida Y, Chen S, Lian J.. Brain microvessels: factors altering their patency after the occlusion of a middle cerebral artery (Wistar rat). Am J Pathol 1994;145:728–740. [PMC free article] [PubMed] [Google Scholar]
- 131. Kloner RA, King KS, Harrington MG.. No-reflow phenomenon in the heart and brain. Am J Physiol Circ Physiol 2018;315:H550–H562. [DOI] [PubMed] [Google Scholar]
- 132. Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA.. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Physiol 2003;285:F191–F198. [DOI] [PubMed] [Google Scholar]
- 133. Flores J, DiBona DR, Beck CH, Leaf A.. The role of cell swelling in ischemic renal damage and the protective effect of hypertonic solute. J Clin Invest 1972;51:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Khanna A, Rossman JE, Fung HL, Caty MG.. Attenuated nitric oxide synthase activity and protein expression accompany intestinal ischemia/reperfusion injury in rats. Biochem Biophys Res Commun 2000;269:160–164. [DOI] [PubMed] [Google Scholar]
- 135. Boros M, Takaichi S, Hatanaka K.. Ischemic time dependent microvascular changes and reperfusion injury in the rat small intestine. J Surg Res 1995;59:311–320. [DOI] [PubMed] [Google Scholar]
- 136. Brunner H, Cockcroft JR, Deanfield J, Donald A, Ferrannini E, Halcox J, Kiowski W, Lüscher TF, Mancia G, Natali A, Oliver JJ, Pessina AC, Rizzoni D, Rossi GP, Salvetti A, Spieker LE, Taddei S, Webb DJ; WorkingGrouponEndothelinsandEndothelialFactorsof theEuropeanSocietyofHypertension. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J Hypertens 2005;23:233–246. [DOI] [PubMed] [Google Scholar]
- 137. Iwakura K, Ito H, Ikushima M, Kawano S, Okamura A, Asano K, Kuroda T, Tanaka K, Masuyama T, Hori M, Fujii K.. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J Am Coll Cardiol 2003;41:1–7. [DOI] [PubMed] [Google Scholar]
- 138. Eitel I, Hintze S, Waha S. D, Fuernau G, Lurz P, Desch S, Schuler G, Thiele H.. Prognostic impact of hyperglycemia in nondiabetic and diabetic patients with ST-elevation myocardial infarction: insights from contrast-enhanced magnetic resonance imaging. Circ Cardiovasc Imaging 2012;5:708–718. [DOI] [PubMed] [Google Scholar]
- 139. Zia MI, Ghugre NR, Roifman I, Strauss BH, Walcarius R, Mohammed M, Sparkes JD, Dick AJ, Wright GA, Connelly KA.. Comparison of the frequencies of myocardial edema determined by cardiac magnetic resonance in diabetic versus nondiabetic patients having percutaneous coronary intervention for st elevation myocardial infarction. Am J Cardiol 2014;113:607–612. [DOI] [PubMed] [Google Scholar]
- 140. Ota S, Tanimoto T, Orii M, Hirata K, Shiono Y, Shimamura K, Matsuo Y, Yamano T, Ino Y, Kitabata H, Yamaguchi T, Kubo T, Tanaka A, Imanishi T, Akasaka T.. Association between hyperglycemia at admission and microvascular obstruction in patients with ST-segment elevation myocardial infarction. J Cardiol 2015;65:272–277. [DOI] [PubMed] [Google Scholar]
- 141. Reinstadler SJ, Stiermaier T, Eitel C, Metzler B, S de W, Fuernau G, Desch S, Thiele H, Eitel I.. Relationship between diabetes and ischaemic injury among patients with revascularized ST-elevation myocardial infarction. Diabetes Obes Metab 2017;19:1706–1713. [DOI] [PubMed] [Google Scholar]
