

Abstract

Plasmon resonances have appeared as a promising method to boost the fluorescence intensity of single emitters. However, because research has focused on the enhancement at low excitation intensity, little is known about plasmon–fluorophore coupling near the point where the dye saturates. Here we study plasmon-enhanced fluorescence at a broad range of excitation intensities up to saturation. We adopt a novel DNA-mediated approach wherein dynamic single-molecule binding provides a controlled particle–fluorophore spacing, and dynamic rebinding circumvents artifacts due to photobleaching. We find that near saturation the maximum photon count rate is enhanced by more than 2 orders of magnitude at the optimal particle–fluorophore spacing, even for a dye with a high intrinsic quantum yield. We compare our results to a numerical model taking into account dye saturation. These experiments provide design rules to maximize the photon output of single emitters, which will open the door to studying fast dynamics in real time using single-molecule fluorescence.
Single-molecule fluorescence is a powerful technique to gain molecular-level insight into biological processes. Single-molecule studies directly reveal molecular properties and their underlying statistics, instead of ensemble observables that obscure underlying distributions and nonsynchronized processes.1,2 Binding kinetics, for example, can be visualized using fluorescently labeled ligands such as DNA or proteins that dynamically bind and unbind to target molecules.3,4 Single-enzyme kinetics have also been revealed by means of fluorogenic substrates, in which single catalytic turnover events can be recorded.5,6 Conformational dynamics have been probed based on fluorescence quenching, fluorescence anisotropy and lifetime,7 and Föster resonance energy transfer (FRET), allowing the fundamental investigation of structure–function relationships.8−10 Indeed, single-molecule sensitivity impacts applications and has enabled crucial advances in medical diagnostics and biosensing, wherein, for example, continuous monitoring of biomolecules and single-molecule sequencing have been demonstrated.11−14
The brightness, or detected photon count rate (PCR), of single fluorophores is essential in many applications but strongly depends on the electronic properties and associated excitation and decay channels of the fluorophore. It is ideal in the detection of biological process to use fluorescent dyes with high PCR and chemical stability to allow highly sensitive single-molecule measurements. However, the PCR of single organic dyes is limited to 106–107 photons per second, of which typically only 1% reaches the detector because of limited collection efficiency.15−17 The maximum PCR is intrinsically limited by saturation, which is the result of the finite decay rate of the fluorophore from the excited state and the presence of long-lived triplet excited states. This limits the maximum excitation rate that can be achieved because one has to “wait” for decay back to the ground state. The limited brightness of single molecules limits the signal-to-noise ratio particularly at short integration times and has prohibited real-time single-molecule studies at submillisecond time scales.
Plasmon resonances have appeared as a promising method to boost the fluorescence intensity of single emitters.18 Since Drexhage first demonstrated that the spontaneous emission rate of fluorescent molecules could be accelerated by coupling to the photonic modes of nearby metal surfaces, much effort has been spent to study the interactions of plasmonic nanostructures and fluorophores.19 Metal nanoparticles stand out with excellent optical properties and have been used to enhance single-molecule fluorescence.20,21 The origin of these fluorescence enhancements is the strong modification of the excitation and decay rates of a molecular dipole close to a particle. The excitation rate is locally increased because of the enhanced electromagnetic field in the vicinity of the particle surface, while the radiative and nonradiative decay rates are strongly modified because of the coupling of the molecular dipole moment to the plasmon modes of the particle. Both excitation and decay-rate modification are strongly dependent on the optical properties of the nanoparticle and the fluorophore.
Most research has focused on the quantification of enhancement factors, which indicate by what factor the total detected fluorescence intensity is enhanced when a fluorophore is brought in proximity to a metal particle.22−24 These studies have shown that the fluorescence of single weakly emitting molecules can be enhanced by thousands of times.25−27 High enhancement factors at low excitation power have mostly been applied to perform single-molecule spectroscopy at high concentrations.28−31 However, a different class of applications involves real-time single-molecule fluorescence measurements of biomolecular dynamics, for which not the fluorescence enhancement but rather the absolute PCR matters. To achieve the highest PCR it is necessary to pump molecules close to or even above fluorescence saturation.32,33 However, little is known about the effect of plasmon–molecule coupling on the saturation dynamics of single dyes, and design rules to achieve maximum absolute photon count rates are not known.
Here we present a single-molecule study of plasmon-enhanced fluorescence of a dye with a high intrinsic quantum yield that is pumped close to saturation. A particular concern is preventing artifacts due to photobleaching, for which we adopt an approach based on low-affinity DNA interactions resulting in controlled residence times of the fluorophore near the particle, continuous refreshment of the dye by rebinding, and a well-controlled spacing between particle and fluorophore. We measure the PCR as a function of excitation power, from which we derive the PCRmax and saturation intensity (Isat). The PCR is strongly modified because of plasmon-fluorophore coupling, while we find thatthe PCRmax is enhanced by more than 2 orders of magnitude at ∼1.5 nm from the particle surface. The experimental findings are in good agreement with numerical simulations that include saturation. These experiments provide mechanistic insight and design rules to optimize the absolute photon output of single emitters. This will open the door to studying microsecond dynamics in real time, e.g., DNA hairpin dynamics and transition paths in protein folding.34−37
To explain the origin of fluorescence saturation we employ
a simplified
model that represents the dye as a three-level system as in Figure 1a, in which the ground
state S0, the first excited singlet state S1 and the triplet state T1 are shown. Upon illumination,
the molecule is excited from S0 to S1 with a
certain excitation rate 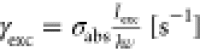 , where σabs is the absorption
cross section of the molecule [m2] and Iexc is the illumination power density [W m–2]. The molecule relaxes from S1 to S0 emitting
photons with a radiative decay rate γr or nonradiatively
with a nonradiative decay rate γnr. The molecule
may transition from S1 to T1 with an intersystem-crossing
rate of γisc and decays from T1 to ground
state S0 with a triplet decay rate γT.
