

Abstract
Organic compounds in the atmosphere vary widely in their molecular composition and chemical properties, so no single instrument can reasonably measure the entire range of ambient compounds. Over the past decade, a new generation of in-situ, field-deployable mass spectrometers has dramatically improved our ability to detect, identify, and quantify these organic compounds, but no systematic approach has been developed to assess the extent to which currently available tools capture the entire space of chemical identity and properties that is expected in the atmosphere. Reduced-parameter frameworks that have been developed to describe atmospheric mixtures are exploited here to characterize the range of chemical properties accessed by a suite of instruments. Multiple chemical spaces (e.g. oxidation state of carbon vs. volatility, and oxygen number vs. carbon number) were populated with ions measured by several mass spectrometers, with gas- and particle-phase α-pinene oxidation products serving as the test mixture of organic compounds. Few gaps are observed in the coverage of the parameter spaces by the instruments employed in this work, though the full extent to which comprehensive measurement was achieved is difficult to assess due to uncertainty in the composition of the mixture. Overlaps between individual ions and regions in parameter space were identified, both between gas- and particle-phase measurements, and within each phase. These overlaps were conservatively found to account for little (<10%) of the measured mass. However, challenges in identifying overlaps and in accurately converting molecular formulas into chemical properties (such as volatility or reactivity) highlight a continued need to incorporate structural information into atmospheric measurements.
Graphical abstract
Multiple reduced-parameter frameworks are coupled to assess the degree to which multi-instrument measurements fully capture the chemical space
Introduction
The atmosphere contains an immense number of organic compounds. This chemical complexity arises not only from the range of atmospheric sources – namely emissions from plants (biogenic emissions) and combustion processes (of biomass, biofuels, and fossil fuels) – but also from subsequent atmospheric oxidation processes, which can lead to the formation of a very large number of oxidized products from a given precursor.1–3 As a result of these complex emissions and oxidation processes, atmospheric organic species span an extremely wide range in chemical formulas, chemical structures, and chemical/physical properties. This diversity in properties includes molecular size (with molecules having from one to tens of carbon atoms), volatility (from volatile species present only in the gas phase to effectively nonvolatile ones present only in the condensed phase), and degree of oxidation (from reduced alkanes to highly oxidized multifunctional species). They also include a wide range of functional groups (including carbonyls, alcohols, acids, nitrates, peroxides, etc.) and carbon skeletons (with branches, rings, and aromatic moieties).
Such chemical complexity poses a major challenge in atmospheric organic chemistry, given the analytical challenges associated with detecting, quantifying, and characterizing such a large number and wide diversity of compounds. Mass spectrometric instruments, which can detect a range of species simultaneously, are well-suited for making such measurements; this is clearly illustrated by the dramatic influence that newly-developed mass spectrometric instruments have had on our understanding of atmospheric chemical composition over the last fifteen years or so (e.g. refs 4 and 5). However, because of the selectivity that can arise from the inlet design and ionization schemes, a single mass spectrometric instrument typically cannot measure all the organic species in a sample. Instead the full chemical characterization of atmospheric organic species can only be made using multiple instruments simultaneously. While this multi-instrument approach maximizes the fraction of organic species measured (and the area in “chemical space” covered), there are complications associated with it. These include the completeness of the measurement suite (whether any species or classes of species are left unmeasured, due to systematic measurement gaps), as well as overlaps among instruments (the extent to which different instruments may measure the same species within the mixture). To our knowledge these have never been systematically explored for modern atmospheric chemistry instrumentation, in particular the new generation of advanced mass spectrometric instruments that are seeing widespread use in the laboratory and field.
In this work we describe and apply an approach for such an assessment, by comparing the range of chemical space occupied by atmospheric organic compounds to the capabilities of a number of instruments. A full description of a given molecule requires the use of several parameters. For example, description of molecules’ chemical formulas require the use of at least three (largely independent) variables, the number of carbon atoms, the number of oxygen atoms, and the number of hydrogen atoms (or more if heteroatoms such as N or S are included); however these values alone provide relatively little information on molecular structure or properties, requiring additional dimensions in chemical space to describe a given species. Because of the challenges in visualization of such multidimensional spaces, a number of recently-developed frameworks have reduced chemical space (and descriptions of organic species) down to two parameters only.6–10 These reduced-parameter descriptions have substantial value for describing properties of interest for a given study or application; at the same time, important chemical information will be lost by the reduction of chemical space to just two dimensions, and so no single framework is sufficient to fully understand the range in chemical properties of a mixture.
Therefore here we utilize multiple two-dimensional frameworks to assess the ability of multi-instrument measurements to fully characterize a complex mixture of atmospheric organic compounds. Specific goals are as follows:
Characterize the strengths and weakness of a given instrument or suite of instruments without assuming prior knowledge about instrument capabilities
Identify “measurement gaps”: regions of chemical spaces that are not accessed by measurements and may represent unmeasured compounds
Identify “overlaps”: individual ions and regions of parameter spaces that are measured by multiple instruments; and
Explore the extent to which gaps, overlaps, and lack of structural information challenge our ability to comprehensively measure complex organic mixtures
We apply this approach to a complex mixture of organic compounds generated in the laboratory, intended to simulate the complexity and chemical properties of ambient atmosphere organic species, by employing a suite of mass spectrometric instruments that are currently used widely in laboratory and field studies. This work thus provides an exploration of the capability of currently available instrumentation to comprehensively characterize ambient atmospheric organic carbon.
Methodology
The frameworks discussed in this work each describe gas- and particle-phase mass with two parameters, in combination using six unique parameters: oxidation state of carbon (OSC), volatility in terms of saturation concentration (C*), carbon number (nC), oxygen number (nO), and ratios of elements, specifically hydrogen-to-carbon (H:C) and oxygen-to-carbon (O:C). Each instrument used in this work accesses these parameters differently, either through direct measurement, or through estimation from measured quantities. Furthermore, some instruments measure individual molecular ions to compile an ensemble composition, while some measure bulk average properties. This yields a complex set of data that must be synthesized, and parameters interconverted, for comprehensive inclusion in frameworks. See Appendix for calculation of (and interconversion between) these parameters. A summary of the data accessed by the instruments brought to bear in this work is shown in Table 1. Operating details are discussed below. Note that none of these instruments provide molecular structure of the measured analytes.
Table 1.
