

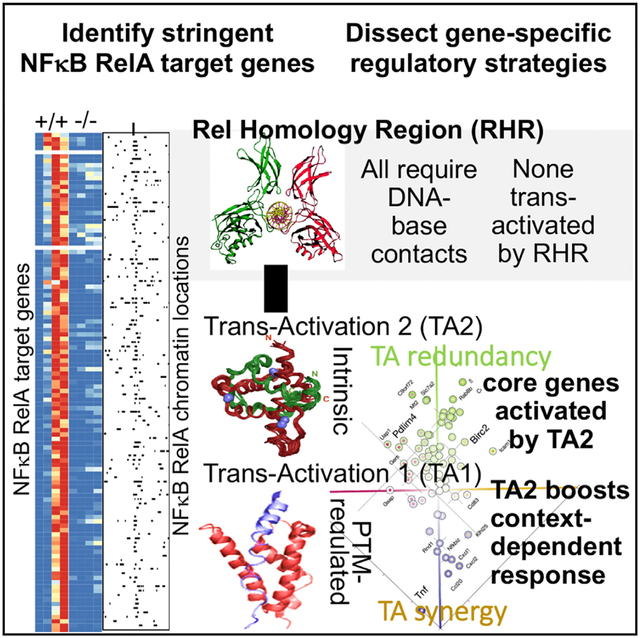
SUMMARY
Nuclear factor κB (NF-κB) RelA is the potent transcriptional activator of inflammatory response genes. We stringently defined a list of direct RelA target genes by integrating physical (chromatin immunoprecipitation sequencing [ChIP-seq]) and functional (RNA sequencing [RNA-seq] in knockouts) datasets. We then dissected each gene’s regulatory strategy by testing RelA variants in a primary-cell genetic-complementation assay. All endogenous target genes require RelA to make DNA-base-specific contacts, and none are activatable by the DNA binding domain alone. However, endogenous target genes differ widely in how they employ the two transactivation domains. Through model-aided analysis of the dynamic time-course data, we reveal the gene-specific synergy and redundancy of TA1 and TA2. Given that post-translational modifications control TA1 activity and intrinsic affinity for coactivators determines TA2 activity, the differential TA logics suggests context-dependent versus context-independent control of endogenous RelA-target genes. Although some inflammatory initiators appear to require co-stimulatory TA1 activation, inflammatory resolvers are a part of the NF-κB RelA core response.
Graphical Abstract
In Brief
Ngo et al. developed a genetic complementation system for NF-κB RelA that reveals that NF-κB target-gene selection requires high-affinity RelA binding and transcriptional activation domains for gene induction. The synergistic and redundant functions of two transactivation domains define pro-inflammatory and inflammation-response genes.
INTRODUCTION
An important concept is molecular biology is the modular domain organization of transcription factors (TFs) (Keegan et al., 1986), typically distinguishing between a DNA-binding domain (DBD) and a separable transcriptional activation domain (TAD) that could be fused to a heterologous DBD. Prominent mammalian TFs, including nuclear factor κB (NF-κB) RelA (Schmitz and Baeuerle, 1991; Schmitz et al., 1994), conform to the modular domain model. Such studies used exogenous reporter genes that provided a convenient assay for TF activity. However, they lacked the physiological context of endogenous regulatory regions, which may involve complex protein-protein interactions and are often considerable distances from the transcription start site. Indeed, subsequent studies provided numerous examples in which the functional distinction between DNA binding and transcriptional activation did not neatly segregate into distinct structural domains, with the DBD providing transcriptional activity (Corton et al., 1998) or, conversely, not being required for TF recruitment to the target gene (Kovesdi et al., 1986).
Given that the regulatory context of endogenous target genes determines a TF’s mode of function, next-generation TF structure-function studies may be considered probes of the regulatory diversity of its endogenous target genes. However, only with the advent of quantitative transcriptomic and epigenomic measurement capabilities enabled by next-generation sequencing has it become feasible to undertake such studies. The present study is leveraging such technological development to dissect the regulatory strategies of inflammatory response genes that are regulatory targets of NF-κB RelA.
The NF-κB family member RelA is a ubiquitously expressed potent transcriptional activator that is induced by exposure to pathogens and inflammatory cytokines to activate the expression of many inflammatory and immune-response genes (Hayden and Ghosh, 2008; Hoffmann and Baltimore, 2006). The signaling mechanisms involved in regulating NF-κB RelA activity have been elucidated in detail (Basak et al., 2012; Mitchell et al., 2016), but there is much more uncertainty about how it controls endogenous target genes. Indeed, although many genes have been identified to be potentially regulated by NF-κB (http://www.bu.edu/NF-kb/gene-resources/target-genes/), there is no database that lists the NF-κB target genes in a particular physiological condition, defined cell type, and stimulus.
RelA’s domain organization is characterized by the Rel homology region (RHR), which mediates dimerization and DNA binding functions (Baeuerle and Baltimore, 1989) and was structurally characterized by X-ray crystallography (Chen et al., 1998a, 2000). However, it is possible that for some endogenous target genes, promoter recruitment of RelA is mediated primarily by protein-protein interactions, for example, via pre-bound CREB-binding protein (CBP) (Mukherjee et al., 2013).
RelA’s C terminus contains two transactivation domains, TA1 and TA2 (Ballard et al., 1992; Moore et al., 1993; Schmitz and Baeuerle, 1991), which interact with transcriptional regulatory factors. TA1 consists of an amphipathic helix characteristic of an acidic activation domain and can interact with MED80 subunit of the Mediator complex, Tfb1/p62 subunit of TFIIH, and CBP via the kinase-inducible domain interacting (KIX) domain (van Essen et al., 2009; Lecoq et al., 2017; Mulero et al., 2019). TA2 also interacts with CBP/p300 but via the TAZ1 domain with sufficient affinity that it could be characterized by nuclear magnetic resonance (Mukherjee et al., 2013; Nyqvist et al., 2019), as well as MED80. The RHR was reported to interact with CBP as well via phosphorylated S276 (Zhong et al., 2002), and knockin mutations of the S276A or S276E mutation showed complex mouse phenotypes and transcriptional defects (Dong et al., 2008). It also remains possible that trans-activation of some target genes is mediated by other sequences within RelA, for example, between RHR and TA2/TA1.
Interestingly, a key difference between TA1 and TA2 is the role of post-translational modification (PTM). Unlike TA2, TA1’s conformation is thought to be regulated (Savaryn et al., 2016) by phosphorylation of any of seven potential sites (S529, Ser535, S536, S543, S547, S550, and S551 in human RelA) altering cofactor interactions (Milanovic et al., 2014) and, hence, transcriptional activation potential (Hochrainer et al., 2013). In particular, S536 is a prominent phospho-acceptor site (Christian et al., 2016), which is a reliable biomarker for RelA activity. This suggests a model in which TA1’s transactivation activity is dependent on a phosphorylation event that integrates the activity of other signaling pathways, such as CKII, CaMKIV, RSK1, TAK1/TAB1, PI3K/Akt, ATM, and IKKε (Bae et al., 2003; Bao et al., 2010; Bird et al., 1997; Bohuslav et al., 2004; Buss et al., 2004a; Haller et al., 2002; Moreno et al., 2010; Sabatel et al., 2012; Sakurai et al., 2003; Wang et al., 2000; Yoboua et al., 2010), whereas TA2’s transactivation potential is intrinsic to its biochemical structure. Remarkably, however, it remains unknown whether NF-κB response genes show differential regulation by TA1 versus TA2; this is an important question because TA2-regulated genes would be expected to provide for a core NF-κB-mediated inflammatory response, whereas TA1-regulated genes are dependent on other signaling pathways as well.
Here, we report the stringent identification of endogenous NF-κB target genes induced by tumor necrosis factor (TNF) in primary fibroblasts and a genetic complementation system for RelA variants to probe their regulatory strategies. We found that all RelA target gene binding was dependent on base-specific contacts within the κB sequence, and all transactivation was dependent on the two C-terminal activation domains; neither the transcriptional activation domain nor did the DNA binding domain proved sufficient for target gene selection or transcriptional activation, respectively. However, we discovered remarkable gene-specific regulatory strategies involving the transcriptional activation domains TA1 and TA2. Although most genes were regulated by both TA1 and TA2, the two transactivations functioned either redundantly or synergistically, forming logical OR-or-AND gates in a gene-specific manner. Although OR gate genes were robustly activated by the constitutive TA2 activity, on AND gate genes TA2’s role is to boost the PTM-regulated TA1 activity. Thus, our study distinguishes NF-κB RelA target genes as being either largely signaling context independent or largely signaling context dependent.
RESULTS
Stringent Identification of TNF-Responsive NF-κB RelA Target Genes
To identify high-confidence NF-κB RelA target genes, we applied two rigorous criteria, namely (1) TNF-induced NF-κB/RelA binding to the target gene’s regulatory regions, and (2) TNF-induced transcriptional activation that is genetically dependent on NF-κB. To examine the genome-wide binding pattern of NF-κB, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) with RelA-specific antibodies during a short course of TNF stimulation of 0, 0.5, and 3 h in wild-type (WT) primary murine embryonic fibroblasts (MEFs). Bioinformatic analysis of the replicate datasets (false-discovery rate [FDR] < 0.01) identified in 9,829 RelA peaks that were TNF-inducible at 0.5 or 3 h (Figure 1A). Overall, these ChIP-enriched RelA binding events were notably enhanced at 0.5 h and largely remained at 3 h (Figure 1B, left panel). We identified the locations of these RelA peaks to be 44% in intergenic regions, 17% within promoter regions (comprising 5 kB upstream of the transcription start site), 36% within intronic regions, and 3% within exonic regions of genes (Figure 1B, right panel). Genome browser tracks of two known NF-κB target genes, Nfkbia and Cxcl10, confirmed easily discernible peaks above a low background, and those peaks were highly inducible in response to TNF and highly reproducible between the two datasets (Figure 1C).