The total decay rate γtot = γr +
γnr + γisc is related to the fluorescence
lifetime, while the quantum yield
, where σabs is the absorption
cross section of the molecule [m2] and Iexc is the illumination power density [W m–2]. The molecule relaxes from S1 to S0 emitting
photons with a radiative decay rate γr or nonradiatively
with a nonradiative decay rate γnr. The molecule
may transition from S1 to T1 with an intersystem-crossing
rate of γisc and decays from T1 to ground
state S0 with a triplet decay rate γT.
The total decay rate γtot = γr +
γnr + γisc is related to the fluorescence
lifetime, while the quantum yield 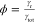 dictates the fraction of excitations that
result in photon emission. The PCR of a fluorescent molecule is then
given by
dictates the fraction of excitations that
result in photon emission. The PCR of a fluorescent molecule is then
given by
| 1 |
where ηcol is the collection efficiency of the setup as a whole, and the saturation intensity is written as38
| 2 |
Equation 1 indicates the nonlinear dependence of PCR
at excitation rates
around and above Isat, whereas eq 2 indicates that Isat is dictated by the total decay rate and
the triplet dynamics of the molecule. As conceptually shown in Figure 1b, with increasing
excitation power density, the PCR of both free-space and plasmon-enhanced
single molecules lose linearity and approach a plateau PCRmax that is expressed as 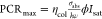 because
because 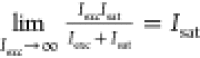 . In the presence of a
nanoparticle, both
the emission and excitation rates in eq 1 are strongly modified. First, the excitation rate
in the vicinity of a single nanoparticle is strongly enhanced because
of the concentrated electromagnetic field induced by the plasmon resonance.
Second, both γr and γnr are strongly
increased because of coupling with plasmon modes in the nanoparticle.
We first assume that triplet dynamics is not affected by plasmon coupling,
but we will return to this point later.
. In the presence of a
nanoparticle, both
the emission and excitation rates in eq 1 are strongly modified. First, the excitation rate
in the vicinity of a single nanoparticle is strongly enhanced because
of the concentrated electromagnetic field induced by the plasmon resonance.
Second, both γr and γnr are strongly
increased because of coupling with plasmon modes in the nanoparticle.
We first assume that triplet dynamics is not affected by plasmon coupling,
but we will return to this point later.
Figure 1.

Photophysics of a single emitter modified by a plasmonic nanoantenna. (a) A three-level Jablonski diagram for a single fluorescent molecule. (b) Cartoon of saturation curves of an emitter in free space (blue) and in the vicinity of a plasmonic nanoantenna. Both the saturation intensity (Isat) and the maximum photon count rate (PCRmax) are modified. Note here that this figure is for illustrative purposes and is not a simulation result.
The net effect of these rate modifications is a modification of the saturation dynamics as illustrated in Figure 1b, showing that the PCR is enhanced at all power densities because of the local field enhancement. Notably, also PCRmax is significantly enhanced because of a modified Isat. The plasmonic modification of the photophysics strongly depends on the optical properties of both the nanoparticle and the fluorophore, and also their relative position and orientation. The effect of nanostructures on fluorescence saturation was first experimentally investigated by Ebbesen et al. to show that nanoapertures enhance the PCR of diffusing dyes on an ensemble-averaged level.39 More recently van Hulst et al. reported that an up to 100-fold decrease in excitation power was required to reach Isat for single light-harvesting complexes coupled to single gold nanoparticles because of near-field enhancement. They also observed a 50-fold enhancement of PCRmax due to modification of the emitter’s decay rates.40
We numerically evaluate the modifications of the radiative and nonradiative rates using the boundary element method (BEM),41 and by combining the numerical simulation results with the three-level photophysical model as in eq 1 we calculate the modification of the saturation dynamics for particles of various dimensions. Near saturation the PCRmax is proportional to the radiative rate enhancement, namely
| 3 |
where the superscript “0” indicates free-space values. Equation 3 indicates that the PCRmax enhancement follows the position and wavelength dependence of the γr enhancement, as shown in Figure 2a. (Numerical simulation results for comparison can be found in the Supporting Information.) In Figure 2b we show the PCRmax modification normalized to free-space as a function of SPR wavelength. We find indeed that the maximum PCRmax enhancement up to ∼150 when the SPR wavelength of the particle coincides with the fluorophore’s emission wavelength, whereas the enhancement is a factor of 3× lower when the SPR is resonant with the excitation wavelength. This is fundamentally different in the low-excitation regime where most other studies have been performed, where the enhancement is dominated by the excitation process. In Figure 2c we show the modification of Isat normalized to its free-space values. A maximum Isat increase of ∼400 is found for a plasmon that is resonant with λem because of the strong wavelength dependence of the decay rates that again exhibit a maximum at λem as in eq 2. This implies that substantially higher excitation power densities are needed to reach the saturation regime for a plasmon-coupled fluorophore, but this is alleviated by the strong near-field enhancement that results in dye saturation at acceptable laser powers.