Summary of instruments and accessible parameters in this work. X = measured directly, O = calculated from measured parameters, -- = not available
| Instrument | Phase | Bulk/ molec. | nC | nO | C* | OSC | H:C O:C | Structure |
|---|---|---|---|---|---|---|---|---|
| PTR | gas | molec. | X | X | O | O | X | -- |
| I- CIMS | gas | molec. | X | X | O | O | X | -- |
| NO3- CIMS | gas | molec. | X | X | O | O | X | -- |
| FIGAERO I- CIMS | part. | molec. | X | X | X | O | X | -- |
| TD-AMS | part. | bulk | O | O | X | O | X | -- |
Instrument descriptions
Four high-resolution time-of-flight mass spectrometers (HR-ToF-MS) were used to measure gas- and particle-phase organic compounds. All four instruments rely on the same detector (HTOF, Tofwerk) with time-resolution of minutes to seconds and mass resolution of ~4,000 Δm/m, with different sampling and collection techniques to target different ranges of volatility and functionality. Pertinent operational details of all instruments are discussed below. Only rough calibrations are necessary to draw the conclusions and produce the figures in this work, so discussion of calibration is limited here to overviews, with references provided for interested readers.
“PTR”:
Proton Transfer Reaction Mass Spectrometer (Ionicon Analytik).11,12 Air was sampled through 1 meter PEEK polymer capillary heated to 80 °C, with an unheated Teflon® filter in line to prevent intrusion of particles to the detector. Analytes are sampled into a “drift tube” to react with H3O+ to yield an ion of the molecular formula plus H+, allowing detection of any analytes with a proton affinity higher than that of water. Many large compounds (> C5) are known to fragment or dehydrate upon ionization clouding the interpretation of ions as analyte molecular formulas.13 Calibration is based on a general reaction rate between analytes and reagent ions, adjusted for transmission losses in the instrument and empirical correction factors determined through introduction of known compounds.14
“I− CIMS”:
Chemical Ionization Mass Spectrometer using iodide as a reagent ion (Aerodyne Research Inc.).15–17 Air was sampled at 1 lpm through 20 cm of 6.35 mm (¼”) O.D. stainless steel tubing extending into the chamber connected to the instrument by a ~1 meter Teflon® line. Added to this sample flow was 9 lpm of pure N2, diluting the sample but decreasing the residence time within the sample line. The instrument sub-sampled 2 lpm of this dilute sample. Iodide ions,, generated in-situ from methyl iodide using a polonium source, were reacted with sample to form ion-molecule clusters that were analyzed and detected by the mass spectrometer at their molecular formula plus I−. This ionization scheme has a theoretically calculable maximum sensitivity based on the ion-molecule reaction rate, but deviation from this “kinetic sensitivity” occurs based on the strength of the bond between the iodide and the analyte within this cluster. A detailed body of work by Iyer, Lee, Lopez-Hilfiker, and co-workers15,18,19 has demonstrated that this bond strength controls the sensitivity of this instrument to an analyte, and that it is empirically measurable by modulating operating parameters. Their approach was used here to calibrate ions detected by I− CIMS in both the gas and particle phase.
“FIGAERO I− CIMS”:
Particle-phase compounds were also measured by I− CIMS using the FIGAERO (Filter Inlet for Gas and AEROsols) inlet.20 Particles were collected onto a Teflon filter and subsequently desorbed by heated N2 and sampled into the ion-molecule reaction chamber, where the analytes are ionized, detected, and calibrated as discussed above. The temperature at which an ion is desorbed and detected correlates with the volatility of the collected mass.20,21 The relationship between FIGAERO desorption temperature and volatility described by Lopez-Hilfiker et al.20 was applied to these data; though uncertainty exists in this relationship, even an error of 1–2 orders of magnitude in estimated vapor pressure of these ions does not change the conclusions of this work.
“NO3− CIMS”:
Chemical Ionization Mass Spectrometer using nitrate as a reagent ion (Aerodyne Research, Inc.).22,23 Air was sampled at 1 lpm through ~10 cm stainless steel tubing, then immediately diluted with 9 lpm of pure N2 as laminar sheath flow to prevent loss to the walls. The centerline flow of sample was reacted with NO3−, generated in-situ through x-ray ionization of nitric acid, to form ion-molecule clusters that are analyzed and detected by the mass spectrometer at their molecular formula plus NO3−. These ions are calibrated to their lower-limit estimated mass loading by assuming they are detected at maximum kinetic sensitivity based on the ion-molecule reaction rate as determined by empirical calibration of malonic acid.
The PTR, I− CIMS, and NO3− CIMS all utilize forms of “soft ionization,” striving to preserve the molecular ion with minimal fragmentation. Analytes in each of these instruments are measured by clustering with a reagent ion, which is ignored in order to accurately describe analytes in terms of their chemical properties and compare analytes between instruments. For example, the ion (I)CxHyOz− measured by I− CIMS was considered the same ion as (NO3)CxHyOz− measured by NO3− CIMS and CxHy+1Oz+ measured by PTR.
“TD-AMS”:
Thermal Denuder – Aerodyne Aerosol Mass Spectrometer (Aerodyne Research Inc.). Air was passed through a heated annular carbon denuder, volatilizing particle-phase mass and removing the evolved gases.24 Particles reaching the AMS were focused into a beam and impacted onto a 600 °C vaporizer.25 Volatilized analytes were ionized by electron impact for analysis by the mass spectrometer. Unlike the soft-ionization mass spectrometers, which provide molecular information about their analytes, this is a bulk measurement technique. The observed ions are fragments, from which particle composition can be determined. Elemental analysis of particles was calculated using the technique of Canagaratna et al.26,27 Denuded particles were compared to particles sampled through a bypass line to determine mass fraction and elemental composition remaining as a function temperature. This property was converted to an approximate volatility based on the work of Faulhaber et al.24 This instrument was used only for measurements of particle properties (elemental composition, volatility) but not mass.