Figure 1. Identifying NF-κB RelA Target Genes in the TNF Response of Murine Embryonic Fibroblasts (MEFs).
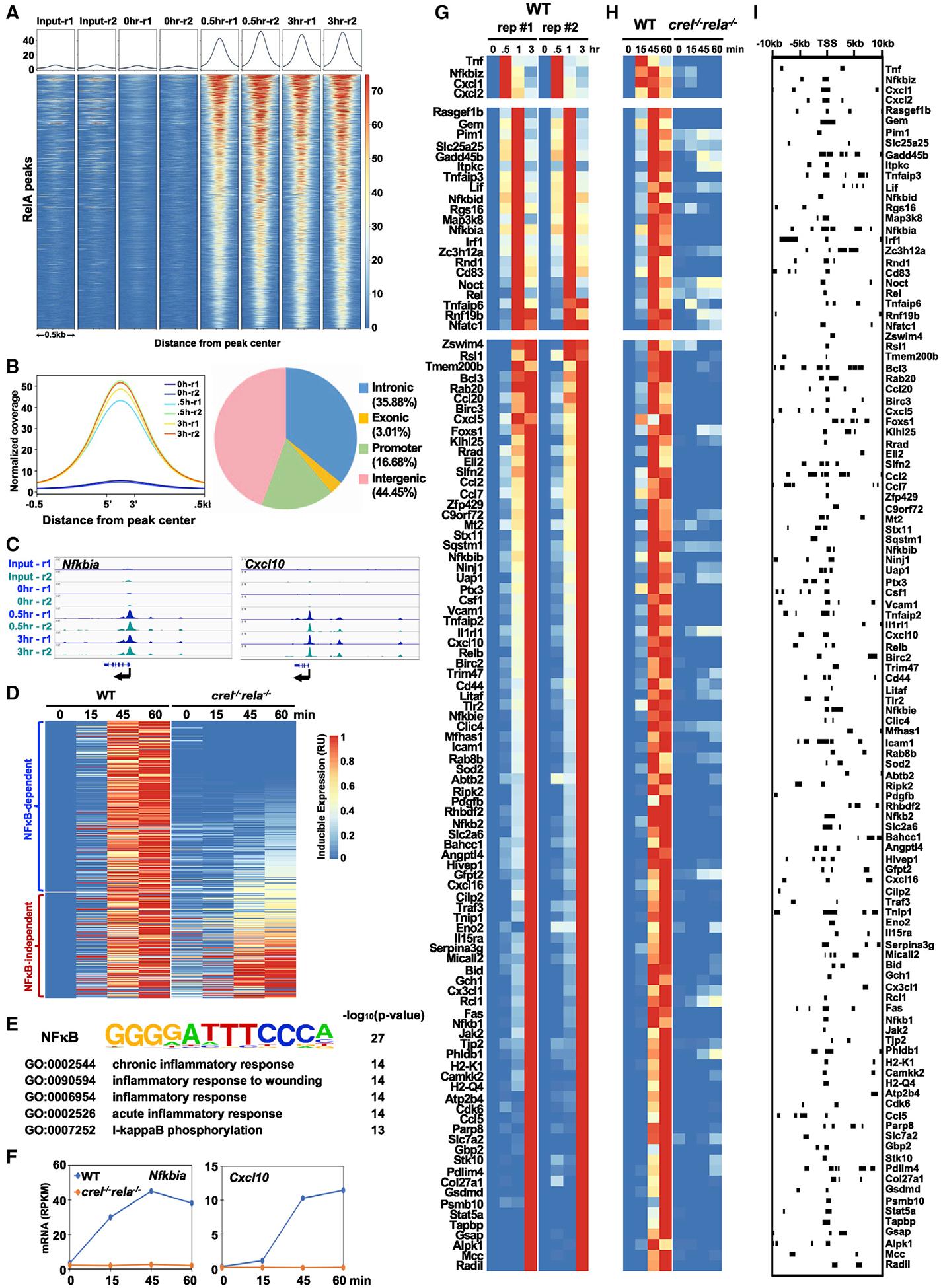
(A) Genome-wide binding of NF-κB RelA ChIP-seq in WT MEFs stimulated with 10 ng/mL of TNF at 0.5 and 3 h; 9,829 RelA-binding events were identified in two independent replicate experiments. The heatmap shows read density along ±0.5 kB of DNA centered around the peak read density for each binding event and ordered by high to low read density.
(B) Left panel: normalized RelA ChIP-seq density over identified peaks within the genome. Right panel: pie chart shows where RelA peaks are found to be located.
(C) Genome browser tracks of two replicates (r1 and r2) of RelA ChIP-seq peaks in WT MEFs for two known NF-κB target genes, Nfkbia and Cxcl10.
(D) Heatmap of the relative induced expression of 419 nascent transcripts measured by caRNA-seq analysis, whose maximal induced expression was ≥1 RPKM and ≥2-fold over basal. Blue to red indicate low to high expression levels, respectively. Of these 419 genes, 229 genes were protein coding and NF-κB dependent, as their maximal expression was <50% in crel−/− rela−/− compared with WT MEFs. The heatmap of gene expression is ordered by high to low NF-κB dependence.
(E) De novo motif and gene ontology analyses for NF-κB-dependent genes identified the most highly enriched motif (within −1.0 to 0.3 kB of the TSS) and GO terms (using Enrichr Ontologies tool [Kuleshov et al., 2016]). For NF-κB-independent genes, top motifs and GO terms showed substantially less significance.
(F) Line plots of chromatin-associated RNA (caRNA) abundance in RPKM for known NF-κB target genes, Nfkbia and Cxcl10.
(G) Heatmap of mature mRNA (polyA+ RNA-seq) relative induced expression, as in (D), for 113 NF-κB target genes, which were defined by the presence of a RelA ChIP-seq peak and TNF-inducible NF-κB-dependent caRNA expression. Two biological replicates of WT MEFs are shown, ordered by their peak time of expression.
(H) Heatmap of the relative induced expression of nascent transcripts determined by caRNA-seq for these 113 NF-κB-dependent genes shown in the same order as in (G).
(I) A map of 297 RelA-binding peaks identified by ChIP-seq for each of the 113 NF-κB target genes within ±10 kB of the TSS, shown in the same order as in (G) and (H).
To assess the NF-κB-dependent transcriptional response to TNF, we performed gene expression analysis of nascent transcripts via chromatin-associated RNA sequencing (caRNA-seq) in WT and crel−/− rela−/− MEF cells (Figure 1D). We chose caRNA-seq analysis rather than polyA+ RNA-seq to ensure that we identified transcriptional induction, rather than changes in mRNA levels mediated by post-transcriptional mechanisms, for example, changes in the mRNA half-life. Previous work has shown that the absence of RelA can be partially compensated for by the NF-κB family member cRel (Hoffmann et al., 2003), necessitating the use of the combination knockout to assess RelA-dependent gene expression. Our 1-h analysis revealed 419 TNF-inducible genes in WT MEFs whose caRNA levels were more than 2-fold induced in WT MEFs at any time point after TNF stimulation relative to the unstimulated (0 h) point (Figure 1D). Setting the maximum reads per kilobase of transcripts per million mapped reads (RPKM) for each gene to 1 and the basal RPKM in unstimulated WT cells to be 0, we plotted data from crel−/− rela−/− MEF cells next to WT data. We found that 259 genes had peak expression values that were less than half in the mutant relative to WT, and 160 genes showed expression that was more than half, leading us to label the former as largely “NF-κB dependent” and the latter largely “NF-κB independent.” Motif-enrichment analysis of the regulatory DNA regions associated with transcription start sites (TSSs) with HOMER (Heinz et al., 2010) revealed a preponderance of κB sites in the NF-κB-dependent genes but no highly significant motif for NF-κB-independent genes (Figure 1E). Focusing on those that are protein coding (229 and 120, respectively), we undertook gene ontology (GO) analysis with Enrichr Ontologies (Kuleshov et al., 2016) for over-represented functional pathways in each category. We found that the top five most highly enriched GO terms (by p value) for the NF-κB-dependent genes described the inflammatory response and NF-κB activation (Figure 1E), whereas the NF-κB-independent genes did not yield highly significant terms. Line graphs of two NF-κB-dependent genes, Nfkbia and Cxcl10 (Figure 1F), exemplify the high degree of NF-κB dependence in their TNF-induced expression.
To develop the list of stringent NF-κB RelA-regulated target genes, we intersected the above-described physical/binding data (within 10 kB of each gene’s TSS) and the genetic/functional data and further ensured that the nascent transcripts were indeed processed into mature mRNAs by performing polyA+ RNA-seq from total RNA isolated from WT MEF cells in response to TNF, extending our time analysis to 3 h (0.5, 1, and 3 h time points). Selecting for peak fold change in response to TNFs of ≥2, an FDR of <0.01 and ≥50% NF-κB dependence of the maximum value in the time course, we generated a list of 113 stringently selected NF-κB/RelA-regulated target genes (Figure 1G). The replicate datasets showed a high level of concordance. The corresponding caRNA-seq data (Figure 1H) indicate that, aside from the very early gene group, the timing of the peak mRNA abundance may not be driven by transcriptional mechanisms but, rather, by post-transcriptional mechanisms, such as mRNA processing (Pandya-Jones et al., 2013) or mRNA half-life (Hao and Baltimore, 2009). Mapping the RelA ChIP-seq data associated with each gene (Figure 1I) identified probable RelA binding events responsible for gene induction but did not reveal a correlation between RelA binding location and transcriptional induction dynamics.
