

Abstract
5-Hydroxymethylcytosine (5hmC) arises from the oxidation of 5-methylcytosine (5mC) by Fe2+ and 2-oxoglutarate-dependent 10–11 translocation (TET) family proteins. Substantial levels of 5hmC accumulate in many mammalian tissues, especially in neurons and embryonic stem cells, suggesting a potential active role for 5hmC in epigenetic regulation beyond being simply an intermediate of active DNA demethylation. 5mC and 5hmC undergo dynamic changes during embryogenesis, neurogenesis, hematopoietic development, and oncogenesis. While methods have been developed to map 5hmC, more efficient approaches to detect 5hmC at base resolution are still highly desirable. Herein, we present a new method, Jump-seq, to capture and amplify 5hmC in genomic DNA. The principle of this method is to label 5hmC by the 6-N3-glucose moiety and connect a hairpin DNA oligonucleotide carrying an alkyne group to the azide-modified 5hmC via Huisgen cycloaddition (click) chemistry. Primer extension starts from the hairpin motif to the modified 5hmC site and then continues to “land” on genomic DNA. 5hmC sites are inferred from genomic DNA sequences immediately spanning the 5-prime junction. This technology was validated, and its utility in 5hmC identification was confirmed.
Graphical Abstract
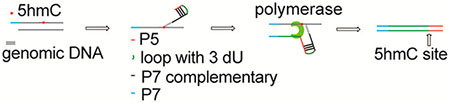
The oxidative derivative of 5-methylcytosine (5mC) catalyzed by 10–11 translocation (TET) enzymes,1–4 5-hydroxymethylcytosine (5hmC), is an oxidative intermediate in TET-mediated oxidation, but it may also have functional roles itself.5–9 We and others have developed different methods for 5hmC mapping.10–15 For instance, TAB-seq14 can detect 5hmC at base resolution but requires a relatively high DNA input (hundreds of ng) and highly active TET enzymes. A selective chemical-labeling method of 5hmC (5hmC-Seal)16 maps 5hmC in a more cost-effective way with much less starting material. Briefly, a modified glucose moiety (6-N3-glucose) is transferred to the hydroxyl group of 5hmC by T4 bacteriophage β-glucosyltransferase (β-GT), forming 6-N3-β-glucosyl-5hmC (N3-5gmC). Utilizing Huisgen cycloaddition (click) chemistry,17 a biotin tag is then coupled to the azide group on the glucose of N3–5gmC for selective, sensitive, and unbiased pull-down of 5hmC-containing DNA.16 By introducing a library construction strategy using engineered Tn5 transposase, we improved the detection limit to 1000 cells (5hmC Nano-Seal).18 However, these methods lack base resolution precision. Building on these existing technologies, we present a new strategy, named Jump-seq, for 5hmC sequencing at nearly base resolution without sequencing the entire genome.
The new strategy takes advantage of the selective chemical labeling of 5hmC along with highly efficient transposase-based DNA fragmentation and adaptor tagmentation.19,20 The procedure is outlined in Figure 1. (1) Genomic DNA is fragmented and tagged by biotin-P7 adapter sequence. (2) 5hmC in genomic DNA is then labeled with a modified azideglucose using β-GT-mediated selective chemical labeling. (3) A hairpin DNA oligonucleotide, with a P5 adapter sequence and a unique sequence carrying an alkyne group, is covalently connected to the azide-modified 5hmC, and the loop part carries three deoxyribose uracils by design. (4) Primer extension starts from the hairpin DNA attached to 5hmC as indicated. Primer extension from the hairpin motif extends to the modified 5hmC site and continues to “land” on surrounding genomic DNA, eventually reaching the P7 adapter installed by transposase. The dU linker in the hairpin motif tethered to 5hmC is cleaved by USER enzyme (NEB). Extension products with P5 and P7 adapters are subsequently amplified and sequenced. (5) 5hmC single sites are inferred from the juncture connecting the hairpin sequence and any genomic DNA sequence.
Figure 1.
