

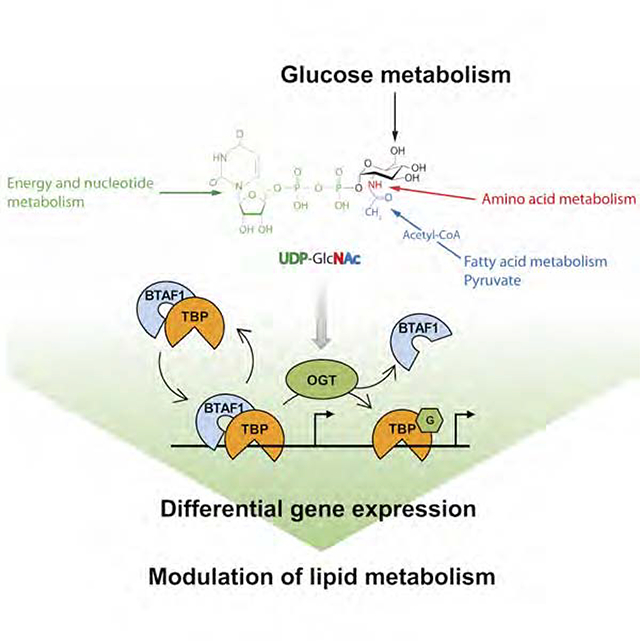
Summary
In eukaryotes, gene expression is performed by three RNA polymerases that are targeted to promoters by molecular complexes. A unique common factor, the TATA-box binding protein (TBP), is thought to serve as a platform to assemble pre-initiation complexes competent for transcription. Herein, we describe a novel molecular mechanism of nutrient regulation of gene transcription by dynamic O-GlcNAcylation of TBP. We show that O-GlcNAcylation at T114 of TBP blocks its interaction with BTAF1, hence the formation of the B-TFIID complex, and its dynamic cycling on and off of DNA. Transcriptomic and metabolomic analyses of TBPT114A CRISPR/Cas9 edited cells showed that loss of O-GlcNAcylation at T114 increases TBP binding to BTAF1 and directly impacts expression of 408 genes. Lack of O-GlcNAcylation at T114 is associated with a striking reprogramming of cellular metabolism induced by a profound modification of the transcriptome, leading to gross alterations in lipid storage.
Keywords: TATA-Box binding protein, TBP, O-GlcNAc, O-GlcNAcylation, BTAF1, B-TFIID, lipid droplets, metabolism, transcription, nutrient sensing
eTOC blurb
Sensing nutrient availability is critical for cells to adapt their metabolism. Hardivillé et al., report a glucose-sensitive regulatory mechanism of basal transcription machinery by O-GlcNAcylation of TBP. T114-O-GlcNAc-TBP impairs B-TFIID complex formation which impacts in return expression of genes involved in lipid metabolism, resulting in gross alteration of lipid storage.
Graphical Abstract
INTRODUCTION
Gene transcription is a conserved cell mechanism performed by three RNA polymerases (RNAP). Each RNAP has a dedicated set of transcription factors that recognize specific core promoters to form the pre-initiation complex (PIC) competent for gene transcription initiation (Koster et al., 2015). Among the vast diversity of transcription factors, the TATA-Box Binding Protein (TBP) is the unique common factor. While TBP is the sole necessary and sufficient factor for PIC assembly and TFIID transcription activity in vitro (Vannini and Cramer, 2012), TBP-associated factors (TAFs) play a pivotal role in core promoter recognition and TBP activity regulation. TBP also interacts dynamically with B-TFIID TATA-Box Binding Protein Associated Factor 1 (BTAF1) and forms the B-TFIID complex (Mousson et al., 2008; Timmers et al., 1992). BTAF1 regulates TBP dynamics on chromatin, controls its correct promoter positioning (de Graaf et al., 2010) and regulates an equilibrium between transcriptionally inactive B-TFIID and active TFIID complex at promoter (Choukrallah et al., 2012). B-TFIID appears to have dual functions by redistributing TBP from TATA-with to TATA-less promoters, hence promoting gene expression driven by the latter (Zentner and Henikoff, 2013). Despite extensive work on how TBP and TAFs interact to initiate transcription, the regulation of their activity by post-translation modifications (PTM) remains largely unexplored.
O-GlcNAcylation is a dynamic PTM finely tuned by two enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). UDP-GlcNAc, the donor substrate of OGT, is synthetized by the hexosamine biosynthetic pathway (HBP) that is considered as a cellular nutrient sensor since alteration in major metabolic pathways (i.e. glucose (Glc), amino acid, fatty acid (FA) and nucleotide metabolism) are reflected by changes in the cellular concentration of UDP-GlcNAc and OGT/OGA activities (Bond and Hanover, 2015; Yang and Qian, 2017). O-GlcNAcylation controls many protein functions and its dysregulation is associated with various metabolic-related diseases (Banerjee et al., 2016; Yang and Qian, 2017). Although, O-GlcNAcylation of many gene-specific transcription factors and its relevance in the RNAPII transcription initiation and elongation have been documented (Bond and Hanover, 2015; Lewis et al., 2016; Resto et al., 2017), its role at the very early steps of pre-initiation complex (PIC) assembly are largely unknown.
Herein, we describe and provide mechanistic insight as to how O-GlcNAcylation of TBP affects interactions with BTAF1. Using CRISPR/Cas9, we generated a homozygous O-GlcNAc mutant at the key TBP-T114 residue, and examined its site-specific functions on gene expression and cell metabolism. These data show a direct link between nutrients, O-GlcNAcylation and gene transcription at the basal mechanism level and in contributing to cell homeostasis.
RESULTS
O-GlcNAcylation of TBP regulates its dynamic interaction with chromatin
We first investigated whether TBP is O-GlcNAcylated by OGT. In vitro O-GlcNAcylation showed that TBP is a very potent substrate for OGT (Figure 1A). Immunoprecipitation (IP) experiments showed that TBP is O-GlcNAcylated in HeLa cells (Figure 1B), confirming that TBP is a substrate of OGT in vivo. O-GlcNAc stoichiometry suggested that TBP O-GlcNAcylation is highly regulated in vivo. Although O-GlcNAcylation of TBP does not impact its intracellular trafficking (Figure S1A), the modification is restricted to chromatin-bound TBP (Figure 1C). OGA inhibition by Thiamet G (TMG) led to increased binding of TBP to chromatin within 2 h (Figure S1B). OGT also precipitated with chromatin. We then postulated that OGT could act on TBP when the latter associates with DNA to regulate TBP’s promoter dynamics. Thus, we performed a strip fluorescent recovery after photobleaching assay (Figures 1D and 1E). After photobleaching, fluorescence displayed a fast and incomplete recovery phase, reflecting the GFP-TBP pool that freely moves within the nucleus, while the second phase corresponds to the chromatin immobilized GFP-TBP pool that exchange between the bleached and non-bleached area at a slower rate. Inhibition of TBP O-GlcNAcylation by the OGT inhibitor Ac4SGlcNAc treatment decreased its chromatin immobilization, while TMG treatment increased it (Figure 1D and S1C). Since O-GlcNAcylation is a nutrient sensor that reacts to Glc level (Bond and Hanover, 2015; Vaidyanathan and Wells, 2014; Yang and Qian, 2017), we investigated chromatin dynamic interactions with TBP under systemic growth conditions in normal (LG, 5.5 mM) or high (HG, 25 mM) Glc. Chromatin-immobilized GFP-TBP was lower under LG compared to HG (Figure 1E and S1D), suggesting that glycemia directly influences TBP DNA binding.
Figure 1: Dynamic Interactions of TBP with chromatin are regulated by O-GlcNAcylation.

(A) Recombinant human TBP is a substrate of OGT in vitro. TBP (◁) was incubated with or without recombinant OGT (∗), [3H]UDP-GlcNAc and calf intestinal alkaline phosphatase (§).
(B) O-GlcNAcylation of TBP in HeLa Cells. Treatment with CpNagJ or incubation with 1 M GlcNAc were used as controls. O-GlcNAcylation of TBP was revealed by CTD110.6 immunoblotting of IPed TBP.
(C) O-GlcNAc moiety is detected only on DNA associated TBP (left panel). HeLa cells were fractionated into cytoplasmic and nuclear extract (CE and NE respectively). NE was subsequently fractionated into DNA-bound protein fraction (Chr) and soluble fraction (SF). Input, non-relevant IPed samples and TBP IPed samples were probed with CTD110.6 and TBP antibodies. Right panel represents loading control and immunoblots probed with the indicated antibodies.
(D) Effect of TMG and Ac4SGlcNAc on TBP dynamic interaction with chromatin. Data are mean, n≥9.
