

SUMMARY
Ovarian cancer is typified by the development of chemotherapy resistance. Chemotherapy resistance is associated with high aldehyde dehydrogenase (ALDH) enzymatic activity, increased cancer “stemness,” and expression of the stem cell marker CD133. As such, ALDH activity has been proposed as a therapeutic target. Although it remains controversial which of the 19 ALDH family members drive chemotherapy resistance, ALDH1A family members have been primarily linked with chemotherapy resistant and stemness. We identified two ALDH1A family selective inhibitors (ALDH1Ai). ALDH1Ai preferentially kills CD133+ ovarian cancer stem-like cells (CSCs). ALDH1Ai induce necroptotic CSC death, mediated, in part, by the induction of mitochondrial uncoupling proteins and reduction in oxidative phosphorylation. ALDH1Ai is highly synergistic with chemotherapy, reducing tumor initiation capacity and increasing tumor eradication in vivo. These studies link ALDH1A with necroptosis and confirm the family as a critical therapeutic target to overcome chemotherapy resistance and improve patient outcomes.
Graphical Abstract
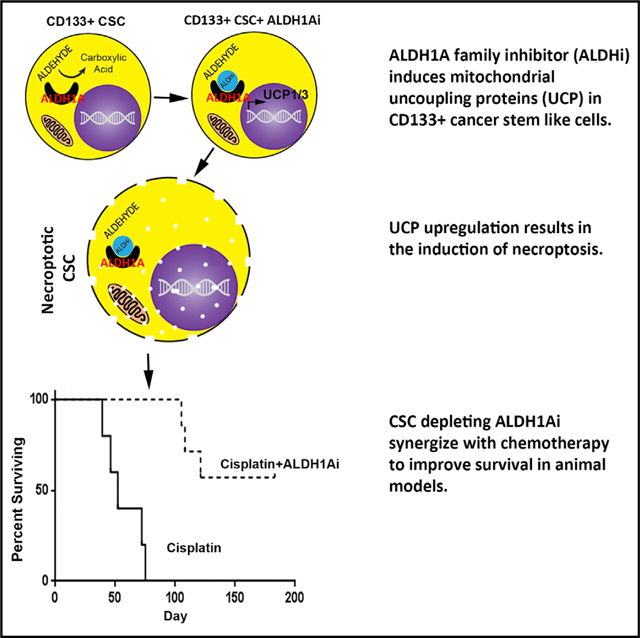
In Brief
Chefetz et al. identify ALDH1A enzyme family inhibitors (ALDH1Ai) that trigger necroptosis in CD133+ ovarian CSCs. Necroptosis is mediated in part by the induction of mitochondrial uncoupling proteins and reduction of oxidative phosphorylation. ALDH1Ai is highly synergistic with chemotherapy, reducing tumor initiation and growth and increasing tumor eradication rates.
INTRODUCTION
The aldehyde dehydrogenase (ALDH) superfamily has 19 protein-coding family members with varying tissue or development expression (Koppaka et al., 2012). ALDHs metabolize endogenous and exogenous aldehydes to their corresponding carboxylic acids to prevent accumulation of toxic aldehydes. ALDH1A enzymes are also involved in the biosynthesis of retinoic acid (RA). Via synthesis of RA, ALDH1A enzymes indirectly regulate RA-mediated transcription of hundreds of genes that regulate normal stem cell proliferation and differentiation (Balmer and Blomhoff, 2002; Napoli et al., 1995).
ALDH enzymes also have a role in cancer. ALDHs mediate resistance to chemotherapy via direct drug metabolism and by regulation of reactive oxygen species (Bretz et al., 2012; Dylla et al., 2008). Many studies have used broad ALDH inhibitors such as N,N-diethylaminobenzaldehyde (DEAB) and disulfiram (DSF) to demonstrate a general role for ALDH in chemotherapy resistance (Kast and Belda-Iniesta, 2009; Morrison et al., 2010; Rezk et al., 2015; Yip et al., 2011). Other studies have used knockdown of select ALDH1 family members to support their role in chemotherapy resistance; ALDH1A1 or ALDH1A3 knockdown increases chemosensitivity in ovarian cancer, melanoma, breast cancer, and lung cancer (Januchowski et al., 2013). ALDH activity is essential to chemotherapy resistance in breast cancer (Raha et al., 2014), and ALDH1A2 and ALDH1A3 are implicated in chemotherapy resistance in mesothelioma (Cortes-Dericks et al., 2014).
In ovarian cancer, in TP53 wild-type patients’ high mRNA expression of ALDH1A2 is associated with poor overall survival (Ma and Zhao, 2016), and ALDHbright cells (cells with high ALDH enzymatic activity based on the ALDEFLUOR assay) are resistant to cisplatin (Silva et al., 2011). ALDH1A1 is upregulated more than 100-fold in ovarian cancer cells selected for taxane resistance in vitro, and ALDH1A1 knockdown reversed that chemotherapy resistance (Steg et al., 2012). Furthermore, paired biopsies taken from patients (Steg et al., 2012) and patient-derived ovarian tumor xenografts (Dobbin et al., 2014) before and after chemotherapy demonstrated increased expression of ALDH1A1 and the cancer stem-like cell (CSC) marker CD133 in posttreatment samples.
ALDH activity identifies CSCs in numerous cancers, including ovarian cancer (Deng et al., 2010; Landen et al., 2010; Silva et al., 2011). Limited numbers of primary human ALDHbright cells initiate tumors, whereas a 50-fold excess of ALDHdim tumor cells cannot. ALDH used in combination with CD133 enriches CSC isolation, with as few as 11 ALDHbrightCD133+ primary human ovarian cancer cells capable of initiating heterogeneous ovarian tumors in mice as well as passage through several generations (Kryczek et al., 2012; Silva et al., 2011). Importantly, we performed single-cell lineage tracing studies with primary human ovarian cancer cells and defined a clear ovarian cancer differentiation hierarchy, with ALDHbrightCD133+ cells serving as multipotent stem cells in both cell lines and primary patient samples (Choi et al., 2015).
Given the role of ALDH1A isozymes in chemotherapy resistance, stem cell biology, and its link with CSC, ALDH1A activity has been proposed as a therapeutic target in cancer. Here, we report the identification of the potent, ALDH1A family-specific inhibitor (ALDH1Ai) 673A; 673A inhibits all three ALDH1A family members (half-maximal inhibitory concentration [IC50] < 350 nM for each) with minimal inhibition of ALDH2 or ALDH3 (IC50 ~15 μM). The 673A treatment leads to the induction of necroptotic cell death preferentially in CD133+ ovarian CSCs. Necroptosis is dependent in part on the induction of mitochondrial uncoupling proteins (UCPs) and is associated with inhibition of oxidative phosphorylation (OXPHOS) in CSCs. Necroptosis in CSCs is associated with chemotherapy synergy, a reduction in tumor initiation capacity, and eradication of tumors in vivo. Together these data strongly support the ALDH1A family as an important therapeutic target in cancer.
RESULTS
ALDH1A Family Members in Ovarian Cancer
ALDH1A family members have been proposed as therapeutic targets in ovarian cancer. However, which ALDH1A enzyme or enzymes to target remains uncertain. Evaluation of ALDH1A family (ALDH1A1, ALDH1A2, or ALDH1A3) member expression in multiple ovarian cancer cell lines, using qRT-PCR revealed that, typically, at least one ALDH1A family member was significantly expressed in each cell line (Figure 1Ai). Similarly, in silico analysis of ALDH1A expression in 30 ovarian cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) data set indicated that at least one, and in many cases, two, ALDH1A isozymes are expressed in each cell line (Figure 1Aii; Table S1). Although ALDH1A1 is expressed in all cell lines tested, ALDH1A3 is the dominant isoform in OVCAR8, OVCAR5, and PEO1.
Figure 1. ALDH1A Family Members in Ovarian Cancer.

(A) qRT-PCR (i) and in silico (ii) CCLE analysis of ALDH gene expression in various ovarian cancer cell lines.
(B) Analysis of ALDH1A family member DNA deletion and amplification or mRNA expression changes in the ovarian cancer TCGA database.
(C) (i) qRT-PCR confirmation of ALDH1A family member mRNA downregulation with siRNA treatment in PEO4 cells. (ii) Cell counts in the indicated cell lines following ALDH family member downregulation.
(D) Cell viability in FACS sorted CD133+ and CD133− from A2780 and Ovsaho 72 h after ALDH1A1 or ALDH1A3 downr0egulation.
Error bars represent SDs. Results are a summary of n = 3 independent experiments with at least three technical replicates. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
Analysis of ALDH1A family members in 316 high-grade serous ovarian cancers (HGSCs) in The Cancer Genome Atlas data set (https://cancergenome.nih.gov/) demonstrated that deep deletions, mRNA downregulation, or missense mutations of ALDH1A family members occur in 0.6% or less of cases (Figure 1B). We identified no cases with two ALDH1A family members deleted, suggesting that at least one ALDH1A family member may be necessary for cancer cell viability.
Given predominant expression of ALDH1A1 and ALDH1A3, we validated and performed siRNA knockdown of ALDH1A1 and ALDH1A3 in three HGSC ovarian cancer cell lines that have a high level of “stemness” based on high expression of CD133. Knockdown of either ALDH1A1 or ALDH1A3 resulted in a significant reduction in cell viability in all three cell lines, with knockdown of the predominant isozyme having the greatest effect (Figure 1Cii). To determine whether ALDH1A or ALDH1A3 were differentially affecting CSC, we performed small interfering RNA (siRNA) knockdown in fluorescence-activated cell sorting (FACS)-isolated CD133+ and CD133− cells from two cell lines with distinct CD133+ cell populations. siRNA knockdown of ALDH1A1 or ALDH1A3 in FACS-sorted CD133+ and CD133− from A2780 and Ovsaho cells was associated with statistically significant preferential depletion of CD133+ CSC in both cell lines (Figure 1D).
Identification of an ALDH1A Family-Specific Inhibitor
The observations above and the literature suggest that the ALDH1A family members could contribute to cancer stemness (Condello et al., 2014; Li et al., 2014; Raghavan et al., 2017; Yip et al., 2011). Given the differential expression of ALDH1A family members, we reasoned that a pan-ALDH1A class inhibitor would have the broadest utility. Because ALDH1A family member knockdown was associated with preferential depletion of CD133+ cells, we evaluated several known ALDH inhibitors for the ability to deplete CD133+ CSCs. Although the ALDH2 inhibitor daidzin had no significant toxicity to ovarian cancer cells or CD133+ CSCs, high doses of the ALDH1A inhibitor DEAB demonstrated preferential depletion of CD133+ cells (Figure 2A).
Figure 2. Identification of an ALDH1Ai, 673A.