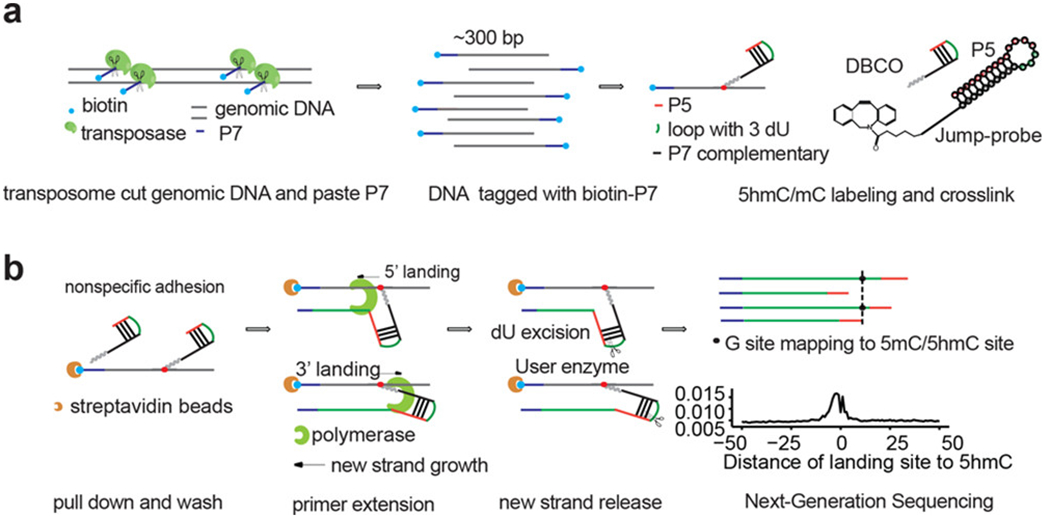
Jump-seq strategy. Genomic DNA is fragmented and tagged with a biotin-P7 adapter by transposase followed by 5hmC labeling with an azide-modified glucose using β-GT. A hairpin DNA (with P5 adapter) carrying an alkyne is added covalently to the modified glucose. After primer extension from the hairpin and cleavage from the tethered hairpin, the newly synthesized strand is subjected to library construction and sequencing. 5hmC single site location is inferred from the polymerases “landing” site pattern that connects the hairpin sequence and any genomic DNA sequence.
We used several spike-in DNA sequences (Table S1) to confirm the specificity and sensitivity of Jump-seq by high-throughput sequencing. The distribution of reads generated from spike-in sequences with one or two 5hmCpG sites suggested a jump pattern that peaked at the true 5hmC site, and jump distance was typically small (less than 10 bp) (Figure 2a,b). For multiple 5hmC sites, if two 5hmC sites are too close, the landing site pattern could influence each other. However, our previous TAB-seq results14 suggested that the majority of 5hmC sites in mouse and human genomes are at least four base pairs away from each other. To calculate the enrichment efficiency, oligos with 72 unmodified CpG sites were combined with a spike-in oligo containing one 5hmCpG site and subjected to 5hmC Jump-seq. 5hmC enrichment was calculated as follows: (5hmC spike-in hmCpG site mapped reads/total hmCpG site number)/(negative background CpG site mapped reads/total CpG site number). The enrichment fold was (22/1)/(0.78/(72)) = 2030.8 (Figure 2c), demonstrating that the Jump-seq signal is enriched 2000-fold at a 5hmC site compared to an unmodified CpG site. To evaluate “jump” effectiveness, the following gradient of 5hmC-modified spike-in was added into 48 ng of mES genomic DNA: 0%, 1%, 5%, 10%, 25%, 50%, 75%, and 100%. The read count of 5hmC Jump-seq was linearly correlated with the 5hmC amount (R2 = 0.94), supporting the Jump-seq strategy as a powerful 5hmC semiquantitative tool (Figure 2d).
Figure 2.