(E) Effect of Glc level on TBP dynamic interaction with chromatin. Data are mean, n≥9.
(F) Increased O-GlcNAcylation by HG or TMG enhances TBP promoter occupancy on GAPDH, H4 and U6 promoter. Data are mean ± SEM, n=3, *p<0.05.
(G) O-GlcNAc stoichiometry of TBP and TBPT114A. Cells were transfected with indicated constructs. Lysates were O-GlcNAc mass tagged and immunoblotted with M2 antibody (left panel). Grey and orange curves represent the densitometry analysis profile of the non-PEGylated and PEGylated TBP constructs, respectively (middle and right panel).
(H) Co-IP of BTAF1 with TBP wild-type and mutant constructs. Samples were immunoblotted with indicated antibodies (top panel). Protein level of TBP constructs in the input (middle panel). Densitometry analysis of the BTAF1:TBP and mutants co-IP (bottom panel). Data are mean ± SEM, n=3, *p<0.05.
(I) Proposed model of the T114 O-GlcNAc regulation of TBP behavior on chromatin through BTAF1 interaction.
See also Figure S1.
Effects of TMG or HG on TBP’s chromatin interactions was further confirmed by ChIP assays on selected class I (45s), class II TATA-with (GAPDH), class II TATA-less (H4) and class III (U6) promoters, with a larger sensitivity shown by RNAP-II and –III selected promoters (Figure 1F). Differences observed between HG and TMG are most likely due to the fact that HG increased the presence the O-GlcNAc moiety on TBP while TMG blocked its removal.
Since BTAF1 regulates TBP-chromatin interaction (de Graaf et al., 2010), we hypothesized that TBP O-GlcNAcylation might regulate its dynamic interaction with chromatin by altering B-TFIID complex formation. We first pulled down transcription PICs on selected promoters. Addition of OGT and UDP-GlcNAc to HeLa cell nuclear extracts led to exclusion of BTAF1 from the PIC (Figure S1E). Of note, OGT and a shorter isoform of OGA (Butkinaree et al., 2008; Kim et al., 2006) were highly and specifically enriched at promoters in our PIC assembly assay. Thus, both OGA and OGT are present at promoters to ensure high O-GlcNAcylation cycling rates on TBP. In vivo, TMG treatment led to decreased BTAF1 pull down with TBP, while Ac4SGlcNAc increased their interaction (Figure S1C, S1D and S1F). All together, these data suggest that a nutrient-sensitive O-GlcNAcylation of TBP regulates its dynamic promoter interaction by inhibiting its association with BTAF1.
To get more insight into the molecular mechanism, we mapped O-GlcNAc sites of TBP including one at the N-terminal domain (NTD) on residue T114 using LC-MS/MS (Figure S1G). Next, we constructed a single point mutant for the T114 O-GlcNAc site and we confirmed that this construct did not alter TBP stability nor its binding to DNA (Figure S1H). We then investigated its O-GlcNAcylation stoichiometry via a mass-tag method using a DBCO-PEG of 4.4 KDa for O-GlcNAc labeling. We detected two forms of TBP: one without O-GlcNAc and one with an apparent molecular weight corresponding to a PTM with two O-GlcNAc moieties (Figure 1G). Densitometry analyses revealed that about 8% of TBP is O-GlcNAcylated. Mutation of the T114 residue to an alanine led to a near complete abolition of TBP O-GlcNAcylation. Although the estimated molecular weight showed that a second O-GlcNAc site exists, our data indicate that T114 is the main O-GlcNAc site on TBP. Next, we investigated the ability of the TBPT114A construct to form the B-TFIID complex. As expected, the TBPR188E construct, known to impair TBP:BTAF1 interaction (Klejman et al., 2005), co-IPed with a very limited amount of endogenous BTAF1 (Figure 1H). In contrast, TBPT114A co-IPed with almost 3-fold higher amount of BTAF1 compared to TBPWT. All together, these data show that the Glc-sensitive O-GlcNAcylation of TBP at the T114 residue inversely correlates with TBP:BTAF1 interaction (Figure 1I).
Mutation of the T114 O-GlcNAc site of TBP induces a major modification of the transcriptome and a profound metabolic reprogramming leading to an accumulation of lipid droplets
Since reduced TBP:BTAF1 interaction affects PIC formation and RNAPII recruitment at selected promoters in vertebrates (Choukrallah et al., 2012), we hypothesized that impaired TBPT114 O-GlcNAcylation and subsequent increased B-TFIID complex formation impacts the transcriptome. Thus, we engineered endogenous TBP gene using CRISPR/Cas9 (Figure S2A). First, we validated that TBP mutation had no effect on its expression level nor its cellular localization (Figure S2B–E). We confirmed that TBP3X-T114A exhibited a higher interaction with BTAF1, an impaired O-GlcNAcylation, and was not responsive to TMG treatment compared to TBP3X-WT (Figure S2F). Of note, TBP3X-T114A co-IPed with similar amounts of negative cofactor 2 alpha (NC2α) compared to TBP3X-WT. Since TBP has been shown to be phosphorylated (Morachis et al., 2011; Segil et al., 1996), we confirmed that the mutation did not alter its phosphorylation. We also confirmed that the increased O-GlcNAc-dependent promoter occupancy of TBP is T114 dependent since the mutant is no longer sensitive to TMG treatment (Figure S2G).
Principal component analysis of the RNA deep sequencing data (Figure S2H) showed that 49.3% of the variation clustered the TBP3X-T114A mutant apart from the two controls (PC1 axis). Only about 14% of the variation (PC2 axis) served to separate the TBPWT from TBP3X-WT cell line, indicating that most of the observed differences within the samples are rather due to the T114 mutation rather than by the presence of the TAG. Differentially expressed genes were consistent when transcription profiles of the TBP3X-T114A were compared to the TBPWT or TBP3X-WT (r=0.94; Figure S2I) and variations were mainly present in TBP3X-T114A rather than in TBP3X-WT (Figure S2J), further confirming that the insertion of the 3X-FLAG has minimal effects. Four hundred and eight transcripts in TBP3X-T114A passed the cut off to be differentially expressed, with a fold change < −2 or > 2 standard deviations (SD) and with a p-value < 0.05 (Figure 2A and Table S1). Gene ontology (GO) analysis showed that 186 dysregulated genes in TBP3X-T114A cells are related to metabolism (Figure S2K). Top hit of network analysis identified 53 genes associated with energy production, lipid metabolism and molecular transport and third hit identified 44 genes related to lipid metabolism, small molecule biochemistry and cell signaling (Figure 2B). Ingenuity pathway analysis (IPA) predicted that the TBP3X-T114A cells have reduced lipolysis, reduced FA metabolism and accumulation of FAs (Figure 2C).
Figure 2: Mutation of the TBP T114 O-GlcNAc site leads to profound metabolic genes reprogramming and triggers accumulation of lipid droplets.

(A) Volcano plot of mRNA transcript differentially express between TBP 3X-T114A and TBP 3X-WT cells. Blue and Green dots represent genes with fold change < −2 or > 2 SD and with a p-value < 0.05. Green dots are the top 5 dysregulated genes from Figure 2C and selected targets showed in Figure 2G and 2H.
(B) Network enrichment in TBP 3X-T114A cells.
(C) IPA representation showing prediction function pathway of lipolysis, FA concentration and metabolism. Pathways identified based on 22 molecules with a p = 7.99×10−5, 9 molecules with a p = 0.001 and 39 molecules with a p = 3.07 ×10−5 respectively.
(D) HeLa cells LD content under HG.
(E) Quantitative analysis of the LDs size (left panel) and number per cell (right panel) of the HeLa TBP WT cell line and its edited counterparts. LDs size data are median with interquartile range (n=3, total of 83 LDs were measured). Number of LDs per cell are mean ± SEM (n=3, total of 30 cells). *p<0.05 compared to both control cell lines.
(F) FA synthesis rate in TBP 3X-WT and TBP 3X-T114A cells. Data are mean ± SD, n=6, *p<0.05.
(G) Protein levels of key mRNAs that were differentially expressed in TBP 3X-T114A compared to TBP 3X-WT and TBP WT cell lines.
(H) Densitometry analysis of immunoblots showed in Figure 2G. Data are mean ± SD, n ≥ 3, *p < 0.05 compared to TBP WT, #p < 0.05 compared to TBP 3X-WT.
(I) DECR2, ACSF2, CPT2 and PLIN2 promoter occupancy of TBP 3X-WT and TBP 3X-T114A under LG treated or not with TMG. Data are mean ± SEM, n=3, *p<0.05, NS: not significant.