- 142. Jensen CJ, Eberle HC, Nassenstein K, Schlosser T, Farazandeh M, Naber CK, Sabin GV, Bruder O.. Impact of hyperglycemia at admission in patients with acute ST-segment elevation myocardial infarction as assessed by contrast-enhanced MRI. Clin Res Cardiol 2011;100:649–659. [DOI] [PubMed] [Google Scholar]
- 143. Reinstadler SJ, Stiermaier T, Eitel C, Saad M, Metzler B, Waha S, De Fuernau G, Desch S, Thiele H, Eitel I.. Antecedent hypertension and myocardial injury in patients with reperfused ST-elevation myocardial infarction. J Cardiovasc Magn Reson 2016;18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Carrick D, Haig C, Maznyczka AM, Carberry J, Mangion K, Ahmed N, May VTY, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Davie A, Mahrous A, Mordi I, Ford I, Radjenovic A, Welsh P, Sattar N, Wetherall K, Oldroyd KG, Berry C.. Hypertension, microvascular pathology, and prognosis after an acute myocardial infarction. Hypertension 2018;72:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Iwakura K, Ito H, Kawano S, Okamura A, Kurotobi T, Date M, Inoue K, Fujii K.. Chronic pre-treatment of statins is associated with the reduction of the no-reflow phenomenon in the patients with reperfused acute myocardial infarction. Eur Heart J 2006;27:534–539. [DOI] [PubMed] [Google Scholar]
- 146. Reindl M, Reinstadler SJ, Feistritzer HJ, Theurl M, Basic D, Eigler C, Holzknecht M, Mair J, Mayr A, Klug G, Metzler B.. Relation of low-density lipoprotein cholesterol with microvascular injury and clinical outcome in revascularized ST-elevation myocardial infarction. J Am Heart Assoc 2017;6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Symons R, Masci PG, Francone M, Claus P, Barison A, Carbone I, Agati L, Galea N, Janssens S, Bogaert J.. Impact of active smoking on myocardial infarction severity in reperfused ST-segment elevation myocardial infarction patients: the smoker’s paradox revisited. Eur Heart J 2016;37:2756–2764. [DOI] [PubMed] [Google Scholar]
- 148. Reinstadler SJ, Eitel C, Fuernau G, de Waha S, Desch S, Mende M, Metzler B, Schuler G, Thiele H, Eitel I.. Association of smoking with myocardial injury and clinical outcome in patients undergoing mechanical reperfusion for ST-elevation myocardial infarction. Eur Heart J Cardiovasc Imaging 2017;18:39–45. [DOI] [PubMed] [Google Scholar]
- 149. Haig C, Carrick D, Carberry J, Mangion K, Maznyczka A, Wetherall K, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Davie A, Mahrous A, Mordi I, Ahmed N, Teng Yue May V, Ford I, Radjenovic A, Welsh P, Sattar N, Oldroyd KG, Berry C.. Current smoking and prognosis after acute ST-segment elevation myocardial infarction: new pathophysiological insights. JACC Cardiovasc Imaging 2019;12:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rawshani A, Rawshani A, Franzén S, Eliasson B, Svensson A-M, Miftaraj M, McGuire DK, Sattar N, Rosengren A, Gudbjörnsdottir S.. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med 2017;376:1407–1418. [DOI] [PubMed] [Google Scholar]
- 151. Murthy VL, Naya M, Foster CR, Gaber M, Hainer J, Klein J, Dorbala S, Blankstein R, Di Carli MF.. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012;126:1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Carli MF, Di Janisse J, Grunberger G, Ager J.. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol 2003;41:1387–1393. [DOI] [PubMed] [Google Scholar]