“SMPS”:
Bulk measurements of particle-phase mass were obtained with a Scanning Mobility Particle Sizer (TSI Incorporated) calibrated by the manufacturer. Particles were classified by diameter and converted to mass concentrations using an assumed density of 1.4 g m−3.28
Chamber oxidation
In this study, a complex mixture of organic compounds (most importantly species spanning a range of degrees of oxidation) was generated by the chamber oxidation of α-pinene, a model biogenic hydrocarbon by the hydroxyl radical (OH). Details of chamber operation are described elsewhere29 so are overviewed only briefly here. Reactions occurred in a temperature-controlled 7.5 m3 Teflon chamber by the in-situ generation of hydroxyl radicals (OH) through the photolysis of nitrous acid (HONO), generated by the reaction of sulfuric acid and sodium nitrite. In the experiment described here, 60 ppb of α-pinene was oxidized in the presence of ammonium sulfate seed aerosol at 20 °C at low (<5%) relative humidity. NO levels were sufficiently high to dominate RO2 chemistry (“high-NOX” conditions). OH concentrations above ambient are maintained such that after approximately 8 hours reaction time, the product mixture was exposed to the equivalent of approximately 24 hours of oxidation in the atmosphere (assuming an average atmospheric [OH] of 2×106 molec cm−3). The focus of this work is on analytical capabilities, so measurements reported here are mostly averages over the entire experiment. The oxidative evolution of the reaction mixture, and subsequent chemical insight, will be the focus of future work.
Results
Distribution of measured ions in common parameter spaces
In order to characterize the capabilities of the instrument suite and the chemical properties of the sample mixture, we place all measured data into a variety of recently-developed reduced-parameter frameworks. Four two-dimensional frameworks are used in this work:
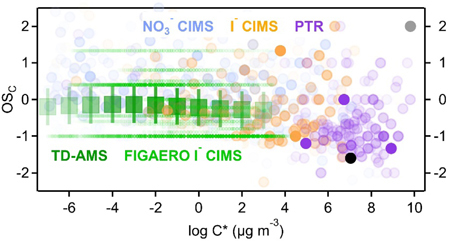
These four spaces, populated with the measurements taken in this work (colored by instrument), are shown in Fig. 1. Panels on the left are populated with individual ions (circles) to demonstrate the extent of coverage in each of these frameworks, while panels on the right show the area in the parameter space that encompasses most of the mass (80%) measured by each instrument (i.e., where a given instrument measures most signal). In dark green markers are TD-AMS measurements; these points are not single compounds, but instead represent average measurements, binned by decades in volatility, with the approximate ranges spanned illustrated by the shown error bars.
Figure 1.

Reduced parameter frameworks populated by ions observed in the sample mixture (α-pinene photolysis products). Left: individual ions as circles, with opacity correlated with fraction of measured instrument mass. Right: ovals that contain 80% of measured instrument mass (optimized as smallest area ellipse that meets this criteria). Four frameworks are shown: (a,b) oxidation state by volatility; (c,d) hydrogen to carbon ratio by oxygen to carbon ratio; (e,f) oxygen number by carbon number; and (g,h) oxidation state by carbon number. In all panels, particle phase measurements shown are the bulk average measurements from the TD-AMS, binned into 10 bins (dark connected green markers) with approximate range shown as error bars based on uncertainty and known constituents, and the FIGAERO I− CIMS (light green oval and dots), using nC,calc estimated from measured volatility. Gas phase measurements shown are from the NO3− CIMS (light blue), I− CIMS (orange), and PTR (purple). Carbon monoxide and dioxide, not measured by these mass spectrometers, are shown in gray and numbered as 1 and 2, respectively, with precursor α-pinene numbered as 3.
Coverage and gaps.
The focus of this work is on the extent to which information may be missing from these parameterizations, and to which these frameworks can elucidate the capabilities and limitations of a given instrument, set of instruments, or measurement technique.
Each instrument accesses a relatively large swath of the area described by all of these frameworks (panels in Fig. 1), but most of the mass is confined to a somewhat smaller region (right panels). For gas-phase compounds, nearly the entire parameter space of oxidation state and volatility (Fig. 1a) is covered by the instruments in this suite. Carbon monoxide and dioxide are not accessed by the mass spectrometers used in this work, but are expected as oxidation products of α-pinene, so are also shown. Each instrument measures most mass in a discrete, mostly unique region (Fig. 1b). Volatile, less oxidized ions are measured by PTR, gases with moderate volatility and moderate oxidation are measured by I− CIMS, while low-volatility, highly-oxidized gases are measured by NO3− CIMS.
A similar conclusion is drawn from the other three parameter spaces: the gas-phase instruments in this suite measure ions in relatively unique regions of the parameter space, with a trend in increasing oxygen content, and increasing carbon number, going from PTR to I− CIMS to NO3− CIMS. These trends in nO and nC are both associated with decreasing volatility and decreasing hydrogen content. The regions occupied by each instrument are consistent with their expected strengths based on their inlet design (potential losses to surfaces) and their ion-molecule formation processes (sensitivity and selectivity). However, even in the absence of such information, these data empirically constrain the capabilities of a given instrument from measurements. The I− CIMS, for instance, is observed to measure moderate volatility gases with 3 to 7 oxygen atoms and fewer than 10 carbon atoms; this characterization emerges without any appreciable prior knowledge about the capabilities of an instrument, a valuable approach in the development of new instrumentation and analytical techniques and in the characterization and validation of an experimental setup.
Attempting to determine the measurement range of a given instrument, or whether a parameter space is fully covered by available instrumentation, unfortunately has a major inherent limitation. Without knowing in advance what chemical species are present (or absent) in a mixture, it is difficult to determine the extent to which a given instrument suite fully characterizes the atmospheric mixture. Top-down measured constraints could aid in estimates of completeness, including a direct measurement of “total suspended carbon” or the total reactivity of the product mixture to an oxidant such as OH. However, these methods often yield disagreement between the observed constraint and the measured concentrations32 and the inherent uncertainty of any measurement makes it almost impossible to rule out the possibility that a small but not insignificant fraction (e.g., 15%) of organic carbon is left unmeasured. Furthermore, these methods do not provide substantial insights into the chemical properties or structures of any unmeasured compounds. Conversely, gaps in measurements can only be identified by placing data in the context of a continuous parameter space in which missing data is apparent. In other words, without an idea of what everything is, it is difficult to determine if everything is being measured.