We then hypothesized that TBP3X-T114A cells should exhibit more lipid droplets (LDs) than TBP3X-WT cells. Staining and quantification of LDs (Figure 2D and 2E) showed that TBP3X-T114A cells contained more LD per cell (18.1 ± 8.1 vs. 4.3 ± 1.9 and 4 ± 2 for the TBPWT and the TBP3X-WT respectively) and of an increase in size (0.62 μm ± 0.15 vs. 0.48 μm ± 0.19 and 0.46 μm ± 0.25 for the TBPWT and the TBP3X-WT respectively). Increased LD content in TBP3X-T114A cells can be explained by ~3-fold increase in FA synthesis compare to TBP3X-wt cells (Figure 2F). We then performed comparative metabolite analysis by 1H-NMR. Levels of many metabolites were increased in the mutant compared to both control cell lines, indicating that lack of T114-O-GlcNAcylation led to profound metabolic reprogramming of HeLa cells (Figure S2L). Several metabolites involved in lipid metabolism were increased and IPA analysis showed that FA synthesis of FA, FA metabolism and concentration of FA would be increased while lipid peroxidation would be inhibited (Figure S2M). Levels of citrate and acetate, lipogenesis substrates, were increased ~3-fold in TBP3X-T114A cells. The levels of glutamine and glutamate were also increased in TBP3X-T114A cells, suggesting that glutaminolysis, which contributes to lipid synthesis via the IDH pathway in tumor cells, was slowed down. Expression of IDH1 was sharply decreased by 5-fold and total IDH activity was 50% reduced in mutant compared to TBP3X-WT (Table S1 and Figure S2O). Overall, 1H-NMR data suggest that the metabolism of HeLa TBP3X-T114A cells is significantly reprogrammed.
Key dysregulated genes related to lipid metabolism and LDs were validated at the protein level by immunoblotting (Figure 2G, 2H and Table S1). Expression of 2,4-Dienoyl-CoA reductase (DECR2) and the Acyl-CoA synthetase family member 2 (ACSF2), enzymes involved in FA beta-oxidation, were decreased in the mutant compared to control by 5.3 and 3.8-fold respectively, and their protein levels were reduced. Similarly, carnitine palmitoyltransferase 2 (CPT2), an enzyme required for activated acyl-CoA transfer to mitochondria, was reduced at both mRNA and protein levels. Interestingly, disruption of CPT2 triggers accumulation of LDs in cell lines, mouse heart and drosophila glial cell (Lin et al., 2018; Pereyra et al., 2017; Schulz et al., 2015). Expression levels of Perilipin 2 (PLIN2), a protein involved in lipid metabolism and a key component of LDs (Conte et al., 2016; Mishra et al., 2004), were increased in TBP3X-T114A by 5.7-fold compared to TBP3X-WT. At protein level, PLIN2 was also increased in the mutant compared to both control cell lines. We then tested the ability of the mutant to bind with the promoter of these genes (Figure 2I). We observed that TBP3X-WT and TBP3X-T114A bind DECR2, ACSF2 and CPT2 promoters at a comparable level. Increased O-GlcNAcylation induced by TMG led to increase promoter occupancy of TBP3X-WT, while TBP3X-T114A was no longer sensitive. Also, TBP3X-T114A showed an increased occupancy at PLIN2 promoter that is not further increase by TMG treatment unlike the TBP3X-WT, confirming that O-GlcNAcylation of TBP regulates its promoter interaction.
LD content is regulated by both Glc-sensitive TBPT114 O-GlcNAcylation and BTAF1 interaction
Since physiological (fasting, 5.5 mM blood Glc level) or low Glc availability leads to increased LD content in cell lines and tissues (Browning et al., 2012; Cabodevilla et al., 2013; Petan et al., 2018; Stannard et al., 2002), we investigated whether these effects were dependent on TBPT114 O-GlcNAcylation. As expected (Figure S1C, S1D and S1F), TMG or HG lowered TBP:BTAF1 interaction compared to LG. Also, Ac4SGlcNAc reversed the effect of HG. In contrast, TBP3X-T114A:BTAF1 interaction was not responsive to any treatments (Figure S3A and S3B). Furthermore, LG increased LD content in TBP3X-WT cells to the level observed in the mutant. While none of the treatments impacted size nor number per cell of LDs in the TBP3X-T114A cells, TMG treatment greatly reversed the effect of LG in TBP3X-WT (Figure S3C and S3D).
To prove that the Glc-sensitive O-GlcNAc-dependent TBP:BTAF1 interaction is involved in regulation of LD content, we downregulated BTAF1 using shRNA. Transcripts of BTAF1 (Table S2) as well as protein level (Figure 3B) were greatly reduced. Downregulation of BTAF1 led to a drastic decrease of LDs in TBP 3X-T114A cells (TBP 3X-T114A,shBTAF1) compared to shRNA control (TBP 3X-T114A,shC; Figure 3A). Quantitative analysis showed that LD content (number and size) were reduced to the level observed in TBP 3X-WT expressing shRNA control cells (TBP 3X-WT,shC; Figure 3C). Gene expression analysis showed that DECR2, ACSF2 and CPT2 expression levels were reversed upon down-regulation of BTAF1 (Figure 3D). IPA analysis of the transcriptome of TBP 3X-T114A,shC cells (Table S2) showed an activation of pathways leading to accumulation of LDs as well as to a decrease of LDs depletion. In contrast, transcriptome analysis of TBP 3X-T114A,shBTAF1 cells compared to shRNA control revealed an activation of pathways involved in LDs depletion and an inhibition of pathways involved in LDs accumulation (Figure 3E). All together, these data show that downregulation of BTAF1 reverts the accumulation of LDs observed upon mutation of the TBPT114 O-GlcNAc site, confirming that the Glc-sensitive O-GlcNAc-dependent B-TFIID complex formation regulates cellular lipid metabolism and storage as LDs.
Figure 3: Downregulation of BTAF1 reverts LDs accumulation in TBP3X-T114 cells.

(A) LDs staining of indicated cell lines under HG.
(B) BTAF1 protein level in TBP 3X-WT and TBP 3X-T114A cells expressing shRNA control (sh C) or shRNA BTAF1 (sh BTAF1).
(C) Quantitative analysis of the LD number per cell (left panel) and size (right panel) of Figure 3A. Number of LD per cell are mean ± SEM (n=3, total of 36 cells per condition) and LDs size data are median with interquartile range (n=3, total of 83 LDs were measured per condition), *p<0.05.
(D) Gene expression level of DECR2, ACSF2, CPT2 and PLIN2 in indicated cell lines. Data are expressed in Log2(FC) compered to TBP 3X-WT,shC cells and shown as mean ± SEM, n=3, *p<0.05, NS: not significant.
(E) IPA representation showing upregulation prediction function pathway of accumulation, quantity and depletion of LDs. Prediction made using transcriptomic data set of TBP 3X-T114A,shC vs. TBP 3X-WT,shC (left panel) and TBP 3X-T114A,shBTAF1 vs. TBP 3X-T114A,shC (right panel).
Hyperglycemia inhibits TBP:BTAF1 interaction in different cell types and tissues
We then investigated the Glc-sensitive O-GlcNAc-dependent B-TFIID complex in non-cancerous cell lines. IP experiments showed that endogenous TBP was O-GlcNAcylated in MCF10A and HEK-293 cells and that this modification was responsive to increased Glc levels (Figure 4A and 4E). Potency of the experimental condition was confirmed by global protein O-GlcNAcylation (Figure 4B and 4F). Also, increased TBP:BTAF1 interaction in LG was reversed by TMG, while reduced B-TFIID complex formation in HG was partially restored by Ac4SGlcNAc in MCF10A (Figure 4C and 4D) and HEK-293 cells (Figure 4G and 4H).
Figure 4: O-GlcNAc dependent TBP interaction with BTAF1 is Glc sensitive in non-cancerous cells.

Data regarding MCF10A (A-D) and HEK-293 cells (E-F) are presented.
(A, E) O-GlcNAcylation of TBP under experimental conditions.
(B, F) Effect of experimental conditions on global O-GlcNAcylation in MCF10A and HEK-293 cells.
(C, G) In situ interaction of TBP:BTAF1 stained by proximity ligation assay (PLA) under LG treated or not with TMG, or under HG treated or not with Ac4SGlcNAc.
(D, H) Quantification of PLA signal. Number of PLA dots per nuclei are shown as median with interquartile range (n=3, total of 63 cells for MCF10A and 78 cells for HEK-293 per condition), *p<0.05.
See also Figure S4.