- 153. Liu Z. Insulin at physiological concentrations increases microvascular perfusion in human myocardium. Am J Physiol Metab 2007;293:E1250–E1255. [DOI] [PubMed] [Google Scholar]
- 154. Williams SB, Goldfine AB, Timimi FK, Ting HH, Roddy M, Simonson DC, Creager MA.. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation 1998;97:1695–1701. [DOI] [PubMed] [Google Scholar]
- 155. Ammar RF, Gutterman DD, Brooks LA, Dellsperger KC.. Impaired dilation of coronary arterioles during increases in myocardial O2 consumption with hyperglycemia. Am J Physiol Metab 2017;279:E868–E874. [DOI] [PubMed] [Google Scholar]
- 156. Barrett EJ, Liu Z, Khamaisi M, King GL, Klein R, Klein BEK, Hughes TM, Craft S, Freedman BI, Bowden DW, Vinik AI, Casellini CM.. Diabetic microvascular disease: an endocrine society scientific statement. J Clin Endocrinol Metab 2017;102:4343–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, Yellon DM.. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol 2008;103:274–284. [DOI] [PubMed] [Google Scholar]
- 158. Paiva M, Riksen NP, Davidson SM, Hausenloy DJ, Monteiro P, Gonçalves L, Providência L, Rongen GA, Smits P, Mocanu MM, Yellon DM.. prevents myocardial reperfusion injury by activating the adenosine receptor. J Cardiovasc Pharmacol 2009;53:373–378. [DOI] [PubMed] [Google Scholar]
- 159. Paiva MA, Gonçalves LM, Providência LA, Davidson SM, Yellon DM, Mocanu MM.. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther 2010;24:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Calvert JW, Gundewar S, Jha S, Greer JJM, Bestermann WH, Tian R, Lefer DJ.. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008;57:696–705. [DOI] [PubMed] [Google Scholar]
- 161. Gundewar S, Calvert J, Jha S, Toedt-Pingel I, Ji S, Nunez D, Ramachandran A, Anaya-Cisneros M, Tian R, Lefer D.. Activation of AMPK by metformin improves left ventricular function and survival in heart failure. Circ Res 2009;104:403–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Techiryan G, Weil BR, Palka BA, Canty JM.. Effect of intracoronary metformin on myocardial infarct size in Swine. Circ Res 2018;123:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Díaz R, Goyal A, Mehta S, Afzal R, Xavier D, Pais P, Chrolavicius S, Zhu J, Kazmi K, Liu L, Budaj A, Zubaid M, Avezum A, Ruda M, Yusuf S.. Glucose-insulin-potassium therapy myocardial infarction. JAMA 2007;298:2399–2405. [DOI] [PubMed] [Google Scholar]
- 164. Sonne DP, Engstrøm T, Treiman M.. Protective effects of GLP-1 analogues exendin-4 and GLP-1(9-36) amide against ischemia-reperfusion injury in rat heart. Regul Pept 2008;146:243–249. [DOI] [PubMed] [Google Scholar]
- 165. Timmers L, Henriques JPS, Kleijn DPV, de DeVries JH, Kemperman H, Steendijk P, Verlaan CWJ, Kerver M, Piek JJ, Doevendans PA, Pasterkamp G, Hoefer IE.. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol 2009;53:501–510. [DOI] [PubMed] [Google Scholar]
- 166. Chang G, Zhang D, Yu H, Zhang P, Wang Y, Zheng A, Qin S.. Cardioprotective effects of exenatide against oxidative stress-induced injury. Int J Mol Med 2013;32:1011–1020. [DOI] [PubMed] [Google Scholar]