An Experimental System to Dissect Regulatory Strategies of NF-κB RelA Target Genes
To characterize the regulatory strategies of NF-κB RelA-target genes by probing them for their requirements of RelA functional domains, we developed an experimental workflow for retroviral complementation of RelA-deficient MEF cells with specifically engineered variants of RelA and performed subsequent transcriptomic analysis in response to TNFs. To prolong the lifespan of primary MEFs (Todaro and Green, 1963), which would otherwise not be amenable for such genetic manipulation, we investigated whether we could use MEFs deficient in p53. Transcripts induced by TNF, interleukin (IL)-1β, or lipopolysaccharide (LPS), revealed similar gene expression patterns between two biological replicates of p53-deficient MEFs and their littermate WT controls (Figure S1A). Pairwise comparisons revealed high reproducibility in inducible gene expression between p53−/− and WT MEFs at each time point (Figures S1B–S1E), justifying the use of p53-deficient MEFs for studies of inflammatory response genes.
To develop the primary MEF RelA complementation system, we bred knockout mouse strains to produce a strain that was not only deficient in NF-κB subunits but also in p53, which allowed production of primary p53−/− crel−/− rela−/− MEF cells from E12.5 embryos (before embryonic lethality of this genotype at E13). After expansion of these cells for about a week, we retrovirally transduced them with RelA-wt (Figure 2A), selected transduced cells with puromycin, and examined complementation by biochemical assays. Immunoblotting revealed RelA expression in reconstituted cells at about the same level as WT control MEFs (Figure S2A). An electrophoretic mobility shift assay (EMSA) with a κB site probe showed strong DNA binding activity after TNF stimulation (Figure S2B).
Figure 2. A RelA Genetic Complementation System for Studying the Control of Endogenous NF-κB RelA Target Genes.
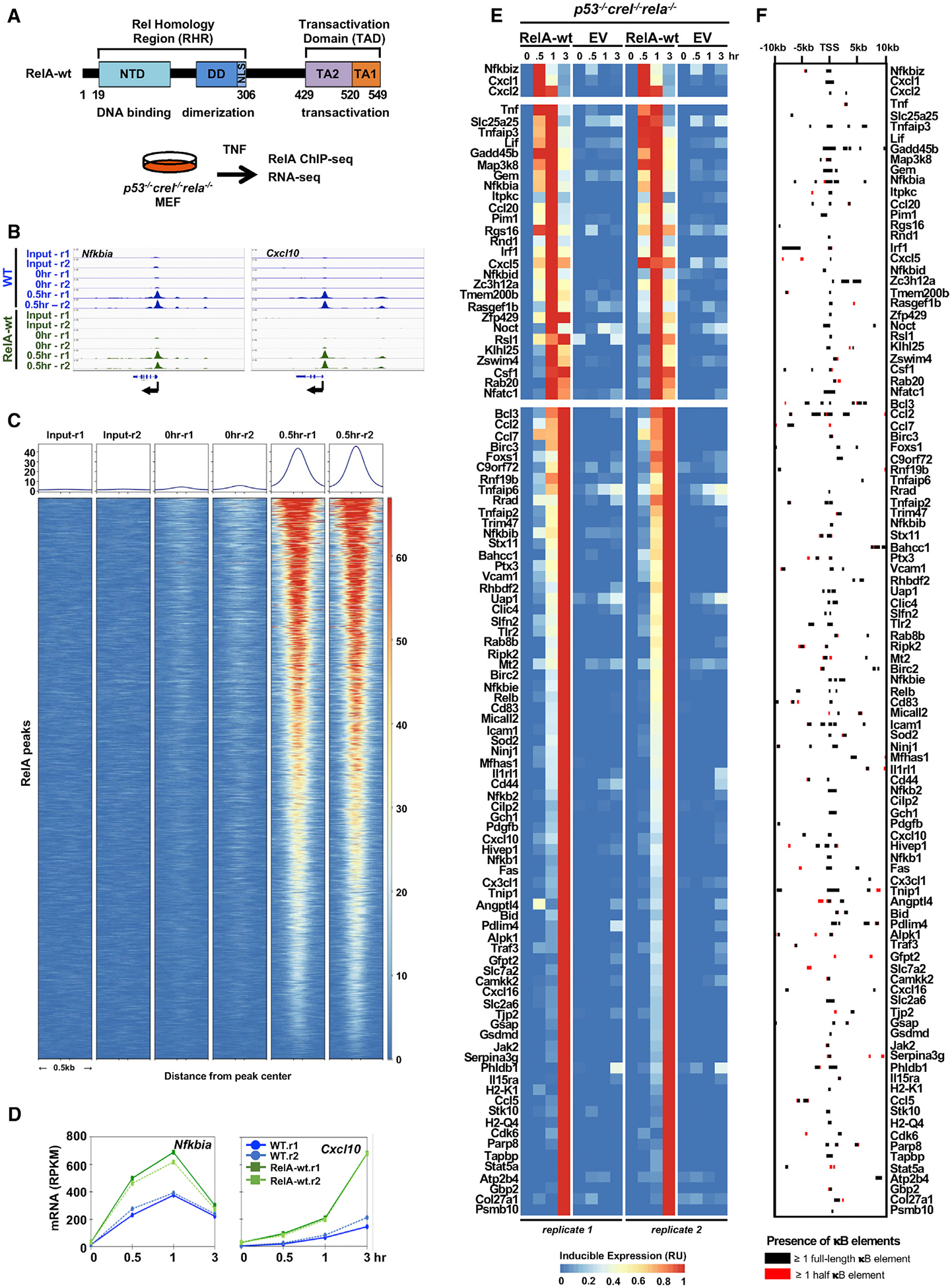
(A) Schematic of the domain structure of the mouse RelA protein (with relevant amino acid numbers) and the primary cells to be complemented and the experimental assays to be performed.
(B) Genome browser tracks of RelA binding events on the Nfkbia and Cxcl10 genes in WT MEFs and p53−/− crel−/− rela−/− MEFs reconstituted with RelA-wt, data from two independent experiments (r1 and r2) are shown.
(C) Heatmap of the read density along ±0.5 kB of RelA ChIP-seq peaks after 0.5 h of TNF treatment for 104 genes identified as NF-κB-dependent genes in WT MEFs and p53−/− crel−/− rela−/− MEFs reconstituted with RelA-wt.
(D) Line plots of mRNA expression in RPKM of Nfkbia and Cxcl10 genes analyzed by polyA+ RNA-seq.
(E) Heatmap of relative induced expression in reconstituted RelA-wt and empty vector (EV) in p53−/− crel−/− rela−/− MEFs, as determined by polyA+ RNA-seq. Two independent experiments are shown.
(F) A map of the NF-κB binding sites (kB DNA sequences) within each of the 223 RelA ChIP-seq peaks identified with an FDR < 0.01 and associated with the 104 NF-κB target genes. Full κB elements refer to 9-, 10-, and 11-bp sequences conforming to the consensus: 5′-GGR(N3–5)YCC-3′, where R = A or G; Y = C or T; N = any nucleotide (A, C, G, or T). Half-κB elements refer to 5′-GGR-3′ and 5′-YCC-3′.
To further test the genetic complementation system, we examined NF-κB RelA binding via RelA ChIP-seq using RelA-wt reconstituted MEFs with and without 0.5 h of TNF stimulation on previously identified endogenous NF-κB target genes (Figure 1G), such as Nfkbia and Cxcl10 (Figure 2B). Importantly, we found that RelA-binding peaks identified in WT MEF controls were indeed also present in the reconstituted RelA-wt cells (Figure 2C).
Next, we examined gene expression responses to TNF in reconstituted MEFs stimulated with TNF using polyA+ RNA-seq. We found high reproducibility in replicate reconstituted MEFs on Nfkbia and Cxcl10 (Figure 2D). Extending our analysis to the 113 NF-κB target genes, a scatterplot analysis demonstrated good correlations in polyA+ RNA expression patterns between WT MEF controls and the reconstituted RelA-wt MEFs at each time-point analysis (Figure S2C). Specifically, we found that 104 genes satisfied the same criteria applied to WTs and crel−/− rela−/− MEFs, namely TNF fold-change over time of ≥2, an FDR of <0.01, and ≥50% NF-κB dependence of the maximum expression value in the time-course (Figure 2E), thereby validating the primary MEF complementation system. In sum, our analysis revealed 104 NF-κB target genes (Table S1) that showed robust RelA DNA binding and RelA-dependent, TNF-inducible expression at both nascent and mature mRNA transcript level.
All TNF-Induced NF-κB Target Genes Require that RelA Makes Base-Specific Contacts within the κB Element
Prior structural and functional studies of NF-κB-DNA complexes demonstrated that NF-κB binds to a variety of κB sites, in which NF-κB recognizes 9–11-bp palindromic κB DNA elements to the sequence 5′-GGR(N3–5YCC)-3′ (Hoffmann et al., 2006). Using that information, we identified all κB DNA elements that were associated with all identifiable RelA ChIP-seq peaks (Figure 1I) for each of the 104 genes (Table S1). Mapping the RelA ChIP-seq data and κB DNA elements associated with each gene within 10 kB of their TSSs (Figure 2F), we found κB DNA elements for 223 RelA binding peaks on 104 genes. Those 223 RelA binding events harbor 617 full-κB and 12,768 half-κB DNA elements with an FDR of <0.01. When the FDR is relaxed to <0.05, then the 234 RelA binding events are identified as harboring 663 full-κB and 14,072 half-κB DNA elements, and the conclusions are confirmed (Figure S2D). Overall, we did not observe a correlation between the relative affinity of the κB site sequence determined by in vitro protein-binding-DNA microarrays (Siggers et al., 2011) and the number of reads within a peak (Figure S2E).