Since O-GlcNAcylation is very abundant in brain and that brain extracellular Glc level correlates with blood Glc level (Jacob et al., 2002), we next challenged our findings in brain of streptozotocin (STZ)-induced diabetic rats. IP and co-IP of TBP showed that TBP O-GlcNAcylation was increased while TBP:BTAF1 was decreased in hyperglycemic rat brain compared to control (Figure S4A). Data mining revealed that pathways leading to LDs accumulation would be inhibited, while pathways leading to LDs depletion would be activated upon hyperglycemia in rat hippocampus (Figure S4C). STZ-induced hyperglycemia led to reduced LD content in rat hippocampus (Figure S4D). Overall, our data suggest that the O-GlcNAc-dependent regulation of TBP:BTAF1 interaction is an ubiquitous mechanism that regulates lipid storage as LDs in response to nutritional environment changes.
DISCUSSION
Herein, we present evidence that increased extracellular Glc concentration is reflected on TBP through PTM modification of its NTD, resulting in modification of LDs metabolism. It was recently shown that knock-down of one element of the TFIID core complex results in enlarged LDs in fat bodies in Drosophila (Fan et al., 2017). Considering that the formation of B-TFIID and TFIID are mutually exclusive (Choukrallah et al., 2012), increased formation of the B-TFIID complex induced by the lack of O-GlcNAcylation of the T114 residue would lead to impaired proper assembly of TBP and TAFs to form the TFIID complex, suggesting that the association of TBP with either BTAF1 or TAFs would control lipid metabolism.
Interestingly, restricted Glc access (physiological, LG or nutrient deprivation) leads to increased LD content in various cell lines and tissues (Browning et al., 2012; Cabodevilla et al., 2013; Petan et al., 2018; Stannard et al., 2002). Independently of Glc level, TBP3X-T114A cells develop a phenotype similar to that seen in its wild-type counterpart under LG. Hence, our data suggest that the lack of LDs observed under increased Glc level is due to a higher TBPT114 O-GlcNAcylation resulting from an increased HBP flux. Therefore, TBP O-GlcNAcylation is a cellular mechanism to adjust cell metabolism to nutritional environment changes.
Increase LDs formation in brain has been hypothesized to protect brain from oxidative stress-induced neurodegeneration (Liu et al., 2015). Our data show that TBPT114 O-GlcNAcylation is correlated with an inhibition of the B-TFIID complex formation and lower LDs, perhaps reducing the protective role of LDs in brain. It is possible that dysregulation of proper O-GlcNAc-dependent formation of B-TFIID could be involved in the etiology of neurodegenerative metabolic related diseases.
Unlike its DNA-binding domain, conservation of the NTD of TBP is restricted to vertebrates (Koster et al., 2015). Although previous work has shown that deletion of the NTD results in growth delay in chicken cells (Um et al., 2001), it had had mild effects in mice. Massive embryo lethally observed at mid gestation in these transgenic mice could be rescued by rearing embryos in immunocompromised mothers, suggesting a defect at placental/maternal interaction (Hobbs et al., 2002). Although dispensable, in vitro studies have shown that the NTD of TBP would be involved in regulation of protein-protein interaction for PIC assembly and initiation of transcription (Bondareva and Schmidt, 2003; Lescure et al., 1994; Mittal and Hernandez, 1997; Zhao and Herr, 2002). NTD of TBP is annotated as intrinsically disordered region (IDPR) and its potential high flexibility has been hypothesized to mask interacting surfaces of the core domain (Davidson, 2003). IDPRs are suspected to be regulatory domains that would undergo a disorder-to-order transition upon PTM affecting protein function(s) (Darling and Uversky, 2018). Here, we highlight that the NTD is a regulatory domain that integrates signaling from the nutrient sensor O-GlcNAcylation to adjust cellular homeostasis. It was suggested that the concomitant phosphorylation of the NTD of TBP and TFIIB by DNA-PK may stimulate the transcription initiation of RNAPII-dependent genes (Chibazakura et al., 1997; Morachis et al., 2011). Conversely, phosphorylation of TBP may result in transcription silencing during mitosis (Segil et al., 1996). Since a close relationship exists between O-GlcNAcylation and phosphorylation (Hardiville and Hart, 2014; Mishra et al., 2011), a complex “bar code” may coexist to fine-tuned TBP activity.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gerald W. Hart (gerald.hart@uga.edu) and/or to the Core C4. Unique reagents generated in this study are available from the lead contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HeLa cells and stable HeLa cells expressing GFP-TBP (a kindly gift of Pr. Timmers, Department of Physiological Chemistry and Netherlands Proteomic center, University Center Utrecht, Utrecht, Netherlands) were grown in DMEM (Dulbecco’s Modified Eagle’s Medium) at 37°C under 5% CO2. The media was complemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate and either 5.5- or 25 mM glucose and replaced every 24 h. Lentiviral transduction was performed following manufacturer’s instruction. Briefly, 0.25*106 cells were plated in 6-well plate. The following day, media was replaced by complete DMEM 25 mM glucose, 5 μg/ml polybrene, and 20 μl of shRNA control or shRNA BTAF1 lentiviral particles (Santa Cruz Biotechnology) at 1*106 IFU were subsequently added. Stably shRNA expressing cells were selected 48 h latter with DMEM 2 μg/ml Puromycin. HEK-293 cells were grown in EMEM (Eagle’s Minimum Essential Medium) at 37°C under 5% CO2. The media was complemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate. For high glucose experiments, cells were grown in the same media complemented to 25 mM glucose. MCF10A cells were grown in MEGM (Mammary Epithelial Cell Growth Medium) at 37°C under 5% CO2; glucose was 6.47 ± 0.17 mM or complemented to 25 mM glucose for high glucose condition. For HEK-293 and MCF10A cells, media was replaced every 48 h.
Animal models
Male Sprague-Dawley rats (Harlan Laboratories) cared and procedures were approved by the Institutional Animal Care and Use Committee of The Johns Hopkins University. Rats were housed in groups of 3 using 12 h dark/light cycles, provided with water and fed ad libitum Hyperglycemia was induced by intraperitoneal injection of a single dose of streptozotosin (STZ; Sigma-Aldrich) as described in (Banerjee et al., 2015). Blood glucose levels were monitored weekly. Animals were sacrificed 21 days post injection. At sacrifice, STZ injected rats exhibited non-fasting blood glucose levels of 503±81 mg.dL−1 compared to 132±15 mg.dL−1 for control animals. After anesthesia, rats were either perfused and brains were fixed in 4% paraformaldehyde, equilibrated in 40% sucrose and embedded in Tissue-Tek O.C.T. compound before storage at −80°C for further cryosectioning to a thickness of 12μm, or brains were dissected and flash frozen in liquid nitrogen for western blot analysis.
METHOD DETAILS
SDS-PAGE and Western Blot
Protein samples were denatured by Laemmli buffer and resolved on NuPAGE Bris-tris Gel (Invitrogen) according the manufacturer’s directions. For western blot, gels were transferred to polyvinylidene difluoride (PVDF) membrane. Anti-PhosphoThreonine western blotting was conducted following strictly manufacturer’s instructions (Giagen). For CDT110.6 antibody, PVDF membranes were blocked for 1 h at room temperature with 5% BSA-TBST. For all the other antibodies, PVDF membranes were blocked for 1 h at room temperature with 5% milk-TBST. Following blocking, PVDF were washed 3 times for 5 min with TBST and incubated overnight at 4°C with primary antibody diluted in 5% BSA-TBST as follow: mouse anti-TBP, 1/5000; rabbit anti-TBP, 1/5000; anti-BTAF1, 1/5000; anti-NC2α, 1/2000; anti-GFP, 1/1000; anti-CPT2, 1/1000; Anti-DECR2, 1/1000; anti-ACSF2, 1/2000; anti-PLIN2, 1/1000; Anti-HSC70, 1/5000; anti-Lamin A/C, 1/5000; anti-GAPDH, 1/10.000; anti-3XFLAG M2, 1/5000; anti-NMNAT1, 1/5000; pan anti-PhosphoThreonine Q7, 1/500; anti-O-GlcNAc CTD110.6, 1/5000; RL2, 1/2000; anti-OGT, 1/5000; and anti-OGA, 1/5000. Then, membranes were washed 3 times 5 min with TBST and incubated for 1 h at room temperature with secondary HRP-antibody. Prior developing, membranes were extensively washed with TBST before developing.
Co-immunoprecipitation
Cells were lysed in CoIP buffer (Tris-HCl 10 mM pH 7.5, KCl 10 mM, EDTA 2 mM, Glycerol 10%, TX-100 0.1%) complemented freshly with 100 μM PUGNAc, 1mM PMSF and 1X of Protease Inhibitor Cocktail 1 (PICA). Two milligrams of lysate was immunoprecepipated by 2 μg of TBP antibody MAB3658 or OGT antibody (AL25, homemade) and 20 μL of 50% slurry magnetic beads (Millipore) overnight at 4°C. Then, beads were washed 5 times with CoIP buffer 100 mM KCl. Precipitated proteins were eluted by 20 μL of Laemmli buffer 2X and resolved by SDS page (NuPAGE 4–12% Bis-tris Gel, Novex).