Figure 2.

Numerical calculations of photophysical modifications in the vicinity of a single gold nanorod with a size of 63 nm × 25 nm. (a) PCRmax modifications normalized to that of a free space emitter PCRmax0, calculated with orientation-averaged radiative and total decay rate modifications as a function of fluorophore position. Fluorophores are assumed to be single-wavelength dipole emitters with a quantum yield of unity at 664 nm (resonant with the particle plasmon). (b) PCRmax modification normalized by the value of a free space fluorophore PCRmax. λexc = 637 nm and λem = 664 nm indicate the excitation and emission wavelengths. The SPR wavelength of the particles was tuned by changing their length while keeping their diameter at 25 nm. (c) Isat modification normalized by the saturation intensity of a free space fluorophore Isat0. For wavelength-dependent calculations, the dipole emitter is placed at 4 nm from the tip, and the results are orientation averaged.
To experimentally study PCRmax and Isat we perform single-molecule fluorescence microscopy on single gold nanorods. The heterogeneous size distribution of the colloidally synthesized gold nanorods allows us to study the SPR wavelength dependence in a single measurement. In Figure 3a we illustrate the detection of single particles and single fluorescent molecules based on an objective-type TIRF configuration. Gold nanorods are first spin-coated on a microscope coverslip, functionalized with a mixture of thiolated single-stranded docking strands and short 10 nt antifouling strands, and mounted in a flow cell. Imager strands fluorescently labeled with ATTO647N with a 9 nt complementary sequence to the docking strands are injected into the flow cell under a continuous flow of 100 μL/min. This results in controlled residence times of the fluorophore near the particle dictated by the affinity (∼1 μM), continuous refreshment of the dye by rebinding, and a well-controlled spacing between particle and fluorophore.
Figure 3.

Single-molecule fluorescence microscopy and single-particle spectroscopy. (a) Schematic illustration of the setup for objective-type total internal reflection fluorescence (o-TIRF) microscopy, with the inset showing the surface functionalization and DNA binding on a single gold nanorod. (b) EMCCD image of immobilized DNA-funtionalized AuNRs immersed in imager solution, with diffraction-limited spots from the one-photon photoluminescence of single AuNRs. (c) Hyperspectral scattering spectrum of a typical single AuNR with a longitudinal SPR wavelength of 640 nm (green) and the ensemble absorption/emission spectra (blue and red) of ATTO 647N in aqueous buffer. (d) The distribution of longitudinal SPR wavelengths measured by single-particle hyperspectral spectroscopy. The inset is a TEM image showinga dried droplet of suspension from which we obtained the size and geometry of the AuNRs.
In Figure 3b we show the detected one-photon photoluminescence of single gold nanorods on the EMCCD camera under 637 nm laser excitation. Camera counts were converted to photon counts by measuring in the photon-counting mode of the EMCCD. The scattering spectra of each particle are measured with hyperspectral imaging under white light illumination in the same field of view.42 A typical scattering spectrum of a particle is shown in Figure 3c, from which the SPR wavelength is acquired after Lorentzian fitting. The particles exhibit a distribution of SPR wavelengths due to size nonuniformity inherited from colloidal synthesis which we exploit here to study plasmon-enhanced fluorescence as a function of spectral overlap between the fluorophore and the plasmon. In Figure 3d we show the distribution of longitudinal SPRs for ∼300 single nanorods in a single field of view.
Upon filling the flow cell with imager solution, we observe fluorescent bursts appearing as point spread functions (PSFs) superposed on the constant one-photon photoluminscence of single gold nanorods. In Figure 4a we show the detected PCR from frame-by-frame summation of 9-by-9 pixels centered around a single gold nanorod, excited by a 637 nm s-polarized CW laser with a power density in the focal plane of 2 × 107 W m–2. The large variation in the fluorescence intensities from these bursts can be attributed to the random binding location of each imager. Because it is expected that the maximally enhanced bursts originate from binding events close to the tip of the nanorods, we average the PCR of the 10 strongest fluorescence events and correlate them with the SPR wavelength of the same particle as shown in Figure 4b. We find that the optimum PCR enhancement occurs near the emission peak of the dye. This indicates that the PCR enhancement is dominated by the modification of the emission process rather than the excitation process. Note that individual particles exhibit a different PCR even for the same plasmon wavelength. This is a result of the random orientation of the particles in the s-polarized excitation field formed by total internal reflection.
Figure 4.