To determine whether DNA-base-specific contacts of the RelA protein are in fact required for recruiting NF-κB to target sites and for activating target genes upon inflammatory stimulation, we generated a triple amino acid mutation (R35A, Y36A, and E39A), depicted in Figure 3A, termed the RelA DNA-binding mutant (RelADB). The mutations abrogate base-specific contacts within the κB sequence (Figure 3B; Chen et al., 1998a). We retro-virally transduced that DNA-binding mutant into primary p53−/− crel−/− rela−/− MEFs and examined the NF-κB by biochemical assays. Immunoblotting confirmed expression of reconstituted RelADB protein, albeit at slightly lower levels (about 75%) than found with RelA-wt (Figure 3C). Co-immunoprecipitation confirmed that the RelADB mutant was able to associate with p50 to form dimeric NF-κB and to interact with p105, IκBα, and IκBβ to form higher-order IκB-NF-κB complexes, characteristic of latent NF-κB in unstimulated cells (Figure S3A). We next assessed the NF-κB DNA-binding activity via EMSA and found that RelADB showed no κB-site-binding activity in response to TNF stimulation, unlike the RelA-wt control (Figure 3D), although immunoblotting of the nuclear extracts confirmed the nuclear presence (Figure S3B). In sum, the RelADB mutant does not support high-affinity NF-κB DNA-binding in vitro.
Figure 3. High-Affinity Binding by RelA Is Required for the TNF-Induced Expression of All NF-κB Target Genes.
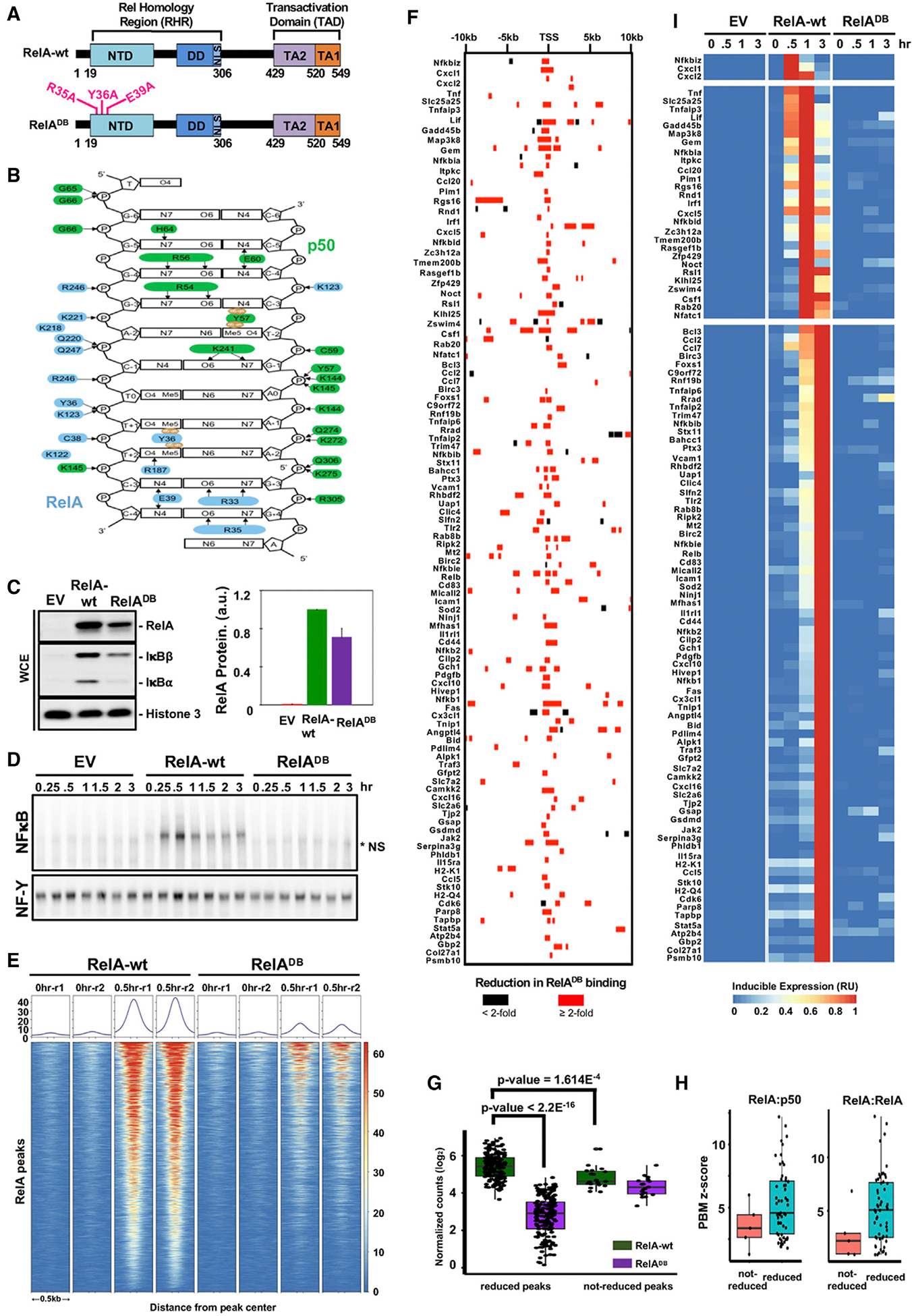
(A) Schematic representation of the targeted mutations in the DNA binding mutant of murine RelA (R35A, Y36A, and E39A).
(B) Schematic of the κB DNA contacts made by RelA:p50 NF-κB heterodimer, adapted from Chen et al. (1998a). The heterodimer structure reveals base-specific contacts made by specific amino acids in the RelA protein, indicating the critical roles of R35, Y36, and E39. Arrows denote hydrogen bonds; brown ovals indicate Van der Waals contacts.
(C) Protein expression of RelA, IκBβ, IκBα, and histone 3 as loading control, by immunoblot in unstimulated cells genetically complemented with indicated EV, RelA-wt, and RelADB. Quantified protein expression is shown on the right. Error bars represent the standard deviation from two independent experiments.
(D) Analysis of nuclear NF-κB activity by EMSA with κB and NF-Y (loading control) probes of nuclear extracts prepared at indicated times after TNF stimulation of genetically complemented cells shown in (C). The data are representative of two independent experiments. *NS, non-specific band.
(E) Heatmap density shows RelA ChIP-seq peaks at 9,829 locations defined in Figure 1A, in genetically complemented (RelA-wt and RelADB) p53−/− crel−/− rela−/− MEFs in response to 0.5 h of TNF stimulation.
(F) A map of RelA binding events associated with 104 NF-κB target genes indicating whether RelA binding is reduced ≥2-fold in the RelADB mutant (red bar).
(G) Quantification and comparison of RelA peaks shown in (F). Averaged normalized counts from two replicates of RelA ChIP-seq experiments in RelA-wt or RelADB expressing MEFs are plotted to compare RelA peaks that are highly dependent on high-affinity binding by RelA versus those that are less dependent. For each category, Mann-Whitney U-test results indicate that peaks less dependent on high-affinity binding by RelA show a lower read count in RelA-wt control cells.
(H) Correlation between RelA ChIP-seq peaks and protein-binding microarray (PBM)-determined z-scores of the strongest-κB binding-site sequences for RelA:p50 and RelA:RelA dimers retrieved from Siggers et al (2011). Of the seven 10-bp κB site sequences within not-reduced RelA peaks; the five with the strongest z-scores were plotted.
(I) Heatmap for the transcriptional TNF response of the 104 target genes in genetically complemented RelA-wt and RelADB cells, analyzed by polyA+ RNA-seq.
We next examined NF-κB RelA binding in vivo by RelA ChIP-seq. On previously identified 9,829 endogenous chromatin sites (Figure 1A), the RelADB mutant showed dramatically diminished TNF-induced RelA binding (Figure 3E). For example, on Nfkbia and Cxcl10 genes, TNF-induced RelA binding was dramatically reduced (Figure S3C). Focusing on the 104 NF-κB target genes (Figure 2E), we evaluated every previously identified RelA binding peak and found that most showed a substantial >2-fold reduction in the RelADB mutant (Figure 3F). Detailed differential RelA ChIP peak analysis (Table S1) revealed that those few RelA peaks that were less affected by the RelADB mutant were generally less prominent in WT cells (Figure 3G), which we interpreted to be indicative of the strong contribution of high-affinity DNA binding ability. Furthermore, they generally showed poorer κB site motifs, as quantified by in vitro protein-DNA microarray studies (Figures 3H and S3D). Overall, our analysis reveals that NF-κB RelA selects most inflammatory-response target genes via direct RelA-DNA interactions via base-specific contacts.
Next, we investigated whether the reduced κB recruitment of the DNA binding mutant results in a diminished transcriptional response from the NF-κB target genes. We performed polyA+ RNA-seq in reconstituted MEFs stimulated with TNF over a 3-h course analysis (0.5, 1, and 3 h). Examining the model target genes, Nfkbia and Cxcl10, we found that the RelADB mutant did not support their gene expression (Figure S3E). Extending the analysis to the 104 NF-κB target genes, we found that the TNF-induced gene expression response was almost entirely abolished in the RelADB mutant cells (Figure 3I). This is also evident in a principal-component analysis (PCA) of the 104 genes (Figure S3F), which shows that the transcriptional response of the mutant largely overlaps with the empty vector control. Only one gene retained 50% transcriptional activation, Rrad (Ras-related glycolysis inhibitor and calcium channel regulator). Close examination revealed some residual RelA binding within the promoter region (Figure S3G), including a κB binding site at −123 bp (5′-GGGAATCCCC-3′), whose 4G stretch could provide sufficient affinity to heterodimeric NF-κB via the p50 binding partner (Cheng et al., 2011).
In sum, our analysis showed that for most genes the triple amino acid mutation abrogates TNF-induced in vivo DNA binding (RelA ChIP data) and target gene expression. Our results suggest that RelA target-gene selection depends on base-specific interactions between RelA and the κB site; our data do not rule out that protein-protein interactions with chromatin-bound factors contribute to binding-site selectivity but indicate that such interactions do not suffice for the activation of inflammatory response NF-κB target genes.
All TNF-Induced NF-κB Target Genes Are Regulated by Two C-Terminal TA Domains
To investigate the role of RelA’s C-terminal region in the transcriptional activation of endogenous NF-κB target genes, we first generated two truncated, variant forms of murine RelA: a full truncation of its C-terminal region (deleting aa 326–549; Δ326–549), referred to as C-termΔ; and a deletion specifically of its two TA domains (deleting residues 429–549; Δ429–549), referred to as TADΔ (Figure 4A).