- 167. Scholte M, Timmers L, Bernink FJ, Denham RN, Beek AM, Kamp O, Diamant M, Horrevoets AJ, Niessen HW, Chen WJ, van Rossum AC, van Royen N, Doevendans PA, Appelman Y.. Effect of additional treatment with EXenatide in patients with an Acute Myocardial Infarction (EXAMI): study protocol for a randomized controlled trial. Trials 2011;12:240.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bernink FJP, Timmers L, Diamant M, Scholte M, Beek AM, Kamp O, Marques KMJ, Denham RN, Chen WJY, Doevendans PA, Rossum AC, Van Royen N, Van Horrevoets AJG, Appelman Y.. Effect of additional treatment with EXenatide in patients with an Acute Myocardial Infarction: the EXAMI study. Int J Cardiol 2013;167:289–290. [DOI] [PubMed] [Google Scholar]
- 169. Roos ST, Timmers L, Biesbroek PS, Nijveldt R, Kamp O, Rossum AC, van Hout GPJ, van Stella PR, Doevendans PA, Knaapen P, Velthuis BK, Royen N, van Voskuil M, Nap A, Appelman Y.. No benefit of additional treatment with exenatide in patients with an acute myocardial infarction. Int J Cardiol 2016;220:809–814. [DOI] [PubMed] [Google Scholar]
- 170. Lewington S, Clarke R, Qizilbash N, Peto R, Collins R.. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002;360:1903.. [DOI] [PubMed] [Google Scholar]
- 171. Panza JA, Quyyumi AA, Brush JE, Epstein SE.. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med 1990;323:22–27. [DOI] [PubMed] [Google Scholar]
- 172. Brush JE, Faxon DP, Salmon S, Jacobs AK, Ryan TJ.. Abnormal endothelium-dependent coronary vasomotion in hypertensive patients. J Am Coll Cardiol 1992;19:809–815. [DOI] [PubMed] [Google Scholar]
- 173. Panza JA, Casino PR, Kilcoyne CM, Quyyumi AA.. Role of endothelium-derived nitric oxide in the abnormal endothelium- dependent vascular relaxation of patients with essential hypertension. Circulation 1993;87:1468–1474. [DOI] [PubMed] [Google Scholar]
- 174. Quyyumi AA, Mulcahy D, Andrews NP, Husain S, Panza JA, Cannon RO.. Coronary vascular nitric oxide activity in hypertension and hypercholesterolemia. Comparison of acetylcholine and substance P. Circulation 1997;95:104–110. [DOI] [PubMed] [Google Scholar]
- 175. Golino P, Maroko PR, Carew TE.. The effect of acute hypercholesterolemia on myocardial infarct size and the no-reflow phenomenon during coronary occlusion-reperfusion. Circulation 1987;75:292–298. [DOI] [PubMed] [Google Scholar]
- 176. Ko YG, Won H, Shin DH, Kim JS, Kim BK, Choi D, Hong MK, Bae JH, Lee S, Lim DS, Jang Y.. Efficacy of early intensive rosuvastatin therapy in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention (ROSEMARY Study). Am J Cardiol 2014;114:29–35. [DOI] [PubMed] [Google Scholar]
- 177. Post S, Post MC, Branden BJ, Van Den Eefting FD, Goumans MJ, Stella PR, Es HW, Van Wildbergh TX, Rensing BJ, Doevendans PA.. Early statin treatment prior to primary PCI for acute myocardial infarction: REPERATOR, a randomized placebo-controlled pilot trial. Catheter Cardiovasc Interv 2012;80:756–765. [DOI] [PubMed] [Google Scholar]
- 178. Kaufmann PA, Gnecchi-Ruscone T, di Terlizzi M, SchäFers KP, LüScher TF, Camici PG, Coronary heart disease in smokers: vitamin C restores coronary microcirculatory function. Circulation 2000;102:1233–1238. [DOI] [PubMed] [Google Scholar]
