

Abstract
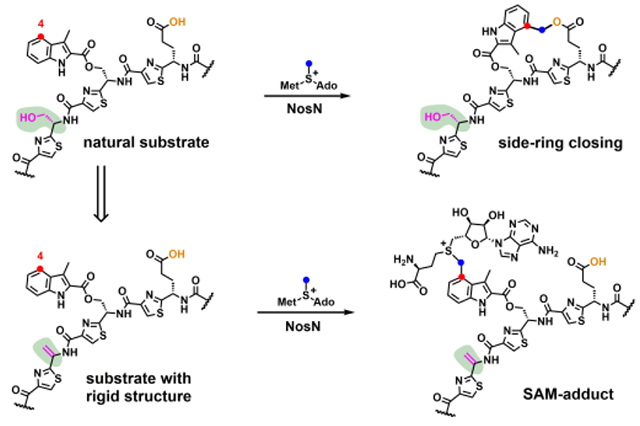
Nosiheptide is a ribosomally synthesized and post-translationally modified thiopeptide natural product that possesses antibacterial, anticancer, and immunosuppressive properties. It contains a bicyclic structure composed of a large macrocycle and a unique side-ring system containing a 3,4-dimethylindolic acid bridge connected to the side chains of Glu6 and Cys8 of the core peptide via ester and thioester linkages, respectively. In addition to the structural peptide, encoded by the nosM gene, the biosynthesis of the side-ring structure requires the actions of NosI, J, K, L, and N. NosN is annotated as a class C radical S-adenosylmethionine (SAM) methylase, but its true function is to transfer a C1 unit from SAM to C4 of 3-methyl-2-indolic acid (MIA) with concomitant formation of a bond between the carboxylate of Glu6 of the core peptide and the nascent C1 unit. However, exactly when NosN performs its function during the biosynthesis of nosiheptide is unknown. Herein, we report the syntheses and use of three peptide mimics as potential substrates designed to address the timing of NosN’s function. Our results show that NosN clearly closes the side ring before NosO forms the pyridine ring, and most likely before NosD/E catalyzes formation of the dehydrated amino acids, although the possibility of a more random process (i.e. NosN acting after NosD/E) cannot be ruled out. Using a substrate mimic containing a rigid structure, we also identify and characterize two reaction-based adducts containing SAM fused to C4 of MIA. The two SAM-adducts are derived from a consensus radical-containing species proposed to be the key intermediate-or a derivative of the key intermediate-in our proposed catalytic mechanism of NosN.
Graphical Abstract
Introduction
Nosiheptide (NOS) is a thiopeptide natural product that is ribosomally synthesized and then post-translationally modified to yield its final structure. It displays potent in vitro activity against a variety of clinically relevant Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus, penicillin-resistant Streptococcus pneumoniae, and vancomycin-resistant enterococci.1–2 However, the poor aqueous solubility and pharmacokinetic properties of NOS and other similar thiopeptide natural products greatly limit their clinical application. Although NOS has been chemically synthesized,3–4 no efforts to use chemical synthesis to improve its pharmacological properties have been reported, most likely because of its highly complex chemical structure. By contrast, the biosynthetic pathway for NOS formation, which relies on the extensive modification of a ribosomally synthesized precursor peptide, offers the potential for relatively facile generation of analogues through semisynthetic and biosynthetic methods.4–5 However, engineering of the NOS biosynthetic pathway requires a thorough understanding of how it is assembled from the initial precursor peptide.
Structurally, NOS is composed of a macrocycle with a fused side-ring structure. The core macrocycle contains multiple thiazoles, dehydrated serine and threonine amino acids, and a central pyridine ring. The side-ring structure is formed by connecting the side chains of Glu6 and Cys8 on the core macrocycle via ester and thioester bonds, respectively, with a 3-methyl-4-hydroxymethyl-2-indolic acid bridge, as shown in Figure 1C. The side-ring structure of NOS is important for its antibiotic properties, and it has been shown that C5’-fluorination on the side-ring leads to an improvement in its antibacterial activity.6
Figure 1.

A) Biosynthetic gene cluster of NOS; B) The amino acid sequence of NosM; and C) The proposed NOS biosynthetic pathway; I. formation of the thiazole rings; II. attachment of MIA to Cys8 of the core peptide; III. NosN-catalyzed formation of the side-ring structure; IV. formation of dehydrated amino acids; V. formation of the central pyridine ring.
The biosynthetic gene cluster of NOS was identified by Liu et al. and contains 16 genes, designated nosA-nosP (Figure 1A).1 The formation of NOS starts from NosM, a ribosomally synthesized precursor peptide of 50 amino acids (aa). As shown in Figure 1B, the first 37 aa of the precursor peptide, termed the leader peptide, primarily provide recognition for some of the downstream enzymes, while the remaining 13 aa, called the core peptide, are post-translationally modified and incorporated into the final product. The biosynthesis of NOS’s core macrocycle is initiated by thiazole formation, which is catalyzed by NosF, G, and H (Figure 1C). NosG and NosH function as a complex to catalyze cyclodehydration of the thiol side chains of target cysteine residues on NosM with the adjacent amide bond, giving thiazoline intermediates that are dehydrogenated by NosF using flavin mononucleotide as the oxidant.7 However, Cys8 is left intact during thiazole formation and is immediately acylated with 3-methyl-2-indolic acid (MIA) by NosI, J, and K.7 MIA, in turn, is derived from L-Trp through the action of NosL, a radical S-adenosylmethionine (SAM) enzyme.8–9 We, and others, have shown that NosJ is an acyl carrier protein, and that NosI activates MIA to afford MIA-AMP before transferring MIA to NosJ.10–11 Next, NosK catalyzes the transfer of MIA from MIA-NosJ to a conserved seryl residue (S102) on itself before transferring it to Cys8 of the thiazole-containing core peptide.7, 10 The actual substrates of the remaining transformations, including dehydration by NosD and E, macrocyclization and pyridine formation by NosO, and the NosN-catalyzed formation of the side-ring structure, have yet to be identified. In the NosD and E reaction, it is likely that the hydroxyl groups on the side chains of Ser1, Ser10, Ser12, Ser13, and Thr3 are activated via glutamylation and dehydrated to give dehydroalanine (Dha) and dehydrobutyrine (Dhb), as described for the similar transformations in the biosynthesis of thiomuracin.12 The Dha derived from Ser1 and Ser10 are then cyclized by NosO, a putative Diels-Alderase, via a [4+2] azacycloaddition to remove the leader peptide and form the core pyridine motif.13–14 NosA, B, and C are the tailoring enzymes in the later stages of the maturation of NOS,15 and NosP is believed to regulate the pathway.16–17
In our previous work, using the tripeptide substrate mimic 1 (Figure 2), we showed that NosN, previously annotated as a methylase, actually catalyzes both the transfer of a C1 unit from SAM and the subsequent formation of the ester linkage of the side-ring system.18 However, the timing of NosN’s function remains undefined. Herein, by synthesizing additional substrate mimics of the NosN reaction, we provide experimental evidence that NosN acts definitely before NosO catalyzes formation of the central pyridine ring and most likely right after NosK appends MIA to the linear peptide. We also succeed in trapping and characterizing a species, which is likely a key intermediate in the reaction or a derivative thereof, supporting our proposed NosN reaction mechanism.
Figure 2.
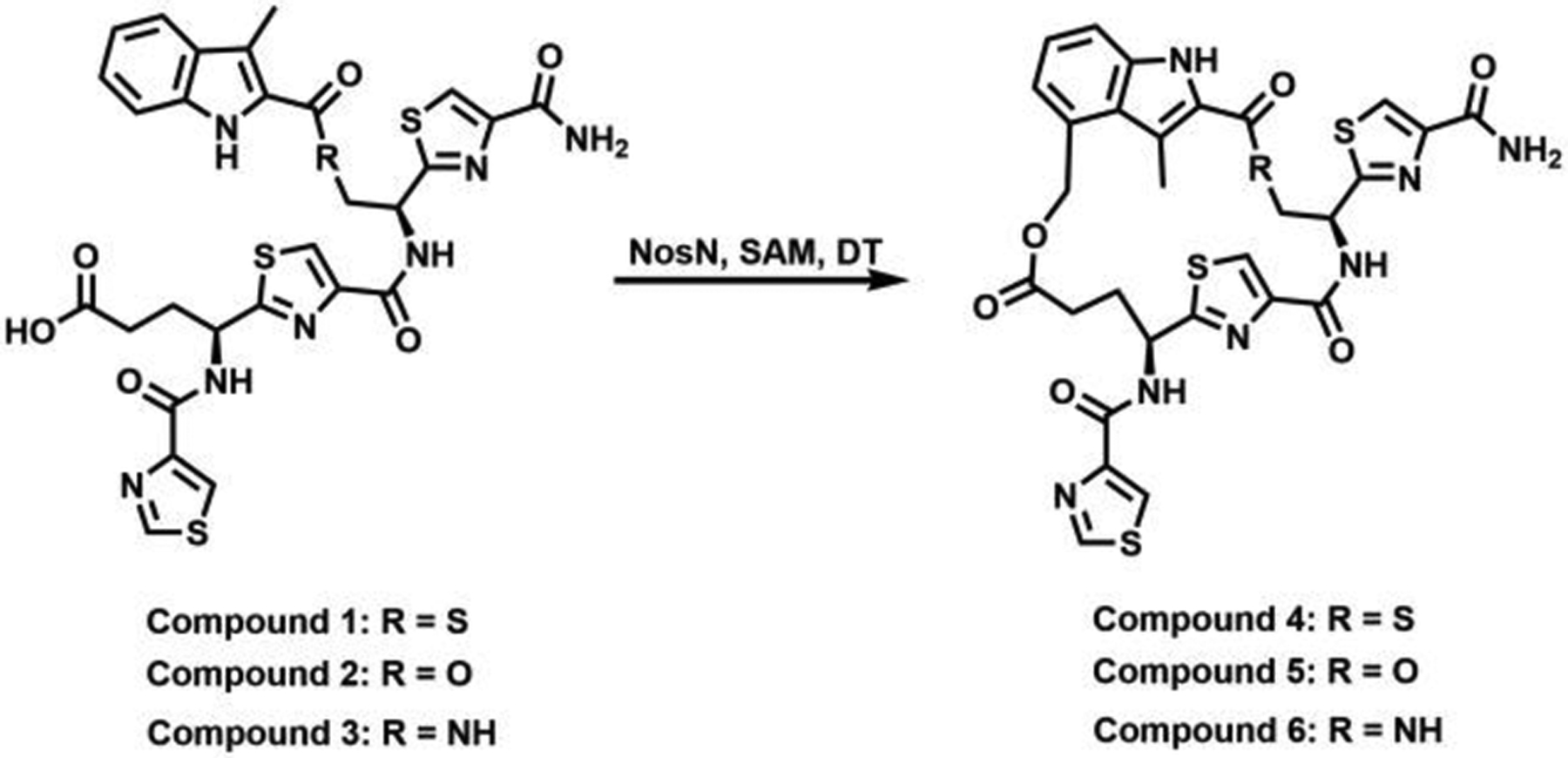
Chemical structures of MIA-S-3mer (1) analogs and their ring-closed products.