Fluorescence time traces and correlation with SPR wavelengths of single AuNRs. (a) Fluorescence time trace showing single imager strands binding to single AuNRs. This AuNR’s SPR wavelength is determined to be 661 nm. (b) SPR wavelengths of ∼130 single AuNRs correlated with the mean photon count rates of the 10 brightest events. The error bars indicate the standard deviation of the count rates. (c) Dark and bright time distributions of imager binding. (d) Mean bright times as a function of power density. The error bars indicate the standard deviation of mean bright times collected from up to 100 single nanorods. The inset illustrates the determination of bright times from on a fluorescence time trace.
In order to separate the signals of binding events from background,
we applied a threshold (∼10× the background noise) to
locate the timestamps of signals. The characteristic bright times
(tbright) and dark times (tdark) of the transient binding events can be extracted
from the time trajectory. tbright and tdark are related to the thermodynamic properties
of the single-molecule DNA interaction and can be directly related
to the (single-site) association rate 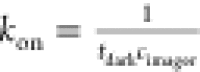 and the dissociation rate
and the dissociation rate 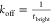 , with cimager being the imager concentration.43 In Figure 4c we show the extracted tbright and tdark histograms and fit single-exponential
distributions for both. We
find tbright = 0.4 ± 0.1 s and tdark = 3 ± 0.3 s, in good agreement with
previous reports where 9 bp duplexes were used.43,44
, with cimager being the imager concentration.43 In Figure 4c we show the extracted tbright and tdark histograms and fit single-exponential
distributions for both. We
find tbright = 0.4 ± 0.1 s and tdark = 3 ± 0.3 s, in good agreement with
previous reports where 9 bp duplexes were used.43,44
Photobleaching of a fluorophore might occur via triplet-mediated mechanisms45 and via higher-order excited states.46 We demonstrate experimentally that the effect of photobleaching is negligible in our experiments because of the refreshment of fluorophores by repeated DNA hybridization. The burst duration is thus controlled by the number of complementary nucleotides. This is evidenced by the fact that tbright is independent of laser power. In Figure 4d we show the mean tbright as a function of power density, collected from time traces of ∼100 single gold nanorods. We find that tbright is 0.4 s independent of power density, which confirms that any nonlinearity in power-dependent PCR is not caused by photobleaching. Fluorescence time traces and histograms of tbright distributions measured under different power densities can be found in the Supporting Information.
Near saturation, the PCR enhancement compared to free-space excitation becomes power-dependent. We therefore determine saturation curves by collecting time trajectories of fluorescence bursts under different excitation power densities ranging from 5.2 × 105 to 2.4 × 107 W m–2. The dynamic range of the power density series is limited by the laser spot size and excitation laser power (see the Supporting Information for more details). All the saturation curves are fitted with eq 1 yielding PCRmax and Isat. In Figure 5 we show the saturation curves of free-space fluorophores (see the Supporting Information for more data regarding the saturation dynamics without an antenna) and plasmon-coupled fluorophores for two gold nanorods with a longitudinal SPR of 670 and 638 nm, resonant with the excitation and emission wavelengths, respectively. We clearly observe a nonlinear dependence of the detected intensity commensurate with fluorescence saturation. As stated above, this nonlinearity is a result of fluorescence saturation instead of photobleaching at increased power density. We find that both PCRmax and Isat are significantly modified, and both are more enhanced for 670 nm than 638 nm plasmon wavelengths.
Figure 5.

Saturation curves of plasmon-enhanced and nonenhanced single-molecule fluorescence. Inset shows the same saturation curves on a linear scale. The saturation intensity of free imagers is determined to be Isat0 = 6.4 × 106 W m–2, and maximum PCR at saturation PCRmax = 8.3 × 103 s–1. For the particle with a SPR of 670 nm, PCRmax670 = 2.3 × 105 s–1 and Isat = 3.5 × 107 W m–2. For the particle with a SPR of 638 nm, PCRmax638 = 5.9 × 104 s–1 and Isat = 3.9 × 107 W m–2.
We now investigate the modified PCRmax and Isat for more particles to reveal the SPR wavelength dependencies. In Figure 6 we show the SPR wavelength-dependent PCRmax and Isat, which are obtained from the fits to the saturation curves. We employ here docking strands with different lengths to explore the dependence of the enhancement on the particle–fluorophore spacing. In Figure 6a,b we show the results for measurements in which 30 nt docking strands are used to fully cover the gold nanorods, and imager strands with ATTO647N labeled at the far-end of the complementary sequence, leading to ∼4 nm time-averaged spacing between fluorophore and particle (see the Supporting Information for the calculation of the time-averaged spacing). Panels c and d of Figure 6 show the results for measurements with 15 nt docking strands and a flipped imager strand with ATTO647N at the near-end of the complementary region, shortening the time-averaged particle–fluorophore spacing to ∼1.5 nm.
Figure 6.

(a and b) Modified PCRmax and Isat of plasmon-enhanced fluorescence for AuNRs coated with
30 nt docking strands (4 nm spacing) and (c and d) for AuNRs
coated with 15 nt docking strands and near-end labeled imager (1.5 nm
spacing). Note here that the simulated Isat* is normalized
to the near-field intensity at the position of the fluorophore to
be able to display both the measured and simulated values on the same y-axis, i.e., 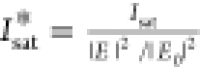 . In all graphs, red dots are experimental
data points, and black dots are simulation results with lines as a
guide to the eye. R2 values are used to
evaluate the fitting quality, and only curves with R2 >0.995 are plotted. See the Supporting Information for full simulation details including the decay
rates used in the simulations.