- 179. Zeiher AM, SchäChinger V, Minners J.. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation 1995;92:1094–1100. [DOI] [PubMed] [Google Scholar]
- 180. Newby DE, Wright RA, Labinjoh C, Ludlam CA, Fox KAA, Boon NA, Webb DJ.. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: a mechanism for arterial thrombosis and myocardial infarction. Circulation 1999;99:1411–1415. [DOI] [PubMed] [Google Scholar]
- 181. Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA, Halvorsen S, Hindricks G, Kastrati A, Lenzen MJ, Prescott E, Roffi M, Valgimigli M, Varenhorst C, Vranckx P, Widimský P, Collet J-P, Kristensen SD, Aboyans V, Baumbach A, Bugiardini R, Coman IM, Delgado V, Fitzsimons D, Gaemperli O, Gershlick AH, Gielen S, Harjola V-P, Katus HA, Knuuti J, Kolh P, Leclercq C, Lip GYH, Morais J, Neskovic AN, Neumann F-J, Niessner A, Piepoli MF, Richter DJ, Shlyakhto E, Simpson IA, Steg PG, Terkelsen CJ, Thygesen K, Windecker S, Zamorano JL, Zeymer U, Windecker S, Aboyans V, Agewall S, Barbato E, Bueno H, Coca A, Collet J-P, Coman IM, Dean V, Delgado V, Fitzsimons D, Gaemperli O, Hindricks G, Iung B, Jüni P, Katus HA, Knuuti J, Lancellotti P, Leclercq C, McDonagh T, Piepoli MF, Ponikowski P, Richter DJ, Roffi M, Shlyakhto E, Simpson IA, Zamorano JL, Chettibi M, Hayrapetyan HG, Metzler B, Ibrahimov F, Sujayeva V, Beauloye C, Dizdarevic-Hudic L, Karamfiloff K, Skoric B, Antoniades L, Tousek P, Terkelsen PJ, Shaheen SM, Marandi T, Niemelä M, Kedev S, Gilard M, Aladashvili A, Elsaesser A, Kanakakis IG, Merkely B, Gudnason T, Iakobishvili Z, Bolognese L, Berkinbayev S, Bajraktari G, Beishenkulov M, Zake I, Lamin HB, Gustiene O, Pereira B, Xuereb RG, Ztot S, Juliebø V, Legutko J, Timóteo AT, Tatu-Chiţoiu G, Yakovlev A, Bertelli L, Nedeljkovic M, Studenčan M, Bunc M, García de Castro AM, Petursson P, Jeger R, Mourali MS, Yildirir A, Parkhomenko A, Gale CP.. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2018;39:119–177.28886621 [Google Scholar]
- 182. Francone M, Bucciarelli-Ducci C, Carbone I, Canali E, Scardala R, Calabrese FA, Sardella G, Mancone M, Catalano C, Fedele F, Passariello R, Bogaert J, Agati L.. Impact of primary coronary angioplasty delay on myocardial salvage, infarct size, and microvascular damage in patients with st-segment elevation myocardial infarction. Insight from cardiovascular magnetic resonance. J Am Coll Cardiol 2009;54:2145–2153. [DOI] [PubMed] [Google Scholar]
- 183. Vijayalakshmi K, Whittaker VJ, Kunadian B, Graham J, Wright RA, Hall JA, Sutton A, De Belder MA.. Prospective, randomised, controlled trial to study the effect of intracoronary injection of verapamil and adenosine on coronary blood flow during percutaneous coronary intervention in patients with acute coronary syndromes. Heart 2006;92:1278–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Su Q, Nyi T, Li L.. Adenosine and verapamil for no-reflow during primary percutaneous coronary intervention in people with acute myocardial infarction. Cochrane Database Syst Rev 2015;5:CD009503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Nazir SA, McCann GP, Greenwood JP, Kunadian V, Khan JN, Mahmoud IZ, Blackman DJ, Been