Experimental Procedures
Materials.
All commercial materials were used as received unless otherwise noted. N-(2-Hydroxyethyl)-piperazine-N’-(2-ethanesulfonic acid) (HEPES) was purchased from Fisher Scientific. Imidazole was purchased from J. T. Baker Chemical Co. Potassium chloride and glycerol were purchased from EMD Chemicals. 2-Mercaptoethanol, 5’-deoxyadenosine (5’-dA), and S-adenosylhomocysteine (SAH) were purchased from MilliporeSigma. Isopropyl β-D-1-thiogalactopyranoside (IPTG) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Gold Biotechnology. 3-Methyl-2-indolic acid (MIA) was purchased from Matrix Scientific. Ni-NTA resin was acquired from Qiagen. Tetrahydrofuran, dichloromethane, acetonitrile, and DMF were obtained from a JC Meyer solvent dispensing system and used without further purification. SiliaFlash 60 silica gel (230–400 mess) for flash chromatography was obtained from Silicycle Inc. All Fmoc-amino acid building blocks, coupling reagents, and resins for solid-phase peptide synthesis were purchased from Chem-Impex International, Inc. SAM was synthesized and purified as described previously.19 Titanium(III) citrate was prepared as described.20 All other chemicals and materials were of the highest grade available and were from MilliporeSigma.
General Methods.
UV-visible spectra were recorded on a Cary 50 spectrometer from Varian (Agilent Technologies, Santa Clara, CA) using the WinUV software package to control the instrument. High-performance liquid chromatography (HPLC) with detection by tandem mass spectrometry (LC-MS/MS) was conducted on an Agilent Technologies 1200 system coupled to an Agilent Technologies 6410 QQQ mass spectrometer. The system was operated with the associated MassHunter software package, which was also used for data collection and analysis. HPLC was also conducted on an Agilent 1100 Series system coupled to an Agilent 1100 Series variable wavelength detector and quaternary pump. NMR spectra of all compounds were collected on a Bruker AV-3-HD-500 instrument. High-resolution ESI mass spectra were collected on a Waters LCT Premier instrument.
Overproduction and Purification of NosN.
NosN was overproduced and purified as previously described.18, 21 The resulting protein was exchanged into storage buffer containing 50 mM HEPES (pH 7.5), 400 mM KCl, 10% glycerol, and 2 mM TCEP. DTT was removed from the storage buffer because of its potential ability to perform a Michael addition onto compound 8.
NosN Reactions and Quantification of Products by LC-MS/MS.
All reactions were conducted in a Coy (Grasslakes, MI) anaerobic chamber maintained under an atmosphere of 95% nitrogen and 5% hydrogen gases. The oxygen concentration was maintained below 1 ppm by palladium catalysts. Reactions were conducted in a volume of 200 μL and contained 50 mM HEPES (pH 7.5), 200 mM KCl, 5% glycerol, 2 mM sodium dithionite or Ti(III) citrate, 50 μM NosN, 1 mM substrate, and 150 μM L-Trp as an internal standard. Reactions were initiated with SAM at a final concentration of 1 mM, and at various times, 15 μL aliquots were removed and added to 30 μL methanol to quench the reaction. Quenched samples were centrifuged at 16000 × g for 10 min before pipetting the supernatant into HPLC autosampler vials. Components of each quenched reaction were separated on a Zorbax extend-C18 RRHT column (4.6 mm × 5 mm, 1.8 μm particle size) equilibrated in 98% solvent A (0.1% formic acid, pH 2.4) and 2% solvent B (acetonitrile). A linear gradient of solvent B from 2% to 10% was applied from 0 to 0.5 min, which was followed by a linear gradient to 40% solvent B from 0.5 to 2.5 min. Solvent B was held constant at 40% from 2.5 to 3.5 min and then increased linearly to 100% from 3.5 to 4.5 min before holding constant from 4.5 to 5.5 min. Solvent B was then decreased linearly to 2% from 5.5 to 6 min and then held constant for 2 min to re-equilibrate the column before subsequent sample injections. Products were detected using electrospray ionization in positive mode (ESI+) and quantified with a multiple reaction monitoring (MRM) method for L-Trp (internal standard), SAH, and 5’-dA. MRM parameters for L-Trp, SAH and 5’-dA have been previously reported.18
Nos I, J, K, N Cascade Reaction.
The overproduction and purification of Nos I, J and K was conducted as previously described.10 NosJ was phosphopantetheinylated to afford its holo form as described previously.10 Reactions containing Nos I, J, and K were conducted in a volume of 500 μL, and contained 50 mM HEPES (pH 7.5), 5 mM MgCl2, 5 mM TCEP, 3 mM ATP, 2.5 mM MIA, 100 μM compound 10, 10 μM NosI, and 15 μM holo-NosJ. Reactions were initiated by addition of NosK to a final concentration of 100 μM and incubated at room temperature for 1 h. Reactions were then concentrated with an Amicon centrifugal filter unit (ultra-0.5 mL, MilliporeSigma) containing a membrane with a 10 kDa molecular weight cutoff (MWCO). 300 μL water was added to the concentrate, and the sample was concentrated again. This operation was repeated two additional times to remove all the small molecules. The concentrate was redissolved in 300 μL water, and 400 μL acetonitrile was added to precipitate the proteins. After pelleting the precipitate by centrifugation, the supernatant was removed and lyophilized to dryness. The residue was cycled into the Coy anaerobic chamber and dissolved in anoxic water to be used in a NosN reaction.
The NosN reaction was conducted in a volume of 100 μL and contained 50 mM HEPES (pH 7.5), 200 mM KCl, 1 mM sodium dithionite, 50 μM NosN and 100 μM compound 11, obtained from the previous Nos I, J, K reaction. The reaction was initiated by adding SAM to a final concentration of 1 mM and was incubated at room temperature for 2 h. The reaction was then diluted with 300 μL water and added to an Amicon centrifugal filter unit (ultra-0.5 mL, 10 kDa MWCO) to concentrate the large molecules. This operation was repeated two additional times to remove all the small molecules. The concentrate was dissolved in 75 μL water, and 100 μL acetonitrile was added to precipitate the proteins. After pelleting the precipitate by centrifugation, the supernatant was removed for analysis by mass spectrometry.
RESULTS
NosN recognizes analogs of MIA-S-3mer (1) as substrates
In our previous investigations of the NosN reaction, we designed a simple substrate analog, MIA-S-3mer (1), that potentially captured the salient features of the normal substrate, but which was also relatively simple to synthesize chemically.18 In the presence of compound 1, which has an MIA group attached to a truncated core tripeptide through a thioester linkage, NosN was observed to catalyze the radical-mediated transfer of a C1 unit from SAM to C4 of the MIA group, affording SAH and a putative electrophilic exocyclic methylene intermediate as co-products. The reactive exocyclic methylene intermediate is then captured by the carboxylate group in compound 1 to generate the ester linkage in the side-ring system. However, the simplicity of the substrate does not unambiguously allow determination of exactly when during the biosynthesis of NOS that NosN functions. To address this issue, it is necessary to synthesize potential substrates exhibiting more complex structures using solid-phase peptide synthesis (SPPS). The thioester linkage, however, is not compatible with SPPS, so we assessed whether the thioester could be replaced by ester or amide linkages by substituting the sulfur atom with oxygen (MIA-O-3mer) (2) or nitrogen (MIA-NH-3mer) (3) (Figure 2). As shown in the black traces in Figures S1C and D, under turnover conditions (1 mM SAM, 2 mM sodium dithionite and 50 μM NosN), NosN is able to convert compounds 2 and 3 to the corresponding ring-closed products, as evidenced by the production of species exhibiting mass-to-charge ratios (m/z) of 680 ([M+H]+) and 679 ([M+H]+), respectively. Those two parent ions can be further isolated and fragmented in the MS to daughter ions exhibiting m/z values of 170 ([M+H]+) and 188 ([M+H]+) (black traces in Figure S2C and D), which are characteristic fragments of the ring-closed products, as was reported previously for compound 1 (also shown in Figure S2B).18 To demonstrate definitively that the methyl carbon of SAM is transferred to the peptide product, reactions were conducted with compounds 2 and 3, in which SAM was replaced with S-adenosyl-[methyl-d3]-methionine (d3-SAM). A mass increase of 2 is observed in the parent ions in the products produced from reactions using d3-SAM, which is consistent with the loss of a single deuterium atom from SAM during the transfer of the C1 unit. The SAM- and d3-SAM-containing reactions are consistent with what was previously observed when using compound 1, as shown in Figure S1B.18 Correspondingly, a mass increase of 2 is also observed in the daughter ions of the products from compounds 2 (Figure S2C, red trace) and 3 (Figure S2D, red trace), as well as in the daughter ions of the product of compound 1 (Figure S2B, red trace). To analyze the NosN reaction with compounds 1, 2 and 3 quantitatively, we quantified the formation of SAH and 5’-dA as function of time in the presence of 50 μM NosN. The absence of an appropriate standard did not allow us to quantify the peptide product directly, but the amount of SAH generated is believed to correlate closely with that product. The reaction is multiphasic, with a burst of SAH (0.3 to 0.5 equiv) and 5’-dA (~0.5 equiv) formation that is followed by at least one additional phase. As shown in Figure S3A, the second phase of SAH formation with compound 2 is noticeably faster than that with compounds 1 and 3, indicating that NosN readily recognizes the ester linkage. In addition, each reaction results in production of excess 5’-dA with respect to SAH as shown in Figure S3B, which likely derives from the abortive cleavage of SAM. Nevertheless, we demonstrate that ester or amide linkages between the tripeptide and MIA can be tolerated by NosN. Because the ester linkage is more compatible with further synthetic manipulations, it was used in place of the thioester linkage in the more complex synthetic peptide mimics.