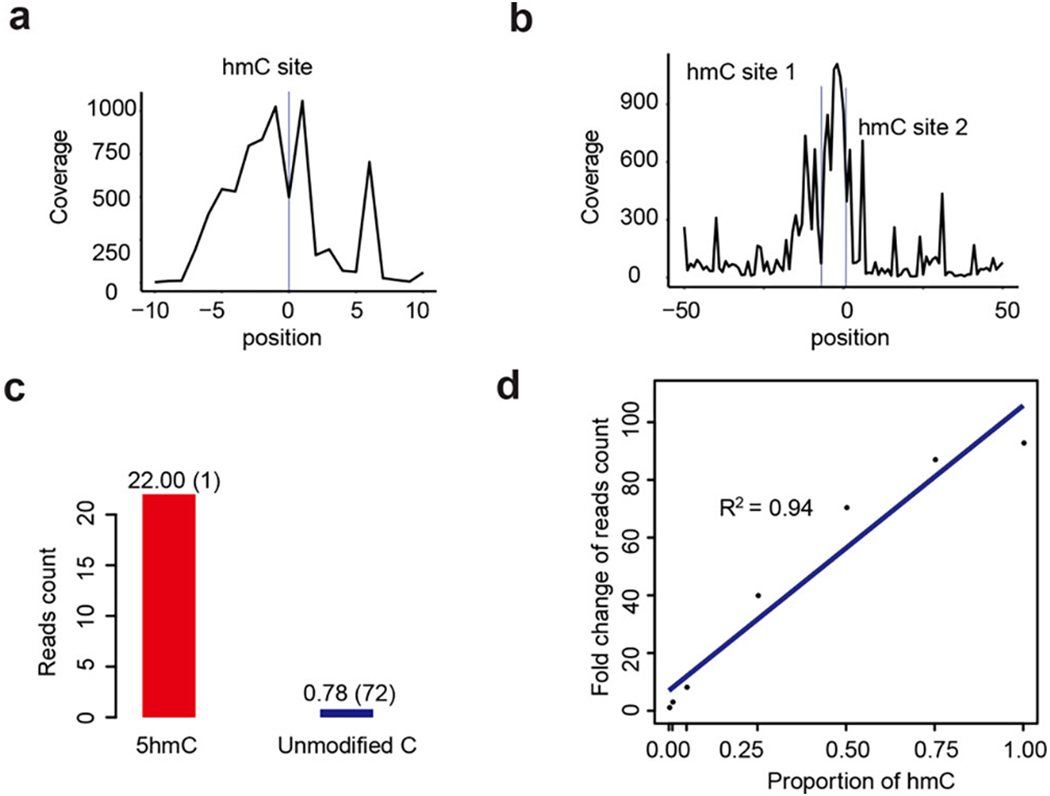
Method validation with spike-in DNA models. Jump-seq shows a nearly base resolution DNA polymerase landing pattern for (a) spike-in with one 5hmC site and (b) spike-in with two 5hmC sites; the y-axis represents reads coverage. (c) The number of reads (in million on y-axis) at 5hmC in spike-in (red) or unmodified C in background DNA fragments (blue) are shown, and total CpG sites are shown in brackets. (d) The fitted regression line is shown for the fold change of number of reads (point) at different 5hmC proportions in the spike-in.
We next sought to create a nearly base resolution map of 5hmC in the whole genome of mESCs with Jump-seq and to compare these data sets with the “gold-standard” base resolution 5hmC maps generated by TAB-seq. We performed Jump-seq on genomic DNA isolated from 400 (2.4 ng), 1000 (6 ng), 2000 (12 ng), 4000 (24 ng), and 8000 (48 ng) mESCs (Figure S2). These results confirmed that this method reveals the locations of 5hmC at nearly base resolution. We observed a unique pattern of primer extension on genomic DNA sequence: “landing” sites of DNA polymerases were distributed around the examined 5hmC sites obtained from TAB-seq, and a “valley” was overlaid on top of the 5hmC sites (Figure S2). A mechanistic explanation for the formation of this “valley” is based on behavior of the polymerases encountering the “gap” (composed of azide glucose and a DBCO linker) between the unique DNA sequence attached to 5hmC and genomic DNA. Polymerases may overcome the obstacle and jump to genomic DNA to continue extension with high efficiency. During this “jump”, some polymerases land 1–10 bases 5′ ahead of the 5hmC site, while others slide back to the genomic strand (1–10 bases toward the 3′) and then extend to the 5′ direction on the genomic template. Less frequently, polymerases may land exactly on the modified 5hmC sites, thus forming a “valley” at the exact 5hmC site. In addition, as double-stranded DNA strands are denatured before jump-probe connection, and “click-based” cross-linking is efficient and unbiased, Jump-seq can reveal the accurate positions of 5hmCs on the Watson and Crick strands of fully hydroxymethylated 5hmCpGs (Figure S3), demonstrating accuracy at nearly the single-base level.