M, Abrams KR, Shipley L, Wilcox R, Adgey AAJ, Gershlick AH.. Strategies to attenuate micro-vascular obstruction during P-PCI: the randomized reperfusion facilitated by local adjunctive therapy in ST-elevation myocardial infarction trial. Eur Heart J 2016;37:1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kelbæk H, Høfsten DE, Køber L, Helqvist S, Kløvgaard L, Holmvang L, Jørgensen E, Pedersen F, Saunamäki K, Backer O, De Bang LE, Kofoed KF, Lønborg J, Ahtarovski K, Vejlstrup N, Bøtker HE, Terkelsen CJ, Christiansen EH, Ravkilde J, Tilsted HH, Villadsen AB, Aarøe J, Jensen SE, Raungaard B, Jensen LO, Clemmensen P, Grande P, Madsen JK, Torp-Pedersen C, Engstrøm T.. Deferred versus conventional stent implantation in patients with ST-segment elevation myocardial infarction (DANAMI 3-DEFER): an open-label, randomised controlled trial. Lancet 2016;387:2199–2206. [DOI] [PubMed] [Google Scholar]
- 187. Carrick D, Oldroyd KG, McEntegart M, Haig C, Petrie MC, Eteiba H, Hood S, Owens C, Watkins S, Layland J, Lindsay M, Peat E, Rae A, Behan M, Sood A, Hillis WS, Mordi I, Mahrous A, Ahmed N, Wilson R, Lasalle L, Généreux P, Ford I, Berry C.. A randomized trial of deferred stenting versus immediate stenting to prevent No- or slow-reflow in acute ST-segment elevation myocardial infarction (DEFER-STEMI). J Am Coll Cardiol 2014;63:2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Belle L, Motreff P, Mangin L, Rangé G, Marcaggi X, Marie A, Ferrier N, Dubreuil O, Zemour G, Souteyrand G, Caussin C, Amabile N, Isaaz K, Dauphin R, Koning R, Robin C, Faurie B, Bonello L, Champin S, Delhaye C, Cuilleret F, Mewton N, Genty C, Viallon M, Bosson JL, Croisille P.. Comparison of immediate with delayed stenting using the minimalist immediate mechanical intervention approach in acute ST-segment-elevation myocardial infarction: the MIMI study. Circ Cardiovasc Interv 2016;9:e003388.. [DOI] [PubMed] [Google Scholar]
- 189. Kim JS, Lee HJ, Yu CW, Kim YM, Hong SJ, Park JH, Choi RK, Choi YJ, Park JS, Kim TH, Jang HJ, Joo HJ, Cho SA, Ro YM, Lim DS.. INNOVATION study (Impact of Immediate Stent Implantation Versus Deferred Stent Implantation on Infarct Size and Microvascular Perfusion in Patients With ST-Segment-Elevation Myocardial Infarction). Circ Cardiovasc Interv 2016;9:e004101.. [DOI] [PubMed] [Google Scholar]
- 190. Sadamatsu K, Yoshida K, Yoshidomi Y, Tokunou T, Tanaka H, Tashiro H, Shikada T, Iwamoto K, Morishige K.. Acute effects of isosorbide dinitrate and nicorandil on the coronary slow flow phenomenon. Am J Cardiovasc Drugs 2010;10:203–208. [DOI] [PubMed] [Google Scholar]
- 191. Roolvink V, Ibáñez B, Ottervanger JP, Pizarro G, van Royen N, Mateos A, Dambrink J-HE, Escalera N, Lipsic E, Albarran A, Fernández-Ortiz A, Fernández-Avilés F, Goicolea J, Botas J, Remkes W, Hernandez-Jaras V, Kedhi E, Zamorano JL, Navarro F, Alfonso F, García-Lledó A, Alonso J, van Leeuwen M, Nijveldt R, Postma S, Kolkman E, Gosselink M, de Smet B, Rasoul S, Piek JJ, Fuster V, van't Hof AWJ.. Early intravenous beta-blockers in patients with ST-segment elevation myocardial infarction before primary percutaneous coronary intervention. J Am Coll Cardiol 2016;67:2705–2715. [DOI] [PubMed] [Google Scholar]