NosN uses the NosK product as a substrate
MIA-NosJ has been proposed by others to be the substrate of NosN.11 In our hand, however, neither methylation of MIA-NosJ nor formation of SAH under turnover conditions with this substrate, which is a common co-product of radical SAM methylases, was observed .18 Given that NosN has been shown to catalyze C1 transfer from SAM and closure of the side-ring system of compound 1, it is likely that the true NosN substrate is one of three potential intermediates generated between the action of NosK and the actions of NosA, B, and C as depicted in Figure S4. Compound 1 represents a minimal structure that is present in three potential intermediates in the NOS biosynthetic pathway, as shown in the structures highlighted in pink in Figure S4. NosN’s ability to close the ring of compound 1, a much smaller peptide mimic of the core peptide, suggested that NosN’s activity might be independent of the leader peptide. Therefore, we truncated most of the leader peptide in our peptide mimics to render their chemical syntheses more facile, and designed and synthesized compounds 7, 8 and 9 (Figure 3) as mimics of three potential substrates for NosN as a strategy to determine when NosN acts during the biosynthesis of NOS.
Figure 3.
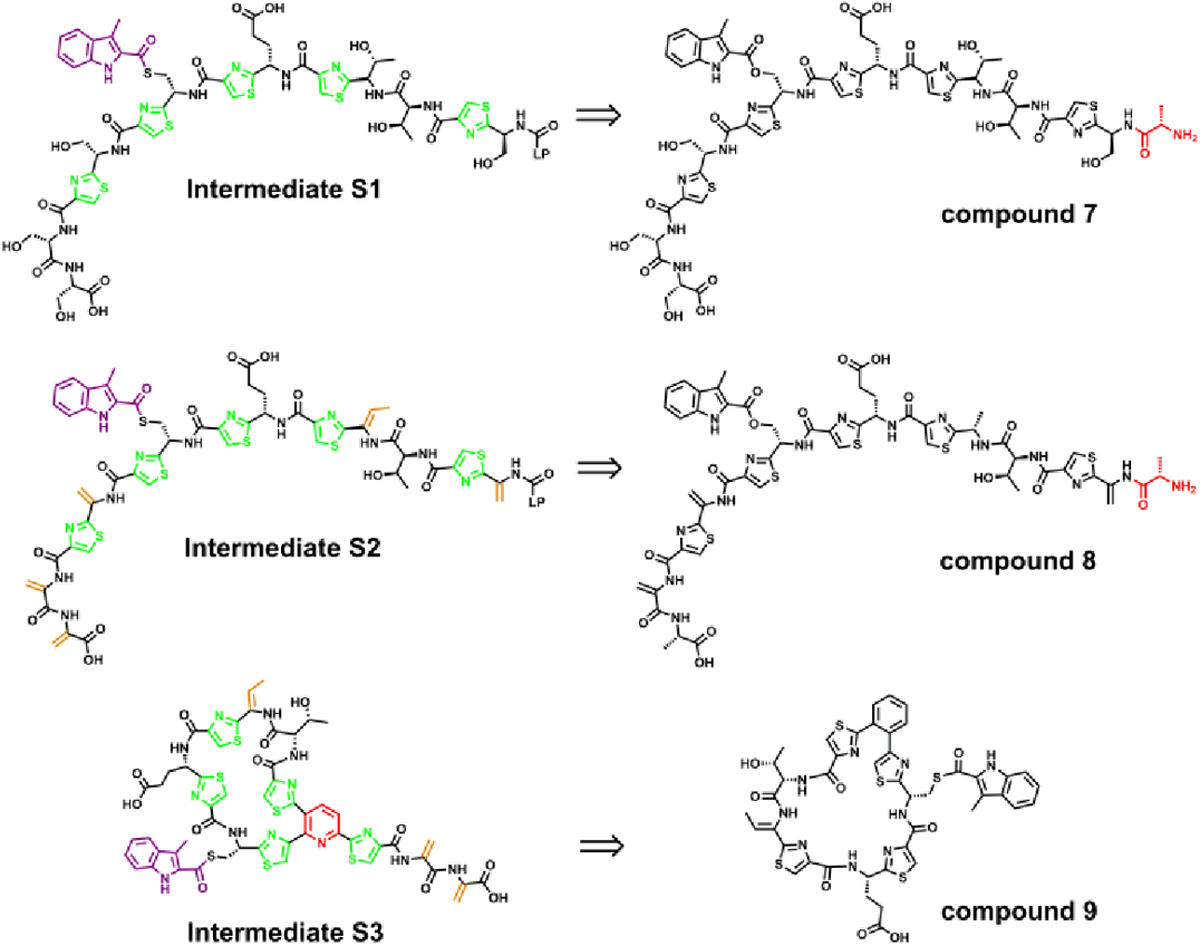
Chemical structures of compounds 7, 8 and 9, designed to identify the true substrate of NosN. The aa residue in red in the chemical structures of compound 7 and 8 corresponds to the added alanine from the leader peptide.
Compound 7 is a mimic of the core peptide structure of the NosK product (intermediate S1 in Figure 3). It also contains an added alanine, which is the very first aa of the leader peptide. Compound 8 mimics intermediate S2 in Figure 3. It is a dehydrated version of compound 7, in which the corresponding serines (except for the serine at the C-terminus) have been converted into Dha. The C-terminal Dha was replaced with an alanine in compound 8, because we observed that the dialkylation-elimination method used to generate Dha from cysteines tended to give an intramolecular cyclized species as the dominant product instead of the desired dehydrated product (as shown in Figure S5).22 Compound 8 was designed to assess whether dehydration by NosD and -E takes place before NosN catalysis, as shown in Figure S4B. Compound 9 is a mimic of intermediate S3 in Figure 3, and was designed to assess whether NosN acts after the NosO-catalyzed formation of the central pyridine ring and core macrocycle as shown in Figure S4C. For ease of synthesis, the central pyridine ring was replaced with a phenyl ring, both of which have similar aromatic, rigid, and planar structures. In addition, the remaining branch on the pyridine ring was removed, also for ease of synthesis. However, the aa within the macrocycle were left intact because they likely affect the overall conformation of the macrocycle.
Because sodium dithionite, the reductant in reactions with compounds 1, 2 and 3, is able to reduce the Dha in compound 8 into alanine,23 giving a series of species that correspond to compound 8 with one, two or three reduced Dha, as shown in Figure S6A,B, we chose Ti(III) citrate as the required low-potential reductant in our reactions with compounds 7, 8 and 9. Ti(III) is not able to reduce the Dha in compound 8 into alanine in the presence or absence of NosN, as shown in Figure S6C,D.20 First, when compound 9 is incubated with NosN (50 μM), SAM (1 mM) and Ti(III) citrate (2 mM) for 1 h, no ring closed product is detected by mass spectrometry as shown in Figure S7A, suggesting that NosN does not operate after NosO forms the pyridine ring and core macrocycle. This result agrees with a previous study, in which NosN was unable to complete side-ring closure of a side-ring-opened variant of NOS produced by a nosN knockout mutant of S. actuosus.1 As shown in Figure 4A,B (red trace), when compound 7 is incubated with NosN (50 μM), SAM (1 mM) and Ti(III) citrate (2 mM) for 1 h, formation of a species exhibiting m/z 1440.8 is observed, which corresponds to the singly charged state ([M+H]+) of the side-ring closed product. Moreover, when SAM is replaced with [13C methyl]SAM in the reaction, a peak with a mass shift of +1 is observed, demonstrating the C1 transfer from SAM to compound 7 (Figure 4B, blue trace). The species with m/z 1440.8 was isolated and fragmented to give characteristic fragments of the side-ring closed product, as shown in Figure 4C (red trace), and a +1 mass unit shift of corresponding ions is observed in the product when using [13C methyl]SAM, as shown in the blue trace. When compound 8 is incubated with NosN (50 μM), SAM (1 mM) and Ti(III) citrate (2 mM) for 1 h, the m/z corresponding to the closed side-ring products is also observed, albeit in lower abundance (Figure S7B, black traces). Again, the transfer of a C1 unit is verified by the mass shift observed when replacing SAM with [13C methyl]SAM in the reaction (Figure S7B, red trace). The formation of SAH and 5’-dA was also monitored in reactions containing compounds 7 and 8. As shown in Figure 5A, the co-product SAH forms more rapidly in the reaction containing compound 7 (black circles) than in the reaction containing compound 8 (red circles), suggesting that NosN recognizes compound 7 as a better substrate than compound 8. Moreover, additional SAH peaks are also observed in the reaction with compound 8, which were also quantified as shown in Figure 5A (green dots). Notably, this SAH peak does not correspond to authentic SAH. We assume the additional SAH is derived from an intermediate that breaks apart into SAH on the mass spectrometer. Therefore, the quantification in Figure 5A (green dots) represents the minimal amount of this species. The characterization of this intermediate is detailed below. Both compounds give rise to similar concentrations of 5’-dA, which surpass those of SAH (Figure 5B), indicating that some 5’-dA is generated through abortive cleavage of SAM. When dithionite is used to replace Ti(III) citrate in reactions, compounds 1 and 7 support multiple turnovers (Figure S8, red and black circles, respectively) — as judged by SAH formation — while compounds 8 and 9 (Figure S8, blue and green circles, respectively) support less than one turnover.