When “empty space” is apparent in a given parameter space, it can be due either to a lack of the ability to measure the compound, or a lack of the compound itself. An example of this issue is that these data lack any measurements of small alkanes and aromatic hydrocarbons, which is apparent from the lack of any detected ions at OSC<2, O:C=0, and H:C>2 (alkanes), or O:C=0 and H:C~1 (aromatics). Neither are formed from α-pinene oxidation, thus neither are detected in this study. However, while aromatics are measurable by the PTR, alkanes are not,14 and the difference between these scenarios cannot be easily determined. All the gas-phase instruments used in this work can be operated with different reagent ions, so missing measurements in a given study can be probed through use of other instrument conditions (e.g. NO+ or O2+ as PTR reagent ions can indeed measure alkanes33). However, the coverage reduced-parameter spaces in Fig. 1 suggest that the suite of instruments used in this work are relatively comprehensive.
A few potential measurement gaps are observed in Fig. 1e (nO vs. nC space), in which no ions are observed with high oxygen number and low carbon number, or at low carbon number and high oxygen number. The former region is largely prohibited by reasonable chemical valence, but the latter might reasonably be expected to be populated. It is not possible to say with certainty from these data whether compounds of nO <~7 and nC > 10 represent a true gap in measurement, or are measurable but not formed in this chemical system.
The multi-framework approach to simultaneously characterize the capabilities of the instrumentation and the properties of organic mixtures is, as with any approach, subject to these uncertainties associated with whether a particular compound class is present. However, this is addressed at least partially by the use of multiple frameworks, as shown in Fig. 1. Populating several different parameter spaces with data from an instrument or instrument suite provides a broader understanding of its capabilities, and more detailed insights into the range of measurements it facilitates. Furthermore, the simplification of data in frameworks can result in multiple molecular formulas collapsing to a single point in a given parameter space which can be resolved by examining other frameworks; for instance, decanol and pinonaldehyde both have the formula C10HxO2, so overlap in the nO vs. nC space despite substantially different chemical properties and structures, but are resolved in the OSC vs. C* space. Coupling several different frameworks therefore reduces the possibility that measurement gaps may be obscured by their simplified parameterization, and is a useful approach to determine the ranges of an instrument suite. From Fig. 1 it is clear that in this highly complex system, few, if any, regions in chemical space are not accessed by this instrument suite, though the extent to which this is achieved in any given set of measurements is entirely dependent on the instruments brought to bear and the diversity of the chemical system.
Overlaps: particle-phase measurements.
The largest clear area of overlap in Fig. 1 is particle-phase mass, which was measured both by its bulk properties (TD-AMS and SMPS) and its molecular composition (FIGAERO I− CIMS). Mass measured by the FIGAERO I− CIMS, though it has significant uncertainty, agrees well with the bulk measurement of particle-phase mass from the SMPS (Fig. 2a), suggesting approximately complete overlap between particle-phase measurements. Furthermore, average measured chemical properties agree well between these measurement techniques (Figs. 2b–2c). Carbon number measurements from the FIGAERO I− CIMS, calculated in this figure from volatility measurement (see Appendix) agrees well with those from the TD-AMS. This agreement exists throughout the duration of the experiment. Time dependence of elemental ratios, measured by both particle-phase instruments, is shown in Fig. 2b. Both O:C and H:C are observed to be in good quantitative agreement, differing by less than 0.05 and 0.10, respectively, and following approximately the same trends throughout the experiment. Furthermore, volatility as calculated from the desorption profile of the FIGAERO particle sample is in excellent agreement with that observed by the TD-AMS, as shown in Fig. 2c (broken lines). This agreement in both composition and volatility is reflected in the frameworks shown in Fig. 1: in all cases the TD-AMS data fall almost entirely within the range of the FIGAERO I− CIMS mass, indicating particle-phase instruments are measuring essentially the same species.
Figure 2.

Comparison of measurements by particle-phase instruments (a) FIGAERO I− CIMS mass vs SMPS mass (assumed density = 1.4 g cm−3), shaded region is uncertainty. (b) Time-series of elemental ratios FIGAERO I− CIMS to TD-AMS. (c) Histograms of volatility distributions, directly measured by FIGAERO I- CIMS (black solid line) and TD-AMS (gray dashed line), and estimated from FIGAERO I− CIMS ions using Eq. 3 as solid green line.
Volatility can also be estimated from the molecular composition measured after heating/volatilization of the particle-phase species within the FIGAERO. However, this calculated distribution (Fig. 2c, solid green line) is substantially different than distributions determined by thermal techniques, suggesting that such speciated measurements do not accurately reflect the volatility of the particle-phase compounds. These volatility distributions imply substantially different distributions of carbon number, as in Fig. 3, which shows the difference between parameters directly obtained from molecular formulas (i.e., nC and nO), versus composition as inferred from volatility and elemental ratios (i.e. nC,calc and nO,calc). Though the general region of coverage is similar between approaches, the majority of the mass is confined to ions with 9 and 10 carbon atoms in their formulas, while the desorption profiles suggest a much broader distribution across compounds with 10 to 20 carbon atoms, consistent with bulk measurements by TD-AMS. This implication of carbon numbers greater than those of the precursor strongly suggests the presence of accretion reactions; previous work34–36 suggests that decomposition of these accretion products into component monomers may occur in thermal analyses of atmospheric organic compounds, accounting for the discrepancy shown in Fig. 3.
Figure 3.

Comparison of distribution of FIGAERO I− CIMS in two frameworks: (a,b) oxygen number by carbon number, and (c,d) oxidation state of carbon by carbon number. Directly measured parameters from molecular formulas (nC,nO) are blue (squares). Parameters calculated from volatility based on Eqs. 2 and 4 (nC,calc,nO,calc) are green (dots). Left: individual ions shown, opacity proportional to measured mass. Right: ovals that contain 80% of measured instrument mass (optimized as smallest area ellipse that meets this criteria).
Overlap between measurements made by the particle-phase techniques consequently appears to be nearly total, with bulk approaches and summed molecular measurements in good agreement in terms of elemental composition and volatility and mass. However, the discrepancy between the parameters inferred through volatility-based and formula-based approaches, even using a single instrument, highlights remaining challenges in understanding the actual molecular structures and identities of compounds present within the particle phase.
Overlaps gas- and particle-phase measurements.