In vitro O-GlcNAcylation
TBP in vitro O-GlcNAcylation were conducted by incubating 1 μg of recombinant HIS-tagged TBP (Sigma-Aldrich), recombinant OGT, 1U of CIP (Neb) with or without 20mM UDP-GlcNAc for 2 h at room temperature. Hot in vitro labeling was conducted as previously described (Sakabe et al., 2010) using 1 μCi of [H3] UDP-GlcNAc per reaction.
Glucose uptake
HeLa TBP WT, TBP 3X-WT and TBP 3X-T114A cells were seeded at 0.25*106 per well in 6-well plate. The following day, cells were washed twice with PBS and incubated for 30 min at 37°C under 5% CO2 in 1 ml of Brebs-Ring buffer (HEPES 20 mM pH 7.4, NaCl 135 mM, KCl 5 mM, MgSO4 1 mM, K2HPO4 5 mM, CaCl2 1 mM) complemented or not with 50 μM of cytochalasin B. Cells were then incubated with Brebs-Rinb buffer 2-deoxyglucose 1 mM complemented with 1μCi of [H3] 2-deoxy-D-glucose (PerkinElmer) for 15min. Cells were subsequently scraped in 250 μL of lysis buffer (HEPES 10 mM pH 7.9, SDS 1%) and samples were cleared by centrifugation. Sample supernatants were counted by liquid scintillation. Counts were background subtracted and normalized to total protein content.
Fatty acids synthesis assay
Cells were platted one day prior to radiolabeling into 6-well plate. Cells were labeled with 1.5 μCi 3H-acetate (New England Nuclear) in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin for 1 hr at 37°C (Bowman et al., 2016). Cells were washed with 1X PBS. Total lipids were extracted with 700 μL of 2:1 chloroform:methanol and 450 μL of 4 mM MgCl2. Cells were transferred to an Eppendorf tube, vortexed, and centrifuged at high speed for 1 min. The aqueous layer was discarded. The remaining organic phase was washed with 450 μl of CHCl3:MeOH (2:1) and 300 μL of MgCl2, vortexed, and centrifuged again. The aqueous layer was removed and equal volumes of the organic phase from each sample was counted by liquid scintillation. Counts were normalized to total protein.
RNA extraction, Genomic DNA extraction, PCR, RT-PCR, cloning and mutagenesis
Total RNA of 2*106 HeLa cells was extracted using RNeasy plus kit (Qiagen) following manufacturer’s instructions. One microgram of total RNA was reverse transcript using GoScript Reverse Transcription System (Promega) following manufacturer’s directions. Full length TBP was cloned from Hela cells cDNA using KOD hot start DNA polymerase (EMD Millipore) with primers TBP_3XFLAG_Fw and TBP_3XFLAG_Rv (Table S3). Amplification fragment was then subjected to recombination reaction with HindIII/XbaI digested p3XFLAG-cmv10 (Sigma-Aldrich) using In-Fusion HD Cloning Kit (Clontech) following manufacturer’s instructions. Clones were screened by HindIII/XbaI restriction mapping and positive clones were sequenced.
TBP mutants have been generated by mutagenesis using KOD hot start DNA polymerase, and adequate primers (Table S3). After 18 cycles of PCR, samples were digested for 2 h at 37°C by DpnI and 1 μl was used for transformation in XL10-Gold Ultracompetent cells (Agilent Technologies). Colonies were screened by sequencing. Positive mutated TBP inserts were subcloned in p3XFLAG-cmv10 to ensure the absence of unwanted mutations in the backbone.
HeLa cells genomic DNA was extracted by phenol/chloroform method using UltraPure Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) following manufacturer’s instructions (Invitrogen). Promoter sequences of 45s, GAPDH, H4 and U6 were PCR amplified by KOD hot start DNA polymerase using appropriate primer sets (Table S3). Amplicon of each target was cloned in pCR8/GW/TOPO TA (Invitrogen) and correct sequences were confirmed by sanger sequencing. Biotinylated fragments were obtained by PCR with corresponding biotinylated primers and pCR8/GW/promoter sequence as template.
For real time RT-PCR, total RNA was extracted and 1μg was reverse transcript as described above. Samples were subsequently analyzed by quantitative PCR on a thermocycler Mx3005p (Stratagene) using Sybr green/ROX master mix and appropriate primer sets (Table S3). Data were normalized to HPRT expression level and expressed as Log2(Fold change) relative to TBP 3X-WT, shC.
In vitro TBP construct production
TBP wild type and O-GlcNAc mutants were in vitro transcribed and translated using PURExpress In vitro Protein Synthesis kit (Neb) according manufacturer’s instructions. Briefly, TBP wild and O-GlcNAc mutant sequences were PCR amplified using KOD hot start polymerase, generic primers 5UTR_TBP3X and 3UTR_TBP (Table S3), and p3XFLAG-TBP wild type or mutant constructs as template. Amplicons were submitted to QIAquick PCR Purification kit (Qiagen) and 500 ng of each construct was used for in vitro protein production.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed using annealed 18 bp oligonucleotides containing a TATA box at central position as probe. Freshly annealed probe was diluted at 2 nM in buffer A (HEPES 20 mM pH 7.9, MgCl2 8 mM, DTT 1 mM). Fresh PURExpress reaction mix (2 μl) was diluted in 8 μl of buffer B (Tris HCl 20 mM pH 7.9, KCl 100 mM, DTT 1 mM, BSA 50 μg/ml, glycerol 20%). Ten microliters of Buffer A-probe was mixed with 10 μl of TBP containing buffer B and incubated for 30 min at 30°C. Reaction mix was subsequently resolved by native electrophoresis on 6% DNA Retardation Gel (Invitrogen) with 0.5X TBE running buffer at 100V for 45 min. Gel was then imaged on a Typhoon 9400 (Amersham Biosciences) using appropriate Alexa 488 wavelength.
Pre -initiation complex capture on immobilized template
Small-scale HeLa cells nuclear extract was conducted as describe in (Folco et al., 2012) in presence of 50 μM PUGNAC in all buffers. Promoter templates were PCR amplified as described above. Amplicons were run on agarose gel and gel purified using QIAquick Gel Extraction Kit (Qiagen). One microgram of PCR amplified template was diluted v/v with 2XBW buffer (HEPES 20 mM pH 7.9, KCl 600 mM, EDTA 1 mM, BSA 25 μg/ml) and rocked at 4°C for 3 h with 50 μl of 1XBW buffer Streptavidin M-280 Dynabeads (Invitrogen) pre-equilibrated in 1XBW buffer. Coupling efficiency was estimated and beads were extensively washed with 1XBW buffer. Beads were subsequently equilibrated in buffer D (HEPES 20 mM pH 7.9, KCl 100 mM, EDTA 0.2 mM, Glycerol 20%, freshly supplemented with DTT 0.5 mM, PMSF 1 mM, PUGNAC 50 μM). One microliter of 50 ng/μl template-bead was rotated end-over-end for 1 h at room temperature with 10 μl of HeLa cells nuclear extract diluted in 14 μl of buffer D supplemented with 6.5 μl of ddH2O, 2 μl of shared salmon sperm DNA (5 μg/ml), 0.5 μl of NP-40 1%, 3 μl of MgCl2 0.1 M and 1 μl of BSA (10 mg/ml). Beads were pelleted using magnetic rack and washed twice with supplemented buffer D. PIC were eluted by Leammli buffer 1X and resolved by SDS-PAGE.
Polyethyleneimine (PEI) precipitation
HeLa cells were lysed with 250 μl per 1*106 cells of PEI precipitation buffer (Tris-HCl 10 mM pH 7.5, NaCl 200 mM, EDTA 2 mM, Glycerol 10%, TX-100 0.1% freshly supplemented with PSMF 1 mM and PUGNAC 50 μM) for 20 min on ice and vortexed every 2 min. Lysates were then sonicated for 5 s and subsequently clarified by centrifugation at 5000 x g for 15 min at 4°C. PEI was added to the supernatant at 0.2% final concentration and incubated for 20 min at 4°C with shaking. PEI:nucleic acid:associated proteins were pulled down at 4°C, for 10 min at full speed. Pellets were washed twice with PEI precipitation buffer 0.2% PEI. Proteins were eluted with 1 M NaCl. Samples were either submitted to western blot either dialyzed with micro dialysis unit 3.5 MWCO against 1 L of dialysis buffer (HEPES 20 mM pH 7.9, KCl 100 mM, EDTA 0.2 mM, Glycerol 20%, freshly supplemented with PMSF 1 mM, PUGNAC 50 μM) at 4°C for 1 h and further used for immunoprecipitation.