Figure 4.
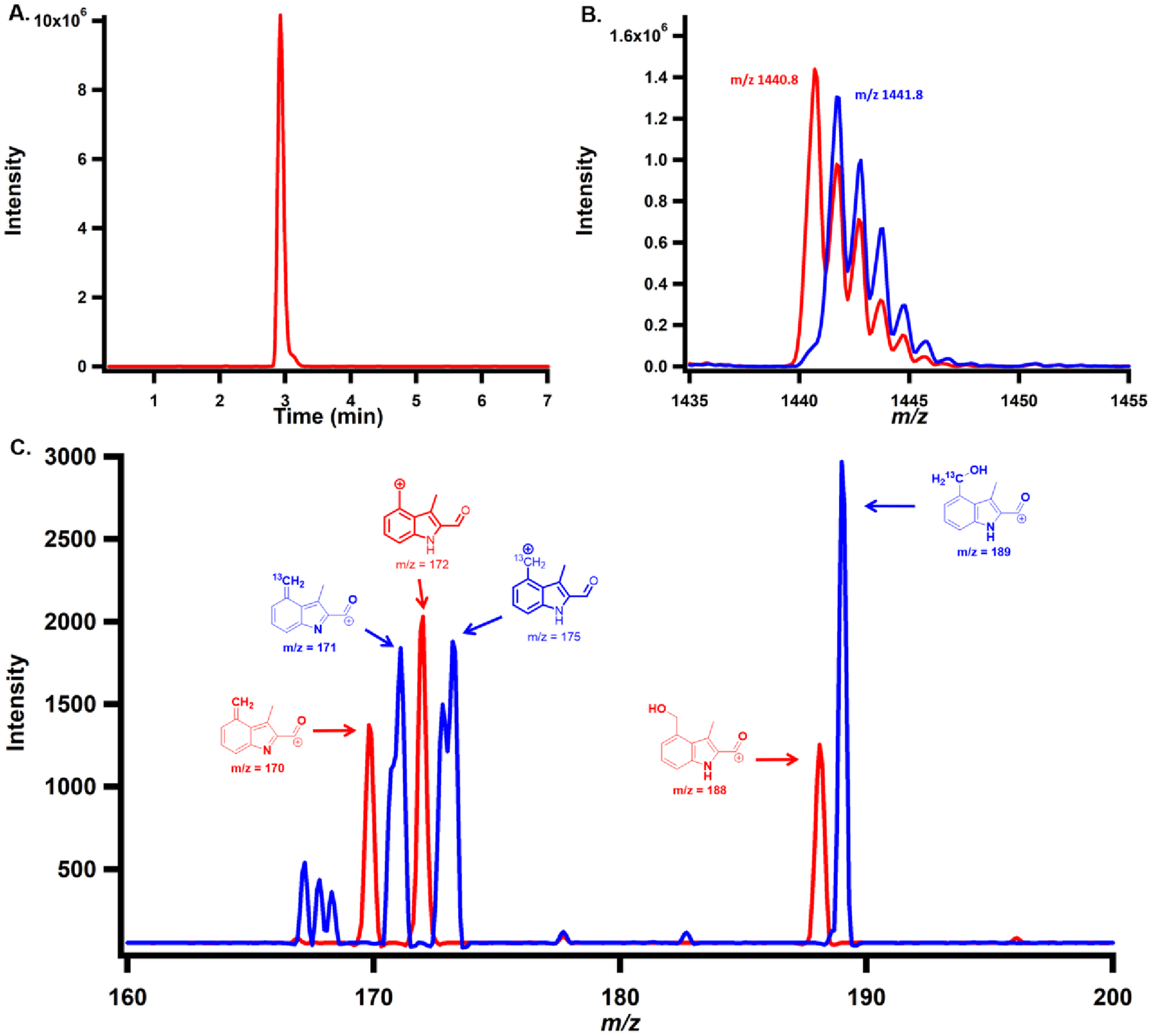
A) Chromatogram displaying the single-ion monitoring (SIM) of the dominant product formed when 1 mM compound 7 is incubated with 1 mM SAM, 50 μM NosN and 2 mM Ti(III) citrate for 1 h. B) MS spectrum of NosN product of compound 7 using SAM (red trace) or [13C methyl]SAM (blue trace). C) MS/MS analysis of NosN product of compound 7 using SAM (red trace) or [13C methyl]SAM (blue trace).
Figure 5.
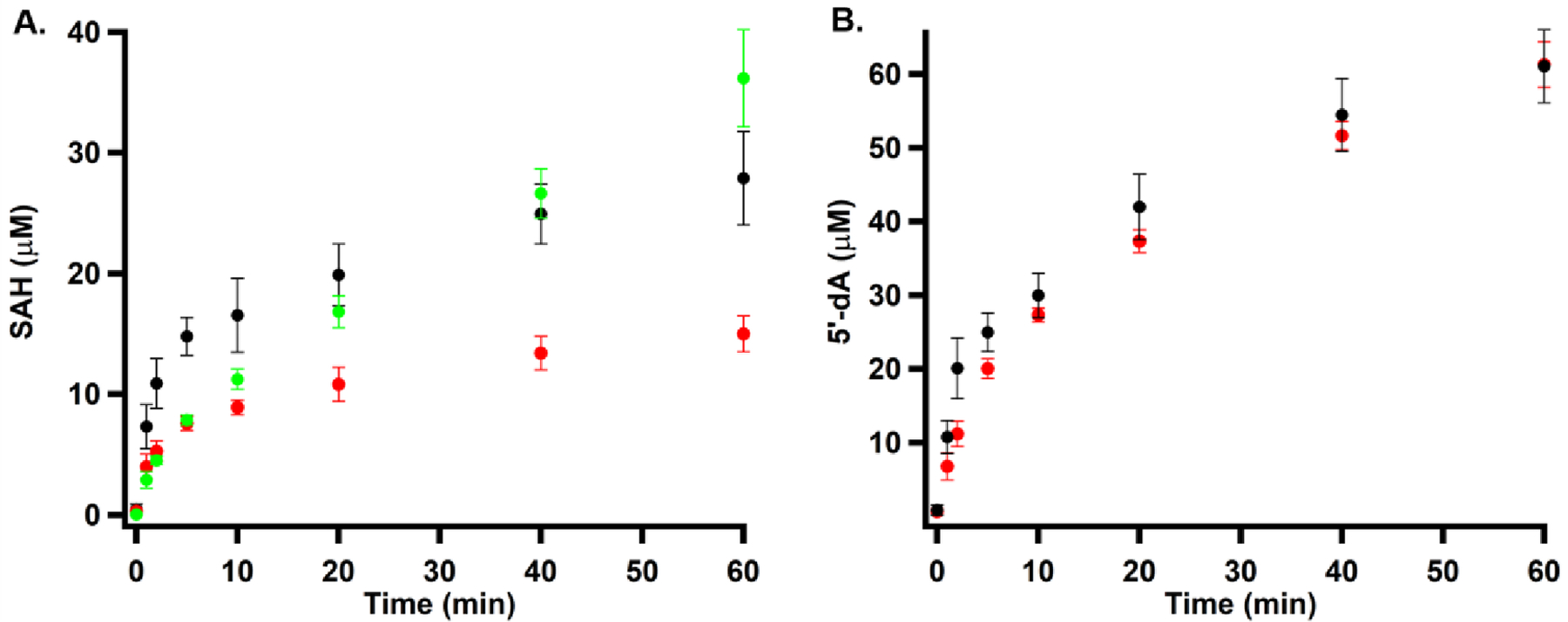
(A) NosN-catalyzed formation of SAH and a SAM adduct in the presence of compound 7 (black circles) and compound 8 (red circles). Green circles represent a SAM-adduct that decomposes to SAH in the mass spectrometer when using compound 8. (B) NosN-catalyzed formation of 5’-dA in the presence of compound 7 (black circles) and compound 8 (red circles). Each reaction contained 1 mM compound 7 or compound 8, 2 mM Ti(III) citrate, and 50 μM NosN, and was initiated with 1 mM SAM.