Figure 4. The RelA C-Terminal Portion Is Required for TNF Induction of All NF-κB Target Genes.
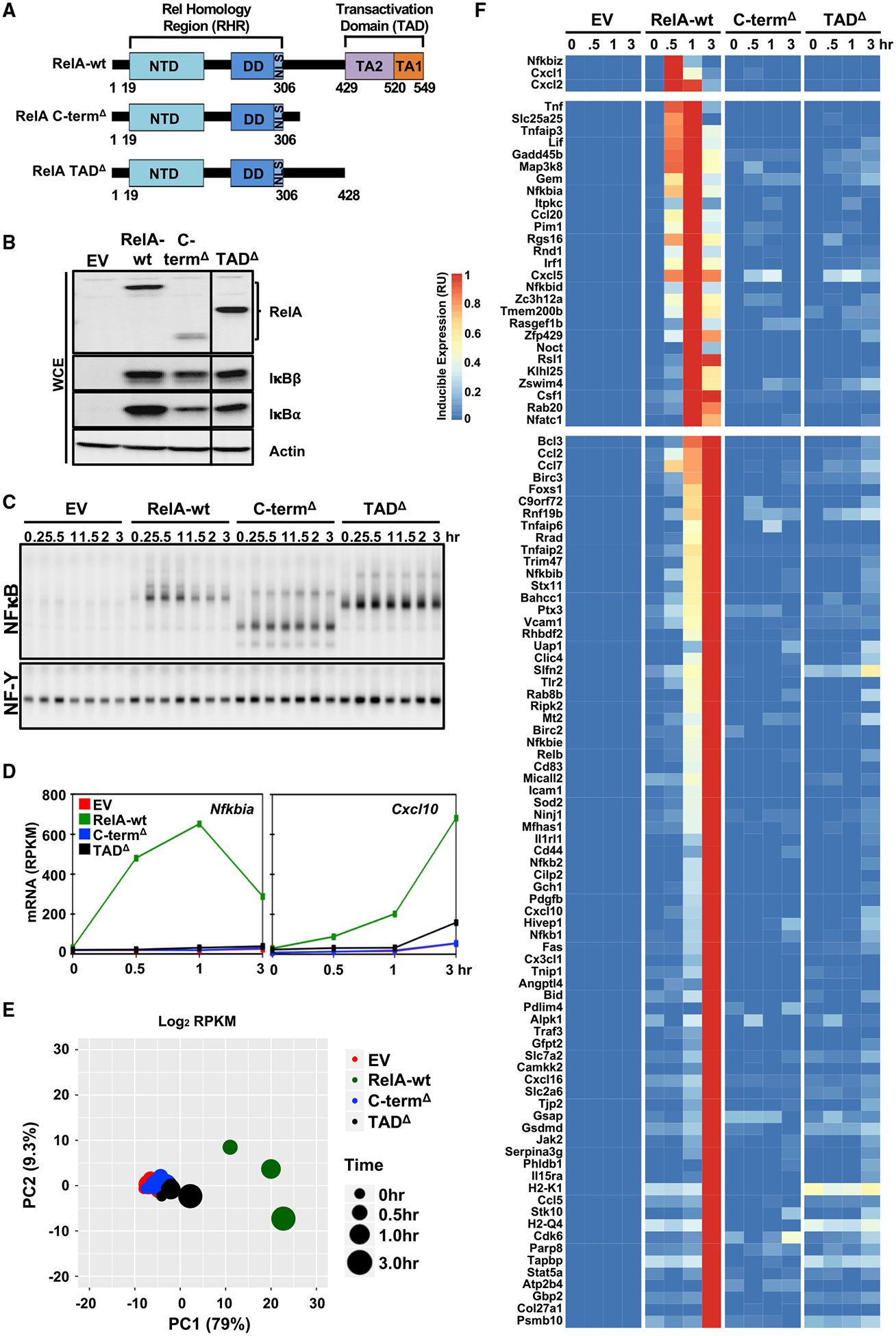
(A) Schematic illustrates the RelA variants to be tested: RelA C-termΔ (aa 1–325) and RelA TADΔ (aa 1–428).
(B) Protein expression of RelA, IκBβ, IκBα, and actin as loading controls in the indicated, unstimulated cells assayed by immunoblotting.
(C) Analysis of nuclear NF-κB activity by EMSA with κB and NF-Y (loading control) probes of nuclear extracts prepared at the indicated times after TNF stimulation of genetically complemented cells shown in (B). The data are representative of two independent experiments.
(D) Line plots of mRNA expression for NF-κB target genes, Nfkbia and Cxcl10, in response to TNF in the same cells.
(E) PCA plot of all 104 NF-κB target genes in response to TNF in the same cells.
(F) Heatmap of the transcriptional response to TNF of the 104 target genes in the same cells. Relative induced expression is shown.
The data shown in (D), (E), and (F) are averaged RPKM from two independent polyA+ RNA-seq experiments.
Immunoblotting of reconstituted MEFs showed close to WT expression of the TADΔ mutant but slightly lower levels of RelA, IκBα, and IκBβ in the C-termΔ mutant (Figure 4B). Nevertheless, NF-κB DNA-binding activity was detected by EMSA at comparable levels to that of reconstituted RelA-wt controls (Figure 4C). Upon stimulation, both mutants showed a normal onset of TNF-inducible binding activity of NF-κB; however, post-induction repression characteristic of TNF-induced NF-κB activity was abrogated, indicative of defective negative-feedback control. Indeed, no TNF-induced Nfkbia mRNA expression was observed in either mutant (Figure 4D).
Extending the analysis to RNA-seq data of the 104 NF-κB target genes, the PCA plot showed that both mutations result in severe deficiency in TNF-induced gene-expression response (Figure 4E), largely equivalent to the empty vector (EV) control. Single-gene analysis by heat-map visualization confirmed that global picture (Figure 4F): none of the 104 genes showed normal TNF induction, with only one gene, Slfn2, being induced to about 50% in the TADΔ mutant. In sum, despite the elevated TNF-induced NF-κB activity in these mutant cells (because of deficient negative feedback control), the data indicate that all transcriptional activation functions of NF-κB require the C-terminal activation domains TA1 and TA2.
To determine whether NF-κB target genes have differential requirements for either TA1 or TA2, we constructed variants of RelA (Figure 5A), a deletion mutant lacking residues 520–549 that define the TA1 (hereafter is referred to as TA1Δ); a double amino acid mutant L449A and F473A, which abrogates the RelA TA2 function of interacting with the TAZ1 domain of transcriptional coactivator, CBP/p300 (Mukherjee et al., 2013), referred to as (TA2CI); and a double mutant (TA2CITA1Δ). (Internal deletion of TA2 appeared to disrupt the function of TA1, which prompted us to design point mutations within the domain.) After reconstitution of p53−/− crel−/− rela−/− MEFs, we confirmed that expression levels of RelA variants and the canonical IκBs were close to those in RelA-wt control (Figure 5B). Further, gel-mobility shift analysis confirmed that those targeted mutations did not interfere with NF-κB dimerization, activation, and DNA binding to κB sites upon TNF stimulation (Figure 5C). However, similar to C-termΔ and TADΔ mutants examined previously, we found that NF-κB DNA-binding activity was persistent over the 3-h time, indicative of a loss of proper negative-feedback control. Indeed, TNF-induced Nfkbia mRNA expression was substantially diminished in single mutants and abrogated in the double mutant (Figure 5D).
Figure 5. RelA Transactivation Domains TA1 and TA2 Both Contribute to the TNF Response of NF-κB Target Genes.
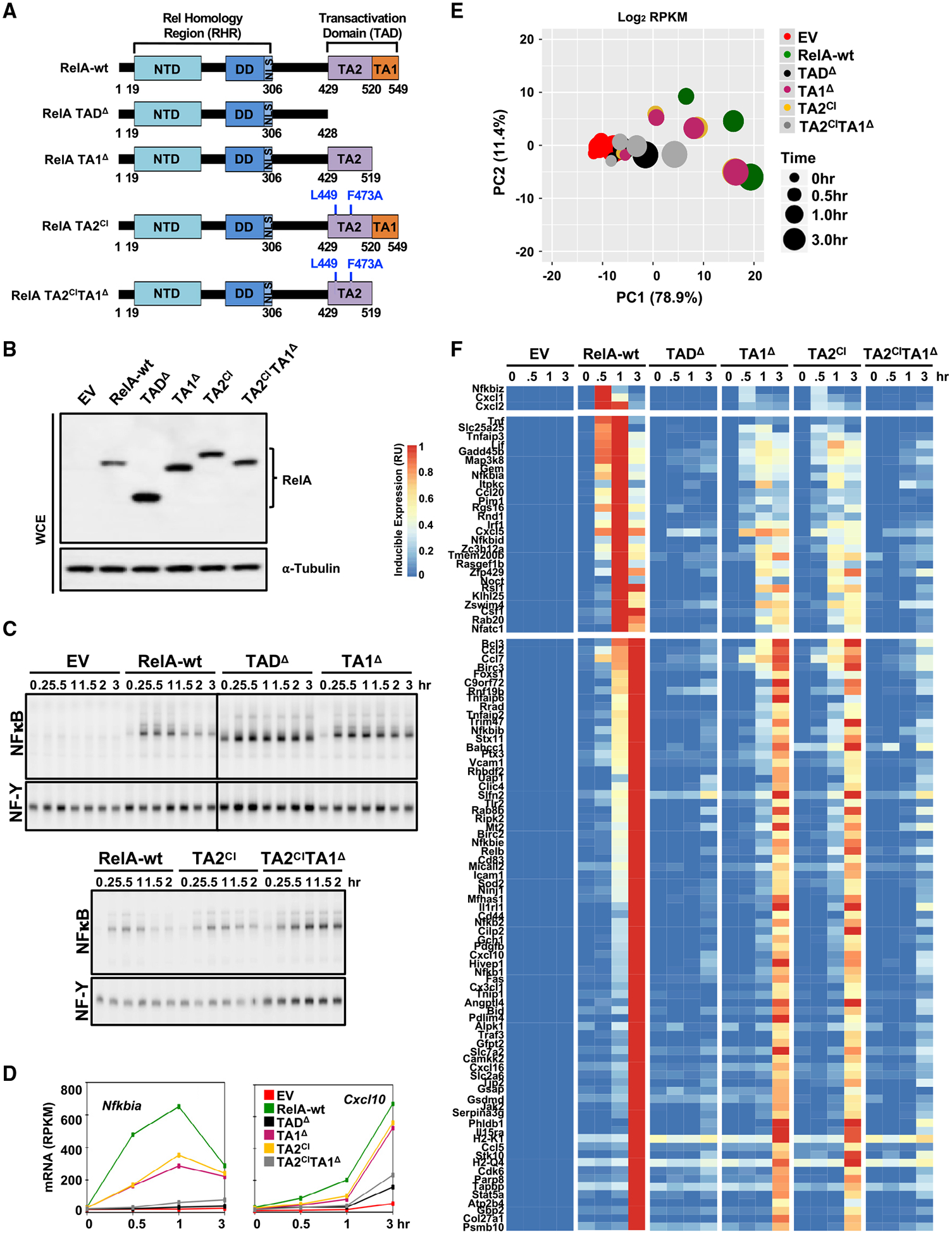
(A) The schematic illustrates the RelA variants to be tested: a deletion mutant of TA1, a CBP-interaction mutant of TA2 (TA2CI) and a combination mutant.