- 192. Ibanez B, Macaya C, Sánchez-Brunete V, Pizarro G, Fernández-Friera L, Mateos A, Fernández-Ortiz A, García-Ruiz JM, García-Álvarez A, Iñiguez A, Jiménez-Borreguero J, López-Romero P, Fernández-Jiménez R, Goicolea J, Ruiz-Mateos B, Bastante T, Arias M, Iglesias-Vázquez JA, Rodriguez MD, Escalera N, Acebal C, Cabrera JA, Valenciano J, Pérez de Prado A, Fernández-Campos MJ, Casado I, García-Rubira JC, García-Prieto J, Sanz-Rosa D, Cuellas C, Hernández-Antolín R, Albarrán A, Fernández-Vázquez F, de la Torre-Hernández JM, Pocock S, Sanz G, Fuster V.. Effect of early metoprolol on infarct size in ST-segment–elevation myocardial infarction patients undergoing primary percutaneous coronary intervention. Circulation 2013;128:1495–1503. [DOI] [PubMed] [Google Scholar]
- 193. Pizarro G, Fernández-Friera L, Fuster V, Fernández-Jiménez R, García-Ruiz JM, García-Álvarez A, Mateos A, Barreiro MV, Escalera N, Rodriguez MD, de Miguel A, García-Lunar I, Parra-Fuertes JJ, Sánchez-González J, Pardillos L, Nieto B, Jiménez A, Abejón R, Bastante T, Martínez de Vega V, Cabrera JA, López-Melgar B, Guzman G, García-Prieto J, Mirelis JG, Zamorano JL, Albarrán A, Goicolea J, Escaned J, Pocock S, Iñiguez A, Fernández-Ortiz A, Sánchez-Brunete V, Macaya C, Ibanez B.. Long-term benefit of early pre-reperfusion metoprolol administration in patients with acute myocardial infarction: results from the Metocard-CNIC trial (Effect of Metoprolol in Cardioprotection during an Acute Myocardial Infarction). J Am Coll Cardiol 2014;63:2356–2362. [DOI] [PubMed] [Google Scholar]
- 194. García-Prieto J, Villena-Gutiérrez R, Gómez M, Bernardo E, Pun-García A, García-Lunar I, Crainiciuc G, Fernández-Jiménez R, Sreeramkumar V, Bourio-Martínez R, García-Ruiz JM, Valle AS, Del Sanz-Rosa D, Pizarro G, Fernández-Ortiz A, Hidalgo A, Fuster V, Ibanez B.. Neutrophil stunning by metoprolol reduces infarct size. Nat Commun 2017;8:14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hausenloy DJ, Barrabes JA, Bøtker HE, Davidson SM, Di LF, Downey J, Engstrom T, Ferdinandy P, Carbrera-Fuentes HA, Heusch G, Ibanez B, Iliodromitis EK, Inserte J, Jennings R, Kalia N, Kharbanda R, Lecour S, Marber M, Miura T, Ovize M, Perez-Pinzon MA, Piper HM, Przyklenk K, Schmidt MR, Redington A, Ruiz-Meana M, Vilahur G, Vinten-Johansen J, Yellon DM, Garcia-Dorado D.. Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol 2016;111:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D.. Strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol 2019;73:89–99. [DOI] [PubMed] [Google Scholar]
- 197. Cung T-T, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit J-F, Jouve B, Motreff P, Tron C, Labeque J-N, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, De Poli F, Morice M-C, Ider O, Dubois-Randé J-L, Unterseeh T, Le Breton H, Béard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M.. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 2015;373:1021–1031. [DOI] [PubMed] [Google Scholar]
- 198. Atar D, Arheden H, Berdeaux A, Bonnet J-L, Carlsson M, Clemmensen P, Cuvier V, Danchin N, Dubois-Rande J-L, Engblom H, Erlinge D, Firat H, Halvorsen S, Hansen HS, Hauke W, Heiberg E, Koul S, Larsen A-I, Le Corvoisier P, Nordrehaug JE, Paganelli F, Pruss RM, Rousseau H, Schaller S, Sonou G, Tuseth V, Veys J, Vicaut E, Jensen SE.. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur Heart J 2015;36:112–119. [DOI] [PubMed] [Google Scholar]