5hmC Jump-seq signals mainly existed in CG dinucleotide contexts (Figure 3b) and showed significant enrichment at enhancers and exons (Figure 3a), which is consistent with a previous study.14 High correlations (r > 0.9) of 5hmC Jump-seq data were observed among replicates at all levels of starting DNA amount (Figure S4). To further validate the new method, we calculated the overlap of mESC 5hmC peaks identified by Jump-seq with published 5hmC-Seal and TAB-seq data (Figure 3c,d). Jump-seq 5hmC peaks showed enrichment of recovered 5hmC sites overlapped by 5hmC-Seal and TAB-seq at all starting DNA levels (Figure 3c,d). 5hmC sites identified by 5hmC Jump-seq data recovered more than 90% 5hmC-Seal 5hmC peaks and approximately 40% TAB-seq 5hmC peaks (Figure 3e), demonstrating high sensitivity. Of note, we called Jump-seq peaks using the 20 bp window. While TAB-seq reveals high-resolution sites (several million single sites in the mammalian genome), 5hmC-Seal peaks are broad and low-resolution (about 50 thousand). Therefore, more overlap between 5hmC-Seal peaks and Jump-seq peaks is expected. Jump-seq 5hmC signals overlapped well with 5hmC-Seal peaks (Figure 3f,g). Jump-seq results performed on mouse ESC genomic DNA showed a base resolution “valley” overlaid on top of the 5hmC sites identified by TAB-seq, proving its accuracy and high resolution (Figure 3h,i).
Figure 3.
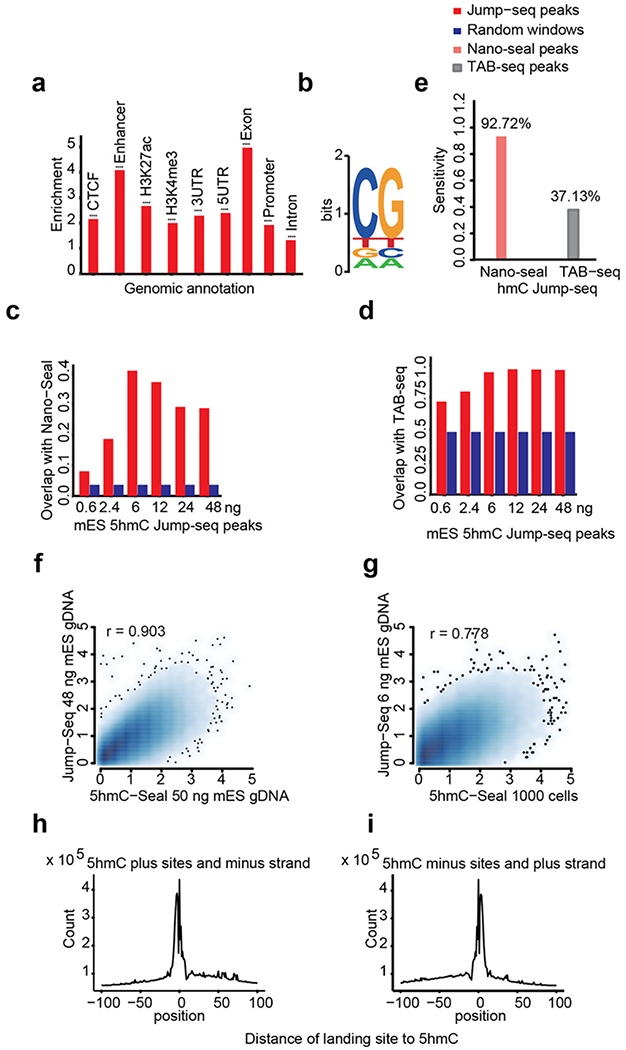
Jump-seq strategy validation. Bam files of 12 replicates of 48 ng 5hmC Jump-seq data were combined to test enrichment and sensitivity. (a) 5hmC signal enrichment at different genomic regions. (b) 5hmC motif. Proportion of 5hmC Jump-seq peaks (red bar) overlapping with 5hmC peaks of (c) 5hmC-Seal and (d) TAB-seq. For each Jump-seq result, the same number of randomly chosen 1 kb windows (blue bar) were compared with 5hmC-Seal and TAB-seq. (e) Sensitivity of Jump-seq as evaluated using previously identified 5hmC sites in TAB-seq. “Enriched” peak windows of 20 bp at FDR 0.05 were called to estimate the proportion of 5hmC-Seal or TAB-seq 5hmC peaks recovered by Jump-seq 5hmC peaks. (f, g) Correlation density plot of 5hmC signal between 5hmC Jump-seq and 5hmC-Seal data. Values of x- and y-axes represent centered and standardized read counts following square-root transformation. (h, i) Read distribution of the Jump-seq strategy. Jump-seq 5hmC sites were overlaid on TAB-seq 5hmC sites. Because the Jump-seq strategy has a complementary strand synthesis step, reads mapped on the plus stand represent the 5hmC sites in the minus strand and vice versa.