(B) Protein expression of RelA and α-tubulin as loading control, in indicated unstimulated cells assayed by immunoblotting with antibodies specific to RelA and α-Tubulin.
(C) Analysis of nuclear NF-κB activity by EMSA with κB and NF-Y (loading control) probes of nuclear extracts prepared at the indicated times after TNF stimulation of genetically complemented cells shown in (B). The data are representative of two independent experiments.
(D) Line plots of mRNA expression for Nfkbia and Cxcl10 genes in response to TNF stimulation in the same cells.
(E) PCA plot of all 104 NF-κB target genes in response to TNF in the same cells.
(F) Heatmap of the transcriptional response of the 104 target genes in the same cells. Relative induced expression is shown.
The data shown in (D), (E), and (F) are the averaged RPKM from two independent experiments of EV, RelA-wt, TADΔ, and TA1Δ and 1 polyA+ RNA-seq replicate of TA2CI and TA2CITA1Δ.
To extend the gene expression analysis to the 104 NF-κB target genes, we analyzed RNA-seq data for all described mutants. PCA revealed that both single mutants showed global deficiencies in the TNF-induced gene expression response, whereas the double mutant appeared almost entirely deficient in transcriptional activation, akin to the complete TADΔ mutant (Figure 5E). Interestingly, visualizing the data at single-gene resolution in the heatmap format (Figure 5F) revealed subtle differences in the functionality of the mutants that warranted a quantitative analysis.
A Model-Aided Analysis Reveals Gene-Specific Logic Gates Formed by RelA TA1 and TA2
For each RelA target gene to assess the functional requirements of TA1 and TA2 quantitatively, we had to address the potentially confounding effect of alterations in the NF-κB activation time course (Figure 5C). We developed a mathematical formalism that considered both the measured time courses of mRNA and the NF-κB DNA-binding activity that drives transcription; hence, an ordinary differential equation (ODE) model of RelA-dependent gene expression was constructed (Figure 6A). The model consists of a single ODE representing the rate of change of the mRNA that resulted from NF-κB-driven synthesis and mRNA decay (see Method Details). NF-κB-driven mRNA synthesis is a function of the amount of NF-κB DNA binding activity and the specific activation activity (kact) of the particular RelA variant.
Figure 6. A Math Model-Aided Analysis Reveals Gene-Specific Requirements for TA1 and TA2.
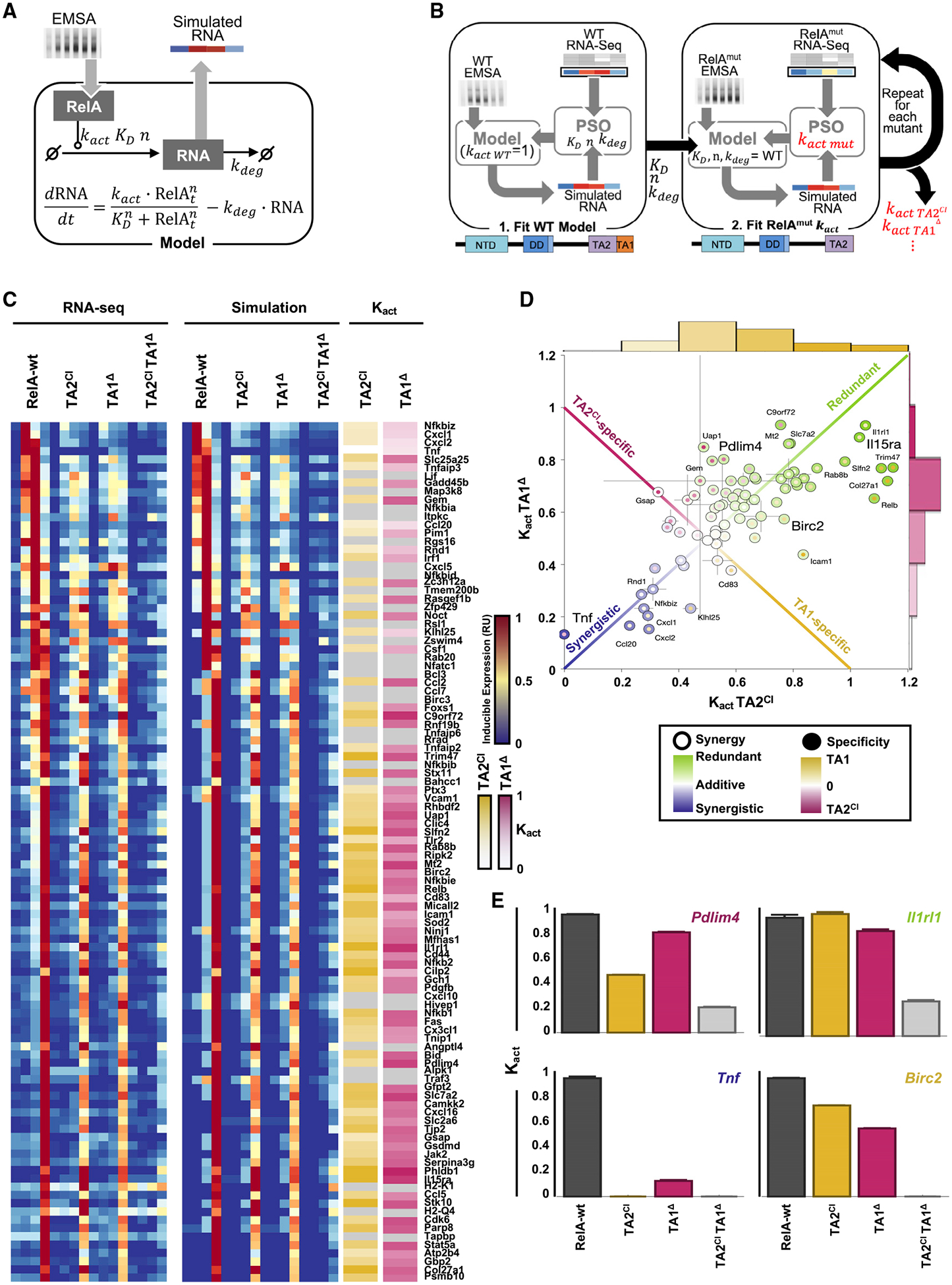
(A) Schematic of the ordinary differential equation (ODE) used to obtain simulated mRNA dynamics from quantified NF-κB activity time courses.
(B) Schematic of the particle swarm optimization (PSO) pipeline used to quantify the activation strengths (kact) for TAD mutants. First KD, nhill, kdeg are fit to RelA-wt data with kact = 1. The identified parameters are then maintained with the new kact fit for each TAD mutant to obtain the remaining activation strength (see Method Details).
(C) Experimental (left, RNA-seq) and simulated best-fit mRNA time-course (middle) heatmaps, along with identified kact for TA2CI (yellow) and TA1Δ (pink). kact is shown relative to RelA-wt with brighter colors indicating closer activity to RelA-wt (no effect of mutation) and white indicating complete loss of activity in the mutant (kact = 0). Gray indicating a fit with ln (dist) < − 2:5 (see Method Details) was not found.
(D) Scatterplot of kact for TA2CI and TA1Δ for 76 genes with a good fit in (C). Center color represents the effect of either a single mutation (from green kact = 1 with no change from RelA-wt and redundancy with respect to a single mutation, to blue kact = 0 where either mutation abrogates gene induction and both domains work synergistically to achieve full gene activation). Border colors represent an off-diagonal effect in which a gene shows a specific loss of induction in TA2CI (pink) or TA1Δ (yellow). Gray lines indicate the standard deviation of kact from three repeated runs of optimization pipeline.
(E) Bar graphs of kact (relative to the mean RelA-wt kact) for RelA-wt, TA2CI, TA1Δ, and TA2CITA1Δ mutants, shown for four example genes arranged to indicate their relative positions in (D). Error bars indicate standard deviations of kact from three repeated runs of optimization pipeline.