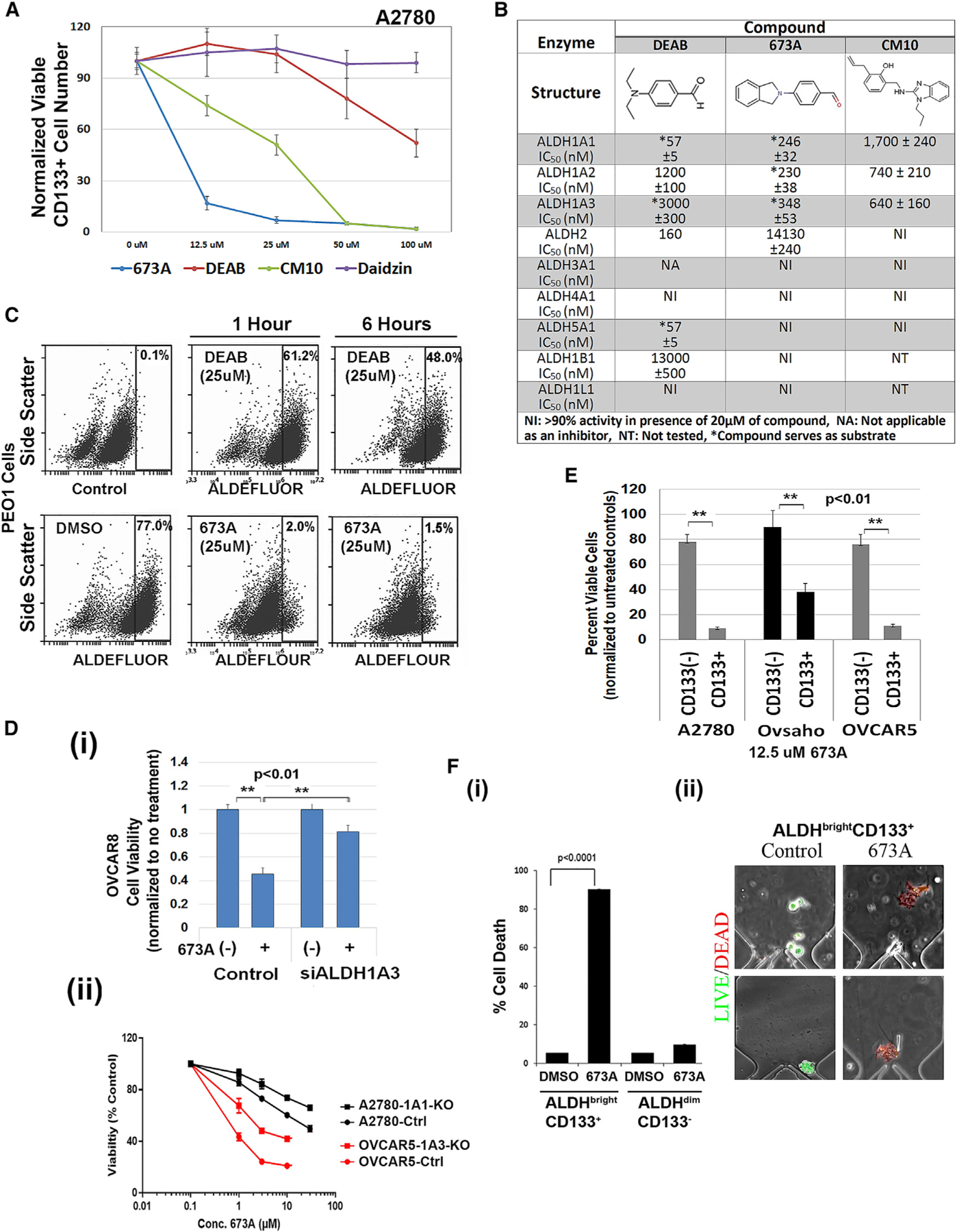
(A) Effect of the indicated ALDH inhibitors on the percentage of viable CD133+ A2780 cells (absolute CD133+ cells in each group are normalized to untreated controls) by FACS 72 h after treatment.
(B) Quantification of in vitro enzymatic inhibition of ALDH family members.
(C) ALDEFLUOR assay of ALDH activity in live PEO1 cells after treatments with 25 μM DEAB or 673A at the indicated times.
(D) Viability of OVCAR8 cell controls or OVCAR8 siALDH1A3 knockdown cells with and without 673 (i), and 673 dose-response curve for the indicated cell lines comparing transfected controls (ctrl) versus either CRISPR knockdown of ALDH1A1 or ALDH1A3 (ii).
(E) Cell viability of FACS-sorted CD133+/− (A2780, Ovsaho, and OVCAR5) cells 72 h after treatment with 12.5 μM 673A.
(F) Quantification (i) of cell death of single CD133+/ALDH+ (A2780) cells (ii) on microfluidic chips 72 h after treatment.
Error bars represent SDs; n = 3 independent experiments with at least triplicate assays. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
We thus screened DEAB analogs for both ALDH inhibitory activity and CD133+ cell depletion. We identified a lead compound, 673A (4-(1,3-dihydro-2H-isoindol-2-yl)benzaldehyde; Figure 2B; complete screen to be described elsewhere). In vitro enzymatic assays revealed that 673A inhibited ALDH1A1 (IC50 246 nM), ALDH1A2 (IC50 230 nM), and ALDH1A3 (IC50 348 nM) with minimal or no inhibition of ALDH2 (IC50 14 μM) or numerous other ALDH family members (Figures 2B and S2A). To confirm inhibition of ALDH activity in live cells, we used PEO1 cells; a HGSC ovarian cancer cell line with very high ALDH activity and a defined CSC pool (Choi et al., 2015). The 673A inhibited PEO1 ALDEFLUOR activity within 1 h of treatment and persisted after 6 h (Figure 2C). At 6 h, at equimolar doses, 673A inhibited 98% of ALDEFLUOR activity versus 40% for DEAB. To evaluate the ability of 673A to preferentially target CD133+ cells, we used cell lines (A2780, OVSAHO, and OvCAR5) with distinct CD133+ cell populations. The 673A was highly effective at preferentially depleting CD133+ CSC in all three cell lines (Figures 2A, 2E, S2B, and S3Bi–S3Biii) with a CSC selective median toxic dose (TD50) of 3–20 μM. Showing selectivity for CSCs over normal cells, 673A demonstrated little or no toxicity to human mesenchymal stem cells or non-malignant MCF10A cells (Figure S3D).
To confirm induction of cell death was ALDH inhibition-mediated, we first used siRNA to knock down the target and assess efficacy. We chose to evaluate siRNA targeting ALHD1A3 in OVCAR8 cells as these cells were most sensitive to siRNA knockdown (Figure 1Cii). ALDH1A3 knockdown resulted in a statistically significant reduction in 673 treatment effect (Figure 2Di). We similarly tested the effect of CRISPR-mediated ALDH1A1 or ALDH1A3 knockout. Given concerns for functional redundancy, we used Ov90 cells, which near-exclusively express ALDH1A1, and OVCAR5 cells, which predominantly express ALDH1A3. Interestingly, ALDH1A1 knockout Ov90 cells were non-viable with all constructs. OVCAR5-ALDH1A3-knockout (KO) cells demonstrated a significant loss in ALDEFLOUR activity (Figure S1Bi) and significant loss in 673 treatment effect (Figure 2Dii;~10× increase in TD50). Given Ov90 cells were non-viable with ALDH1A1 KO, we generated A2780-ALDH1A1-KO cells. Likely related to persistent expression of ALDH1A3, these cells maintained significant ALDEFLUOR activity (Figure S1Bii) and had a less-pronounced reduction (~33× increase in TD50) in 673 treatment effect.
To confirm on-target activity of 673A, we evaluated a second ALDH1Ai, CM10. Previously identified, CM10 represents a completely independent chemical series (Morgan and Hurley, 2015). Similar to 673A, CM10 inhibits ALDH1A1, 1A2, and 1A3 (IC50 1700, 740, and 640 nM, respectively) but does not inhibit any of the other ALDH family members tested (Figure 2B). Similar to 673A, CM10 inhibits ALDEFLUOR activity in live cells (Figure S2D), and preferentially depletes CD133+ cells (Figures 2A and S2E). PubChem searches suggested CM10 and 673A (active in 17 of 673 and 11 of 556 assays, respectively) had limited and non-overlapping, potential off-target activity (Tables S2 and S3). Together, this suggests that CD133 cell depletion is an on-target effect of the ALDH1Ai.
To determine whether 673A was inducing cell death in CD133+ CSC, we FACS-sorted the highly distinct CD133+/− A2780, Ovsaho, and OVCAR5 cells. We observed clear-preferential 673A-mediated death of CD133+ cells (Figure 2E). Similarly, following a single 12.5-μM dose of 673A, IncuCyte real-time cell imaging, and the Cytotox green assay, we observed significant increases in cell death (Figure S4A). Finally, to evaluate this at the single-cell level, FACS-isolated ALDH+CD133+ A2780 cells were loaded into a single-cell microfluidic culture and treated with vehicle or 673A. Although >90% of vehicle-treated cells were viable, >90% of 673A-treated cells died (Figure 2Fi). Dying 673A-treated cells appeared swollen and then fragmented (Figure 2Fii). Taken together, these data suggest that the ALDH1A family inhibition preferentially induces cell death in CD133+ ovarian CSCs.
ALDH1A Inhibition Induces Necroptosis in Ovarian CSCs
We next evaluated the mechanism of cell death. Unlike controls of cisplatin (which induces apoptosis) and shikonin (a compound known to induce apoptosis and necroptosis), 673A-treated CD133+ cells revealed no significant early or late induction of annexin-V (Figure 3Ai). In addition, IncuCyte real-time imaging confirmed no annexin-V positivity for 72 h after treatment, whereas cell death was confirmed by Cytotox dead-cell stain (Figure S4A). Pretreatment of cells with the pan-caspase inhibitor Z-VAD-FMK or the caspase-3 inhibitor Z-DEVD-FMK did not protect cells from 673A-induced cell death (Figures 3Aii–3Aiii). These data suggest that 673A induces nonapoptotic, caspase-independent cell death.
Figure 3. 673A Triggers Necroptosis in Ovarian CSCs.
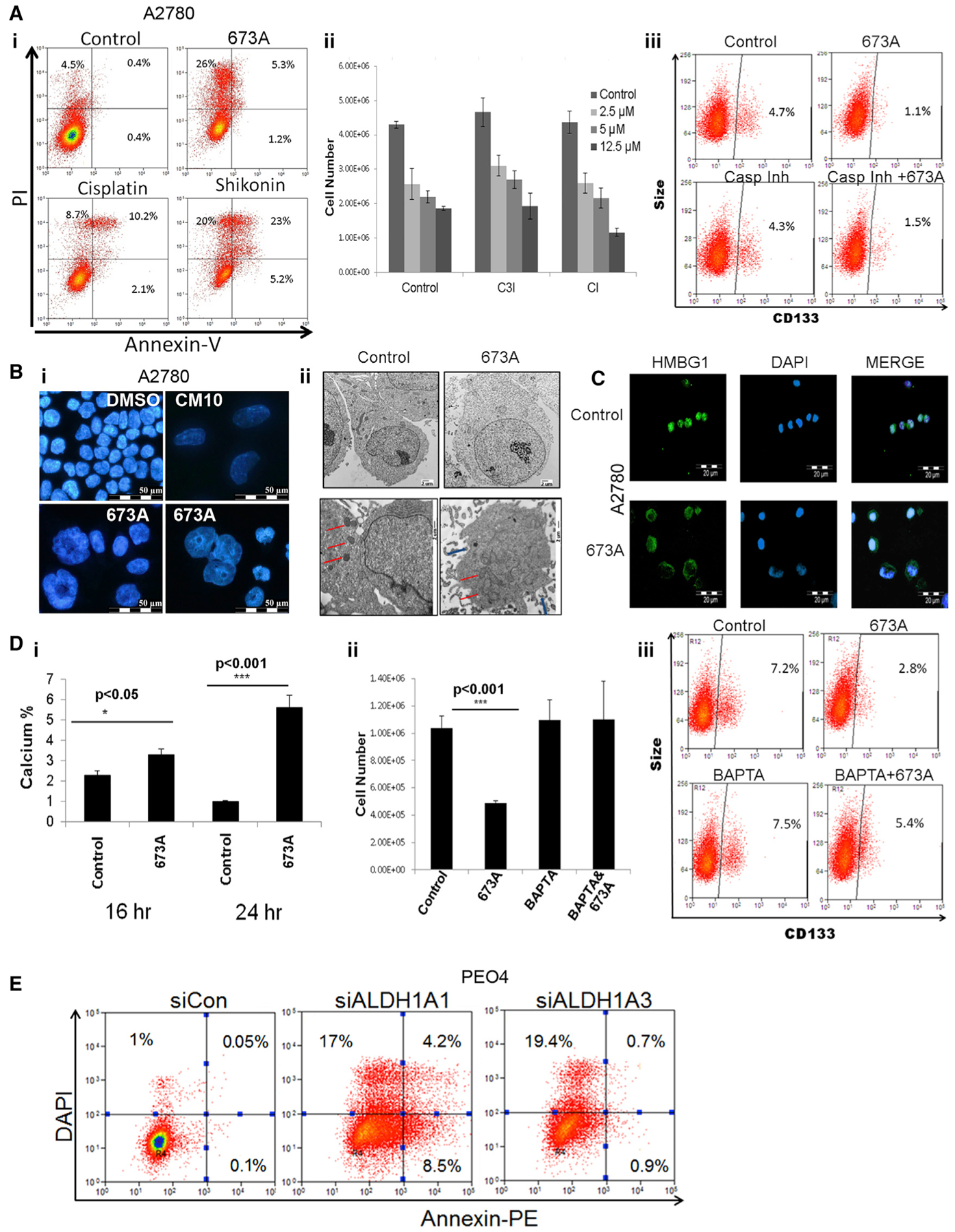
(A) (i) FACS analysis of annexin-V and PI stain as indicators of apoptosis in FACS-sorted CD133+ (A2780) cells after treatment with 12.5 μM 673A, 1 μg/ml cisplatin, or 12.5 μM shikonin. (ii) Cell counts of FACS-sorted CD133+ A2780 cells after pretreatment with pan-caspase inhibitor (CI; 20 μM) and caspase 3 inhibitor (C3I; 20 μM) plus 72-h treatment with the indicated concentrations of 673A. (iii) FACS analysis of CD133+ expression after treatments with CI (20 μM) and 673A (12.5 μM).