. In all graphs, red dots are experimental
data points, and black dots are simulation results with lines as a
guide to the eye. R2 values are used to
evaluate the fitting quality, and only curves with R2 >0.995 are plotted. See the Supporting Information for full simulation details including the decay
rates used in the simulations.
In the above results, we find that the PCRmax enhancement
matches well with theory, with the calculated 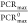 appearing as the upper limit in the experiments
because these events originate from particles that are aligned with
the excitation laser polarization. Particles with a longitudinal plasmon
that is resonant with the emission of the dye enhance PCRmax by nearly 300 fold, an effect dominated by the enhancement of the
radiative rate as described before.
appearing as the upper limit in the experiments
because these events originate from particles that are aligned with
the excitation laser polarization. Particles with a longitudinal plasmon
that is resonant with the emission of the dye enhance PCRmax by nearly 300 fold, an effect dominated by the enhancement of the
radiative rate as described before.
However, the predicted modification of Isat does not follow the measured modification. In Figure 6b,d we show the correlation between the longitudinal SPR and Isat, here plotted as the excitation laser power density required (in other words, Figure 2f divided by the orientation averaged wavelength-dependent near-field intensity at the location of the dye). We hypothesize that this could be due to plasmonic modification of triplet state dynamics. To understand this, it is useful to examine the dependence of Isat enhancement in the form
| 4 |
In the simulations
we have treated the term 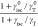 appearing in both eqs 3 and 4 as
unity, following Ebbesen et al.38 Considering
the fact that the PCRmax enhancement (and thus γr/γr0) closely follows the prediction, this could indicate that plasmonic
modification of triplet dynamics, or specifically the ratio γisc/γT. An Isat enhancement up to 40 as we measured may therefore be the result
of a ∼10-fold modification of the ratio γisc/γT. Modification of triplet dynamics has been reported,
but experimental studies are limited to a select number of cases47−51 that indeed report modest modifications, in line with our observation.
A quantitative investigation of triplet modifications requires a temporal
resolution that is not accessible in our current camera-based setup
but could be further investigated using, for example, fluorescence
correlation spectroscopy (FCS).46,51,52
appearing in both eqs 3 and 4 as
unity, following Ebbesen et al.38 Considering
the fact that the PCRmax enhancement (and thus γr/γr0) closely follows the prediction, this could indicate that plasmonic
modification of triplet dynamics, or specifically the ratio γisc/γT. An Isat enhancement up to 40 as we measured may therefore be the result
of a ∼10-fold modification of the ratio γisc/γT. Modification of triplet dynamics has been reported,
but experimental studies are limited to a select number of cases47−51 that indeed report modest modifications, in line with our observation.
A quantitative investigation of triplet modifications requires a temporal
resolution that is not accessible in our current camera-based setup
but could be further investigated using, for example, fluorescence
correlation spectroscopy (FCS).46,51,52
Our experiments indicate that the maximum photon count rate of a dye with an intrinsically high quantum yield can be enhanced by more than 100-fold by plasmon coupling, enabling single-molecule studies with superior signal-to-noise ratio particularly for short integration times. We employed our numerical model to explore how PCRmax and Isat depend on the size of the particle and photophysical properties of different types of emitters (see the Supporting Information). These simulations indicate that particles with a width in the range of 20–30 nm exhibit the highest PCRmax enhancement. A reduced enhancement of PCRmax for smaller particles is due to a reduced radiative efficiency of the plasmon, whereas larger particles suffer from line broadening due to radiation damping.
At a fixed particle size, the intrinsic radiative rate of the emitter then determines PCRmax. This indicates that certain classes of emitters might be more suitable to further push PCRmax. Comparing emitters with various intrinsic radiative rates (see the Supporting Information), we find that PCRmax up to 107 s–1 is feasible for emitters with γr0 = 109 s–1, corresponding to organic fluorophores, enzyme cofactors, and fluorescent proteins that typically have nanosecond fluorescence lifetimes.15 Semiconductor and carbon dots typically exhibit 1–10 ns lifetimes, whereas lanthanides and transition metal–ligand complexes with 100–1000 ns lifetimes are expected to yield > 100 times lower plasmon-enhanced PCRmax compared to organic fluorophores.53 Organic fluorophores as we used here thus provide the highest PCRmax, albeit at the expense of photostability compared to other emitters.
In summary, we presented a single-molecule study of plasmon-enhanced fluorescence of a dye with a high intrinsic quantum yield that is pumped close to saturation, where we focused on modification of the saturation dynamics by single metal nanoparticles using reversible single-molecule DNA hybridization. We have addressed the concern of photobleaching by employing low-affinity DNA interactions resulting in controlled residence times of the fluorophore near the particle, continuous refreshment of the dye by rebinding, and a well-controlled spacing between particle and fluorophore. We have measured the PCR as a function of excitation power, from which we derive the PCRmax and saturation intensity (Isat). The PCR was strongly modified because of plasmon coupling with a PCRmax that was enhanced by more than 2 orders of magnitude at ∼1.5 nm from the particle surface. The experimental findings were in good agreement with numerical simulations that include saturation. These experiments provide design rules to optimize the absolute photon output of single emitters. We envision that this will open the door to studying microsecond dynamics using real-time single-molecule fluorescence, e.g., DNA hairpin dynamics and transition paths in protein folding.