Further, in some regions, gas- and particle-phase measurements overlap, providing a rough picture of the areas of chemical space in which these phases can co-exist. This is observed for I− CIMS measurements across both phases, which likely co-exist due to continuous partitioning between phases as expected in the region of volatility (C* = 1 to 1000 μg m−3) where overlap exists (semivolatile organic species). There is even overlap between measurements of lower-volatility species by particle-phase instruments and the gas-phase measurements by the NO3− CIMS. These highly oxidized, high-carbon-number species are likely formed in the gas phase, and sampled by the CIMS before rapid condensational losses to particles or chamber walls.5 These overlaps in chemical space between gas- and particle-phase measurements are not true overlaps in measurements, but rather provide a valuable opportunity to explore the chemistry and dynamics of a system. For example, though most of the gas-phase mass has 10 carbon atoms or fewer, a number of ions (particularly in NO3− CIMS data) are observed with carbon backbones larger than the precursor; such species can be formed only through oligomerization or accretion reactions. This region of parameter space is also occupied by particle-phase measurements, suggesting a potential formation pathway for this mass through gas-phase reactions followed by condensation.
Overlaps: gas-phase measurements.
While most mass measured by each gas-phase instrument exists in a region of parameter space that is reasonably unique for most frameworks (Fig. 1), there are notable regions of overlap. This includes overlap between PTR and I− CIMS, as well as between I− CIMS and NO3− CIMS. However, even these areas of overlap in chemical space do not mean that the same species is being measured by more than one instrument, since most molecular formulas do not unambiguously represent a single isomer. Thus an ion measured by one instrument is not necessarily from the same compound as the same ion measured by a different instrument. Determination of whether identical ions are from a single chemical species requires the introduction of another dimension; here we examine the similarity in time dependence of such ions. The probability that each instrument measures the same isomer can be qualitatively inferred from the correlation in time evolution between ions, with a high correlations suggesting measurement of the same compound, and low (or negative) correlations coefficient suggesting the ions are in fact from different species.
Results from this analysis are shown in Fig. 4, which shows the same data from Fig. 1g (OSC vs nC space) but highlights molecular formulas that are measured by more than one instrument. There is a reasonably large number of multiply-measured ions; these species are colored by the correlation coefficient of the two time dependences. This approach also accounts for any PTR ions that undergo dehydration subsequent to ionization, since dehydration leads to no change in OSC or nC. However, this approach does not take into account fragmentation of the carbon skeleton within the PTR, since that process is complex and poorly understand. Consequently, due to potential fragmentation and to different instrument sensitivity to isomers, being measured as the same ion in multiple instruments is neither necessary nor sufficient to indicate an overlap in measurement of a molecular species. It is therefore exceedingly difficult to accurately identify all overlapping measurements. Nonetheless, the correlation coefficients between the same ion measured by multiple instruments are generally low, suggesting relatively little overlap among instruments.
Figure 4.

(a) Distribution of all measured gas-phase ions in the oxidation state by volatility framework. Ions measured by a single instrument a hollow light grey circles Ions that are measured by multiple instruments shown as larger solid circles colored by correlation (r) between ion measured by each instrument; red: same molecular formula, blue: PTR formula is dehydrated molecular formula of other instrument. Three examples of ions measured by two instruments are labelled and shown as (b), (c), and (d), colored as in Fig. 1 and (e), pie chart of measured mass of each gas-phase instrument. Mass potentially measured by multiple instruments in red (dark red: both instruments have same formulas; pink: PTR formula is dehydrated I− CIMS formula); all overlapping ions included regardless of correlation coefficient, so represents the upper limit of overlap.
This is shown in more detail in Fig 4b-d, which shows the time dependences of few representative cases. In Fig. 4b, an ion measured by both I− and NO3− CIMS (C8H10O5) are of a similar magnitude and tightly correlated, and thus may be the same species; however they diverge at the end of the experiment, so it may be a combination of a few species, one or more of which overlap. Figure 4c, on the other hand, shows constant close correlation and similar magnitude, though the PTR ion is the dehydrated formula of the I− CIMS ion. In contrast, Fig. 4d shows two ions of the same formula that behave completely differently and are likely different isomers. While the variety of cases and varying degrees of overlap stymie a detailed understanding of overlaps, a reasonable or conservative (upper-limit) estimate of multiply-measured mass can be calculated by assuming all overlapping formulas are instances of multiply-measured compounds regardless of correlation coefficient. Including dehydrated formulas, less than 10% of observed mass is measured by more than one instrument (Fig. 4e), so overlaps between ions are not a major source of uncertainty in a total accounting of product mass.
Limitations of molecular formula-based approaches
By definition, mass spectrometric measurement approaches provide molecular formulas; however a given formula does not necessarily correspond to an individual compound. While a multi-framework approach provides substantial detail about the measurement setup and the system being measured, all of these parameterizations are inherently simplified by excluding any explicit structural information. This introduces some inherent biases and inadequacies to this approach. This limitation must be considered head-on in the interpretation of any reduced-parameter frameworks, as it can limit certain applications of these frameworks and/or introduce biases.
As an example, all of the compounds in the Master Chemical Mechanism (MCM) formed in the OH-initiated oxidation of α-pinene37,38 are shown in Fig. 5 as a function of compound carbon number and the number of unique isomers of each molecular formula. Figure 5a shows only those isomers that have unique functionality (e.g. structures shown in blue), while Fig. 5b separates out constitutional isomers (compounds with the same functional groups in different locations, for example those shown in green). Molecular formulas containing fewer carbon atoms can necessarily have fewer chemically reasonable structures, which is borne out in the increasing numbers of predicted isomers as nC increases. The complexity of C9 and C10 compounds is therefore substantially higher than that of C3 and smaller compounds, with roughly twice as many isomers per carbon number. Several molecular formulas predicted to be formed in reasonably high concentrations are comprised of two or three unique functionalities. In some cases, such as quantifying atmospheric carbon, the simplification introduced by collapsing these structures onto a single formula may be helpful. However, for many applications or studies, the difference between these functionalities may be critical. The example given in Fig. 5, for instance, includes a hydroperoxide, an aldehyde, and an acylperoxynitrate with the same formula; these all have substantially different properties and reactivities, and thus should not be lumped together for model treatments. Importantly, these isomeric structures likely have very different volatilities; this highlights the shortcomings of the relatively simple formula-based approach used in this work (and elsewhere) to interconvert between parameters. Thus it is important that chemical structures (and not just formulas) are determined in future analytical measurements.
Figure 5.