To assess further whether compound 7 mimics the true substrate of NosN, compound 10 was synthesized to be used in a cascade reaction involving incubation with NosI, J, and K simultaneously and then NosN (Figure 6). Compared to compound 7, which only has the first aa of the leader peptide, compound 10 has a full leader peptide consisting of 37 aa. It has been previously demonstrated that NosK accepts compound 10 as a substrate.7 When compound 10 is incubated with NosI, J, and K in a reaction containing MIA and ATP, it becomes acylated on its Cys8, affording compound 11 (calcd 1347.3072 [M+4H]4+; found 1347.3126 [M+4H]4+; Er 4.0 ppm as shown in Figure 6B). Further, when compound 11 is incubated with 50 μM NosN, 1 mM sodium dithionite and 1 mM SAM, an m/z corresponds to a side-ring closed product (calcd 1350.3072 [M+4H]4+; found 1350.3095 [M+4H]4+; Er 1.7 ppm; as shown in Figure 6C) is observed. On the basis of these observations, we suggest that NosN can recognize NosK’s product—which contains MIA attached to Cys8 and no dehydrated serines and threonines—as substrate, indicating that NosN likely closes the side ring of NOS after NosK appends MIA and before NosD/E form dehydrated aa. The fact that NosN can also recognize compound 8, which mimics the structure of peptide proceeded by Nos D/E, also raises the possibility that the timing of NosN’s function is more random. This possibility requires more experimental data to address definitively.
Figure 6.
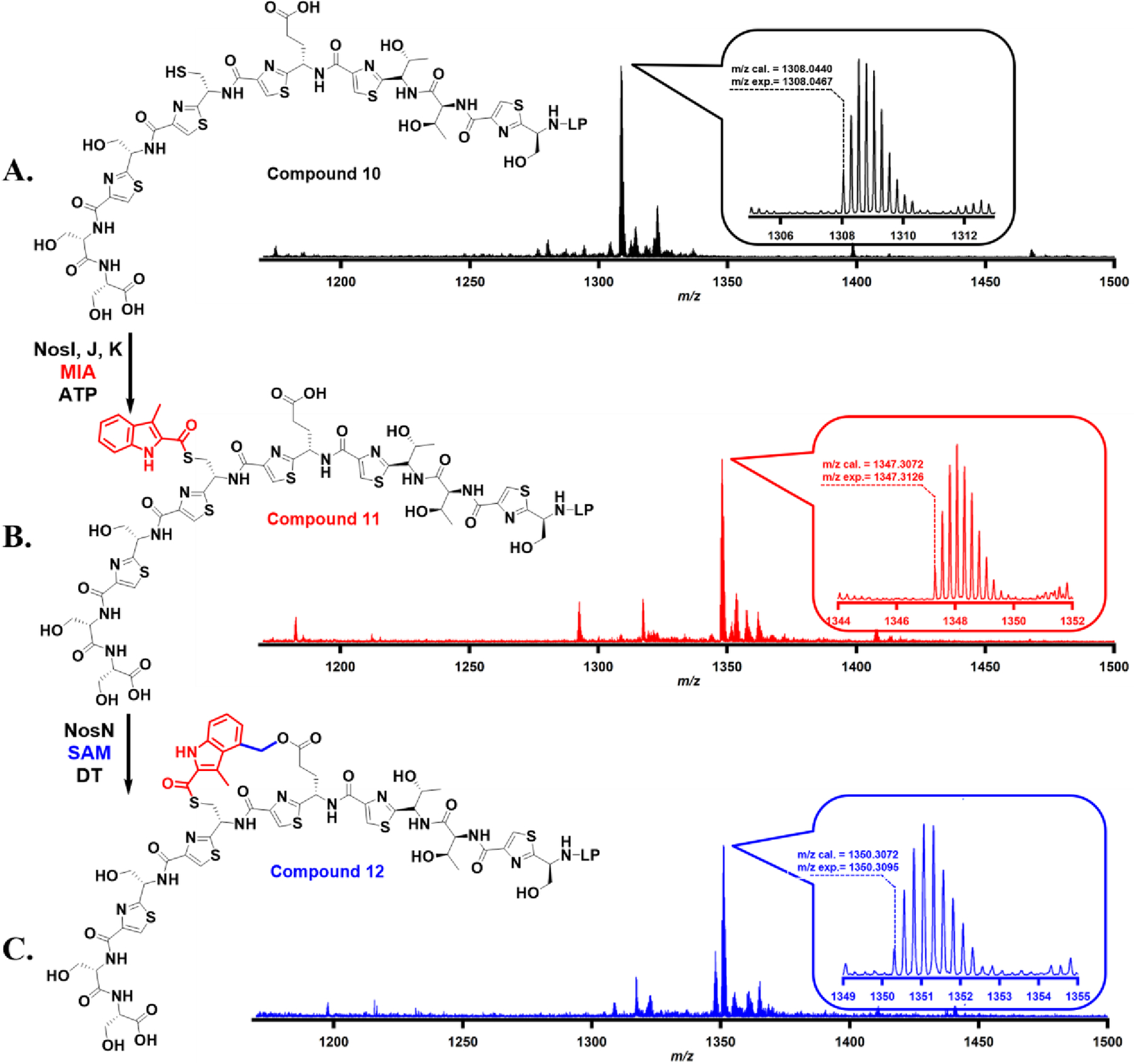
(A). Accurate mass of synthetic compound 10; (B). Accurate mass of compound 11, which is the acylated product of compound 10 in the NosI, J, and K reaction; (C). Accurate mass of compound 12, which is the side-ring closed product of compound 11 upon catalysis by NosN. The detailed reaction conditions are shown in supporting information.
Characterization of off-pathway SAM-adduct intermediates
Intriguingly, as shown in the blue trace in Figure 7A, two additional peaks exhibiting retention times of 3.5 and 4.1 min are observed by MRM while monitoring SAH (transition from 385 to 136) in the NosN assay using 1 mM compound 8, 1 mM SAM, 2 mM Ti(III) citrate and 50 μM NosN for 1 h. These peaks are absent in the reactions conducted under the same conditions but using compound 7 instead of compound 8 (Figure 7A, red trace). Careful analysis of the additional SAH peaks leads to the discovery of two species exhibiting m/z 1724.5 ([M+H]+, namely, SAM adduct I) and m/z 1726.5 ([M+H]+, namely, SAM adduct II) for the singly charged states as shown in Figure 7B and C. The doubly charged state of the two species (m/z = 862.9 and 863.9, [M+2H]2+) is also observed at much higher intensities, as shown in Figure S9. The areas of the two species exhibit a ratio of SAM adduct I to SAM adduct II of roughly 6:1. The m/z of SAM adduct I is equal to the sum of the m/z of the substrate (m/z 1328) and the m/z of SAM (m/z 399), suggesting that it might be an SAM adduct intermediate. SAM adduct II, which exhibits a 2-mass-unit increase, might be a SAM adduct I derivative with a reduced double bond.
Figure 7.
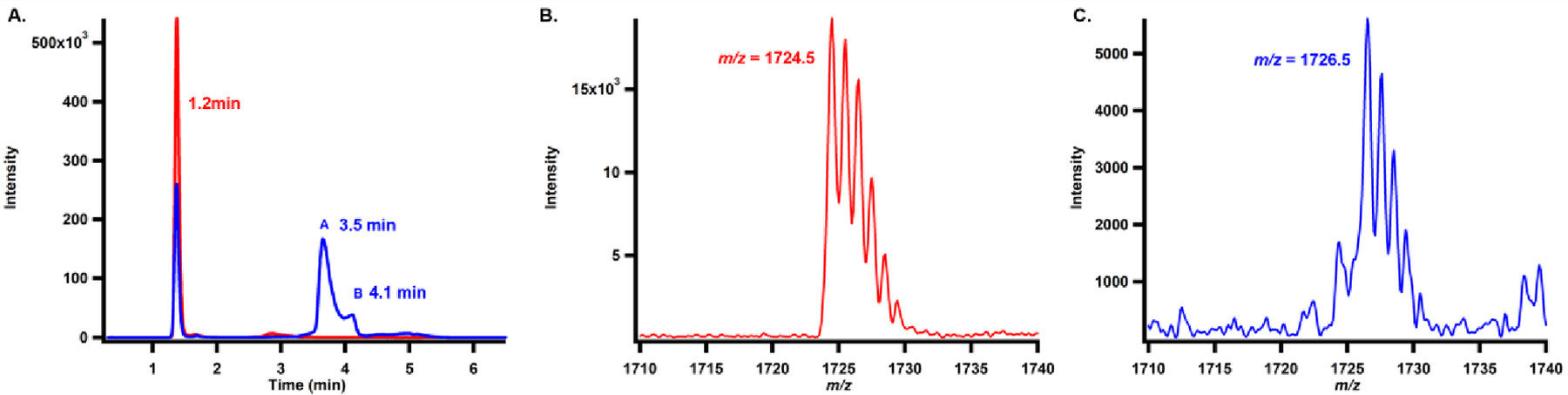
Characterization of SAM-adducts. A). MRM scan of SAH (transition of 358 to 136) in the NosN assay on compound 7 (red trace) and compound 8 (blue trace). The peaks at 1.2 min correspond to the SAH produced by methylation. The peaks at 3.5 and 4.1 min correspond to products that can be fragmented to SAH; B). MS analysis of SAM-adduct I; C). MS analysis of SAM-adduct II. Each reaction contained 1 mM compound 7 or 8, 2 mM Ti(III) citrate, 50 μM NosN, and was initiated with 1 mM SAM and incubated for 1h.