(B) Immunofluorescence (IF) DAPI nuclear stain (i) and TEM images (5,800×) (ii) of FACS-sorted CD133+ A2780 cells 24 h after treatment with 673A (12.5 μM) (red arrows, mitochondria; blue, rupture of plasma membrane).
(C) IF of HMBG1 localization 24 h after 673A treatment; DAPI was used for nuclear stain.
(D) Intracellular calcium level, using Fluo-4 FACS assay after 673A treatments (i). Cell counts (ii) and CD133 FACS analysis (iii) of A2780 cells after pretreatments with 20 μM BAPTA and treatments with 673A.
(E) FACS of annexin-V-PE/DAPI in PEO4 cells after ALDH1A family member downregulation using siRNA (48 h).
Error bars represent SDs n = 3 independent experiments with at least triplicate assays. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
Evaluation of 673A-treated cells stained with DAPI demonstrated clear nuclear swelling and loss of nuclear content consistent with necroptosis (Figure 3Bi). Similarly, transmission electron microscopy (TEM) revealed numerous morphologic features of cells undergoing necrosis: enlarged cell volume, enlarged mitochondria, rupture of plasma membrane with release of cellular content, and the appearance of large vacuoles (Figures 3Bii and S4B). Suggesting that this is programmed cell necrosis, or necroptosis, immunofluorescence revealed clear nuclear-to-cytoplasm translocation of the high mobility group-1 protein (HMGB1) with the 673A treatment (Figure 3C) (Gdynia et al., 2010). CM10 similarly induced a cellular/nuclear swelling necroptotic phenotype (Figure 3Bi), indicating that this is an on-target ALDH-dependent event.
Intracellular calcium (Ca) accumulation can trigger necroptosis. As assessed by the calcium indicator CM-H2DCF, 673A treatment of CD133+ cells was associated with a 1.5- and 6-fold increase in Ca after 16 and 24 h after 673A treatment, respectively (Figure 3Di). Indicating a requirement for Ca in 673A-driven cell death, pretreatment of CD133+ (A2780) cells with the Ca chelator 1,2-bis (2-aminophenoxy)ethane-N,N,N′,N′,-tetraacetic acid (BAPTA) before 673A treatment prevented 673A-induced cell death, based on total cell and CD133+ cell numbers (Figures 3Dii–3Diii). Indicating that 673A induction of necroptosis is an ALDH-dependent knockdown of ALDH1A3-induced cell death in PEO4 cells without significant induction of annexin-V (Figure 3E). siRNA knockdown (KD) of ALDH1A1 resulted in modest increases in annexin-V.
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) can propagate ligand-activated necroptosis (Linkermann et al., 2012). Suggesting 673A-induced necroptosis is RIPK1 independent, RIPK1 downregulation did not rescue cells from 673A-induced necroptosis (Figures 4Ai and S2Ci). Similarly, although RIPK1 is strongly expressed in most ovarian cancer cell lines (Figure S4Cii), pretreatments with the RIPK1 inhibitor Nec-1 did not rescue cells from 673A-induced cell death (Figures 4Aii–4Aiii).
Figure 4. Role of RIPKs in ALDH Inhibition-Induced Cell Death.

(A) Cell counts following 72 hr transfection of RIPK1 siRNA with and without 12.5-μM 673A treatment (i). Cell counts of FACS-sorted CD133+ A2780 (ii) and OVCAR8 (iii) cells after pretreatment with RIPK1 inhibitor Nec-1 and treatments with or without 673A.
(B) Cell counts after 72 h transfection of RIPK3 or control siRNA with or without 12.5 μM of 673A (i). Cell counts of FACS-sorted CD133+ A2780 cells 72 h after pretreatment with 5 μM of MLKL inhibitor necrosulfonamide (NSA) plus treatment with the indicated doses of 673A (ii).
(C) A western blot of PGAM5 protein immunoprecipitated (IP) with FADD, DRP1, and RIPK3 antibodies with or without 673A treatment in CD133+ (A2780).
(D) A western blot analysis of pDRP1, DRP1, and PGAM5 after 673A treatments. Note that controls were on the same blot but were separated, thus the gel break. Exposures for controls and the remaining gel are identical.
(E) A western blot analysis of DRP1 protein in mitochondrial and cytoplasmic fractions (i) and MLKL protein in membranous and cytoplasmic cellular fractions (ii) after 12.5-μM 673A treatment.
Error bars represent SDs n = 3 independent experiments with at least triplicate assays. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
RIPK1-independent necroptosis can be driven by RIPK3. Interestingly RIPK3 KD alone (Figure S4Ciii) resulted in a 70% reduction in cell number (Figure 4Bi). This made it difficult to interpret the effect of RIPK3 downregulation on 673A-mediated CSC death (Figure 4Bi). Similarly, high doses of necrosulfonamide (NSA), an inhibitor of the RIPK3 immediate downstream target MLKL also reduced cell numbers. However, low doses of NSA partly rescued cells from 673A-induced CD133+ cell death in A2780 and OVCAR8 cells, suggesting a potential role for RIPK3/MLKL in 673A-triggered death (Figures 4Bii and S4D). We also evaluated the effect of 673A on necroptosome formation. Numerous proteins have been linked with the formation of a necroptosome, including RIP3K, MLKL, DRP1, FADD, and PGAM5 (Wang et al., 2012). PGAM5 acts as a convergence point in RIPK3-dependent and -independent necroptosome formation, resulting in MLKL translocation to cell membranes (Cai et al., 2014) and DRP1 translocation to the mitochondria to increase membrane permeability (Wang et al., 2012). To determine whether 673A treatment induced the formation of the necroptosome complex, we independently immunoprecipitated FADD-, DRP1-, and RIPK3-associated protein complexes in control and 673A-treated cells and performed western blots for PGAM5. For all three immunoprecipitated proteins, we observed increased association of PGAM5 in 673A-treated cells versus control cells (Figure 4C). Eight and 12 h after 673A treatment, we also observed dephosphorylation of DRP1 and increased PGAM5 expression with the appearance of the PGAM5-S splice variant (Figure 4D). Similarly, we observed a significant increase both in the mitochondrial association of DRP1 (Figure 4Ei) and in the proportion of MLKL protein localized to the cell membrane fraction after 673A treatments (Figure 4Eii). Taken together, these data suggest 673A induces calcium-dependent necroptosis.
ALDH1A Family Inhibition Induces the Expression of the Mitochondrial UCPs
ALDH1A family members regulate the biosynthesis of RA to indirectly regulate RA-mediated gene expression. Indicating gene expression may have a role in 673A-mediated CD133+ cell death, 673A-induced CD133+ cell death is prevented by the transcription inhibitor flavopiridol (Figure 5A). qRT-PCR evaluation of mRNA expression of known RA target genes (Balmer and Blomhoff, 2002) in bulk cells after 673A treatment showed a significant increase in the expression of the mitochondrial UCPs UCP1 and UCP3 (Figure 5Bi). Importantly and potentially explaining the preferential sensitivity of CD133+ cells to ALDH1A family inhibition, qRT-PCR of FACS-isolated cells demonstrated that UCP1 and UCP3 were preferentially induced by 673A in CD133+ cells versus CD133− cells (Figure 5Bii).
Figure 5. 673A Induces Expression of the Mitochondrial Uncoupling Proteins.
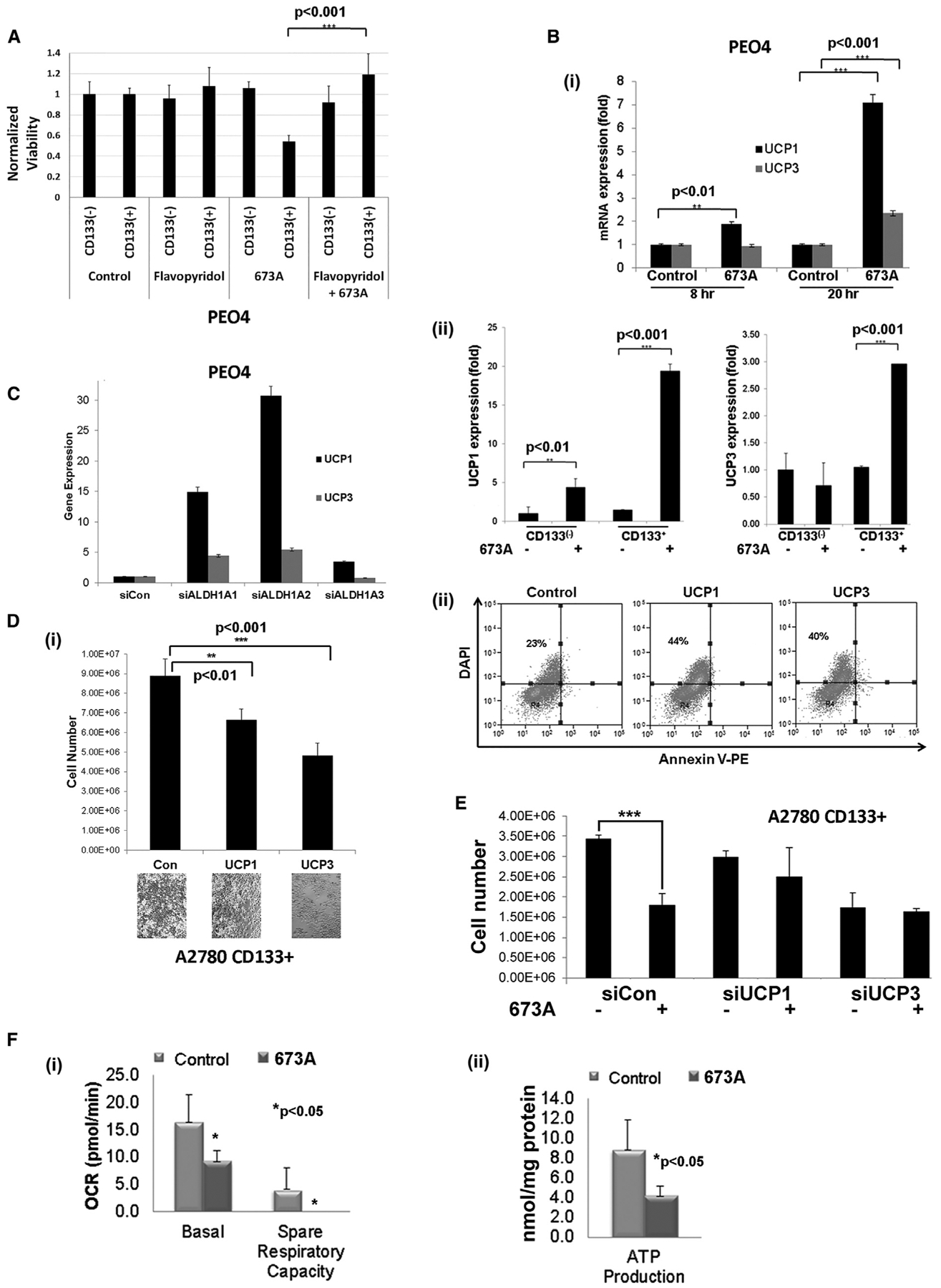
(A) Count of viable cells of CD133+ and CD133− PEO4 cells after 36-h treatment with transcription inhibitor flavopiridol ± 673A.
(B) qRT-PCR analysis of uncoupling protein 1 (UCP1) and UCP3 mRNA expression in total PEO4 cells (i) and FACS-sorted CD133+/− PEO4 cells (ii) after treatments with 12.5 μM of 673A.
(C) qRT-PCR analysis of UCP1/3 expression 48 h after siRNA downregulation of ALDH1A family members in PEO4 cells.
(D) Cell counts (i) and FACS analysis (ii) of annexin-V/DAPI after 48 h of UCP1 and UCP3 overexpression in FACS-sorted CD133+ A2780 cells.
(E) Cell counts of cells transfected with UCP1 and UCP3 siRNA and treated with 12.5 μM 673A (at 72 h).
(F) Quantification of basal and spare respiratory capacity (i) and ATP production in primary HGSC cells (ii) after treatment with 12.5 μM 673A as assessed by Seahorse.