Given the DNA binding activity measured for each RelA TA mutant, we could identify the activation rate (kact) for each gene that optimally accounts for the measured mRNA profile (Figure 6B). Other parameters (KD, nhill, kdeg) were also fit but assumed to be the same for all RelA variants because they represent intrinsic properties of the gene promoter of mRNA. Particle-swarm optimization was used to minimize the distance between simulated RNA time courses and the measured RNA-seq data for each of the 104 genes, and the model was able to recapitulate the experimental findings indicating that altered activation strengths associated with each TA mutant could explain the altered RNA-seq results (Figure 6C). A stringent criterion (ln(dist) < − 2:5, see Method Details) was applied to exclude 28 genes with unsatisfactory fits. For the remaining genes, a wide range of activation strengths (kact) values were identified for the TA1Δ and TA2CI mutants, from ~1 (in which TA mutation did not affect gene induction strength relative to RelA-wt) to 0 (where TA mutation of a single TA domain entirely abrogated gene expression) (Figure 6C).
Our hope for this analysis was to identify differences in relative dependence on TA1 versus TA2 among the 104 RelA target genes, and we found modest specificity: for example, inflammatory genes Cxcl2, Cd83, and Icam1 had greater TA1 dependence, whereas Pdlim4 (Src inhibitor) and Gsap (protease) showed a higher degree of TA2 dependence. However, for many genes, the optimally parameterized kact values for TA1Δ and TA2CI mutants were correlated (Figure 6C) with marked differences in the degree of the decrease associated with both mutants (Figure 6D). For example, Cxcl1, Cxcl2, and Tnf, showed a substantial decrease in expression when mutating either TA domain (Figures 6D and 6E, bottom left), whereas Il15ra and Il1rl1 expression remained normal when either TA domain was removed (Figures 6D and 6E, top right). On the former class of genes, the inability of either TA domain to function effectively on its own suggests that TA1 and TA2 function synergistically, whereas, on the latter class of genes, they seem to function redundantly or independently. Thus, the unexpected conclusion from this analysis is that RelA target genes differ whether they impose an AND gate or an OR gate logic on the two TA domains within RelA. Given that the activity of TA1 is thought to be dependent on the phosphorylation status of serines in its amphipathic helix, an AND-versus-OR gate logic determines distinct regulatory role for TA2: in the latter case, TA2 may be primary driver of transcriptional activation, but in the former case, TA2 merely assists or boosts TA1-driven gene activation.
A Knockin Mouse Reveals that Regulatory Strategies of RelA Target Genes Are Generally Conserved in Different Cell Types but Are Often Stimulus Specific
The complementation of RelA-deficient primary MEF cells with mutated variants of RelA allowed us to study the effects of numerous specific mutations on the TNF response. To dissect regulatory strategies of endogenous target genes in other cell types and in response to other stimuli, we engineered a mouse strain that harbors the previously described RelA TA2CITA1Δ variant as a knockin mutation, termed RelATA (Figure S4A). Homozygous RelATA/TA mice are not embryonically lethal, unlike the RelA−/− mice, but are sickly and smaller than their heterozygous littermates (Figure S4B), whose genotype was verified by PCR (Figure S4C). To examine whether RelA target genes showed similar or differential regulatory strategies in other cell types and other inflammatory stimuli, we produced extensive RNA-seq data by stimulating WT and RelATA/TA primary MEFs and bone-marrow-derived macrophages (BMDMs) with TNF or Toll-like 4 receptor ligand LPS.
We first identified genes that were activated in both MEFs and BMDMs by a stringent log2 fold-change >2 threshold (Figure 7A). We found 37 such genes in response to TNF and 481 in response to LPS stimulation (Figure 7A). After peak-normalizing the data for each gene, we plotted the relative-expression data from WT and RelATA/TA MEFs and BMDMs in order of induction peak time in WT MEFs and subsequent hierarchical clustering (Figure 7B). We found that most gene-activation events were diminished in both mutant MEFs and BMDMs. We evaluated the phenotype by plotting the percentage of expression in RelATA/TA cells relative to the WT peak expression at the same time point for each gene (Figure 7C). In MEFs, most genes were TA dependent (28/37), and most of those (25/28) were also TA dependent in BMDMs. In response to LPS, most genes were TA independent in both MEFs and BMDMs, given the strong activation of NF-κB-independent signaling pathways of IRF3/ISGF3 and MAPK p38. However, of the 154 genes that were identified as TA dependent in MEFs, ~90% showed an equivalent degree of TA dependence in BMDMs. These data suggest that regulatory strategies of RelA target genes are generally conserved in different cell types when induced by the same stimulus.
Figure 7. A Knockin Mouse Reveals that the Dual TAD Requirement Pertains to Other Cell Types and Stimuli.
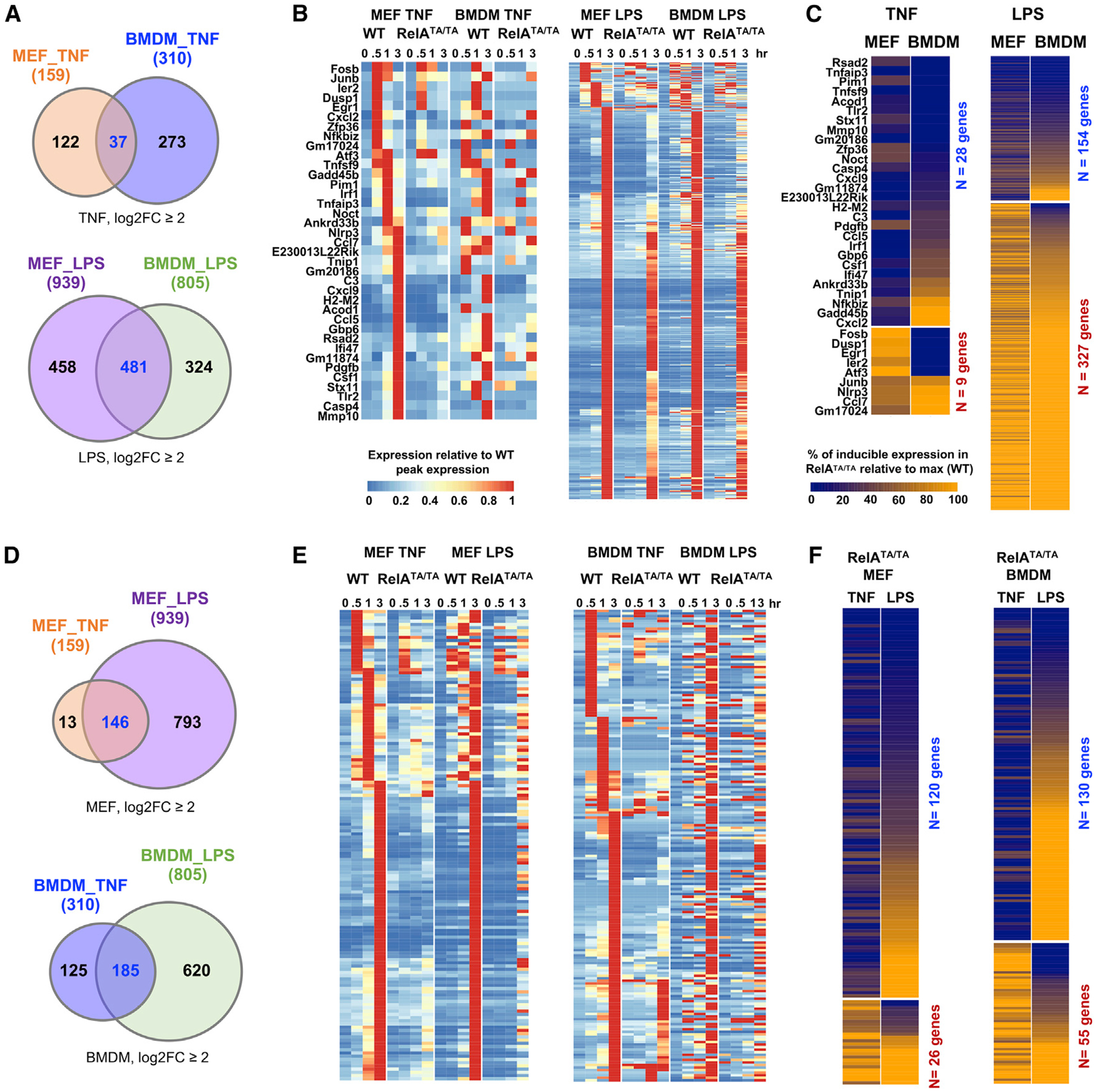
(A) Venn diagrams display overlap in induced transcriptional response in MEFs and BMDMs. Induced genes satisfy two criteria: log2 fold-change ≥2 and maximum RPKM >1, by polyA+ RNA-seq during a 3-h time-course analysis. In response to TNF, 159 genes are induced in WT MEFs and 310 are induced in WT BMDMs (left panel). In response to LPS, 939 genes are induced in WT MEFs and 805 induced genes in WT BMDMs (right panel).
(B) Heatmaps of relative expression of the induced genes in WT and RelATA/TA mutant MEFs or BMDMs in response to TNF (left panel) or LPS (right panel). Gene names and data plotted in heatmaps are listed in Tables S2A and S2B for TNF and LPS, respectively.
(C) RelA-TAD dependence of induced genes: percentage of peak expression in RelATA/TA mutant versus WT. Genes are rearranged by their TAD dependence in TNF stimulation and further re-arranged in each category by LPS response. Genes induced by TNF in MEFs and BMDMs (left panel). Gene names and data plotted in heatmap are listed in Table S2C. Genes induced by LPS in MEFs and BMDMs. Gene names and data plotted in heatmap are listed in Table S2D (right panel).
(D) Venn diagrams displays overlap in induced transcriptional response to TNF and LPS. In WT MEFs, 159 genes were induced by TNF and 939 by LPS (left panel). In WT BMDMs, 310 genes were induced by TNF and 805 were induced by LPS (right panel).
(E) Heatmaps of relative expression of TNF- versus LPS-induced genes in WT and RelATA/TA mutant MEFs (left panel) or BMDMs (right panel). Gene names and data plotted in the heatmap are listed in Tables S3A and S3B for MEFs and BMDMs, respectively.