Acknowledgments
P.Z. and Y.W. acknowledge financial support from The Netherlands Organization for Scientific Research (NWO VIDI). We thank Marcel Verheijen for TEM imaging; Solliance and the Dutch province of Noord-Brabant are acknowledged for funding the TEM facility. This work is part of the research programme of the Foundation for Fundamental Research on Matter (FOM) which is financially supported by The Netherlands Organisation for Scientific Research (NWO). We thank Lex Dedding for useful discussions regarding bright-time measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c00155.
Sample preparation, single-molecule fluorescence microscopy and single-particle spectroscopy, calibration of TIRF excitation power density, free-space fluorescence saturation of single molecules, derivation of single-molecule photon count rate, fluorescence timetraces as a function of power density and SPR wavelength, and numerical simulations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sauer M.; Hofkens J.; Enderlein J.. Handbook of Fluorescence Spectroscopy and Imaging: From Single Molecules to Ensembles; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Hinterdorfer P.; Oijen A., Eds. Handbook of Single-Molecule Biophysics; Springer US: New York, NY, 2009. [Google Scholar]
- Elenko M. P.; Szostak J. W.; van Oijen A. M. Single-Molecule Imaging of an in Vitro-Evolved RNA Aptamer Reveals Homogeneous Ligand Binding Kinetics. J. Am. Chem. Soc. 2009, 131, 9866–9867. 10.1021/ja901880v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen A. M. Single-molecule approaches to characterizing kinetics of biomolecular interactions. Curr. Opin. Biotechnol. 2011, 22, 75–80. 10.1016/j.copbio.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Lu H. P.; Xun L.; Xie X. S. Single-Molecule Enzymatic Dynamics. Science 1998, 282, 1877–1882. 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- Lu H. P. Sizing up single-molecule enzymatic conformational dynamics. Chem. Soc. Rev. 2014, 43, 1118–1143. 10.1039/C3CS60191A. [DOI] [PubMed] [Google Scholar]
- Zheng D.; Lu H. P. Single-Molecule Enzymatic Conformational Dynamics: Spilling Out the Product Molecules. J. Phys. Chem. B 2014, 118, 9128–9140. 10.1021/jp5014434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney S. A.; Joo C.; Ha T. Analysis of Single-Molecule FRET Trajectories Using Hidden Markov Modeling. Biophys. J. 2006, 91, 1941–1951. 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenkamp B.; Schmid S.; Doroshenko O.; Opanasyuk O.; Kühnemuth R.; Rezaei Adariani S.; Ambrose B.; Aznauryan M.; Barth A.; Birkedal V.; et al. Precision and accuracy of single-molecule FRET measurementsa multi-laboratory benchmark study. Nat. Methods 2018, 15, 669–676. 10.1038/s41592-018-0085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C.; Ha T. Single-Molecule FRET with Total Internal Reflection Microscopy. Cold Spring Harbor Protocols 2012, 2012, pdb.top072058. 10.1101/pdb.top072058. [DOI] [PubMed] [Google Scholar]
- Rogers M. L.; Boutelle M. G. Real-Time Clinical Monitoring of Biomolecules. Annu. Rev. Anal. Chem. 2013, 6, 427–453. 10.1146/annurev.anchem.111808.073648. [DOI] [PubMed] [Google Scholar]
- Fabrizio E. D.; Schlücker S.; Wenger J.; Regmi R.; Rigneault H.; Calafiore G.; West M.; Cabrini S.; Fleischer M.; Hulst N. F. v.; et al. Roadmap on biosensing and photonics with advanced nano-optical methods. J. Opt. 2016, 18, 063003. 10.1088/2040-8978/18/6/063003. [DOI] [Google Scholar]
- Taylor A. B.; Zijlstra P. Single-Molecule Plasmon Sensing: Current Status and Future Prospects. ACS Sensors 2017, 2, 1103–1122. 10.1021/acssensors.7b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A.; Kloosterman W. P.; Hestand M. S. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 2019, 37, 72–85. 10.1016/j.tibtech.2018.07.013. [DOI] [PubMed] [Google Scholar]
- Moerner W. E.; Fromm D. P. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum. 2003, 74, 3597–3619. 10.1063/1.1589587. [DOI] [Google Scholar]
- Klehs K.; Spahn C.; Endesfelder U.; Lee S. F.; Fürstenberg A.; Heilemann M. Increasing the Brightness of Cyanine Fluorophores for Single-Molecule and Superresolution Imaging. ChemPhysChem 2014, 15, 637–641. 10.1002/cphc.201300874. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Halle J.; Wojdyr M.; Sahoo D.; Scheblykin I. G. Quantitative measurement of fluorescence brightness of single molecules. Methods Appl. Fluoresc. 2014, 2, 035003. 10.1088/2050-6120/2/3/035003. [DOI] [PubMed] [Google Scholar]