Number of isomers of a given molecular formula included in the Master Chemical Mechanism produced by the oxidation of α-pinene as a function of carbon number. Top: number of isomers with different functional groups, conformers ignored. Bottom: number of isomers with different conformation. Average of possible isomers at each carbon number shown as black line, weighted by modeled concentrations in simulated experiment shown as dashed line. Structures shown as examples.
Nonetheless, certain frameworks can provide some insight into chemical structure. Parameterization by elemental ratios (as in Fig. 1c,d) is often used in the study of atmospheric organic carbon oxidation to deduce the average change in functional groups by changes in H:C and O:C. This chemical space provides some structural information: for instance, ions with O:C > 2.0 must include oxygen atoms not bonded to carbon, such as nitrate or peroxide groups (which can in turn be distinguished by the presence of nitrogen). These details provide chemical insights that may be useful in studying mechanisms and reaction pathways. There is substantial overlap in the regions of this parameter space accessed by each instrument, however, so it is difficult to draw significant meaning from the distribution of ions in this framework. None of these frameworks, though, provide true comprehensive structural information to inform chemical properties and functionality, as such detail is simply unavailable from a mass spectrometer without additional axes of separation.
Recent advances have begun to bring structural information to these data, though still only roughly. Overlap of measured ions across multiple instruments can provide some degree of separation due to selectivity of each ionization scheme; for instance, between hydroxyacetone and propionic acid, both C3H6O2, PTR is more sensitive to the former, while I− CIMS is an order of magnitude more sensitive to the latter.15 More generally, varying the operating parameters of a single instrument can provide some degree of separation of isomers. Recent work has shown that modulating CIMS operating parameters such as tuning voltages can decompose some ion-molecule clusters but not others,19 providing possible leverage with which to probe structure. Other approaches are to couple mass spectrometers with other techniques that introduce additional dimensions of separation; these include volatility-resolved approaches (implemented here for TD-AMS and FIGAERO-CIMS, but not for gas-phase instruments), ion mobility spectrometry, and gas chromatography. These techniques of course come with additional complexities or limitations, but still represent important potential approaches for adding structural information to the purely molecular-formula-based information that mass spectrometry provides.
Conclusions
The primary goal of this work is to develop an approach to apply reduced-parameter descriptive spaces to assess the extent to which an instrument of suite of instruments is able to measure all of the compounds in a chemical system. By comparing the regions of coverage of available instruments across multiple frameworks, considering the expected range of analytes in the system, and critically assessing potentially gaps and overlaps, it is clear that current instrumentation accesses nearly the entire range of chemical space of these frameworks. However, a lack of structural information or unambiguous identification of compounds and isomers remains a critical gap in most parameterizations which will likely require continued advances in instrumentation, most importantly techniques that provide information on the analytes beyond simply their chemical formulas.
A substantial uncertainty in assessing the completeness of current measurement capabilities is a lack of comprehensive understanding of atmospheric composition. Without knowing the full range of compounds present in the system it is difficult to assess whether all compounds are accessed by available instrumentation, while the full range of compounds is uncertain without complete measurements. For example, had the experiment used here been the OH-initiated oxidation of methane, the instruments that measure multifunctional gas-phase species (I− and NO3− CIMS) and those that measure particle-phase species (TD-AMS and FIGAERO-CIMS) would have measured nothing. Thus the vast majority of all the areas in chemical spaces shown in Fig. 1 (with the exception of volatile C1 compounds) would appear as gaps. It is therefore important in designing a similar study or applying these frameworks to consider both the expected range of products as well as that covered by the instrument suite. However, by populating common frameworks using measurements from a complex, atmospherically relevant mixture, these data suggest that the capabilities of current instrumentation span the entire range of chemical properties expected in the atmosphere. Exploring additional chemical systems and incorporating additional analytical techniques (such as measurements of molecular structure) will allow future applications of this multi-framework approach to provide more complete assessments of the current strengths and measurement gaps associated with available instrumentation.
Acknowledgements
This work was made possible primarily by the NSF Postdoctoral Research Fellowship program (AGS-PRF 1433432) support of GIVW, who operated the TD-AMS, analysed its data, and synthesized all data into the unified frameworks shown and authored the manuscript in close partnership with JHK. PM operated the NO3− CIMS and I− CIMS with support from JBN, and analysed the data they collected in cooperation with MRC, JTJ, and DRW. REO operated the chamber. LS and DAK provided the PTR and assisted with its operation in cooperation with PKM, AC, and AHG, who analyzed the PTR data.
Appendix
Measurement of parameters and interconversion among parameters
Elemental ratios are measured by all instruments either directly from molecular ions, or through bulk average composition determined by fragment ions. From these ratios, the average oxidation state of carbon atoms in a compound (OSC) can be estimated as described by Kroll et al., which assumes that all nitrogen in the system is present as organic nitrates:
| Eq. (1) |
OSC is a function of carbon bonding within the molecule, so cannot be directly measured without structural information; it is consequently a calculated parameter for all mass spectrometers in this work.
Volatility is described in this work as a compound’s saturation concentration, C*, the gas-phase concentration, in units of μg m−3, at which a compound is evenly split between the gas and particle phase. This parameter is related to vapor pressure by temperature (T), the average molecular weight of the absorbing phase (MW), and the ideal gas constant (R) and is commonly used as a descriptive parameter for organic compounds in models.39,40
Most instruments in this work measure either bulk volatility or molecular formulas of individual ions, but in most cases not both (the one exception is FIGAERO I- CIMS, discussed below). As shown in Table 1, few instruments measure both nC and C*. Consequently, to place all data into a given framework, conversion must occur between parameters for one or another instrument. Conversion between elemental composition and volatility is possible based on the previous work of Daumit et al.,41 who demonstrate that carbon number can be calculated from measured volatility and vice versa as:
| Eq. (2) |
| Eq. (3) |
where elemental ratios can be either measured (e.g., AMS fragments) or calculated (e.g. nH/nC). The terms b0, bC, b=0, and b-OH are the group contribution factors of the zero order term, carbon number, carbonyl, and hydroxyl group, respectively, in the SIMPOL method parameterization for estimation and are equal to 1.79, −0.438, −0.935, and −2.23, respectively, at 20 °C and MW = 200 g mol−1. Nitrate groups impact volatility to approximately the same degree as hydroxyl groups (b-NO3 = −2.48), so are roughly accounted for in this work by treating NO3 groups as a single OH group. The oxygen-to-carbon ratio in Eqs. 2 and 3 is therefore modified as O:C’ to exclude nitrate oxygens (i.e., (nO-2*nN)/nC). A calculated oxygen number can be derived as:
| Eq. (4) |
Observed vs. calculated parameters from FIGAERO I− CIMS
The FIGAERO I− CIMS is the only instrument in this work that provides both direct measurement of volatility (from desorption profiles) and measurement of individual ions for molecular composition. Consequently, composition can be reported as either directly measured (e.g., nC), or calculated from Eq. 2 and 4 (e.g., nC,calc). Previous work34,35 has shown that thermal desorption may decompose low-volatility mass into smaller fragments, so direct measurements of molecular composition are likely biased toward lower carbon numbers. FIGAERO I− CIMS data shown in Fig. 1 is therefore placed into each framework using the parameters calculated from desorption profiles. Measurements of a single ion appear as streaks in these frameworks indicating desorption over a given temperature range. In the nO by nC framework, this desorption presents as a streak with a slope of the O:C of a given ion.