LC-MS/MS was used to further characterize the two species. After careful optimization of collision energies, SAM adduct I can be isolated and fragmented into daughter ions exhibiting m/z 385.3 and m/z 1340.6 (Fragment I) (Figure S10A), while SAM adduct II can be isolated and fragmented into daughter ions exhibiting m/z 385.3 and m/z 1342.6 (Fragment II) (Figure S10B). The 2-mass-unit difference between Fragment I and II (as compared in Figure S10G) suggests the presence of a reduced double bond in Fragment II. When the same experiments are repeated using [13C methyl]SAM, as shown in Figure S10C, D, H, and I, Fragments I and II shift by 1 mass unit, respectively, while m/z 385.3 is retained, indicating that the 13C atoms are incorporated into Fragment I and II. Further, when [13C carboxyl]SAM is used in reactions, no changes are observed in Fragments I and II. By contrast, a one mass unit increase (m/z 386.3) is observed in the SAH fragments of both the [13C carboxyl]SAM adduct I and the [13C carboxyl]SAM adduct II (Figure S10E, F, and J). On the basis of the analysis of the putative SAM adducts by mass spectrometry, we assign the structures of SAM adduct I and SAM adduct II as they are shown in Figure 8. We believe that the two species are generated from a consensus aryl radical intermediate (Figure 9) via either a one-electron oxidation or a one-electron reduction of the intermediate, which we detail further below.
Figure 8.
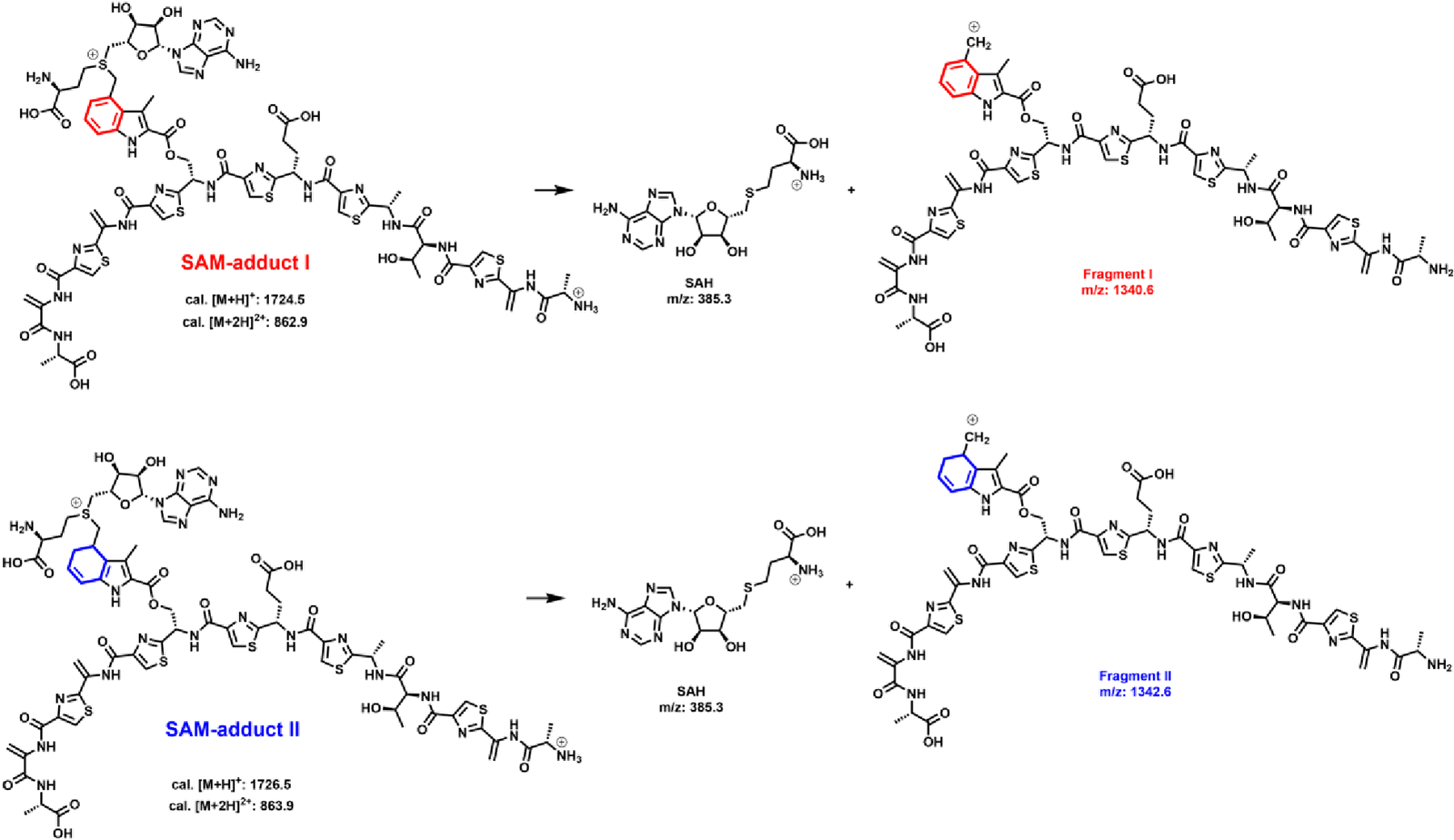
Putative chemical structures of SAM-adduct I and SAM-adduct II, and fragments I and II.
Figure 9.
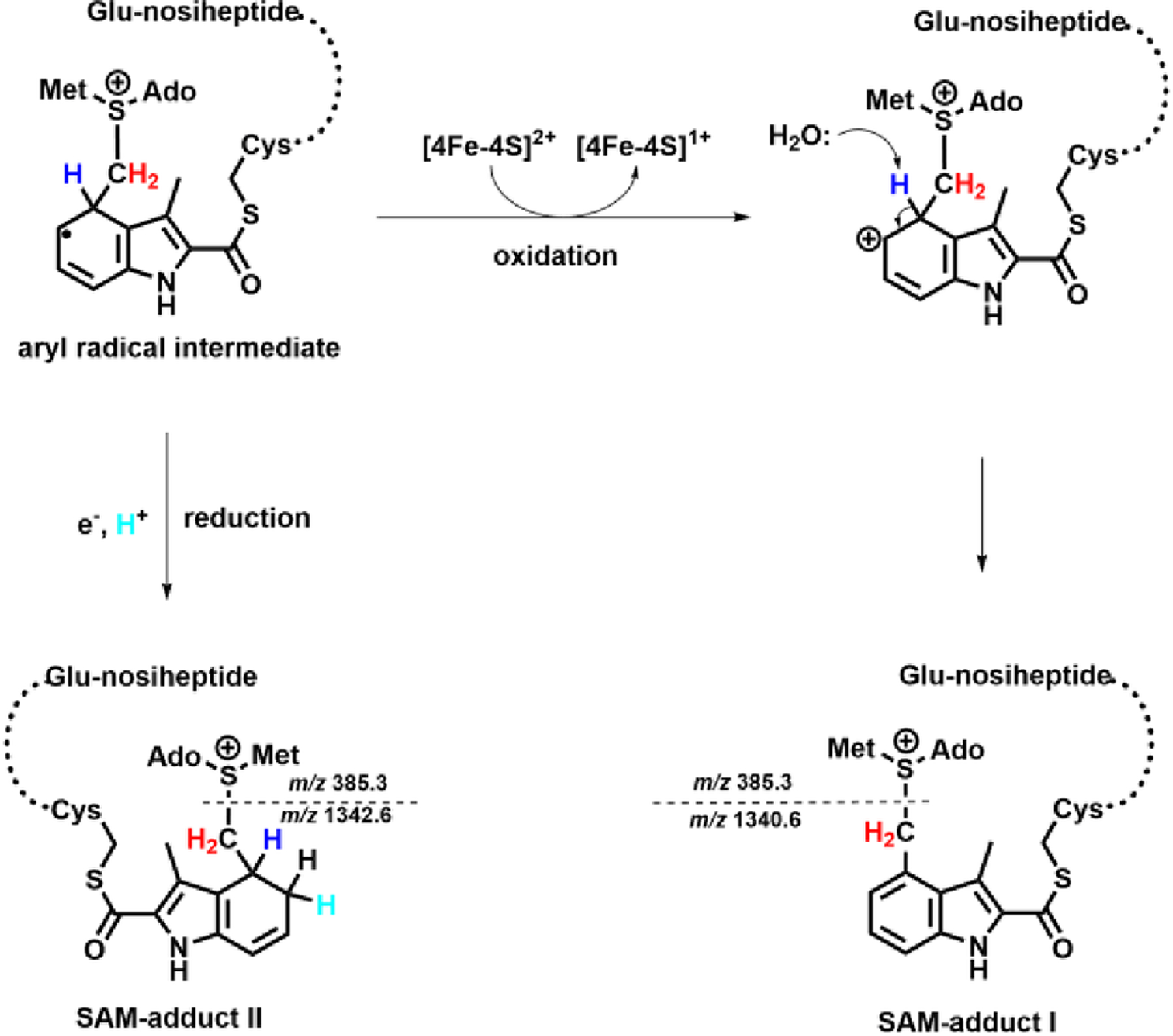
Oxidative or reductive pathways to form SAM-adduct I and II.
To exclude the possibility that the SAM adduct observed in the NosN reaction using compound 8 as substrate is an intermediate which can be converted into the product, we conducted a large scale NosN reaction using compound 8 and isolated the SAM adduct. The HPLC trace for the purification of the SAM adduct is shown in Figure S11A. We collected the peak from 11.1–11.8 min, and then the fractions were lyophilized and redissolved in buffer. The resulting purified SAM adduct was treated without NosN (Figure S11B) or with NosN (Figure S11C) under turnover condition; however, the two sets of reactions show no conversion of SAM adduct to the ring closed product. This experiment confirms that the SAM adduct we observed is an on-pathway adduct, but not a direct intermediate, most likely because of quenching of the radical before proton abstraction takes place.