Error bars represent SDs n = 3 independent experiments with at least triplicate assays. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
UCP1 protein is aberrantly regulated in ALDH1A1-KO mice (Kiefer et al., 2012), supporting UCP induction as an on-target/ALDH inhibition-mediated effect of 673A. Further supporting that UCP1/UCP3 induction is a result of ALDH1A family inhibition, siRNA KD of the various ALDH1A family members similarly resulted in the induction of UCP1 and UCP3 (Figure 5C), with KD of ALDH1A1 and ALDH1A2 having the most prominent effects. Forced UCP3 expression reduced cell numbers 2-fold, whereas UCP1 expression reduced cell numbers ~30% compared with a control (Figure 5Di). Cells with UCP1 or UCP3 overexpression demonstrated no upregulation of annexin-V (Figure 5Dii). Indicating that UCP proteins are important for 673A-trig-gered necroptosis, siRNA downregulation of UCP1 and UCP3 before 673A treatments rescued CD133+ from ALDH inhibition-triggered cell death (Figure 5E).
As mitochondrial UCPs have a critical role in regulating OXPHOS, we next determined OXPHOS capacity with Seahorse after 673A treatments. Analysis of primary cells from HGSC patients after 673A treatment resulted in decreased OXPHOS capacity, associated with a significant decrease in basal and spare respiratory capacity and ATP production (Figures 5Fi and 5Fii).
The Effect of ALDH1A Inhibition on Tumor Stemness and Chemotherapy Resistance
We next examined the biologic effect of 673A-mediated preferential depletion of CSCs. In general, 673A had minimal effect on bulk cell viability in multiple cell lines tested (TD50 of 40–80 μM; Figure S3A). This is consistent with 673A-target CSCs, which make up a small percentage of the cells in most cell lines. Because CSCs have been implicated in chemotherapy resistance, we evaluated combination therapy with 673A and cisplatin chemotherapy, using cellular recovery assays with chemotherapy-resistant SKOv3 cells. Although 673A has limited activity as a single agent versus bulk SKOv3 (Figure S3Ai), 673A was highly synergistic with chemotherapy; 9 days after treatment with cisplatin or 673A alone, there was 100% cellular recovery, and combined treatment of cisplatin and 673A led to a 30-fold reduction in cell number 9 days after treatment (Figure 6A). No therapeutic benefit was noted with the addition of the ALDH2 inhibitor daidzin to cisplatin (Figure 6A). Given these observations, we evaluated combination indexes of 673A and cisplatin in primary human ovarian cancer cells, and 673A and adriamycin in triple-negative breast cancer cell lines. The 673A was highly synergistic with chemotherapy in both cases (Figure 6Aii) (combination index range 0.2–0.9). Similarly, we assessed the ability of 673A to radiosensitize FACS-sorted CD133+/− A2780 cells or ALDH+/− SKOv3 cells. CD133+ cells appeared more radio-therapy resistant at baseline but were significantly radiosensitized by 673A (radiation enhancement ratio [RER] = 1.4; Figures 6Bi and 6Biii). ALDH+ cells, although not inherently more resistant than ALDH− cells at baseline, were also sensitized to radiation therapy by 673A (RER = 1.5; Figures 6Bii–6Biii). Tumor sphere growth assays were used to evaluate the effect of 673A on CSCs in primary patient samples. A single 12.5 μM dose of 673A resulted in an average 19-fold reduction in tumor sphere formation of primary cells (Figure 6C).
Figure 6. The Effect of ALDH Inhibition on Tumor Stemness.
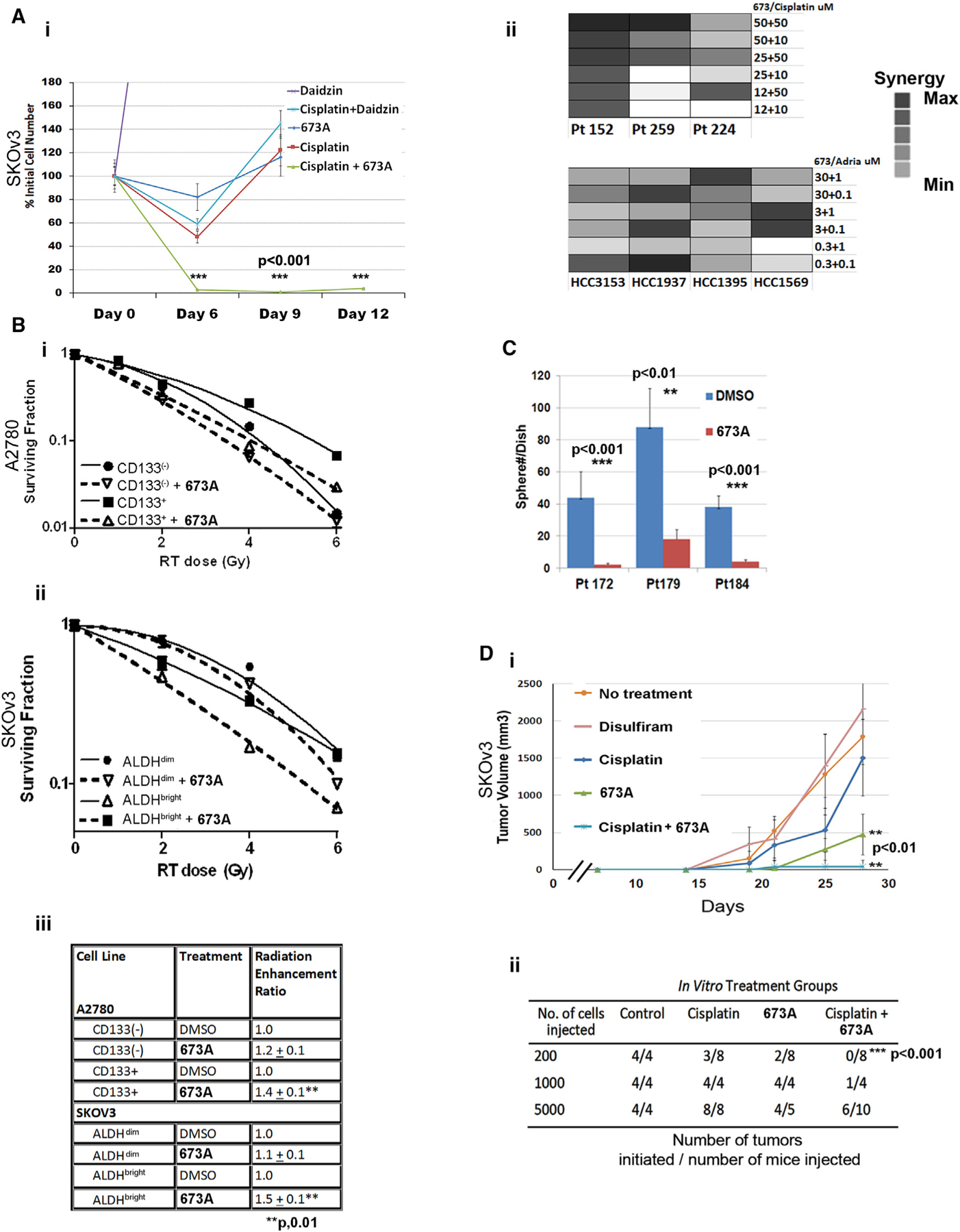
(A) (i) Cell counts of SKOv3 cells over time after treatment with 1 μg/mL cisplatin alone or in combination with ALDH inhibitors. (ii) Synergy factor calculations upon treatment of primary ovarian high grade serous (HGS) samples in spheres (Pt152, Pt259, and Pt224) and breast cancer cell lines (HCC3153, HCC1937, HCC1395, and HCC1569).
(B) Surviving fraction after radiation of FACS-isolated CD133+/− A2780 (i) and ALDH+/− SKOv3 (ii) cells after DMSO or 673A treatment and subsequent radiation, and table of radiation-enhancement ratios (iii).
(C) Sphere formation capacity in primary HGS cells with 673A.
(D) Tumor growth (i) and tumor initiation capacity (ii) of 5,000 FACS-sorted viable (annexin-V−/PI−) cells after single-agent and combined therapy with cisplatin and ALDH inhibitors. Number of established tumors was evaluated 3 months following cell injection.
Error bars represent SDs; n = 3 independent experiments with at least triplicate assays. Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
Next, we assessed the effect of 673A on tumor-initiation capacity. Chemotherapy-resistant SKOv3 cells were treated in vitro with cisplatin (1 μg/ml), DSF, 673A (12.5 μM), or cisplatin and 673A. Forty-eight hours after treatment, live (propidium iodide [PI]− and annexin-V−) cells were FACS-isolated, and 5,000 cells were injected into mice. Although treatment with DSF had no effect on cancer growth, 673A demonstrated a 4–5-fold reductions in tumor growth versus no treatment or cisplatin therapy (Figure 6Di). Furthermore, with a limiting dilution assay, 673A alone was able to reduce tumor initiation ~50% and, when combined with cisplatin therapy, eliminated tumor initiation capacity (Figures 6Di and 6Dii).
673A Synergizes with Chemotherapy In Vivo to Eradicate Cancer in Mice
We next evaluated the anti-tumor effect of 673A in vivo with Ovsaho, HEY-1, and A2780 cell-line xenografts. Three days after cancer cell inoculation, the mice (n = 10 per treatment group) were treated daily with vehicle or 673A (20 mg/kg) for 21 days. The 673A treatment reduced tumor burden 40%–60% (Figure 7Ai) and significantly decreased tumor cell Ki67 expression (Figure 7Bi–ii). H&E analysis of 673A-treated tumors revealed necrotic morphology (Figure 7B). We next assessed combination therapy on OVCAR8 cells, which are ~50% CD133+. We chose a lower dose of 673A because OVCAR8 CD133+ cells are very sensitive in vitro. Combined treatment of 673A at 4 mg/kg (n = 10) daily with three weekly doses of cisplatin reduced tumor burden3.5-fold versus cisplatin only (Figure 7C). qRT-PCR analysis of tumors revealed a significant upregulation of UCP1 and UCP3 in 673A-treated tumors (Figure 7D). Hematologic, renal, and liver function tests revealed no additional toxicity with the addition of 673A to cisplatin (Table S4). Next, we determined the ability of 673A to eliminate “minimal residual disease.” CaoV3 cells (1 × 105) were injected subcutaneously in mice. After 7 days, the mice were treated with cisplatin, 673A, or combination therapy, as above, for 3 weeks, and tumor development over the next 6 months was evaluated. Ninety percent of mice treated with cisplatin developed tumors. In contrast, only 50% of mice treated with 673A alone and 40% of mice treated with cisplatin and 673A developed tumors (Figure 7E). In a parallel study that used intraperitoneal (i.p.) injection of 5 × 104 FACS-sorted ALDH+ SKOv3 cells, 100% of mice treated only with cisplatin died, whereas 62% of mice treated with cisplatin and 673A were disease free at 6 months (Figure 7F). We also examined the effect of 673A with the LSL-K-rasG12D/+PtenloxP/loxP genetic mouse model of endometrioid ovarian cancer (Dinulescu et al., 2005). Two weeks after intra-bursal adeno-CRE virus-mediated tumor initiation, mice were treated with vehicle, carboplatin (25 mg/kg) weekly, or a combination of carboplatin weekly and 673A (20 mg/kg) daily for 7 weeks. Although only 16% of mice treated with carboplatin alone were tumor free after therapy, 27% of animals treated with combined carboplatin and 673A therapy were tumor free at the conclusion of the experiment (Figure 7G). Next, we examined the effect of combined 673A and carboplatin or 673A and cisplatin treatment with two chemotherapy-resistant patient-derived xenograft (PDX) models. The addition of 673A to either carboplatin or cisplatin not only prevented tumor growth but also induced tumor regression, reducing final tumor weight more than 2-fold (Figure 7H).
Figure 7. 673A Efficacy In Vivo.