Notes and references
- 1.Goldstein AH and Galbally I, Environ. Sci. Technol, 2007, 41, 1514–1521. [DOI] [PubMed] [Google Scholar]
- 2.Kroll JH, Smith JD, Che DL, Kessler SH, Worsnop DR and Wilson KR, Phys. Chem. Chem. Phys, 2009, 11, 8005–8014. [DOI] [PubMed] [Google Scholar]
- 3.Aumont B, Szopa S. and Madronich S, Atmos. Chem. Phys, 2005, 5, 2497–2517. [Google Scholar]
- 4.Jimenez J-L, Canagaratna MR, Donahue NM, Prevot ASH, Zhang Q, Kroll JH, DeCarlo PF, Allan JD, Coe H, Ng NL, Aiken AC, Docherty KS, Ulbrich IM, Grieshop AP, Robinson AL, Duplissy J, Smith JD, Wilson KR, Lanz VA, Hueglin C, Sun YL, Tian J, Laaksonen A, Raatikainen T, Rautiainen J, Vaattovaara P, Ehn M, Kulmala M, Tomlinson JM, Collins DR, Cubison MJ, Dunlea EJ, Huffman JA, Onasch TB, Alfarra MR, Williams PI, Bower K, Kondo Y, Schneider J, Drewnick F, Borrmann S, Weimer S, Demerjian K, Salcedo D, Cottrell L, Griffin RJ, Takami A, Miyoshi T, Hatakeyama S, Shimono A, Sun JY, Zhang YM, Dzepina K, Kimmel JR, Sueper D, Jayne JT, Herndon SC, Trimborn AM, Williams LR, Wood EC, Middlebrook AM, Kolb CE, Baltensperger U. and Worsnop DR, Science, 2009, 326, 1525–1529. [DOI] [PubMed] [Google Scholar]
- 5.Ehn M, Thornton JA, Kleist E, Sipilä M, Junninen H, Pullinen I, Springer M, Rubach F, Tillmann R, Lee BH, Lopez-hilfiker FD, Andres S, Acir I-H, Rissanen M, Jokinen T, Schobesberger S, Kangasluoma J, Kontkanen J, Nieminen T, Kurtén T, Nielsen LB, Jørgensen S, Kjaergaard HG, Canagaratna MR, Maso MD, Berndt T, Petäjä T, Wahner A, Kerminen V-M, Kulmala M, Worsnop DR, Wildt J. and Mentel TF, Nature, 2014, 506, 476–479. [DOI] [PubMed] [Google Scholar]
- 6.Kroll JH, Donahue NM, Jimenez J-L, Kessler SH, Canagaratna MR, Wilson KR, Altieri KE, Mazzoleni LR, Wozniak AS, Bluhm H, Mysak ER, Smith JD, Kolb CE and Worsnop DR, Nat. Chem, 2011, 1–7. [DOI] [PubMed] [Google Scholar]
- 7.Donahue NM, Epstein SA, Pandis SN and Robinson AL, Atmos. Chem. Phys, 2010, 11, 3303–3318. [Google Scholar]
- 8.Cappa CD and Wilson KR, Atmos. Chem. Phys, 2012, 12, 9505–9528. [Google Scholar]
- 9.Heald CL, Kroll JH, Jimenez J-L, Docherty KS, DeCarlo PF, Aiken AC, Chen Q, Martin ST, Farmer D. and Artaxo P, Geophys. Res. Lett, 2010, 37. [Google Scholar]
- 10.Pankow JF and Barsanti KC, Atmos. Environ, 2009, 43, 2829–2835. [Google Scholar]
- 11.Graus M, Müller M. and Hansel A, J. Am. Soc. Mass Spectrom, 2010, 21, 1037–1044. [DOI] [PubMed] [Google Scholar]
- 12.Jordan A, Haidacher S, Hanel G, Hartungen E, Märk L, Seehauser H, Schottkowsky R, Sulzer P. and Märk TD, Int. J. Mass Spectrom, 2009, 286, 122–128. [Google Scholar]
- 13.Tani A, Environ. Control Biol, 2013, 51, 23–29. [Google Scholar]
- 14.de Gouw JA and Warneke C, Mass Spectrom. Rev, 2007, 26, 223–257. [DOI] [PubMed] [Google Scholar]
- 15.Lee BH, Lopez-hilfiker FD, Mohr C, Kurtén T, Worsnop DR and Thornton JA, Environ. Sci. Technol, 2014, 48, 6309–6317. [DOI] [PubMed] [Google Scholar]
- 16.Bertram TH, Kimmel JR, Crisp T. a., Ryder OS, Yatavelli RLN, Thornton J. a, Cubison MJ, Gonin M. and Worsnop DR, Atmos. Meas. Tech, 2011, 4, 1471–1479. [Google Scholar]
- 17.Yatavelli RLN, Lopez-hilfiker FD, Wargo JD, Kimmel JR, Cubison MJ, Bertram TH, Jimenez J-L, Gonin M, Worsnop DR and a Thornton J, Aerosol Sci. Technol, 2012, 46, 1313–1327. [Google Scholar]
- 18.Iyer S, Lopez-hilfiker FD, Lee BH, a Thornton J. and Kurtén T, J. Phys. Chem. A, 2016, 120, 576–587. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-hilfiker FD, Iyer S, Mohr C, Lee BH, D’Ambro EL, Kurtén T. and a Thornton J, Atmos. Meas. Tech, 2016, 9, 1505–1512. [Google Scholar]
- 20.Lopez-Hilfiker FD, Mohr C, Ehn M, Rubach F, Kleist E, Wildt J, Mentel TF, Lutz a., Hallquist M, Worsnop D. and Thornton JA, Atmos. Meas. Tech, 2014, 7, 983–1001. [Google Scholar]