- Turunen P.; Rowan A. E.; Blank K. Single-enzyme kinetics with fluorogenic substrates: lessons learnt and future directions. FEBS Lett. 2014, 588, 3553–3563. 10.1016/j.febslet.2014.06.021. [DOI] [PubMed] [Google Scholar]
- Drexhage K. H. Influence of a dielectric interface on fluorescence decay time. J. Lumin. 1970, 1–2, 693–701. 10.1016/0022-2313(70)90082-7. [DOI] [Google Scholar]
- Khatua S.; Orrit M. Probing, Sensing, and Fluorescence Enhancement with Single Gold Nanorods. J. Phys. Chem. Lett. 2014, 5, 3000–3006. 10.1021/jz501253j. [DOI] [PubMed] [Google Scholar]
- Busson M. P.; Rolly B.; Stout B.; Bonod N.; Bidault S. Accelerated single photon emission from dye molecule-driven nanoantennas assembled on DNA. Nat. Commun. 2012, 3, 962. 10.1038/ncomms1964. [DOI] [PubMed] [Google Scholar]
- Kinkhabwala A.; Yu Z.; Fan S.; Avlasevich Y.; Müllen K.; Moerner W. E. Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nat. Photonics 2009, 3, 654–657. 10.1038/nphoton.2009.187. [DOI] [Google Scholar]
- Regmi R.; Al Balushi A. A.; Rigneault H.; Gordon R.; Wenger J. Nanoscale volume confinement and fluorescence enhancement with double nanohole aperture. Sci. Rep. 2015, 5, 15852. 10.1038/srep15852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flauraud V.; Regmi R.; Winkler P. M.; Alexander D. T. L.; Rigneault H.; van Hulst N. F.; García-Parajo M. F.; Wenger J.; Brugger J. In-Plane Plasmonic Antenna Arrays with Surface Nanogaps for Giant Fluorescence Enhancement. Nano Lett. 2017, 17, 1703–1710. 10.1021/acs.nanolett.6b04978. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Khatua S.; Zijlstra P.; Yorulmaz M.; Orrit M. Thousand-fold Enhancement of Single-Molecule Fluorescence Near a Single Gold Nanorod. Angew. Chem., Int. Ed. 2013, 52, 1217–1221. 10.1002/anie.201208125. [DOI] [PubMed] [Google Scholar]
- Khatua S.; Paulo P. M. R.; Yuan H.; Gupta A.; Zijlstra P.; Orrit M. Resonant Plasmonic Enhancement of Single-Molecule Fluorescence by Individual Gold Nanorods. ACS Nano 2014, 8, 4440–4449. 10.1021/nn406434y. [DOI] [PubMed] [Google Scholar]
- Puchkova A.; Vietz C.; Pibiri E.; Wünsch B.; Sanz Paz M.; Acuna G. P.; Tinnefeld P. DNA Origami Nanoantennas with over 5000-fold Fluorescence Enhancement and Single-Molecule Detection at 25 M. Nano Lett. 2015, 15, 8354–8359. 10.1021/acs.nanolett.5b04045. [DOI] [PubMed] [Google Scholar]
- Laurence T. A.; Ly S.; Bourguet F.; Fischer N. O.; Coleman M. A. Fluorescence Correlation Spectroscopy at Micromolar Concentrations without Optical Nanoconfinement. J. Phys. Chem. B 2014, 118, 9662–9667. 10.1021/jp505881z. [DOI] [PubMed] [Google Scholar]
- Punj D.; de Torres J.; Rigneault H.; Wenger J. Gold nanoparticles for enhanced single molecule fluorescence analysis at micromolar concentration. Opt. Express 2013, 21, 27338–27343. 10.1364/OE.21.027338. [DOI] [PubMed] [Google Scholar]
- Punj D.; Mivelle M.; Moparthi S. B.; van Zanten T. S.; Rigneault H.; van Hulst N. F.; García-Parajó M. F.; Wenger J. A plasmonic /‘antenna-in-box/’ platform for enhanced single-molecule analysis at micromolar concentrations. Nat. Nanotechnol. 2013, 8, 512–516. 10.1038/nnano.2013.98. [DOI] [PubMed] [Google Scholar]
- Punj D.; Regmi R.; Devilez A.; Plauchu R.; Moparthi S. B.; Stout B.; Bonod N.; Rigneault H.; Wenger J. Self-Assembled Nanoparticle Dimer Antennas for Plasmonic-Enhanced Single-Molecule Fluorescence Detection at Micromolar Concentrations. ACS Photonics 2015, 2, 1099–1107. 10.1021/acsphotonics.5b00152. [DOI] [Google Scholar]
- Wenger J. Fluorescence Enhancement Factors on Optical Antennas: Enlarging the Experimental Values without Changing the Antenna Design. Int. J. Opt. 2012, 2012, 828121 10.1155/2012/828121. [DOI] [Google Scholar]
- Wenger J.Fluorescence spectroscopy enhancement on photonic nanoantennas. arXiv (physics) 2017, 1709.06749. https://arxiv.org/abs/1709.06749. [Google Scholar]