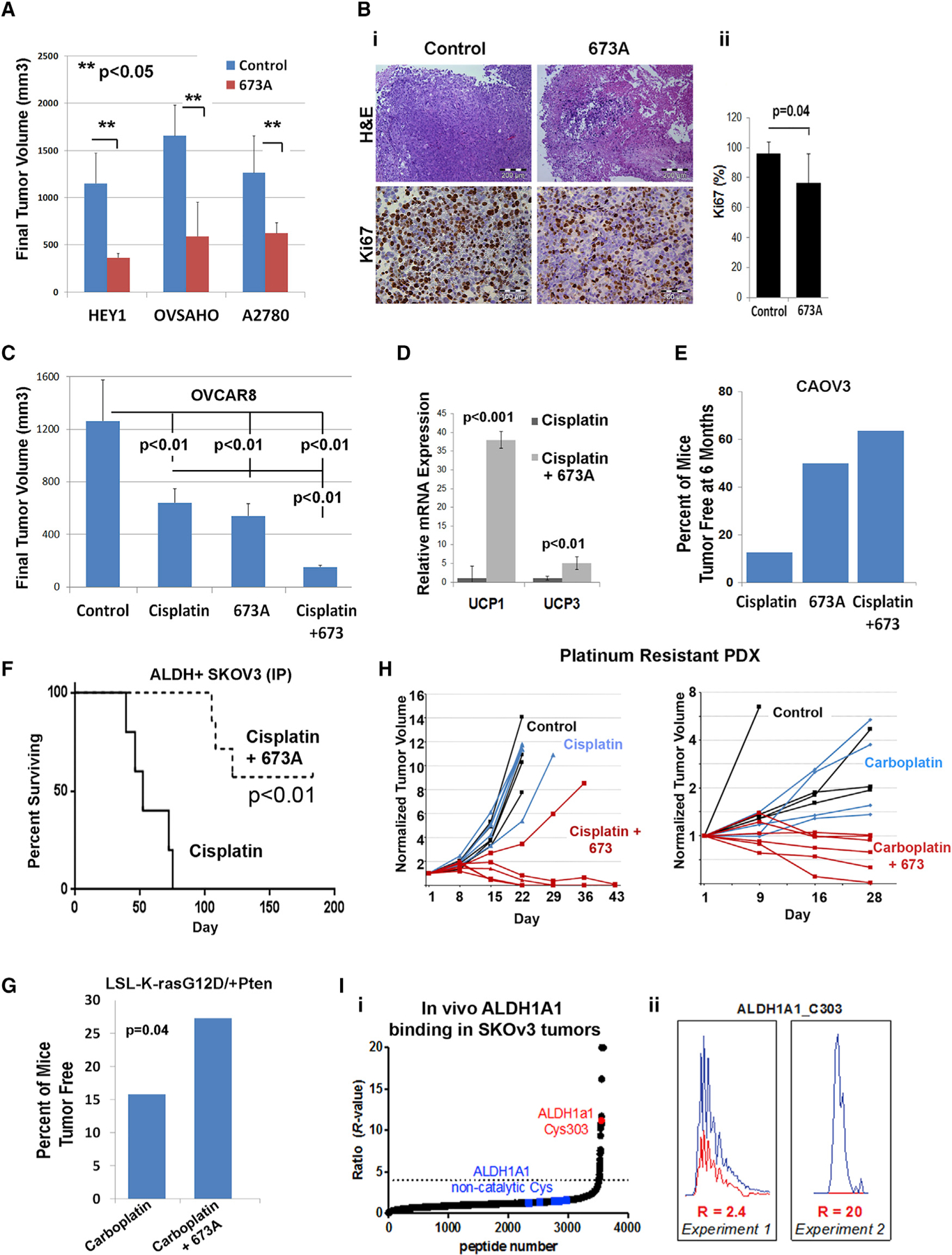
(A) Tumor volumes for HEY-1, Ovsaho, and A2780. Error bars represent means ± SD; n = 10 mice.
(B) (i) H&E and Ki67 images of Ovsaho tumors. (ii) Quantification of Ki67. Scale bars represent 200 μm for H&E and 100 μm for Ki67.
(C) Volumes for OVCAR8 tumors. Error bars represent means ± SD; n = 10 mice; p < 0.01.
(D) qRT-PCR analysis of UCP1 and UCP3 expression in OVCAR8 tumors; n = 3 mice.
(E) Percentage of mice that were tumor free 6 months after inoculation with 1 × 105 CAOV3 cells and treatment with 21 days of 673A (20 mg/kg for 21 days), cisplatin (1 mg/kg once a week for 3 weeks), or both. Fraction surviving is indicated in the parentheses.
(F) Survival studies during 6 months after i.p. inoculation of 1 × 104 FACS-sorted ALDH+ SKOv3 cells.
(G) Percentage of mice that were tumor free after 9 weeks of treatments in an LSL-K-rasG12D/+PtenloxP/loxP endometrioid ovarian cancer model.
(H) Growth curves of platinum-resistant PDX; n = 5 mice/group for two independent PDX models. The 673A was given daily for 21 days at 20 mg/kg; cisplatin or carboplatin were given weekly.
(I) (i) Combined waterfall plot from four biological replicates (mean R values) of ALDH1A inhibition in SKOv3 tumors in vivo. (ii) Representative of inhibition of ALDH1a1_C303 in two data sets in SKOv3 tumors in vivo.
Data are presented as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.005.
Finally, we confirmed on-target activity in vivo. Immunohistochemistry (IHC) demonstrated a reduction of ALDH1A1-expressing cells in responding 673A-treated tumors (Figure S5Ai). In addition, ALDEFLUOR activity in splenocytes of animals being treated with 673A was decreased 3-fold (Figure S5Aii). To further confirm inhibition of ALDH in tumors in vivo, we used isotopic tandem orthogonal proteolysis-activity based protein profiling (isoTOP-ABPP) and click chemistry (Speers and Cravatt, 2004; Weerapana et al., 2010) to evaluate ALDH1A enzyme activity. The 673A-treated tumors demonstrated clear protection of the catalytic residue Cys303 in ALDH1A1, whereas no binding to noncatalytic residues was detected (Figures 7Ii–ii and S5B). Finally, evaluation of tumors that progressed on 673A treatment demonstrated upregulation of ALDH activity, suggesting on-target resistance mechanisms (Figure S5Aiii).
DISCUSSION
In this study, we identified a selective, pan-ALDH1A family inhibitor, 673A. This ALDH1Ai preferentially induced necroptosis in ovarian CSCs, and, with chemotherapy, 673A eliminates chemotherapy-resistant tumors. This study links ALDH enzymatic activity with the necroptosis pathway. Furthermore, it adds to the growing literature supporting ALDH enzymes as important therapeutic targets in cancer and demonstrates the efficacy of selectively targeting the ALDH1A family.
ALDH as a Therapeutic Target
ALDH1A isozymes are known to have a direct role in resistance to the chemotherapeutics ifosfamide and cyclophosphamide (Pinto et al., 2009). ALDH activity has also been broadly linked with resistance to chemotherapeutics such as paclitaxel and doxorubicin, which are not directly metabolized by ALDH isozymes (Bretz et al., 2012; Dylla et al., 2008). Numerous preclinical studies have tested the ability of ALDH inhibitors to reverse chemotherapy resistance. DSF has anti-cancer activity in vitro and in vivo (Irving and Daniel, 1987; Kast and Belda-Iniesta, 2009; Lin et al., 2011; Morrison et al., 2010; Rezk et al., 2015; Yip et al., 2011). Dimethyl ampal thiolester (DIMATE), an ALDH1 and ALDH3 inhibitor, depletes leukemia stem cells but, importantly, does not affect hematopoietic stem cells (Venton et al., 2016). More recently a potent ALDH1A1 selective inhibitor and a pan-ALDH1A inhibitor were found to synergize with chemotherapy (Huddle et al., 2018; Yang et al., 2018), whereas an ALDH-targeted pro-drug effectively eliminated ALDHbright melanoma cells (Sarvi et al., 2018).
Clinical work also supports development of ALDHi. A phase IIb trial in patients with advanced lung cancer demonstrated a statistically significant improvement in the overall survival of patients treated with DSF and chemotherapy versus chemotherapy alone, with ~10% of DSF-treated patients being disease free at 3 years (Nechushtan et al., 2015). Similarly, a small study of high-risk breast cancer patients receiving adjuvant chemotherapy and sodium diethyldithiocarbamic acid (a primary active metabolite of DSF) revealed a trend toward increased overall and disease-free survival (Dufour et al., 1993). These studies suggest not only the efficacy but also the safety of inhibiting ALDH activity in patients. Consistent with the safety of targeting ALDH1A family members, ALDH1A1 is dispensable for the function of murine stem cells (Levi et al., 2009).
Unfortunately, DSF is not an ideal ALDH inhibitor for cancer therapy: DSF has a very short half-life in vivo and numerous active metabolites that are associated with off-target effects, increased toxicity, and dose-limiting specificity (Agarwal et al., 1986; Malcolm et al., 2008; Petersen, 1992). In addition, although DSF can partially reverse chemotherapy resistance in ovarian cancer cells, it has limited efficacy in ovarian cancer in vivo (Rezk et al., 2015). Our analysis of ovarian cancer suggests that different cancers may express different ALDH1A family members. Indeed, ALDH1A1, ALDH1A2, and ALDH1A3 are all reported to be expressed in ovarian cancer (Saw et al., 2012). We observed that siRNA KD or CRISPR KO of one ALDH1A was associated with reduced, but not complete, loss of response to ALDH1Ai. We believe this is likely related to expression of compensatory ALDH1A family members. Dual KD/KO experiments will be necessary to address this, but these are technically challenging because of difficulties in maximizing KD of multiple genes without increasing toxicity related to nucleic acid concentrations. Multi-KO CRISPR is an alternative but is more time consuming, requires multiple selection methods or steps, and has the risk of off-target genome editing (Fu et al., 2013).
Given strong support in the literature for a therapeutic role of ALDH1A enzymes in chemotherapy resistant and stemness, variable expression of different ALDH1A family members in cancers, and the potential redundancy of function for these ALDH1A family members, we developed a pan-ALDH1A family member inhibitor. We found that the ALDH1A pan-family inhibitor 673A effectively reversed chemotherapy resistance. The 673A was effective in cell lines that predominantly expressed either ALDH1A1 or ALDH1A3. Importantly, in several in vivo models of ovarian cancer, 673A significantly enhanced chemotherapeutic activity, resulting in tumor eradication.
The Effect of Targeting CSCs
Although there remains controversy regarding CSCs, ALDH enzymatic activity, either alone or in combination with other stem cell markers, is arguably the best-characterized CSC marker. In ovarian cancer, ALDH+/CD133+ ovarian cancer CSCs meet the definition of CSCs put forth by Pattabiraman and Weinberg (2014); ALDH+/CD133+ ovarian cancer cells demonstrate both (1) increased ability to initiate tumors (Silva et al., 2011) and (2) a clear ability to undergo asymmetric division to both self-renew and produce non-self-daughter cells (Choi et al., 2015). The 673A preferentially induced cell death in CD133+ ovarian cancer cells. Despite targeting less than 10% of the cells in most cancer cell lines, 673A had a significant effect on tumor growth in vivo. As a single agent, 673A could reduce tumor growth >50%. As noted above, when combined with chemotherapy, 673A increased tumor eradication rates in multiple in vivo models of ovarian cancer. We believe this is both via the direct targeting of CD133+ CSCs and the reversal of ALDH1A-driven chemotherapy resistance. Taken together, these data support a critical role for CSCs, whether inherent or because of de-differentiation, as a therapeutic target.
ALDH1A Inhibition and Necroptosis
Treatment of cancer cells with the pan-ALDH1A inhibitor (ALDH1Ai) 673A results in (1) increased formation of necroptosomes, (2) translocation of HMBG1 to the cytoplasm, (3) translocation of mixed lineage kinase domain-like psuedokinase (MLKL) to the membrane, (4) reduced phosphorylation of DRP1 with translocation to the mitochondria, and (5) clear organelle and cellular swelling and leak of intracellular contents. This is all consistent with programmed cell necrosis. Our study, thus, links ALDH activity and necroptosis.
ALDH1Ai-driven necroptosis is dependent on active transcription and translation. ALDH1Ai induced high levels of UCP1 and UCP3 mRNA preferentially in CD133+ cells. This is consistent with ALDH1A1-KO mice, which show induction of UCP1 (Onda and Yoshikawa, 2011). Although mostly studied in thermogenesis and fatty acid metabolism in adipogenesis, increasing evidence links UCP proteins with cancer. UCP3 has been shown to decrease ATP production in HeLa cells and inhibit skin carcinogenesis by blocking AKT activation (Nowinski et al., 2015). UCP1/2/3 overexpression has also been linked with induction of autophagy death in breast cancer cells (Sanchez-Alvarez et al., 2013). We found that 673A treatment resulted in strong induction of UCP expression, a reduction in ATP, and ultimately, necroptosis. Exactly how UCPs are linked with the induction of necroptosis remains to be determined. Induction of UCPs appears critical for ALDH1Ai-induced death because UCP KD partly rescued cancer cells from ALDH1Ai-induced death. Interestingly, CD133+ ovarian cancer cells express higher levels of UCP mRNAs at baseline. This higher level of UCP expression may explain the increased sensitivity of CD133+ cells to ALDH1Ai-induced death.