The present study reported a cost-effective Jump-seq method to achieve bisulfite-free, nearly base resolution sequencing of 5hmC at the whole genome scale. Of note, a similar cross-linking and primer extension strategy, named TOP-seq,21 has been reported to selectively tag the unmethylated CG sites in the genome with 50–500 ng of genomic DNA to infer the methylation state. The Jump-seq strategy excels in directly amplifying 5hmC sites with less than 50 ng of DNA input. With 24 ng of genomic DNA (4000 cells) or more, Jump-seq achieved nearly base resolution 5hmC mapping with modest sequencing depth (about 1/30 that of TAB-seq). The linear correlation between Jump-seq read count and 5hmC amount indicated that it is a useful semiquantitative method for assessing 5hmC levels (Figure 2e). Jump-seq is also compatible with locus-specific 5hmC detection and quantification. By choosing suitable locus-specific primers, Jump-seq could be readily integrated with locus-specific qPCR and microarray for broader clinical utility. Lastly, the tethering of DNA or modifications for lineal amplification provides a strategy that could be broadly applied in detecting other nucleic acid modifications.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Pieter Faber and the University of Chicago Genomics Facility for sequencing support. The authors also thank Dr. Kai Chen and Mr. Zhike Lu for discussion as well as Mr. Wu Tong for manuscript editing. This work was supported by the US National Institutes of Health (R01 HG006827 and P01 NS097206 to C.H.). L.H. is supported by Chicago Fellows Program, Chicago Biomedical Consortium (CBC) postdoctoral award and Leukemia & Lymphoma Society Special Fellow Award. S.G. is supported by National Key R&D Program of China (2016YFA0100400), the National Natural Science Foundation of China (31771646), and Shanghai Rising-Star Program (17QA1404200). X.H. is supported by NIH 2018R01 MH (Grant MH110531). C.H. is a Howard Hughes Medical Institute Investigator. The sequencing data reported in this paper have been deposited into the Gene Expression Omnibus (GEO) under accession number GSE127906.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b02512.
Experimental details and supporting figures including structures, Click reaction, primer extension efficiency, reads distributions, Jump-seq distribution patterns, correlation density plots, and sequences (PDF)
The authors declare the following competing financial interest(s): A patent application has been filed for the technology by the University of Chicago.