Immunofluorescence and LDs staining
HeLa cells were seeded at 2*104 cells per well on poly-D-lysine coated 8-well Culture Slide (Falcon). The flowing day, cells were washed twice with PBS and fixed in PBS-4% paraformaldehyde for 20 min at room temperature. Crosslinking was stop by two washes with PBS and 5 min incubation at room temperature with PBS-0.1 M glycine following by another two washes with PBS. Cells were then permeabilized with PBS 0.5% Triton X-100 for 10 min at room temperature. Prior blocking with PBS-0.05% Tween 20–5% BSA for 1 h at room temperature, cells were washed two times for 5 min with PBS. Primary antibodies anti-TBP (mouse, 1/1000; rabbit 1/500), anti-3XFLAG M2 (1/1000) or anti-BTAF1 (mouse, 1/10) were incubated overnight at 4°C in blocking buffer. Following this step, cells were washed 3 times for 10 min with PBS 0.05% tween 20 and incubated for 1 h at room temperature with Alexa Fluor conjugated secondary antibodies (1/500) (Invitrogen). Slides were washed 3 times for 10 min with PBS-0.05% tween and coverslips were mounted using Duolink In Situ Mounting Medium with DAPI (Sigma-Aldrich). In situ TBP:BTAF1 interaction were detected using Duolink proximity ligation assay (PLA) following strictly manufacturer instructions at the exception that amplification step was conducted for 180 min. Cells were prepared and primary antibodies were used as described above. LDs were stained with Bodipy 493/503 (Thermo Fisher Scientific) on live cells following manufacturer’s instructions. Slides were subsequently fixed with paraformaldehyde and mounted as described before. Cryosections were incubated in PBS-1 mg/ml Bodipy 493/503 for 30 min at room temperature. Prior mounting and image the same day, slices were washed 3 times for 10 min in PBS. Images were captured on a Zeiss 510-Meta confocal microscope.
Fluorescence recovery after photobleaching (FRAP) assay
FRAP assay were performed on live cells using a Zeiss 510-Meta confocal microscope using X40 oil immersion objective lens. HeLa cells expressing GFP-tagged TBP were seeded at 3*105 in poly-D-Lysine coated glass bottom 35 mm dishes (MatTek) and treated or not with 1 μM of TMG for 2 h, or overnight with 5 μM of Ac4SGlcNAc. FRAP assay was conducted and data processed as described in (de Graaf et al., 2010) with minor modifications. Briefly, a strip (8*1 μm) spanning the nucleus was scanned every 21 ms for 15 s. Strip was photo-bleached at 100% laser power after 2.1s (100 scans) for 2 iterations. For each experiment, at least 10 nuclei were monitored per experimental condition. Raw data were normalized to fluorescence intensity before bleaching and fluorescence intensity just after bleaching as follow Inorm,t = It/Ipre-bleached.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay has been conducted as described in (Hardiville et al., 2014). Briefly, 5*106 paraformaldehyde crosslinked HeLa cells were lysed and immunoprecipitated with 5 mg of TBP antibody. After extensive washes, precipitated chromatin was de-crosslinked and purified using QIAquick PCR purification Kit (Qiagen). Samples were analyzed by quantitative PCR on a thermocycler Mx3005p (Stratagene) using Sybr green/ROX master mix and primer sets design for qPCR to amplified promoter of interest or a region 1700 pb upstream +1 at GAPDH promoter as unspecific control (Table S3).
LC-MS/MS analysis
In vitro O-GlcNAcylated TBP, as described above, was resolved by SDS-PAGE and transfer onto PVDF. Membrane was stained with red Ponceau and TBP band were cut out. PVDF piece was extensively washed with ddH2O and equilibrated in digestion buffer (Tris 50 mM pH 8, CaCl2 0.5 mM). Ten nanograms per milliliter of elastase was added to the reaction mix and incubated overnight at 37°C. Following this step, thermolysin was added to the reaction mix at 10 ng/ml and incubated for 5 h at 70°C. Peptides were eluted by several cycles of 5 min in water bath sonication with 20 μl 0.1%TFA.
Digested TBP was submitted to an LTQ-Orbitrap Velos (Thermo Fisher Scientific) attached to an EASY nanoHPLC system (Thermo Fisher Scientific) for tandem mass spectrometry analysis of peptides. Loaded peptides were trapped on a 2 cm trap column (YMC gel ODS-A S-10 μm), fractionated with a 75 μm x 15 cm column [Magic C18 AQ, 5 μm, 100 Å (Michrom Bioresources)], and then electrosprayed through a 15-μm emitter (New Objective). A reversed-phase solvent gradient, which consisted of solvent A (0.1% formic acid) with increasing levels of solvent B (0.1% formic acid, 90% acetonitrile) over a period of 60 min, was used for peptide separation. The LTQ Orbitrap Velos was operated with alternating CID/HCD mode, with 2.0 kV as spray voltage, full MS survey scan range of 350–1800 m/z, data dependent MS/MS analysis of top 8 precursors with minimum signal of 2,000, 30 s dynamic exclusion limit, and isolation width of 1.2. Precursor and the fragment ions were analyzed at 30,000 and 15000 resolutions, respectively.
Tandem mass spectra were analyzed with Mascot v.2.2.2 (MatrixScience) using the customized TBP database, elastase/thermolysin as enzyme, missed cleavage 2, precursor mass tolerance 15 ppm, and fragment mass tolerance 0.03 Da. Carboamidomethylation (Cys) was set as fixed modification and deamidation (Asn/Gln) was set as variable modifications. All MS/MS spectra identifying O-GlcNAc peptides and sites were manually annotated with reference to Protein Prospector (http://prospector.ucsf.edu/prospector/mshome.htm), according to previously described rules (Tabb et al., 2006). The O-GlcNAc diagnostic ion (+204.08) and the corresponding fragments (i.e., 186.07, 168.06, 138.05, and 126.05) were used to confirm the presence of O-GlcNAc on peptides.
O-GlcNAc Mass Tag
O-GlcNAc mass tag was conducted according (Darabedian et al., 2018) with the following modification. HeLa cells were lysed with HEPES 10 mM pH 7.9, NaCl 300 mM, SDS 1% and sonicated for 5 s. lysates were clarified by centrifugation at 4°C for 15 min at full speed. Two-hundred micrograms of total protein was then subjected to GalNAz labeling using O-GlcNAc Click It kit (Invitrogen) according manufacturer’s instructions. Samples were then reduced and alkylated. Fifty micrograms of labeled protein was then incubated either with 10 mM of homemade 4.4 kDa DBCO-PEG or with equivalent volume of DMSO for 1 h at room temperature. Prior to western blot, samples were further purified by chloroform/methanol precipitation. Amino-dPEG12-Tris(m-dPEG24)3 (Quanta Biodesign) was resuspended in methylene chloride under argon and incubated with 3 M excess of DBCO-NHS Ester (Click chemistry Tools) with gentle shaking overnight at room temperature. Reaction mixture was dried down and DBCO-dPEG12-Tris(m-dPEG24)3 product was resuspended in water and filtered. Solution was vacuum dried in speedvac and resuspended at 50 mM in DMSO.
Genome editing
Genomic sequences of TBP have been edited in HeLa cells using GeneArt CRISPR Nuclease Vector (Invitrogen) following manufacturer’s instructions. We disrupted an anti-TBP antibody (mTBP) epitope by inserting a 3X-FLAG at the NTD and we engineered the codon for T114 to code for an Ala, and the codon for S115 with a silent mutation to create a NheI restriction site to track wild type TBP and its edited counterpart. Annealed double-strained DNA gDNA_TBP_E2 or gDNA_TBP_T114 (Table S3) targeting TBP exon 2 and exon 3 respectively were cloned into GeneART CRISPR Nuclease OFP Vector. HeLa cells were seeded at 0.25*106 per well in 6-well plate and transfected the following day using Lipofectamin 3000 (Invitrogen) with 1 μg of GeneART CRISPR vector encoding gRNA targeting exon 1 of TBP and 1.5 μg of a single strain donor DNA (ssODN_E2_3XFLAG) as template for homology directed repair (HDR) and insertion of the 3XFLAG (Table S3). 48 h post transfection the cells were sorted at one cell per well in 96-well plate based on their OFP fluorescence. Clones were screened by PCR on crude genomic DNA using OneTaq Quick-load (NEB) following manufacturer’s instructions with forward primer P1 and reverse primer P2. Positive clones were re-checked by RT-PCR with the forward primer P1 and reverse P3 (Table S3).