Several observations suggest that the necroptosis observed is an on-target effect: (1) siRNA KD of ALDH family members demonstrated similar induction of UCPs and CD133+-selective cell death; (2) ALDH inhibition in vivo resulted in drastic reduction in ALDH1A stain in sensitive tumors, whereas resistant tumors demonstrated strong induction of ALDH activity; (3) an ALDH inhibitor from a completely independent chemical series, CM10, has the same effects on cells; and (4) 673A and CM10 have very little to no activity in common in off-target assays as reported by PubChem (CID 969428 and 889692).
In conclusion, we have discovered that pan-ALDH1A inhibition can trigger calcium-dependent necroptotic cell death preferentially in CSCs. Our results suggest that induction of necroptosis in CSC may represent a powerful therapeutic approach.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ron Buckanovich (buckanovichrj@mwri.magee.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAIL
Ovarian cancer cell lines: PEO1, PEO4, OvCar8, OvCar3, OvCar8, A2780, SKoV3, Ovsaho and Kuramochi were used in this study.
Primary samples: primary ovarian cancer tumors were obtained after surgery and dissociated to single cell suspension. Single cell suspension was used for spheroid studies.
LSL-K-ras G12D/+ Pten loxP/loxP was established by Dr. Romero, OBGYN department, University of Chicago
PDX model of ovarian cancer was established by Dr. Charles Landen, OBGYN department, University of Virginia and Dr. Buckanovich University of Michigan.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice from Jackson labs #00557 were used for all subcutaneous and IP injections
METHOD DETAILS
Cell culture and reagents
PEO1, PEO4, OvCar8, and Kuramochi were used in experiments that assessed 673A effect on CD133 cells due to very high CD133+ expression. A2780, Ovsaho, and OVCAR5 cells were used in experiments which required very distinctive CD133+ population. SKoV3 were used in experiments that required very bright ALDH+ population. OvCar5 and OvCar8 used due to their high ALDH1A3 expression.
A2780, OvCar8, and PEO1 ovarian cancer cells were provided by Dr. S. Murphy (Duke University). SKOv3, A2780, CaoV3, OvCar8, OvCar5 Ovsaho, PEO4, and Kuramochi cells were maintained in RPMI-1640 with 10% FBS and 1% Pen/Strep. Ovsaho and Kuramochi cells were purchased from JCRB cell bank. PEO1 cells were maintained as above with 0.2% pyruvate. 673A was purchased from ChemBridge Corporation. The compounds and reagents used were Z-VAD-FMK (InvivoGen), BAPTA (Tocris), necrostatin-1 (Tocris), necrosulfonamide (Tocris), cisplatin (SICOR Pharmaceuticals), acetaldehyde, propionaldehyde, NAD+, and buffers for ALDH activity (Sigma Aldrich). The kits used were Fluo Calcium Indicator CM-H2DCF (Invitrogen), SuperScript II First-Strand Synthesis SuperMix (Invitrogen), Amaxa Cell Line Nucleofector Kit (Lonza), MTT assay (Invitrogen), and Annexin/PI Apoptosis Detection Kit (BD PharMingen), all according to manufacturers’ instructions.
Growth curves and cell viability assay
Cells (7,500 cells/well) were plated in a 96-well plate. After 24 hr, the medium was replaced with fresh growth medium with various treatments. Growth curves were constructed by imaging plates, using the IncuCyte system (Essen Instruments), where the growth curves were built from confluence measurements acquired during round-the-clock kinetic imaging. Cell viability was determined using MTT assay (Invitrogen). Cell number was assessed by direct counting using a Countess automated cell counter (Invitrogen).
Protein preparation
For total protein extraction, cell pellets were lysed on ice in 1 × phosphate-buffered saline with 1% NP40, 0.1% SDS, and freshly added 20 mg/ml protease inhibitor cocktail and 2 mM phenylmethylsulfonyl fluoride (Sigma Chemical). Protein fractionations were performed using subcellular protein fractionation kits (ThermoFisher Scientific). Protein concentration was determined by BCA protein assay (Pierce Biotechnology).
Western blotting
Western blots were performed as previously described (Coffman et al., 2016). The following antibodies were used: UCP1 (R&D), UCP3 (Abcam), PGAM5 (Gentex), DRP1 (Cell Signaling), pDRP1 (GeneTex), TOM20, GAPDH, ACTB, and Na1K-ATPase1 (Cell Signaling). Specific protein bands were visualized using enhanced chemiluminescence (Pierce Biotechnology).
Flow cytometric analysis and fluorescence-activated cell sorting (FACS)
FACS was performed as previously described (Silva et al., 2011) with anti-human CD133 (Miltenyi Biotec 1:100), then PI or DAPI, followed by incubation with ALDEFLUOR per protocol (Stem Cell Technologies). FACS was performed using the Astrios (Beckman Coulter) or FACS Accuri (BD).
Hanging drop culture for spheroid formation from OvCSCs
FACS-sorted OvCSCs were harvested, and separated from culture media (RPMI 1640 supplemented with 1X fetal bovine serum and 0.1X antibiotics and antimycotics), using high-speed centrifugation. Cells were resuspended in serum-free growth medium (SFM; DMEM/F12 supplemented with 5 ng/ml bFGF, 5 ng/ml EGF, 1X B27 supplement, 1X insulin-transferrin-selenium supplement, 1X nonessential amino acids, and 1X antibiotics and antimycotics). Cell counts were obtained using a hemocytometer and adjusted to a concentration of 10 OvCSCs per 20 μl of serum-free medium. Spheroids were initiated using 10 OvCSCs per spheroid, following protocols established previously. Spheroids were maintained in SFM for a period of 7 days and imaged using live phase contrast microscopy to follow spheroid formation.
Drug treatment on OvCSC spheroids in hanging drop array culture
OvCSC spheroids were initiated with 10 OvCSCs per 20 μl hanging drop in 384-well hanging drop plates. Spheroids were allowed to aggregate and form a 3D microtissue over a period of 7 days. For drug treatment, a 10X stock of drug was prepared independently, and 2 ml was added to the 20ml hanging drops containing spheroids to yield the desired 1X drug concentration. Spheroids were treated with cisplatin, ALDH inhibitor compound 673A, or cisplatin + compound 673A at the indicated concentrations. The effect of drug treatment on spheroids was determined at 72 hr using the CellTiter-Glo 3D metabolic assay to determine viability after drug treatment. Control untreated spheroids were maintained for the same duration in hanging drop culture. At least 20 spheroids (technical replicates) were assayed per experiment, with 3–5 biological replicates.
Radiation survival assays
FACS-sorted cells were seeded at clonal densities, followed by drug and radiation treatments (0–6Gy) and incubation for 10–12 days for colony formation, as previously described (Morgan et al., 2010). Drug cytotoxicity (in the absence of radiation) was calculated as the ratio of surviving drug-treated cells relative to untreated control cells. Survival data were corrected for drug cytotoxicity, and survival curves were fitted using the linear quadratic. Radiation enhancement ratios were calculated as the ratio of the mean inactivation dose under control conditions divided by the mean inactivation under drug treatment conditions, where a value significantly greater than 1 indicates radiosensitization. Radiation was performed using a Philips RT250 (Kimtron Medical) at a dose rate of ~2 Gy/min. Results are from three independent experiments.
Patient tumor specimens
Informed consent was obtained from all patients before tissue procurement. All studies were performed with the approval of the Institutional Review Board of the University of Michigan. Tumors were mechanically dissected into single-cell suspensions and isolated on a Ficoll gradient as previously described (Silva et al., 2011). For ascites, studies of cell pellets were collected by centrifugation, and red cells were lysed using ACK buffer (Lonza), washed, passed through a 40-μm filter, then passed four times through a standard hub pipetting needle to isolate single cells. Human tumor sphere assays were performed as previously described (McLean et al., 2011).
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed as previously described (Chefetz et al., 2013). Briefly, PCR reactions were set up in triplicates with the SYBR Green Master Mix (Applied Biosystems). HPRT was used as an internal control. All primers are designed as intron-exon spanning primers; primer sequences are found in Table S5. All PCR reactions were validated by the presence of a single peak in the melt curve analysis. All products were resolved by 2.5% agarose gel electrophoresis to confirm DNA fragments of expected size.
Immunohistochemistry (IHC)
IHC was performed as previously described (Silva et al., 2011). Tumors were stained at the histology core at the University of Michigan using EDTA-based antigen retrieval and mouse anti-ALDH antibody (BD Biosciences, clone 44/ALDH; 1:100) or anti-Ki67 antibody (Abcam 15580, 1:2,000). For stain quantification, 8–10 sections from 4 to 5 tumors per treatment group were analyzed. For Ki67, cells were counted from 10 low-power fields (× 100) per section by three people. Counts were then compared using a 2-sided Student’s t test.
Isolation of proteome and quantification of protein labeling
For proteomic analysis the tumors were thawed on ice, transferred into 1.5-mL Eppendorf tubes, washed with cold PBS (2 × 1.0 mL), and homogenized in cold PBS (1.0 mL) with zirconium oxide beads (1 scoop) using a Bullet Blender at 4°C (Next Advance Bullet Blender with air cooling; 3 min, speed setting #8). The proteome was separated from the debris using a table-top centrifuge (4°C, 6000 g, 3 min). The homogenization was repeated two additional times, and the combined proteome was further separated into soluble and particulate fractions by ultracentrifugation (100,000 × g, 45 min). The soluble proteomic fraction was analyzed by isoTOP-ABPP.
isoTOP-ABPP IA-alkyne labeling and click chemistry
Samples (500 μL, 2.0 mg/mL) were labeled with 100 μM iodoacetamide alkyne (1, IA-alkyne, 5 μL of 10 mM stock in DMSO) for 1h at ambient temperature and then conjugated to isotopically labeled, TEV-cleavable tags (TEV tags) using copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Reagents for the CuAAC reaction were pre-mixed prior to their addition to the proteome samples. In brief, TEV tags (light or heavy, 10 μL of 5 mM stocks in DMSO, final concentration = 100 μM), tris(benzyltriazolylmethyl)amine ligand (TBTA; 30 μL of 1.7 mM stock in DMSO: t-butanol 1: 4, final concentration = 100 μM), tris(2-carboxyethyl)phosphine hydro-chloride (TCEP; 10 μL of fresh 50 mM stock in water, final concentration = 1 mM), and Cu(OAc)2 (10 μL of 50 mM stock in water, final concentration = 1 mM) were combined in a separate Eppendorf tube, vortexed and spun down using table centrifuge. “Heavy” CuAAC reaction mixture (55 μL) was added to the DMSO-treated control samples and “light” CuAAC reaction mixture (55 μL) was added to compound-treated samples, and the reaction was allowed to proceed for 1h. Upon completion, “heavy” and “light” samples were combined pairwise in a 15-mL conical falcon tube kept on ice, containing 4 mL of cold methanol (pre-chilled at −80° C), 1 mL CHCl3, and 1 mL H2O. Eppendorf tubes from the reaction mixtures were washed with an additional 1mL of H2O each, and the washed fractions were transferred to the same falcon tube (final ratios MeOH: CHCl3: H2O = 4: 1: 4). Following centrifugation (5,000 × g, 10 min, 4°C), CHCl3 and aqueous layers were aspirated without perturbing the protein disk formed, which was resuspended in cold methanol (2 mL) by sonication. CHCl3 (1 mL) was then added, the mixture was vortexed, and the proteins were pelleted by centrifugation (5,000 × g, 10 min, 4°C). The resulting pellets were solubilized in 1.2% SDS in PBS (1 mL) with sonication and heating (95°C, 5 min).
isoTOP-ABPP streptavidin enrichment
SDS-solubilized samples were diluted with 4 mL PBS, streptavidin-agarose beads were added for enrichment (final concentration: 0.2% SDS in PBS), and the bead mixture was rotated for 3 h at ambient temperature. For each sample, 100 mL of streptavidin-agarose bead slurry (Fisher) was washed in 10 mL PBS and resuspended in 1 mL PBS prior to addition to the sample. After incubation, the beads were pelleted by centrifugation (2,000 × g, 2 min) and were extensively washed to remove nonspecifically binding proteins (2 × 10 mL 0.2% SDS in PBS, 2 × 10 mL PBS, and 2 × 10 mL water).