REFERENCES
- (1).Tahiliani M; Koh KP; Shen Y; Pastor WA; Bandukwala H; Brudno Y; Agarwal S; Iyer LM; Liu DR; Aravind L; Rao A Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324 (5929), 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hu L; Li Z; Cheng J; Rao Q; Gong W; Liu M; Shi YG; Zhu J; Wang P; Xu Y Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 2013, 155 (7), 1545–55. [DOI] [PubMed] [Google Scholar]
- (3).Hu L; Lu J; Cheng J; Rao Q; Li Z; Hou H; Lou Z; Zhang L; Li W; Gong W; Liu M; Sun C; Yin X; Li J; Tan X; Wang P; Wang Y; Fang D; Cui Q; Yang P; He C; Jiang H; Luo C; Xu Y Structural insight into substrate preference for TET-mediated oxidation. Nature 2015, 527 (7576), 118–22. [DOI] [PubMed] [Google Scholar]
- (4).Crawford DJ; Liu MY; Nabel CS; Cao XJ; Garcia BA; Kohli RM Tet2 Catalyzes Stepwise 5-Methylcytosine Oxidation by an Iterative and de novo Mechanism. J. Am. Chem. Soc 2016, 138 (3), 730–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wu H; D’Alessio AC; Ito S; Wang Z; Cui K; Zhao K; Sun YE; Zhang Y Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011, 25 (7), 679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kriaucionis S; Heintz N The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324 (5929), 929–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shi DQ; Ali I; Tang J; Yang WC New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet 2017, 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kohli RM; Zhang Y TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502 (7472), 472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ito S; D’Alessio AC; Taranova OV; Hong K; Sowers LC; Zhang Y Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466 (7310), 1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schutsky EK; DeNizio JE; Hu P; Liu MY; Nabel CS; Fabyanic EB; Hwang Y; Bushman FD; Wu H; Kohli RM Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat. Biotechnol 2018, 36, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mooijman D; Dey SS; Boisset JC; Crosetto N; van Oudenaarden A Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat. Biotechnol 2016, 34 (8), 852–6. [DOI] [PubMed] [Google Scholar]
- (12).Zeng H; He B; Xia B; Bai D; Lu X; Cai J; Chen L; Zhou A; Zhu C; Meng H; Gao Y; Guo H; He C; Dai Q; Yi C Bisulfite-Free, Nanoscale Analysis of 5-Hydroxymethylcytosine at Single Base Resolution. J. Am. Chem. Soc 2018, 140 (41), 13190–13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Booth MJ; Branco MR; Ficz G; Oxley D; Krueger F; Reik W; Balasubramanian S Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336 (6083), 934–7. [DOI] [PubMed] [Google Scholar]
- (14).Yu M; Hon GC; Szulwach KE; Song CX; Zhang L; Kim A; Li X; Dai Q; Shen Y; Park B; Min JH; Jin P; Ren B; He C Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149 (6), 1368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu Y; Siejka-Zielinska P; Velikova G; Bi Y; Yuan F; Tomkova M; Bai C; Chen L; Schuster-Bockler B; Song CX Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol 2019, 37, 424. [DOI] [PubMed] [Google Scholar]
- (16).Song CX; Szulwach KE; Fu Y; Dai Q; Yi C; Li X; Li Y; Chen CH; Zhang W; Jian X; Wang J; Zhang L; Looney TJ; Zhang B; Godley LA; Hicks LM; Lahn BT; Jin P; He C Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 2011, 29 (1), 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed 2001, 40 (11), 2004–2021. [DOI] [PubMed] [Google Scholar]
- (18).Han D; Lu X; Shih AH; Nie J; You Q; Xu MM; Melnick AM; Levine RL; He C A Highly Sensitive and Robust Method for Genome-wide 5hmC Profiling of Rare Cell Populations. Mol. Cell 2016, 63 (4), 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Adey A; Shendure J Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012, 22 (6), 1139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Picelli S; Bjorklund AK; Reinius B; Sagasser S; Winberg G; Sandberg R Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014, 24 (12), 2033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Stasevskij Z; Gibas P; Gordevicius J; Kriukiene E; Klimasauskas S Tethered Oligonucleotide-Primed Sequencing, TOP-Seq: A High-Resolution Economical Approach for DNA Epigenome Profiling. Mol. Cell 2017, 65 (3), 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.