Exon 3 of TBP was targeted to edit the codon of the T114 to an alanine, following the procedure described above, with the GeneART CRISPR vector encoding gRNA targeting exon 3 and ssODN_E3_T114A (Table S3) as template for HDR. Clones were screened by PCR on genomic DNA using forward primer P1 and reverse primer P3. Prior agarose gel electrophoresis, PCR samples were submitted to digestion with NheI for 1 h at 37°C. Positive homozygote clones were re-checked by RT-PCR with primers P1 and P3 to confirm the insertion of the 3XFLAG as well as the presence of the new NheI restriction site. Primers P1 to P3 are describe in Table S3 and Figure S2A. Finally, at every step, positive homozygote clones were confirmed by sanger sequencing on genomic DNA as well as cDNA and clones expressing similar level of TBP compared to the wild type were selected to engineer TBP sequence further.
Metabolites extraction and 1H-NMR spectroscopy
HeLa TBP WT, TBP3X-WT, TBP3X-T114A cells were grown under 25 mM glucose HeLa cells. The Day of the experiment, culture media was removed, and each 10 cm plate was washed two times with PBS. Cells were quenched and scraped in 1 ml of ice-cold methanol, transferred to 2 ml Eppendorf tubes, snap frozen in liquid nitrogen for 10 min, thawed on ice for 10 min, vortexed and centrifuged at 12,000 x g for 10 min at 4°C. Supernatant was transferred to a 2 ml Eppendorf tube. Freeze-thaw and vortex cycles were repeated with %80:%20 methanol:water. Supernatants from each step were pooled dried in speed-vac overnight. Pellet was stored at −80°C for protein quantification. Dried samples were reconstituted with 20 mM phosphate buffer containing 0.1 mM TMSP as an internal reference, 0.1 mM NaN3, and pH was adjusted to pH 7.4+/− 0.1. 1H spectra of the cell extracts were recorded on a Bruker Avance III 500MHz (Bruker Instrument, Germany) NMR spectrometer, operating at 499.9 MHz and equipped with room temperature quadrupole nuclei probe. Typical 1H spectra were acquired using Bruker pulse sequence (noesyprld). Acquisition parameters were set as follows: spectral width of 8012.820 with a 64 K data points, 256 scans, with a relaxation delay of 7 s for a total collection time of 48 min. Samples were automatically tuned and match, and shimmed to TMSP signal. Spectrum were exported into Bruker format and were processed with Chenomx NMR Suit 8.2 Professional (Chenomx Inc, Edmonton, Alberta, Canada). TMSP signal (0.0 ppm) was used as a reference peak, spectra manually phase corrected and spline function was applied for the baseline correction. Metabolites were profiled and quantified using built-in Chenomx 500 MHz library. Metabolite concentrations (μM) were normalized to total protein content (μg).
Total RNA-sequencing and transcriptome analysis
HeLa TBP WT, TBP3X-WT, TBP3X-T114A, TBP3X-WT, sh C, TBP3X-T114A, sh C, TBP3X-WT, sh BTAF1, and TBP3X-T114A, sh BTAF1 cells were grown under 25 mM glucose. RNAs were extracted using RNeasy plus kit (QIAGEN) following manufacturer’s instructions. Library preparation, sequencing and data analysis were performed at the Johns Hopkins University Deep Sequencing and Microarray Core Facility. RNA-Seq libraries were constructed using TruSeq stranded Total mRNA library kit (Illumina, Inc.) and 75 cycle runs were performed in multiplex on the NextSeq 500 platform. Reads were aligned to Homo Sapiens genome GRCh38.p11 and NCBI June 2016 transcriptome with the CLCGenomicsServer 8.5.3 (QIAGEN Bioinformatics), where gene expression levels were calculated as fragments per kilobase of exon per million reads mapped (FPKM). FPKM values were imported into the Partek Genomics Suite 6.6 (Partek Inc St. Louis MO, USA) for statistical analysis. Values were log converted and quantile normalized, then compared between biologic classes using a two-tailed one-way ANOVA to determine differential expression. Further analyses and graphic presentation were performed using the Spotifire DecisionSite with Functional Genomics 9.1.2 (TIBCO Spotfire Boston MA, USA). Genes were considered differentially expressed if their log2 fold changes exceeded the mean (no-change) by greater than 2 standard deviations up or down, and that the mean of the three replicate samples was statistically different with a p-value lower than 0.05. Gene ontology of the differentially expressed genes was analyzed with Spotfire’s Gene Ontology Browser, and pathway analysis conducted using Ingenuity Pathway Analysis (IPA, QIAGEN Bioinformatics).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were repeated at least 3 independent times and are biological replicates unless stated otherwise. Statistical analyses were performed using Prim 7 (GraphPad). One- or two-way ANOVA analysis was applied depending on experimental design, except for Figure 2F, Figure S4A and S4D for which unpaired student’s t-test was used. Number of replicates, meaning of the error bars and other relevant statistical considerations are described in the corresponding figure legend. P values <0.05 were considered significant.
Quantification and analysis of RNA-seq experiments are described in the corresponding section of Method Details.
For experiments that were not quantified, images shown are representative of at least three independent biological replicates except for experiments in Figure S1E that were performed twice.
DATA AND CODE AVAILABILITY
RNA-seq data generated during this study have been deposited at NCBI Gene Expression Omnibus under accession number GSE106231 and GSE130309. Original image files are accessible at Mendeley data (http://dx.doi.org/10.17632/jghzhggkbz.1).
Supplementary Material
Table S1: List of differentially express transcripts of more than two standard variation between the TBP WT, TBP 3X-WT and TBP 3X-T114A cells, Related to Figure 2.
Table S2: List of gene expression profile of TBP 3X-WT, shC, TBP 3X-T114A, shC and TBP 3X-T114A, shBTAF1 cells, Related to Figure 3.
Table S3: List of oligonucleotides used in the study, Related to STAR Methods.
Highlights.
TBP is modified by the nutrient sensor O-GlcNAc
T114-O-GlcNAc-TBP regulates B-TFIID complex formation
Dysregulation of T114-O-GlcNAc-TBP results in large changes in the transcriptome
Lack of T114-O-GlcNAc-TBP results in cellular lipid droplets accumulation
Acknowledgements
We thank Pr. Timmers and Dr. De Graaf (Molecular Cancer Research, University Medical Center Utrecht, STR3.217, Utrecht, Netherlands) for providing us with the GFP-TBP-expressing HeLa cell line. We are grateful to G. Yildirir and the NHLBI P01HL107153 Core C4 for producing and providing us CTD110.6, RL2, OGT and OGA antibodies. We would like to thank the JHMI Deep Sequencing and Microarray Core facility for performing and analyzing mRNA deep sequencing. We thank Corentin Spriet (TISBio UGSF FRABio) for his help in image analysis. We express our gratitude to Marlène Motuaire (UGSF-UMR 8576) for her help with the tissue culture. We thank Isabel González-Mariscal (UGC Endocrinologia y Nutricion, HRUM, IBIMA, Malaga, Spain) for reviewing our manuscript. The authors are supported by NIH R01GM116891, R01DK61671, P01HL107153, R01NS072241 and R01DK116746. S.H. is currently funded by “Région Hauts-de-France” and is recipient of GEFLUC award.