Discussion
The construction of the NOS’s side-ring requires the action of NosI, NosJ, NosK, NosL and NosN. It has been established that NosI, J, and K work together to activate and transfer MIA to Cys8 of intermediate I in Figure 1C, which is generated via the action of NosF, G, and H on the precursor peptide NosM.7 NosL and NosN are radical SAM enzymes. NosL catalyzes a complex rearrangement of tryptophan to give MIA,8 while, in a previous study, we demonstrated that NosN catalyzes a C1 transfer from SAM to C4 of compound 1 and also completes formation of the side-ring via nucleophilic attack by Glu6 of the peptide substrate onto a reactive electrophilic intermediate.18 However, compound 1 is a minimal consensus substructure that is contained in all three potential NosN substrates during the maturation of the NOS side-ring, raising the question, when exactly does NosN perform its reaction? To address this question, we designed and synthesized compounds 7, 8, and 9 as probes to identify the true substrate of NosN.
We first demonstrated that NosN can tolerate both ester and amide bonds that connect MIA to the core peptide. In fact, in a thiopeptide natural product that is very similar to NOS, nocathiacin, MIA is appended to the core peptide via an ester linkage.24 This result enabled the synthesis of a number of larger peptide analogs by SPPS, which would be cumbersome with the much more labile thioester linkage. Three mimics were synthesized and used in NosN reactions: one that mimics the peptide substrate after the addition of NosK, 7; one that mimics the peptide substrate after dehydration of target serines and a threonine, 8; and one that mimics the peptide substrate after NosO-catalyzed pyridine ring formation, 9. NosN was able to transfer a C1 unit and complete side-ring formation on both linear peptide mimics 7 and 8, but not the cyclic peptide mimic 9. To assess whether 7 or 8 is the preferred NosN substrate, formation of SAH and 5’-dA was quantified as a function of time in NosN reactions. The extent of 5’-dA formation is reflective both of chemistry that is taking place on the substrate as well as the nonproductive abortive cleavage of SAM. By contrast, in our model for NosN catalysis, SAH is only formed through chemical steps involving the MIA substrate. In reactions with 7, NosN catalyzed a relatively rapid burst of SAH followed by a slower exponential phase. The burst may reflect the extent to which the Fe/S cluster of NosN is reduced by Ti(III) citrate. Moreover, in a cascade reaction involving NosI, J, K and a full-length compound 7-derived peptide substrate containing appropriate thiazole rings and MIA connected to Cys8, NosN was able to transfer a C1 unit to MIA and complete formation of NOS’s side ring. By contrast, in reactions with compound 8, NosN catalyzed formation of SAH as well as formation of two adducts. One adduct contained SAM connected to C4 of MIA through a methylene bridge originating from SAM’s methyl group, while the second adduct, though similar to the first, was reduced by two electrons. Our putative assignment of the true substrate of NosN as deriving from compound 7 is based on the aberrant chemistry observed with compound 8 in the presence of Ti(III) citrate, as well as the significantly greater activity observed in the presence of compound 7 as compared to compound 8 when dithionite is used as the required reductant, although a more random process (i.e. NosN acting after NosD/E) cannot be completely ruled out.
Our working hypothesis for the pathway of NOS biosynthesis is illustrated in Figure 1C. NosM is first processed by NosF, G, and H to form the thiazole rings in NOS. Next, NosI, J, and K collaborate to append MIA to the thiazole-containing peptide. NosN then transfers a C1 unit from SAM to MIA, generating a reactive electrophilic intermediate, which is trapped by Glu6 of the peptide, which completes the synthesis of NOS’s side ring. Although not yet verified experimentally, the downstream transformations should include dehydration by NosD and NosE followed by a NosO-catalyzed [4+2] azacycloaddition to afford the major macrocycle. Finally, NOS biosynthesis is completed upon the action of the tailoring enzymes NosA, NosB, and NosC.
Our proposed NosN reaction mechanism is depicted in Figure 10. The 5’-dA•, obtained from the reductive cleavage of SAM, abstracts a hydrogen atom from the methyl group of another simultaneously bound molecule of SAM. This conclusion is supported by the incorporation of a single deuteron into 5’-dA when using d3-SAM in reactions.18 The resulting methylene radical adds to C4 of MIA to give an aryl radical intermediate. A general base abstracts the C4 proton with concomitant elimination of SAH to give intermediate II with an exocyclic methylene group. Previous studies on 2-hydroxyglutaryl-CoA dehydratase have suggested that the pKa value of the proton adjacent to an enoxyradical is on the order of 14,25 supporting the feasibility of such a deprotonation by an active site base. A similar deprotonation event is proposed in the mechanisms of RlmN and Cfr, two class A RS methylases.26 The radical intermediate II loses an electron — most likely back to the Fe-S cluster — and the resulting electrophilic methide-like intermediate III traps Glu6 to complete the side-ring of NOS. The observation of SAM adducts I and II using a substrate variant (compound 8) is strongly in support of our proposed mechanism, and contradicts previously proposed mechanisms that invoke methylthioadenosine as an intermediate in the reaction.27–28 Compound 8 contains three dehydroalanines in place of the corresponding serines found in compound 7. The planar structure of dehydroalanine residues, due to rotational restriction imposed by the double bonds, greatly reduces the flexibility of compound 8. Therefore, the positioning of compound 8 in the active site of NosN may restrict access by the active site base for proton abstraction upon radical addition to MIA. In the absence of prompt proton abstraction, the radical intermediate degrades via either oxidative or reductive pathways. In the oxidative pathway, the resulting carbocation rearomatizes to give SAM adduct I. In the reductive pathway, a direct single-electron reduction — most likely from the artificial reductant used in the reaction — is followed by protonation to give SAM adduct II. Owing to the poor stability of the carbanion as well as the destruction of the aromaticity of the product, the reductive pathway is likely unfavorable, which is consistent with the lower concentration of SAM adduct II observed in the assay. The 5-(2’-Deoxyuridinyl)methyl radical, a key intermediate in thymine oxidative reactions that are mediated by reactive oxygen species, also suffers both oxidative and reductive fates in the absence of molecular oxygen.29
Figure 10.
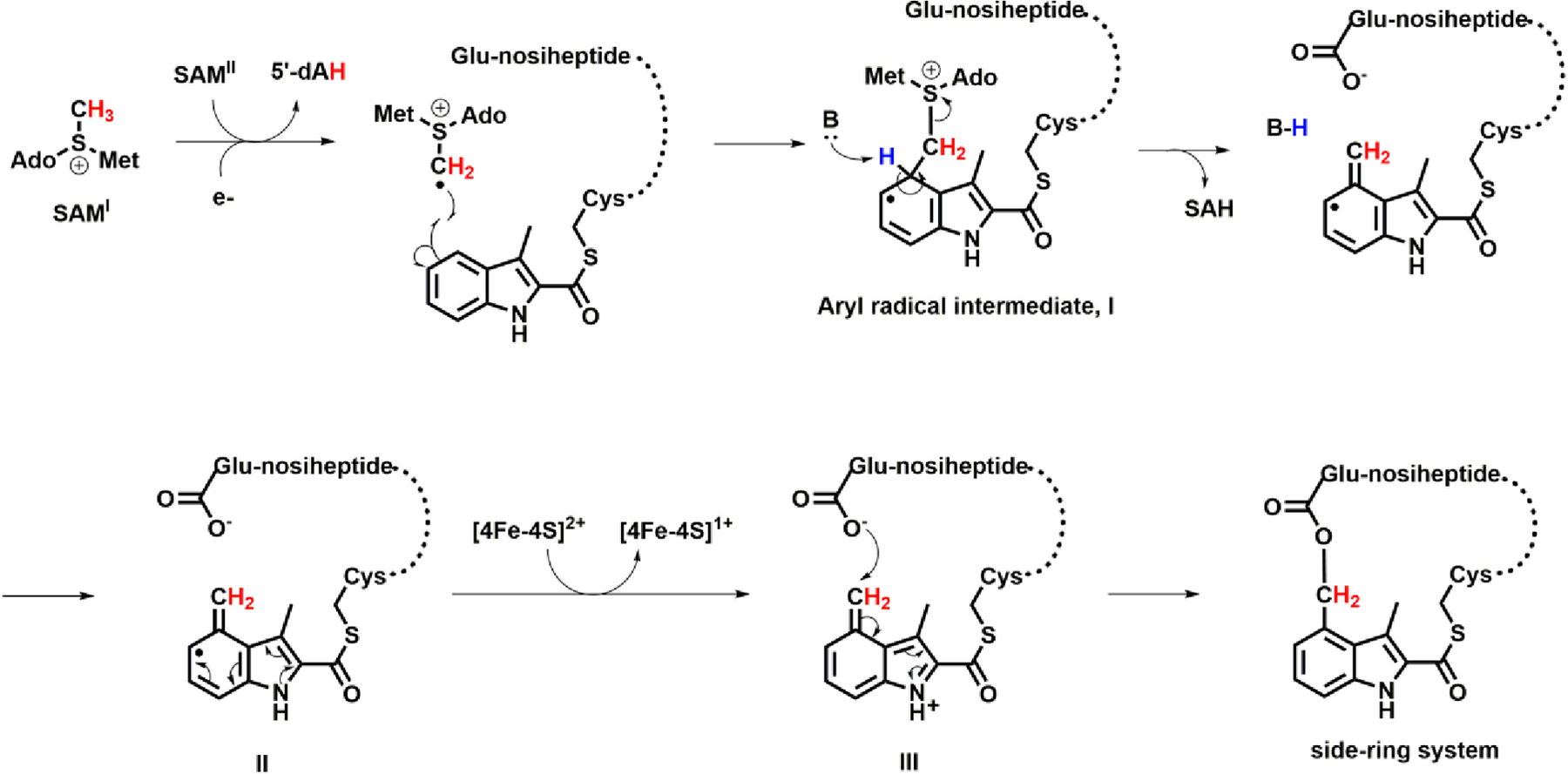
Proposed mechanism of NosN.