isoTOP-ABPP trypsin and TEV digestion
The beads were transferred to Eppendorf tubes with 1 mL water (2 × 0.5 mL), pelleted by centrifugation (4,000 × g, 2 min), and resuspended in PBS containing 6 M urea (0.5 mL). DTT (25 μL of a fresh 200-mM stock in water, final concentration = 10 mM) was then added, and the beads were incubated at 65°C for 15 min. Iodoacetamide (25 μL of a 400-mM stock in water, final concentration = 20 mM) was then added, and the final mixture was incubated at 37°C for 30 min, with shaking. Following the incubation, the mixture was diluted with PBS (0.9 mL), and the beads were pelleted by centrifugation (2,000 × g, 2 min) and resuspended in PBS containing 2 M urea (200 μL). Trypsin (Promega, sequencing grade; 2 μg in 6 μL of trypsin buffer containing 1 mM CaCl2) was added to the mixture, and the digestion was allowed to proceed overnight at 37°C, with shaking. The beads were pelleted by centrifugation (2,000 × g, 2 min) and the tryptic digest was removed by aspiration. The beads were then extensively washed (1 × 1 mL 2 M urea, 3 × 1 mL PBS, 3 × 1 mL water) and transferred to a new Eppendorf tube in 1 mL water (2 × 0.5 mL). The beads were then washed one more time with 200 μL TEV buffer (50 mM Tris, pH 8, 0.5 mM EDTA, 1 mM DTT) and resuspended in 140 μL TEV buffer. TEV protease (4 μL, 80 μM) was added, and the digestion was allowed to proceed at 30°C with rotation overnight. Following the overnight digestion, the beads were pelleted by centrifugation, TEV digest was separated from the beads using Micro Bio-Spin columns with centrifugation (800 × g, 0.5 min), and the beads were washed once with water (100 μL). The samples were then acidified to a final concentration of 5% (v/v) formic acid and stored at −80°C prior to analysis.
isoTOP-ABPP liquid chromatography-mass spectrometry (LC-MS) analysis
Samples were pressure loaded onto 250-μm (inner diameter) fused silica capillary columns packed with C18 resin (Aqua 5 μm, Phenomenex). Samples were analyzed by multidimensional liquid chromatography tandem mass-spectrometry (MudPIT) using an LTQ-Velos Orbitrap mass spectrometer (Thermo) coupled to an Agilent 1200-series quaternary pump. The peptides were eluted onto a biphasic column with a 5-μm tip (100 μm fused silica), packed with C18 (10 cm) and bulk strong cation exchange resin (3 cm, SCX, Phenomenex), in a five-step MudPIT experiment, using 0%, 30%, 60%, 90%, and 100% salt bumps of 500-mM aqueous ammonium acetate and a 5%–100% gradient of buffer B in buffer A (buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; buffer B: 5% water, 95% acetonitrile, 0.1% formic acid) as previously described (Weerapana et al., 2007). Data were collected in data-dependent acquisition mode with dynamic exclusion enabled (20 s, repeat of 2). One full MS (MS1) scan (400–1800 m/z) was followed by 30 MS2 scans (ITMS) of the nth most abundant ions.
isoTOP-ABPP peptide and protein identification
The MS2 spectra data were extracted from the raw file using RAW Xtractor (version 1.9.9.2; available at http://fields.scripps.edu/downloads.php). MS2 spectra data were searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, nonredundant variant of the Human UniProt database (release-2012_11). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146) and up to one differential modification for either the light or heavy TEV tags (+464.28595 and +470.29976, respectively). Peptides were required to have at least one tryptic terminus and to contain the TEV modification. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide falsepositive rate below 1%.
isoTOP-ABPP R value calculation and processing
The heavy/light isoTOP-ABPP ratios (R values) for each unique peptide (DMSO/compound treated) were quantified with in-house CIMAGE software (Weerapana et al., 2010) using default parameters (3 MS1 acquisitions per peak and signal-to-noise threshold set to 2.5). Site-specific engagement of cysteine residues was assessed by blockade of IA-alkyne probe labeling. A maximal ratio of 20 was assigned for peptides that showed a ≥ 95% reduction in MS1 peak area in the compound-treated proteome (light TEV tag) compared to the control DMSO-treated proteome (heavy TEV tag). Ratios for unique peptide sequences were calculated for each experiment; overlapping peptides with the same modified cysteine (e.g., different charge states, elution times, or tryptic termini) were grouped together, and the median ratio is reported as the final ratio (R). Additionally, ratios for peptide sequences containing multiple cysteines were grouped together. Biological replicates of the same treatment were averaged if the standard deviation was below 60% of the mean; otherwise, for cysteines with at least one R value < 4 per treatment, the lowest value of the ratio set was taken. For cysteines in which all R values were > 4, the average was reported. The peptide ratios reported by CIMAGE were further filtered to ensure the removal or correction of low-quality ratios in each individual dataset. The quality filters applied were the following: removal of half-tryptic peptides; removal of peptides with R = 20 and only a single MS2 event triggered during the elution of the parent ion; manual annotation of all the peptides with ratios of 20.
ALDH enzymatic activity
ALDH enzyme in vitro inhibition was assessed for each ALDHi as described (Morgan and Hurley, 2015). ALDHi (10 nm-20 μM,) were combined with propionaldehyde (200 μM) and NAD+ (100 μM) as substrates, and the increase in absorbance due to ALDH-mediated production of NADH with propionaldehyde metabolism was measured.
Transmission electron microscopy
FACs isolated CD133+ cells treated with vehicle or 673A (25 μM) for 18 hours, washed with PBS, fixed with glutaraldehyde, then processed, stained, and cut at the U of M microscopy core. Images were obtained through a Philips CM-100 transmission electron microscope.
Transfections
UCP1 and UCP3 [transOMIC MGC premier expression-ready cDNA clone BC008392; and MGC premier expression-ready cDNA clone BC096] plasmids were transiently transfected into cell with a 4D nucleofector (Lonza). For ALDH KD studies we used ALDH1A1 NC siRNA as described in the Key Resources Table. For ALDH1A3 we used Dharmacon siRNA A-009082-15-0005. For UCP siRNA studies, we used 1 × 104 cells were plated in a regular growth medium and, 24 hr later, were transfected using Lipofectamine RNAiMAX reagent (Invitrogen).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti ALDH1A1 | Novus | NBP1–89152 |
| Anti ALDH1A3 | Novus | NBP1–91657 |
| Anti UCP1 | R&D | MAB6158 |
| Anti UCP3 | Abcam | Ab3477 |
| Anti RIPK1 | Cell Signaling | 3493 |
| Anti RIPK3 | Cell Signaling | 95702 |
| Anti PGAM5 | GeneTex | 119887 |
| Anti p-DRP1 | GeneTex | 50911 |
| Anti DRP1 | Cell Signaling | 5391 |
| Anti β-tubulin | Cell Signaling | 2128P |
| Anti TOM20 | Cell Signaling | 13929S |
| Anti ACTB | Cell Signaling | 3700 |
| Anti GAPDH | Cell Signaling | 5174 |
| Anti Na1K-ATPase1 | Cell Signaling | 3010 |
| Anti FADD | Sigma-Aldrich | 06711 |
| CD133/2-APC | Miltenyl Biotec | 130-113-184 |
| Bacterial and Virus Strains | ||
| Suboloning Efficiency DH5α Competent Cells | Invitrogen | Cat#18265017 |
| Biological Samples | ||
| Patient-derived xenografts PDXs | This paper | Charles Landen lab |
| Patient tumor samples | This paper | Buckanovich lab |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pan-Caspase inhibitor Z-VAD-FMK | InvivoGen | tlrl-vad |
| Caspase –3 Inhibitor Z-DEVD-FMK | R&D | FMK004 |
| BAPTA | Tocris | #2786 |
| Necrostatin-1 | Tocris | 2324 |
| 673A | Chembridge | #6737540 |
| CM10 | Morgan and Hurley, 2015 | Hurley Lab |
| Necrosulfonamide | Tocris | 5025/10 |
| Critical Commercial Assays | ||
| Aldefluor kit | Stem cell | 01700 |
| Fluo Calcium Indicator CM-H2DCF | Invitrogen | F1242 |
| Superscript III First strand synthesis SuperMix | Invitrogen | 18080400 |
| Deposited Data | ||
| CCLE cell line analysis-ALDH1A expression | This paper | Table S1 |
| 673A PubChem search | This paper | Table S2 |
| CM10 PubChem search | This paper | Table S3 |
| Experimental Models: Cell Lines | ||
| Ovsaho, Kuramochi | JCRB bank | JCRB bank |
| PEO1, PEO4, OvCAR8, OvCAR5, OvCAR8, SKOV3 | ATCC | |
| A2780 | Dr. S. Muphy | Duke |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson labs | 00557 |
| LSL-K-rasG12D/+ptenloxP/loxP | Romero lab | Romero lab |
| Oligonucleotides | ||
| siALDH1A1 | Sigma | EHU028501 |
| siALDH1A2 | Sigma | EHU1355351 |
| siALDH1A3 | Sigma | EHU060321 |
| siUCP1 | Dharmacon | 007636–01 |
| siUCP3 | Dharmacon | 007638–01 |
| siRIPK1 | Dharmacon | 004445 |
| siRIPK3 | Dharmacon | 003534 |
| ALDH1A1-LV17 | ABM | #K0070517 |
| ALDH1A3-LV18 | ABM | #K0070718 |
| Recombinant DNA | ||
| UCP1 plasmid | TransOmics | clone BC008392 |
| UCP3 plasmid | TransOmics | Clone BC096 |
CRISPR-Cas ALDH KO
Three viruses for ALDH1A1 or ALDH1A3 (human sgRNA CRISPR All-in-One Lentivirus (Applied Biological Materials Inc (Richmond, BC, Canada)) were used to transduce A2780 and OV90 cells (ALDH1A1) or OVCAR5 cells (ALDH1A3) at an MOI of 5 and puromycin selected for 7 days. KO was confirmed using western blotting (anti-ALDH1A1 from Novus (Centennial, Co) NBP1–89152, anti-ALDH1A3 Novus NBP1–91657 both at 1:1000). ALDH1A1-LV17 (cat#K0070517) most efficiently deleted ALDH1A1 expression. ALDH1A3 LV18 (cat#K0070718) effectively eliminated ALDH1A3 expression.
Animal studies
Experiments were performed with approval of the University Committee on Use and Care of Animals at the University of Michigan, the University of Chicago, or the University of Virginia. Cell line xenografts: 7.5 × 104 A2780 cells or 15,000 CD133+ (A2780) cells, 5 × 106 OVCAR8, 5 × 105 HEY-1, or 1 × 105 CaOV3 cells were injected, in 100 μl of Matrigel (BD Biosciences), subcutaneously into the axillae of 8-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Three days after inoculation, the mice were treated with an IP injection of (1) DMSO 40 μl, (2) cisplatin 500–1,000 μg/kg weekly, (3) 673A 4–20 mg/kg daily, or (4) a combination of cisplatin followed by 673A (n = 10 mice per treatment group) for 3 weeks. Tumors were measured using calipers and tumor volume (L × W × W/2) calculated. For IP tumors, 0.5 × 104 FACS-sorted ALDH+ SKOv3 cells were injected IP in 250 mL PBS. Mice were treated as above and then monitored for 6 months. For the LSL-K-rasG12D/+PtenloxP/loxP genetic mouse model of ovarian cancer, tumor was initiated as described (Dinulescu et al., 2005), with 2.5 × 107 plaque-forming units injected under the bursa of the right ovary, the left ovary serving as control. Treatment was started as indicated two weeks after adenovirus injection. 7 weeks after injection of the virus, the mice were sacrificed and tumor burden assessed.
Patient-derived xenografts
Two patient-derived xenograft (PDX) models were developed as described (Dobbin et al., 2014). One from a patient with chemotherapy resistant disease, one treated in mice to generate platinum resistance. PDX cells were implanted and allowed to grow until 1cm3; then, treated with either: (1) Vehicle, (2) 75 mg/kg carboplatin + 20 mg/kg paclitaxel injected IP weekly, or (3) 75 mg/kg carboplatin + 20 mg/kg paclitaxel injected IP weekly + 2.23 mg/kg 673A injected IP daily. Tumors were monitored as above.
Seahorse mitochondrial flux analyses
A Seahorse XFe96 flux analyzer (Seahorse Biosciences) was used to determine the oxidative consumption rate (OCR). 30,000 cells/well were allowed to adhere overnight, then treated with 673A. One hour before analysis, medium was replaced with XF medium with 10 mM glucose or 1 mM pyruvate and incubated for 1 h in a CO2-free 37°C incubator. The basal respiration of the cells was measured, followed by injection of oligomycin (1 μM), and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 2 μM) to determine maximal respiration. Rotenone (1 μM) and antimycin A (1 μM) were injected to measure the nonmitochondrial respiration. The mitochondrial OCR was calculated by extracting the nonmitochondrial respiration from the basal respiration.