Footnotes
Declaration of Interests
G.W.H. receives a share of royalties received by Johns Hopkins University on sales of the CTD110.6 antibody, which is managed by JHU.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Rahman O, Sasvari-Szekely M, Ver A, Rosta K, Szasz BK, Kereszturi E, and Keszler G (2012). Altered gene expression profiles in the hippocampus and prefrontal cortex of type 2 diabetic rats. BMC Genomics 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee PS, Lagerlof O, and Hart GW (2016). Roles of O-GlcNAc in chronic diseases of aging. Mol Aspects Med 51, 1–15. [DOI] [PubMed] [Google Scholar]
- Banerjee PS, Ma J, and Hart GW (2015). Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A 112, 6050–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, and Hanover JA (2015). A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol 208, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareva AA, and Schmidt EE (2003). Early vertebrate evolution of the TATA-binding protein, TBP. Mol Biol Evol 20, 1932–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CE, Zhao L, Hartung T, and Wolfgang MJ (2016). Requirement for the Mitochondrial Pyruvate Carrier in Mammalian Development Revealed by a Hypomorphic Allelic Series. Molecular and cellular biology 36, 2089–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Baxter J, Satapati S, and Burgess SC (2012). The effect of short-term fasting on liver and skeletal muscle lipid, glucose, and energy metabolism in healthy women and men. J Lipid Res 53, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkinaree C, Cheung WD, Park S, Park K, Barber M, and Hart GW (2008). Characterization of beta-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J Biol Chem 283, 23557–23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodevilla AG, Sanchez-Caballero L, Nintou E, Boiadjieva VG, Picatoste F, Gubern A, and Claro E (2013). Cell survival during complete nutrient deprivation depends on lipid droplet-fueled beta-oxidation of fatty acids. J Biol Chem 288, 27777–27788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibazakura T, Watanabe F, Kitajima S, Tsukada K, Yasukochi Y, and Teraoka H (1997). Phosphorylation of human general transcription factors TATA-binding protein and transcription factor IIB by DNA-dependent protein kinase--synergistic stimulation of RNA polymerase II basal transcription in vitro. Eur J Biochem 247, 1166–1173. [DOI] [PubMed] [Google Scholar]
- Choukrallah MA, Kobi D, Martianov I, Pijnappel WW, Mischerikow N, Ye T, Heck AJ, Timmers HT, and Davidson I (2012). Interconversion between active and inactive TATA-binding protein transcription complexes in the mouse genome. Nucleic Acids Res 40, 1446–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte M, Franceschi C, Sandri M, and Salvioli S (2016). Perilipin 2 and Age-Related Metabolic Diseases: A New Perspective. Trends Endocrinol Metab 27, 893–903. [DOI] [PubMed] [Google Scholar]
- Darabedian N, Thompson JW, Chuh KN, Hsieh-Wilson LC, and Pratt MR (2018). Optimization of Chemoenzymatic Mass Tagging by Strain-Promoted Cycloaddition (SPAAC) for the Determination of O-GlcNAc Stoichiometry by Western Blotting. Biochemistry 57, 5769–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AL, and Uversky VN (2018). Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front Genet 9, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson I (2003). The genetics of TBP and TBP-related factors. Trends Biochem Sci 28, 391–398. [DOI] [PubMed] [Google Scholar]
- de Graaf P, Mousson F, Geverts B, Scheer E, Tora L, Houtsmuller AB, and Timmers HT (2010). Chromatin interaction of TATA-binding protein is dynamically regulated in human cells. J Cell Sci 123, 2663–2671. [DOI] [PubMed] [Google Scholar]
- Fan W, Lam SM, Xin J, Yang X, Liu Z, Liu Y, Wang Y, Shui G, and Huang X (2017). Drosophila TRF2 and TAF9 regulate lipid droplet size and phospholipid fatty acid composition. PLoS Genet 13, e1006664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Lei H, Hsu JL, and Reed R (2012). Small-scale nuclear extracts for functional assays of gene-expression machineries. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiville S, Escobar-Ramirez A, Pina-Canceco S, Elass E, and Pierce A (2014). Delta-lactoferrin induces cell death via the mitochondrial death signaling pathway by upregulating bax expression. Biometals 27, 875–889. [DOI] [PubMed] [Google Scholar]
- Hardiville S, and Hart GW (2014). Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab 20, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs NK, Bondareva AA, Barnett S, Capecchi MR, and Schmidt EE (2002). Removing the vertebrate-specific TBP N terminus disrupts placental beta2m-dependent interactions with the maternal immune system. Cell 110, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob RJ, Fan X, Evans ML, Dziura J, and Sherwin RS (2002). Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: no evidence for protective adaptation by the blood brain barrier. Metabolism 51, 1522–1524. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kang DO, Love DC, and Hanover JA (2006). Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydr Res 341, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejman MP, Zhao X, van Schaik FM, Herr W, and Timmers HT (2005). Mutational analysis of BTAF1-TBP interaction: BTAF1 can rescue DNA-binding defective TBP mutants. Nucleic Acids Res 33, 5426–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MJ, Snel B, and Timmers HT (2015). Genesis of chromatin and transcription dynamics in the origin of species. Cell 161, 724–736. [DOI] [PubMed] [Google Scholar]
- Lescure A, Lutz Y, Eberhard D, Jacq X, Krol A, Grummt I, Davidson I, Chambon P, and Tora L (1994). The N-terminal domain of the human TATA-binding protein plays a role in transcription from TATA-containing RNA polymerase II and III promoters. EMBO J 13, 1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Burlingame AL, and Myers SA (2016). Human RNA Polymerase II Promoter Recruitment in Vitro Is Regulated by O-Linked N-Acetylglucosaminyltransferase (OGT). J Biol Chem 291, 14056–14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Lv D, Zheng Y, Wu M, Xu C, Zhang Q, and Wu L (2018). Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. Onco Targets Ther 11, 3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. (2015). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R, Emancipator SN, Miller C, Kern T, and Simonson MS (2004). Adipose differentiation-related protein and regulators of lipid homeostasis identified by gene expression profiling in the murine db/db diabetic kidney. Am J Physiol Renal Physiol 286, F913–921. [DOI] [PubMed] [Google Scholar]
- Mishra S, Ande SR, and Salter NW (2011). O-GlcNAc modification: why so intimately associated with phosphorylation? Cell Commun Signal 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal V, and Hernandez N (1997). Role for the amino-terminal region of human TBP in U6 snRNA transcription. Science 275, 1136–1140. [DOI] [PubMed] [Google Scholar]
- Morachis JM, Huang R, and Emerson BM (2011). Identification of kinase inhibitors that target transcription initiation by RNA polymerase II. Oncotarget 2, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousson F, Kolkman A, Pijnappel WW, Timmers HT, and Heck AJ (2008). Quantitative proteomics reveals regulation of dynamic components within TATA-binding protein (TBP) transcription complexes. Mol Cell Proteomics 7, 845–852. [DOI] [PubMed] [Google Scholar]
- Pereyra AS, Hasek LY, Harris KL, Berman AG, Damen FW, Goergen CJ, and Ellis JM (2017). Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J Biol Chem 292, 18443–18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petan T, Jarc E, and Jusovic M (2018). Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resto M, Kim BH, Fernandez AG, Abraham BJ, Zhao K, and Lewis BA (2017). O-GlcNAcase is an RNA polymerase II elongation factor coupled to pausing factors SPT5 and TIF1beta. J Biol Chem 292, 16524–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K, Wang Z, and Hart GW (2010). Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 107, 19915–19920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JG, Laranjeira A, Van Huffel L, Gartner A, Vilain S, Bastianen J, Van Veldhoven PP, and Dotti CG (2015). Glial beta-oxidation regulates Drosophila energy metabolism. Sci Rep 5, 7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segil N, Guermah M, Hoffmann A, Roeder RG, and Heintz N (1996). Mitotic regulation of TFIID: inhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev 10, 2389–2400. [DOI] [PubMed] [Google Scholar]
- Stannard SR, Thompson MW, Fairbairn K, Huard B, Sachinwalla T, and Thompson CH (2002). Fasting for 72 h increases intramyocellular lipid content in nondiabetic, physically fit men. Am J Physiol Endocrinol Metab 283, E1185–1191. [DOI] [PubMed] [Google Scholar]
- Tabb DL, Friedman DB, and Ham AJ (2006). Verification of automated peptide identifications from proteomic tandem mass spectra. Nat Protoc 1, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers HT, Meyers RE, and Sharp PA (1992). Composition of transcription factor B-TFIID. Proc Natl Acad Sci U S A 89, 8140–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um M, Yamauchi J, Kato S, and Manley JL (2001). Heterozygous disruption of the TATA-binding protein gene in DT40 cells causes reduced cdc25B phosphatase expression and delayed mitosis. Mol Cell Biol 21, 2435–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan K, and Wells L (2014). Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. J Biol Chem 289, 34466–34471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, and Cramer P (2012). Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol Cell 45, 439–446. [DOI] [PubMed] [Google Scholar]
- Yang X, and Qian K (2017). Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, and Henikoff S (2013). Mot1 redistributes TBP from TATA-containing to TATA-less promoters. Mol Cell Biol 33, 4996–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, and Herr W (2002). A regulated two-step mechanism of TBP binding to DNA: a solvent-exposed surface of TBP inhibits TATA box recognition. Cell 108, 615–627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of differentially express transcripts of more than two standard variation between the TBP WT, TBP 3X-WT and TBP 3X-T114A cells, Related to Figure 2.
Table S2: List of gene expression profile of TBP 3X-WT, shC, TBP 3X-T114A, shC and TBP 3X-T114A, shBTAF1 cells, Related to Figure 3.
Table S3: List of oligonucleotides used in the study, Related to STAR Methods.
Data Availability Statement
RNA-seq data generated during this study have been deposited at NCBI Gene Expression Omnibus under accession number GSE106231 and GSE130309. Original image files are accessible at Mendeley data (http://dx.doi.org/10.17632/jghzhggkbz.1).