- 21.Lopez-hilfiker FD, Mohr C, Ehn M, Rubach F, Kleist E, Wildt J, Mentel TF, Carrasquillo a. J., Daumit KE, Hunter JF, Kroll JH, Worsnop DR and Thornton J. a, Atmos. Chem. Phys, 2015, 15, 7765–7776. [Google Scholar]
- 22.Krechmer JE, Coggon MM, Massoli P, Nguyen TB, Crounse JD, Hu W, Day DA, Tyndall GS, Henze DK, Rivera-Rios JC, Nowak JB, Kimmel JR, Mauldin RL, Stark H, Jayne JT, Sipilä M, Junninen H, Clair J. M. St., Zhang X, Feiner PA, Zhang L, Miller DO, Brune WH, Keutsch FN, Wennberg PO, Seinfeld JH, Worsnop DR, Jimenez JL and Canagaratna MR, Environ. Sci. Technol, 2015, 49, 10330–10339. [DOI] [PubMed] [Google Scholar]
- 23.Jokinen T, Sipilä M, Junninen H, Ehn M, Lönn G, Hakala J, Petäjä T, Mauldin RL, Kulmala M. and Worsnop DR, Atmos. Chem. Phys, 2012, 12, 4117–4125. [Google Scholar]
- 24.Faulhaber a. E., Thomas BM, Jimenez J-L, Jayne JT, Worsnop DR and Ziemann PJ, Atmos. Meas. Tech, 2009, 2, 15–31. [Google Scholar]
- 25.DeCarlo PF, Kimmel JR, Trimborn A, Northway MJ, Jayne JT, Aiken AC, Gonin M, Fuhrer K, Horvath T, Docherty KS, Worsnop DR and Jimenez J-L, Anal. Chem, 2006, 78, 8281–8289. [DOI] [PubMed] [Google Scholar]
- 26.Aiken AC, DeCarlo PF, Kroll JH, Worsnop DR, Huffman JA, Docherty KS, Ulbrich IM, Mohr C, Kimmel JR, Sueper D, Sun Y, Zhang Q, Trimborn AM, Northway M, Ziemann PJ, Canagaratna MR, Onasch TB, Alfarra MR, Prevot ASH, Dommen J, Duplissy J, Metzger A, Baltensperger U. and Jimenez J-L, Environ. Sci. Technol, 2008, 42, 4478–4485. [DOI] [PubMed] [Google Scholar]
- 27.Canagaratna MR, Jimenez J-L, Kroll JH, Chen Q, Kessler SH, Massoli P, Hildebrandt Ruiz L, Fortner E, Williams LR, Wilson KR, Surratt JD, Donahue NM, Jayne JT and Worsnop DR, Atmos. Chem. Phys, 2015, 15, 253–272. [Google Scholar]
- 28.Kuwata M, Zorn SR and Martin ST, Environ. Sci. Technol, 2012, 46, 787–794. [DOI] [PubMed] [Google Scholar]
- 29.Hunter JF, Carrasquillo AJ, Daumit KE and Kroll JH, Environ. Sci. Technol, 2014, 48, 10227–34. [DOI] [PubMed] [Google Scholar]
- 30.Donahue NM, Epstein S. a., Pandis SN and Robinson AL, Atmos. Chem. Phys, 2010, 11, 3303–3318. [Google Scholar]
- 31.van Krevelen DW, Fuel, 1950, 29, 269–284. [Google Scholar]
- 32.Nölscher AC, Butler T, Auld J, Veres P, Muñoz a., Taraborrelli D, Vereecken L, Lelieveld J. and Williams J, Atmos. Environ, 2014, 89, 453–463. [Google Scholar]
- 33.Amador-Muñoz O, Misztal PK, Weber R, Worton DR, Zhang H, Drozd G. and Goldstein AH, Atmos. Meas. Tech, 2016, 9, 5315–5329. [Google Scholar]
- 34.Lopez-hilfiker FD, Mohr C, D’Ambro EL, Lutz A, Riedel TP, Gaston CJ, Iyer S, Zhang Z, Gold A, Surratt JD, Lee BH, Kurtén T, Hu W, Jimenez J-L, Hallquist M. and A Thornton J, Environ. Sci. Technol, 2016, 50, 2200–2209. [DOI] [PubMed] [Google Scholar]
- 35.Isaacman-VanWertz G, Yee LD, Kreisberg NM, Wernis R, a Moss J, V Hering S, de Sa SS, Martin ST, Alexander LM, Palm BB, Hu W, Campuzano-Jost P, Day D. a., Jimenez J-L, Riva M, Surratt JD, Viegas J, Manzi A, Edgerton ES, Baumann K, Souza R, Artaxo P. and Goldstein AH, Environ. Sci. Technol, 2016, 50, 9952–9962. [DOI] [PubMed] [Google Scholar]
- 36.Tobias HJ, Docherty KS, Beving DE and Ziemann PJ, Environ. Sci. Technol, 2000, 34, 2116–2125.s [Google Scholar]
- 37.Jenkin ME, Saunders SM and Pilling MJ, Atmos. Environ, 1997, 31, 81–104. [Google Scholar]
- 38.Saunders SM, Jenkin ME, Derwent RG and Pilling MJ, Atmos. Chem. Phys, 2003, 3, 161–180. [Google Scholar]
- 39.Donahue NM, Robinson AL, Stanier CO and Pandis SN, Environ. Sci. Technol, 2006, 40, 2635–2643. [DOI] [PubMed] [Google Scholar]
- 40.Dzepina K, Volkamer R, Madronich S, Tulet P, Ulbrich IM, Zhang Q, Cappa CD, Ziemann PJ and Jimenez J-L, Atmos. Chem. Phys, 2009, 9, 5681–5709. [Google Scholar]
- 41.Daumit KE, Kessler SH and Kroll JH, Faraday Discuss, 2013, 165, 181. [DOI] [PubMed] [Google Scholar]