- 199. Engstrøm T, Kelbæk H, Helqvist S, Høfsten DE, Kløvgaard L, Clemmensen P, Holmvang L, Jørgensen E, Pedersen F, Saunamaki K, Ravkilde J, Tilsted H-H, Villadsen A, Aarøe J, Jensen SE, Raungaard B, Bøtker HE, Terkelsen CJ, Maeng M, Kaltoft A, Krusell LR, Jensen LO, Veien KT, Kofoed KF, Torp-Pedersen C, Kyhl K, Nepper-Christensen L, Treiman M, Vejlstrup N, Ahtarovski K, Lønborg J, Køber L.. Effect of ischemic postconditioning during primary percutaneous coronary intervention for patients with ST-segment elevation myocardial infarction: a randomized clinical trial. JAMA Cardiol 2017;2:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Sluijter JPG, Davidson SM, Boulanger CM, Buzás EI, de Kleijn DPV, Engel FB, Giricz Z, Hausenloy DJ, Kishore R, Lecour S, Leor J, Madonna R, Perrino C, Prunier F, Sahoo S, Schiffelers RM, Schulz R, Van Laake LW, Ytrehus K, Ferdinandy P.. Extracellular vesicles in diagnostics and therapy of the ischaemic heart : position paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res 2018;114:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Loyer X, Vion A, Tedgui A, Boulanger CM, Loyer X, Vion A, Tedgui A, Boulanger CM.. Microvesicles as cell–cell messengers in cardiovascular diseases. Circ Res 2014;114:345–353. [DOI] [PubMed] [Google Scholar]
- 202. Lawson C, Vicencio JM, Yellon DM, Davidson SM.. Microvesicles and exosomes : new players in metabolic and cardiovascular disease. J Endocrinol 2016;228:57–71. [DOI] [PubMed] [Google Scholar]
- 203. Boulanger CM, Scoazec A, Ebrahimian T, Henry P, Mathieu E, Tedgui A, Mallat Z.. Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation 2001;102:2649–2652. [DOI] [PubMed] [Google Scholar]
- 204. Porto I, Biasucci LM, Maria GL, De Leone AM, Niccoli G, Burzotta F, Trani C, Tritarelli A, Vergallo R, Liuzzo G, Crea F.. Intracoronary microparticles and microvascular obstruction in patients with ST elevation myocardial infarction undergoing primary percutaneous intervention. Eur Heart J 2012;33:2928–2938. [DOI] [PubMed] [Google Scholar]
- 205. Giricz Z, Varga ZV, Baranyai T, Sipos P, Pálóczi K, Kittel Á, Buzás E, Ferdinandy P.. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 2014;68:75–78. [DOI] [PubMed] [Google Scholar]
- 206. Ma F, Liu H, Shen Y, Zhang Y, Pan S.. Platelet-derived microvesicles are involved in cardio-protective effects of remote preconditioning. Int J Clin Exp Pathol 2015;8:10832–10839. [PMC free article] [PubMed] [Google Scholar]
- 207. Liu M, Wang Y, Zhu Q, Zhao J, Wang Y, Shang M, Liu M, Wu Y, Song J, Liu Y.. Protective effects of circulating microvesicles derived from ischemic preconditioning on myocardial ischemia/reperfusion injury in rats by inhibiting endoplasmic reticulum stress. Apoptosis 2018;23:436–448. [DOI] [PubMed] [Google Scholar]
- 208. Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G, Bulluck H, Hausenloy DJ, Berry C, Stiermaier T, Camici PG, Eitel I.. Optimized treatment of ST-elevation myocardial infarction. Circ Res 2019;125:245–258. [DOI] [PubMed] [Google Scholar]