QUANTIFICATION AND STATISTICAL ANALYSIS
Synergy calculation
The drug combination studies were conducted using the Chou-Talalay median effects method, with serial dilutions of both drugs administered at various doses. The combination index (CI), a measure of the quantitative determination of drug interactions, was calculated from the drug dose versus cell viability data using CompuSyn software (http://www.combosyn.com). CI < 1 is synergistic and CI > 1 is antagonistic, with CI = 1 being additive.
Statistical analysis
Data are presented as mean ± SD or SE with *p < 0.05, **p < 0.01, ***p < 0.005. Overall survival studies were performed with the method of Kaplan and Meier, using analysis of variance with the Bonferroni correction. For in vitro assays, except where indicated, results are a summary of triplicate technical replicates from three independent experiments. Student’s t test or ANOVA were used to compare results.
Supplementary Material
Highlights.
We identified ALDH1A family inhibitors (ALDH1Ai) that target CD133+ ovarian CSCs
ALDH1Ai triggers calcium-dependent cell-programmed necrosis
ALDH1Ai induces mitochondrial uncoupling proteins affecting cellular metabolism
ADH1Ai overcomes chemotherapy resistance to increase tumor eradication
ACKNOWLEDGMENTS
The work was supported by the U.S. Department of Defense (grant OC130322) and the NIH (grant 5R01CA214567). Individual investigator support was as follows: Ann and Sol Schreiber Mentored grant and Liz Tilberis early career OCRA awards, the Marsha Rivkin Ovarian Center pilot award, and a WeRoc research grant from Foundation for Women’s Cancer (to I.C.); University of Chicago-NIH CCSG (CA014599) and Ovarian Cancer SPORE (grant P50CA046592) (to I.R.); NIH (R01-AA18123 and R21-CA198409) (to T.D.H.); NIH (R01GM101171) and U.S. Department of Defense (OC140123) (to D.B.L.); and NIH (ES024872) and SU2C (SU2C-AACR-IRG-02-16) (to K.A.B.).
DECLARATION OF INTERESTS
R.J.B., S.D.L., and T.D.H. have patents related to the use of ALDHi as cancer therapy. R.J.B. is a co-founder of Tradewind Bioscience; however, none of the work described in this study is related to, based on, or supported by the company. T.D.H. holds significant financial equity in SAJE Pharma, LLC.; however, none of the work described in this study is related to, based on, or supported by the company.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.032.
REFERENCES
- Agarwal RP, Phillips M, McPherson RA, and Hensley P (1986). Serum albumin and the metabolism of disulfiram. Biochem. Pharmacol 35, 3341–3347. [DOI] [PubMed] [Google Scholar]
- Balmer JE, and Blomhoff R (2002). Gene expression regulation by retinoic acid. J. Lipid Res 43, 1773–1808. [DOI] [PubMed] [Google Scholar]
- Bretz N, Noske A, Keller S, Erbe-Hofmann N, Schlange T, Salnikov AV, Moldenhauer G, Kristiansen G, and Altevogt P (2012). CD24 promotes tumor cell invasion by suppressing tissue factor pathway inhibitor-2 (TFPI-2) in a c-Src-dependent fashion. Clin. Exp. Metastasis 29, 27–38. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, and Liu ZG (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Ingram PN, Yang K, Coffman L, Iyengar M, Bai S, Thomas DG, Yoon E, and Buckanovich RJ (2015). Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc. Natl. Acad. Sci. USA 112, E6882–E6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefetz I, Alvero AB, Holmberg JC, Lebowitz N, Craveiro V, Yang-Hart-wich Y, Yin G, Squillance L, Gurrea Soteras M, Aldo P, and Mor G (2013). TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 12, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman LG, Choi YJ, McLean K, Allen BL, di Magliano MP, and Buckanovich RJ (2016). Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 7, 6916–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello S, Morgan CA, Nagdas S, Cao L, Turek J, Hurley TD, and Matei D (2014). β-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Dericks L, Froment L, Boesch R, Schmid RA, and Karoubi G (2014). Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDH(high)CD44(+) phenotype and sphere-forming capacity. BMC Cancer 14, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Yang X, Lassus H, Liang S, Kaur S, Ye Q, Li C, Wang LP, Roby KF, Orsulic S, et al. (2010). Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS ONE 5, e10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinulescu DM, Ince TA, Quade BJ, Shader SA, Crowley D, and Jacks T (2005). Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med 11, 63–70. [DOI] [PubMed] [Google Scholar]
- Dobbin ZC, Katre AA, Steg AD, Erickson BK, Shah MM, Alvarez RD, Conner MG, Schneider D, Chen D, and Landen CN (2014). Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 5, 8750–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour P, Lang JM, Giron C, Duclos B, Haehnel P, Jaeck D, Jung JM, and Oberling F (1993). Sodium dithiocarb as adjuvant immunotherapy for high risk breast cancer: a randomized study. Biotherapy 6, 9–12. [DOI] [PubMed] [Google Scholar]
- Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, et al. (2008). Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 3, e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, and Sander JD (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gdynia G, Keith M, Kopitz J, Bergmann M, Fassl A, Weber ANR, George J, Kees T, Zentgraf H-W, Wiestler OD, et al. (2010). Danger signaling protein HMGB1 induces a distinct form of cell death accompanied by formation of giant mitochondria. Cancer Res. 70, 8558–8568. [DOI] [PubMed] [Google Scholar]
- Huddle BC, Grimley E, Buchman CD, Chtcherbinine M, Debnath B, Mehta P, Yang K, Morgan CA, Li S, Felton J, et al. (2018). Structure-based optimization of a novel class of aldehyde dehydrogenase 1A (ALDH1A) Subfamily-selective inhibitors as potential adjuncts to ovarian cancer chemotherapy. J. Med. Chem 61, 8754–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving CC, and Daniel DS (1987). Influence of disulfiram on the metabolism of the urinary bladder carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine in the rat. Carcinogenesis 8, 1309–1315. [DOI] [PubMed] [Google Scholar]
- Januchowski R, Wojtowicz K, and Zabel M (2013). The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharmacother 67, 669–680. [DOI] [PubMed] [Google Scholar]
- Kast RE, and Belda-Iniesta C (2009). Suppressing glioblastoma stem cell function by aldehyde dehydrogenase inhibition with chloramphenicol or disulfiram as a new treatment adjunct: an hypothesis. Curr. Stem Cell Res. Ther 4, 314–317. [DOI] [PubMed] [Google Scholar]
- Kiefer FW, Vernochet C, O’Brien P, Spoerl S, Brown JD, Nallamshetty S, Zeyda M, Stulnig TM, Cohen DE, Kahn CR, and Plutzky J (2012). Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med 18, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, and Vasiliou V (2012). Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev 64, 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, Banerjee M, Mao Y, Kotarski J, Wicha MS, et al. (2012). Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 130, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen CN Jr., Goodman B, Katre AA, Steg AD, Nick AM, Stone RL, Miller LD, Mejia PV, Jennings NB, Gershenson DM, et al. (2010). Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther 9, 3186–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi BP, Yilmaz OH, Duester G, and Morrison SJ (2009). Aldehyde dehydrogenase 1a1 is dispensable for stem cell function in the mouse hematopoietic and nervous systems. Blood 113, 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Xiang Y, Xiang L, Xiao Y, Li F, and Hao P (2014). ALDH maintains the stemness of lung adenoma stem cells by suppressing the Notch/CDK2/CCNE pathway. PLoS ONE 9, e92669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, Kachhap SK, Shim JS, Liu JO, Nelson WG, et al. (2011). Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 71, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, and Krautwald S (2012). Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761. [DOI] [PubMed] [Google Scholar]
- Ma YM, and Zhao S (2016). Prognostic values of aldehyde dehydrogenase 1 isoenzymes in ovarian cancer. OncoTargets Ther. 9, 1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm R, Olive MF, and Lechner W (2008). The safety of disulfiram for the treatment of alcohol and cocaine dependence in randomized clinical trials: guidance for clinical practice. Expert Opin. Drug Saf 7, 459–472. [DOI] [PubMed] [Google Scholar]
- Morgan CA, and Hurley TD (2015). Development of a high-throughput in vitro assay to identify selective inhibitors for human ALDH1A1. Chem. Biol. Interact 234, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, et al. (2010). Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 70, 4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, and Buckanovich RJ (2011). Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest 121, 3206–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BW, Doudican NA, Patel KR, and Orlow SJ (2010). Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 20, 11–20. [DOI] [PubMed] [Google Scholar]
- Napoli JL, Boerman MH, Chai X, Zhai Y, and Fiorella PD (1995). Enzymes and binding proteins affecting retinoic acid concentrations. J. Steroid Biochem. Mol. Biol 53, 497–502. [DOI] [PubMed] [Google Scholar]
- Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI, Nisman B, Peretz T, and Peylan-Ramu N (2015). A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic nonsmall cell lung cancer. Oncologist 20, 366–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowinski SM, Solmonson A, Rundhaug JE, Rho O, Cho J, Lago CU, Riley CL, Lee S, Kohno S, Dao CK, et al. (2015). Mitochondrial uncoupling links lipid catabolism to Akt inhibition and resistance to tumorigenesis. Nat. Commun 6, 8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda T, and Yoshikawa H (2011). Neoadjuvant chemotherapy for advanced ovarian cancer: overview of outcomes and unanswered questions. Expert Rev. Anticancer Ther 11, 1053–1067. [DOI] [PubMed] [Google Scholar]
- Pattabiraman DR, and Weinberg RA (2014). Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discov 13, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen EN (1992). The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr. Scand. Suppl 369, 7–13. [DOI] [PubMed] [Google Scholar]
- Pinto N, Ludeman SM, and Dolan ME (2009). Drug focus: pharmacogenetic studies related to cyclophosphamide-based therapy. Pharmacogenomics 10, 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S, Mehta P, Ward MR, Bregenzer ME, Fleck EMA, Tan L, McLean K, Buckanovich RJ, and Mehta G (2017). Personalized medicine-based approach to model patterns of chemoresistance and tumor recurrence using ovarian cancer stem cell spheroids. Clin. Cancer Res 23, 6934–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M, and Settleman J (2014). The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell sub-population. Cancer Res. 74, 3579–3590. [DOI] [PubMed] [Google Scholar]
- Rezk Y, Yang K, Bai S, Mclean K, Johnston C, Reynolds RK, and Buckanovich RJ (2015). Disulfiram’s antineoplastic effects on ovarian cancer. J. Cancer Ther 6, 1196–1205. [Google Scholar]
- Sanchez-Alvarez R, Martinez-Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, Sartini M, Rubin E, Lisanti MP, and Sotgia F (2013). Mitochondrial dysfunction in breast cancer cells prevents tumor growth: under-standing chemoprevention with metformin. Cell Cycle 12, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvi S, Crispin R, Lu Y, Zeng L, Hurley TD, Houston DR, von Krieg-sheim A, Chen CH, Mochly-Rosen D, Ranzani M, et al. (2018). ALDH1 Bio-activates nifuroxazide to eradicate ALDHHigh melanoma-initiating cells. Cell Chem. Biol 25, 1456–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw YT, Yang J, Ng SK, Liu S, Singh S, Singh M, Welch WR, Tsuda H, Fong WP, Thompson D, et al. (2012). Characterization of aldehyde dehydrogenase isozymes in ovarian cancer tissues and sphere cultures. BMC Cancer 12, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, Ginestier C, Johnston C, Kueck A, Reynolds RK, et al. (2011). Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 71, 3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers AE, and Cravatt BF (2004). Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol 11, 535–546. [DOI] [PubMed] [Google Scholar]
- Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, and Landen CN (2012). Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res 18, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton G, Pérez-Alea M, Baier C, Fournet G, Quash G, Labiad Y, Martin G, Sanderson F, Poullin P, Suchon P, et al. (2016). Aldehyde dehydrogenases inhibition eradicates leukemia stem cells while sparing normal progenitors. Blood Cancer J. 6, e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, and Wang X (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Speers AE, and Cravatt BF (2007). Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)—a general method for mapping sites of probe modification in proteomes. Nat. Protoc 2, 1414–1425. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomics. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S-M, Martinez NJ, Yasgar A, Danchik C, Johansson C, Wang Y, Baljinnyam B, Wang AQ, Xu X, Shah P, et al. (2018). Discovery of Orally Bioavailable, Quinoline-Based Aldehyde Dehydrogenase 1A1 (ALDH1A1) Inhibitors with Potent Cellular Activity. J. Med. Chem 61, 4883–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, and Wang W (2011). Disulfiram modulated ROS- MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 104, 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.