In conclusion, we have tested three mimics of three potential biosynthetic intermediates during the biosynthesis of NosN, and we proposed the timing and substrate specificity of NosN. We also observed two adduct species that support our proposed reaction mechanism. Under the global antibiotic resistance crisis and urgent need for antibiotics with novel mechanisms of action, our work sheds light on the possible bioengineering of NOS, which is difficult to access solely by chemical synthesis.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Anne Stanley and Dr. Suja Rani Maddukuri at Penn State College of Medicine core facilities for assistant with solid phase peptide synthesis. We also thank Dr. Philip Smith from the Penn State Metabolomics Facility for the acquisitions of accurate mass data.
Funding Sources
This work was supported by NIH (GM-122595 and AI-133318 to S.J.B.). S.J.B. is an investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- 5’-dA
5’-deoxyadenosine
- 5’-dA•
5’-deoxyadenosyl radical
- aa
amino acid
- Dha
dehydroalanine
- Dhb
dehydrobutyrine
- LC-MS
liquid chromatography with detection by mass spectrometry
- MIA
3-methyl-2-indolic acid
- MRM
multiple reaction monitoring
- NOS
nosiheptide
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SIM
single-ion monitoring
- SPPS
solid-phase peptide synthesis
Footnotes
Supporting Information
Figures S1-S11. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Yu Y; Duan L; Zhang Q; Liao RJ; Ding Y; Pan HX; Wendt-Pienkowski E; Tang GL; Shen B; Liu W, Nosiheptide Biosynthesis Featuring a Unique Indole Side Ring Formation on the Characteristic Thiopeptide Framework. Acs Chem Biol 2009, 4 (10), 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haste NM; Thienphrapa W; Tran DN; Loesgen S; Sun P; Nam SJ; Jensen PR; Fenical W; Sakoulas G; Nizet V; Hensler ME, Activity of the thiopeptide antibiotic nosiheptide against contemporary strains of methicillin-resistant Staphylococcus aureus. J Antibiot 2012, 65 (12), 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wojtas KP; Riedrich M; Lu JY; Winter P; Winkler T; Walter S; Arndt HD, Total Synthesis of Nosiheptide. Angew Chem Int Edit 2016, 55 (33), 9772–9776. [DOI] [PubMed] [Google Scholar]
- 4.Just-Baringo X; Albericio F; Alvarez M, Thiopeptide Engineering: A Multidisciplinary Effort towards Future Drugs. Angew Chem Int Edit 2014, 53 (26), 6602–6616. [DOI] [PubMed] [Google Scholar]
- 5.Lin Z; He QL; Liu W, Bio-inspired engineering of thiopeptide antibiotics advances the expansion of molecular diversity and utility. Curr Opin Biotech 2017, 48, 210–219. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q; Li YX; Chen DD; Yu Y; Duan LA; Shen B; Liu W, Radical-mediated enzymatic carbon chain fragmentation-recombination. Nat Chem Biol 2011, 7 (3), 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu YP; Du YA; Zhang F; Liao RJ; Zhou SX; Peng C; Guo YL; Liu W, Thiolation Protein-Based Transfer of Indolyl to a Ribosomally Synthesized Polythiazolyl Peptide Intermediate during the Biosynthesis of the Side-Ring System of Nosiheptide. J Am Chem Soc 2017, 139 (50), 18186–18189. [DOI] [PubMed] [Google Scholar]
- 8.Bhandari DM; Fedoseyenko D; Begley TP, Mechanistic Studies on Tryptophan Lyase (NosL): Identification of Cyanide as a Reaction Product. J Am Chem Soc 2018, 140 (2), 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XY; Zhu WY; Liu YJ, Tryptophan lyase (NosL): mechanistic insights into amine dehydrogenation and carboxyl fragment migration by QM/MM calculations. Catal Sci Technol 2017, 7 (13), 2846–2856. [Google Scholar]
- 10.Badding ED; Grove TL; Gadsby LK; LaMattina JW; Boal AK; Booker SJ, Rerouting the Pathway for the Biosynthesis of the Side Ring System of Nosiheptide: The Roles of NosI, NosJ, and NosK. J Am Chem Soc 2017, 139 (16), 5896–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding W; Ji WJ; Wu YJ; Wu RZ; Liu WQ; Mo TL; Zhao JF; Ma XY; Zhang W; Xu P; Deng ZX; Tang BP; Yu Y; Zhang Q, Biosynthesis of the nosiheptide indole side ring centers on a cryptic carrier protein NosJ. Nat Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson GA; Zhang ZG; Tietz JI; Mitchell DA; van der Donk WA, In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin. J Am Chem Soc 2015, 137 (51), 16012–16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cogan DP; Hudson GA; Zhang ZG; Pogorelov TV; van der Donk WA; Mitchell DA; Nair SK, Structural insights into enzymatic [4+2] aza-cycloaddition in thiopeptide antibiotic biosynthesis. P Natl Acad Sci USA 2017, 114 (49), 12928–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wever WJ; Bogart JW; Baccile JA; Chan AN; Schroeder FC; Bowers AA, Chemoenzymatic Synthesis of Thiazolyl Peptide Natural Products Featuring an Enzyme-Catalyzed Formal [4+2] Cycloaddition. J Am Chem Soc 2015, 137 (10), 3494–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y; Guo H; Zhang Q; Duan LA; Ding Y; Liao RJ; Lei C; Shen B; Liu W, NosA Catalyzing Carboxyl-Terminal Amide Formation in Nosiheptide Maturation via an Enamine Dealkylation on the Serine-Extended Precursor Peptide. J Am Chem Soc 2010, 132 (46), 16324–16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li JJ; Li Y; Niu GQ; Guo H; Qiu YP; Lin Z; Liu W; Tan HR, NosP-Regulated Nosiheptide Production Responds to Both Peptidyl and Small-Molecule Ligands Derived from the Precursor Peptide. Cell Chem Biol 2018, 25 (2), 143–+. [DOI] [PubMed] [Google Scholar]
- 17.Wu XR; Jin L; Zhang H; Tong RN; Ma M; Chen YJ, Identification of truncated form of NosP as a transcription factor to regulate the biosynthesis of nosiheptide. Faseb J 2018, 32 (1), 453–+. [DOI] [PubMed] [Google Scholar]
- 18.LaMattina JW; Wang B; Badding ED; Gadsby LK; Grove TL; Booker SJ, NosN, a Radical S-Adenosylmethionine Methylase, Catalyzes Both C1 Transfer and Formation of the Ester Linkage of the Side-Ring System during the Biosynthesis of Nosiheptide. J Am Chem Soc 2017, 139 (48), 17438–17445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwig DF; Booker SJ, Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine. Biochemistry 2004, 43 (42), 13496–13509. [DOI] [PubMed] [Google Scholar]
- 20.Zehnder AJB; Wuhrmann K, Titanium(Iii) Citrate as a Nontoxic Oxidation-Reduction Buffering System for Culture of Obligate Anaerobes. Science 1976, 194 (4270), 1165–1166. [DOI] [PubMed] [Google Scholar]
- 21.Wang B; LaMattina JW; Badding ED; Gadsby LK; Grove TL; Booker SJ, Chapter Nine - Using Peptide Mimics to Study the Biosynthesis of the Side-Ring System of Nosiheptide In Methods in Enzymology, Academic Press: 2018; Vol. 606, pp 241–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison PM; Foley PJ; Warriner SL; Webb ME, Chemical generation and modification of peptides containing multiple dehydroalanines. Chem Commun 2015, 51 (70), 13470–13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camps F; Coll J; Guitart J, Regiospecific Reduction of Unsaturated Conjugated Ketones with Sodium Dithionite under Phase-Transfer Catalysis. Tetrahedron 1986, 42 (16), 4603–4609. [Google Scholar]
- 24.Ding Y; Yu Y; Pan HX; Guo H; Li YM; Liu W, Moving posttranslational modifications forward to biosynthesize the glycosylated thiopeptide nocathiacin I in Nocardia sp ATCC202099. Mol Biosyst 2010, 6 (7), 1180–1185. [DOI] [PubMed] [Google Scholar]
- 25.Smith DM; Buckel W; Zipse H, Deprotonation of enoxy radicals: Theoretical validation of a 50-year-old mechanistic proposal. Angew Chem Int Edit 2003, 42 (16), 1867–1870. [DOI] [PubMed] [Google Scholar]
- 26.Silakov A; Grove TL; Radle MI; Bauerle MR; Green MT; Rosenzweig AC; Boal AK; Booker SJ, Characterization of a Cross-Linked Protein Nucleic Acid Substrate Radical in the Reaction Catalyzed by RlmN. J Am Chem Soc 2014, 136 (23), 8221–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding W; Li YZ; Zhao JF; Ji XJ; Mo TL; Qianzhu HC; Tu T; Deng ZX; Yu Y; Chen FE; Zhang Q, The Catalytic Mechanism of the Class C Radical S-Adenosylmethionine Methyltransferase NosN. Angew Chem Int Edit 2017, 56 (14), 3857–3861. [DOI] [PubMed] [Google Scholar]
- 28.Ding W; Wu YJ; Ji XJ; Qianzhu HC; Chen F; Deng ZX; Yu Y; Zhang Q, Nucleoside-linked shunt products in the reaction catalyzed by the class C radical S-adenosylmethionine methyltransferase NosN. Chem Commun 2017, 53 (37), 5235–5238. [DOI] [PubMed] [Google Scholar]
- 29.Lin GJ; Li L, Oxidation and Reduction of the 5-(2-Deoxyuridinyl)methyl Radical. Angew Chem Int Edit 2013, 52 (21), 5594–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.