- Visser E. W.; Horáček M.; Zijlstra P. Plasmon Rulers as a Probe for Real-Time Microsecond Conformational Dynamics of Single Molecules. Nano Lett. 2018, 18, 7927–7934. 10.1021/acs.nanolett.8b03860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B.; Eaton W. A. Protein folding studied by single-molecule FRET. Curr. Opin. Struct. Biol. 2008, 18, 16–26. 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia M. B.; Borgia A.; Best R. B.; Steward A.; Nettels D.; Wunderlich B.; Schuler B.; Clarke J. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature 2011, 474, 662–665. 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M. Protein folding and misfolding. Nature 2003, 426, 884–890. 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Wenger J.; Gérard D.; Dintinger J.; Mahboub O.; Bonod N.; Popov E.; Ebbesen T. W.; Rigneault H. Emission and excitation contributions to enhanced single molecule fluorescence by gold nanometric apertures. Opt. Express 2008, 16, 3008. 10.1364/OE.16.003008. [DOI] [PubMed] [Google Scholar]
- Gérard D.; Wenger J.; Bonod N.; Popov E.; Rigneault H.; Mahdavi F.; Blair S.; Dintinger J.; Ebbesen T. W. Nanoaperture-enhanced fluorescence: Towards higher detection rates with plasmonic metals. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 77, 045413. 10.1103/PhysRevB.77.045413. [DOI] [Google Scholar]
- Wientjes E.; Renger J.; Cogdell R.; van Hulst N. F. Pushing the Photon Limit: Nanoantennas Increase Maximal Photon Stream and Total Photon Number. J. Phys. Chem. Lett. 2016, 7, 1604–1609. 10.1021/acs.jpclett.6b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenester U.; Trügler A. MNPBEM A Matlab toolbox for the simulation of plasmonic nanoparticles. Comput. Phys. Commun. 2012, 183, 370–381. 10.1016/j.cpc.2011.09.009. [DOI] [Google Scholar]
- Beuwer M. A.; Prins M. W. J.; Zijlstra P. Stochastic Protein Interactions Monitored by Hundreds of Single-Molecule Plasmonic Biosensors. Nano Lett. 2015, 15 (5), 3507–3511. 10.1021/acs.nanolett.5b00872. [DOI] [PubMed] [Google Scholar]
- Jungmann R.; Steinhauer C.; Scheible M.; Kuzyk A.; Tinnefeld P.; Simmel F. C. Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett. 2010, 10, 4756–4761. 10.1021/nl103427w. [DOI] [PubMed] [Google Scholar]
- Schnitzbauer J.; Strauss M. T.; Schlichthaerle T.; Schueder F.; Jungmann R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]
- Vogelsang J.; Kasper R.; Steinhauer C.; Person B.; Heilemann M.; Sauer M.; Tinnefeld P. A Reducing and Oxidizing System Minimizes Photobleaching and Blinking of Fluorescent Dyes. Angew. Chem., Int. Ed. 2008, 47, 5465–5469. 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- Eggeling C.; Volkmer A.; Seidel C. A. M. Molecular Photobleaching Kinetics of Rhodamine 6G by One- and Two-Photon Induced Confocal Fluorescence Microscopy. ChemPhysChem 2005, 6, 791–804. 10.1002/cphc.200400509. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Aslan K.; Previte M. J. R.; Malyn S. N.; Geddes C. D. Metal-Enhanced Phosphorescence: Interpretation in Terms of Triplet-Coupled Radiating Plasmons. J. Phys. Chem. B 2006, 110, 25108–25114. 10.1021/jp065261v. [DOI] [PubMed] [Google Scholar]
- Stefani F. D.; Vasilev K.; Bocchio N.; Gaul F.; Pomozzi A.; Kreiter M. Photonic mode density effects on single-molecule fluorescence blinking. New J. Phys. 2007, 9, 21–21. 10.1088/1367-2630/9/2/021. [DOI] [Google Scholar]
- Chaudhuri D.; Li D.; Sigmund E.; Wettach H.; Höger S.; Lupton J. M. Plasmonic surface enhancement of dual fluorescence and phosphorescence emission from organic semiconductors: effect of exchange gap and spin–orbit coupling. Chem. Commun. 2012, 48, 6675. 10.1039/c2cc31843a. [DOI] [PubMed] [Google Scholar]
- Gill R.; Tian L.; van Amerongen H.; Subramaniam V. Emission enhancement and lifetime modification of phosphorescence on silver nanoparticle aggregates. Phys. Chem. Chem. Phys. 2013, 15, 15734. 10.1039/c3cp50407g. [DOI] [PubMed] [Google Scholar]
- Wenger J.; Cluzel B.; Dintinger J.; Bonod N.; Fehrembach A.-L.; Popov E.; Lenne P.-F.; Ebbesen T. W.; Rigneault H. Radiative and Nonradiative Photokinetics Alteration Inside a Single Metallic Nanometric Aperture. J. Phys. Chem. C 2007, 111, 11469–11474. 10.1021/jp0726135. [DOI] [Google Scholar]
- Eggeling C.; Widengren J.; Rigler R.; Seidel C. A. M. Photobleaching of Fluorescent Dyes under Conditions Used for Single-Molecule Detection: Evidence of Two-Step Photolysis. Anal. Chem. 1998, 70, 2651–2659. 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.