(F) NF-κB dependence of induced genes: percentage of peak expression in RelATA/TA mutant versus WT. Genes induced by MEFs by TNF and LPS (left panel). Gene names and data plotted in heatmap are listed in Table S3C. Genes induced by BMDMs by TNF and LPS (right panel). Gene names and data plotted in heatmap are listed in Table S3D.
We then asked whether regulatory strategies of NF-κB target genes may be stimulus specific. To that end, we identified 146 genes that were induced by both TNF and LPS in MEFs and 185 that were induced by both stimuli in BMDMs (Figure 7D). Heatmap visualization of the gene expression time courses revealed that most genes induced rapidly by TNF peaked later in response to LPS (Figure 7E, left and right panels). Examining their RelA TA dependence by collapsing the data to the peak time point, we found that, of the 120 MEF genes that were TA-dependent in response to TNF, only 60% showed an equivalent degree of TA dependence in response to LPS, and 15% were entirely TA independent (Figure 7F, left and right panels). Similarly, of the 26 genes whose TNF induction was TA independent, half showed TA dependence in response to LPS. In BMDMs, the lack of equivalence was even more pronounced: only 25% showed an equivalent degree of TA dependence (or independence) in response to TNF and LPS, and more than 40% showed entirely the opposite regulatory requirements for RelA’s TA domains.
We also assessed the 104 RelA target genes identified previously in the primary MEF RelA complementation system that were expressed in the knockin MEFs and BMDMs in response to TNF and LPS stimulations (Figure S5A). We found that all of those genes were induced in MEFs by >2-fold but not all of them were induced in BMDMs (grayed out rows marked by an asterisk), indicative of cell-type-specific epigenomes or enhancer landscapes. In response to TNF, induction of all 104 genes was almost entirely abrogated in the RelATA/TA MEFs (left panel), whereas in response to LPS, a small subset of genes remained induced.
We wondered whether compensation by cRel, which was documented in RelA−/− MEFs (Hoffmann et al., 2003), was affecting our results. We thus bred the RelATA knockin strain to a crel−/− background and produced RNA-seq data from crel−/− RelATA/TA and crel−/− MEFs in response to TNF and LPS stimulations. We found that all 104 target genes were induced by both TNF and LPS in crel−/− control MEFs (Figure S5A). Interestingly, in response to LPS, those genes that showed induction in the mutant, by and large, also showed induction in the mutant in the crel−/− background. Further, expanding our view to all genes induced by both TNF and LPS by log2 fold change >2 (and maximum RPKM >1), we found 146 genes in WT MEFs; of which, 145 also satisfied these stringent criteria induced in crel−/− MEF controls. Our results with crel−/− RelATA/TA and RelATA/TA MEFs demonstrate that the lack of RelA TA dependence is generally not due to cRel compensation (Figure S5C).
The stimulus-dependent requirement of RelA TAs suggested that gene regulatory strategies are stimulus specific. To investigate whether that could be explained by the induction of distinct combinations of TFs, we performed motif-enrichment analysis. We found that regulatory regions of genes that were RelA TA dependent in response to both TNF and LPS were highly enriched for κB sites (Figure S5C, right panel). In contrast, genes that were TA dependent in the TNF, but not in the LPS, condition, showed enrichment also of interferon response factor (IRF) and PU.1:IRF8 binding sites, akin to previously identified the IRF-NF-κB OR gate genes (Cheng et al., 2017). Further, the 26 genes that showed RelA TA-independent induction in response to both TNF and LPS did not show enrichment to any stimulus-induced TFs. To further examine whether these TA-independent genes are also NF-κB independent for their transcriptional response, we looked at their TNF response in p53−/− crel−/− rela−/− MEFs reconstituted with empty vectors and found that almost all showed substantial TNF induction (Figure S5D); whether the data for two outliers is reliable may require additional experiments, but generally, our results indicate that NF-κB’s capacity to induce inflammatory-response genes depends on RelA’s transactivation domain.
In sum, our transcriptomic analysis using newly developed RelATA/TA mice revealed that regulatory strategies of RelA target genes were generally conserved between MEFs and BMDMs when the same stimulus was used but that, in either cell type, RelA TA dependence observed in response to TNF was not always correlated with dependence in response to LPS, which also induces IRF family members. These data indicate that some NF-κB target genes are capable of engaging distinct regulatory strategies to different stimuli depending on the availability of stimulus-induced TFs (Cheng et al., 2017, 2015; Ourthiague et al., 2015).
DISCUSSION
In this study, we reveal gene-specific regulatory strategies of endogenous RelA target genes in primary cells by probing their stimulus-induced expression with engineered RelA variants. We stringently identified target genes by both functional and physical binding criteria, developed an experimental system to test mutational variants in primary cells, and systematically analyzed both chromatin DNA recruitment and transcriptional trans-activation at single-gene resolution. A key transcriptional activation domain mutation was produced as a knockin mouse mutant line to examine other cell types and stimuli, and a mathematical modeling approach enabled a quantitative analysis to reveal gene-specific logic gates formed by the two TA domains.
We first showed that RelA recruitment and transcriptional activation of all endogenous NF-κB target genes induced by TNF are dependent on DNA base-specific contacts. Akin to previous X-ray structural analysis of the NF-κB-DNA complex (Chen et al., 1998b), we observed that some RelA binding events only contain half sites. Importantly, in our RelADB mutant analysis, the p50 dimerization partner may still contribute to DNA binding but that appears to be insufficient. Although the gene-expression response to TNF is rapid, future studies may address whether the dependence on DNA base-specific contacts also pertains to longer-term expression dynamics mediated by NF-κB.
Our studies further indicate that all endogenous target genes induced by TNF require the C-terminal region of RelA that harbors two transactivation domains TA1 and TA2. That is remarkable given that S276 was reported to play critical roles in recruiting CBP/p300 (Zhong et al., 1998), and knockin mutant mice harboring the S276A mutation showed striking animal phenotypes and alterations in the expression of many genes, including some known NF-κB target genes (Dong et al., 2008). Our analysis suggests that these mechanisms are insufficient for TNF-induced NF-κB target gene activation and may support the notion that the phenotype of S276 mutation may be due to alterations at larger time and epigenetic scales (Cheng et al., 2008).
By further probing target genes for the roles of the two C-terminal activation domains TA1 and TA2, we found remarkable gene specificity. Target genes did not differ very much in whether they were more highly regulated by TA1 versus TA2, but they differed in whether they required either just TA or required both TAs for gene activation. In other words, some NF-κB target genes allow TA1 and TA2 to function independently, forming a logical OR gate, and some NF-κB target genes require the synergistic function of TA1 and TA2, with the TAs forming a logical AND gate. These distinct regulatory strategies may be mediated by distinct mechanistic requirements for the recruitment of molecular co-activators. For example, genes that exhibit an OR gate logic, are activated sufficiently by recruiting CBP/p300 via TA2, but genes exhibiting an AND gate logic need not only recruit CBP/p300 but also additional co-activators via TA1.
The functionality of TA1 is thought to be regulated by PTMs, particularly phosphorylation, just as the transactivation potential of the NF-κB protein cRel is modulated by TNF-induced serine phosphorylation of S460 and S471 in a gene-specific manner (Starczynowski et al., 2005). In contrast, TA2 is thought to have intrinsic transactivation activity via a high-affinity interaction with CBP/p300. However, within TA2, S467 was also reported as a potential phosphorylation site (Buss et al., 2004b; Geng et al., 2009; Mao et al., 2009; Schwabe and Sakurai, 2005). To investigate whether S467 has a role in the TNF induction of NF-κB target genes in fibroblasts, we constructed the S467A variant within full-length RelA, TA1-deleted RelA, or the CBP-interaction mutant TA2CI (Figure S6A). Gel-mobility shift analysis showed TA2S467A did not alter the DNA-binding activity of NF-κB compared with each respective parent construct (Figure S6B; cf. Figure 5C), and protein expression levels of those RelA variants and the canonical IκBα and IκBβ were normal (Figure S6C). We then undertook polyA+ RNA-seq analysis, and PCA revealed only minimal alteration in the TNF-induced gene expression response with S467A in the WT or TA2CI context (Figure S6D). Furthermore, in a heatmap with single-gene resolution (Figure S6E), we observed very little effect by the S467A mutation. Only chemokines Cxcl1, Cxcl2, and Ccl20, which are rapidly and highly induced, showed a slight reduction but no enhancement. Our data thus confirm that an intrinsic CBP/p300 interaction affinity (probed with the TA2CI mutation) is the primary mode of function of the TA2 domain. This contrasts with how TA1’s functionality is enhanced by phosphorylation events mediated by coordinated kinases, such as CKII and IKKε (O’Shea and Perkins, 2008; Buss et al., 2004a; Wang et al., 2000).
What are the implications of this regulatory distinction of TA1 versus TA2 for the transcriptional control of NF-κB target genes, which differ in how they employ TA1 and TA2? Considering NF-κB target genes that are regulated redundantly by TA1 OR TA2, we would expect them to respond to all inflammatory stimuli that activate the IKK-NF-κB axis because TA2 is effective for its induction, and regulated TA1 activity may merely provide some modulation. In contrast, NF-κB target genes that are regulated synergistically by TA1 AND TA2 will be strongly activated only by those inflammatory stimuli that also induce the phosphorylation of TA1 serines that potentiate its acidic activation function and recruit other necessary regulatory cofactors for the synergistic activation to enhance their transcription through RelA (Blair et al., 1994; van Essen et al., 2009). Alternatively, TA1 phosphorylation may be a function of the microenvironmental context defined, thus rendering the activation of these gene’s tissue context dependent. We note, for such genes controlled by an AND gate of TA1 and TA2, the role of TA2 is effectively to amplify the effect of phosphorylation-mediated regulation of TA1 in activity, rendering them more responsive to these PTMs then if they were regulated by only TA1.
To address why some genes may require the synergistic activity of two TAs, whereas others do not, we first analyzed the content of nucleosome-destabilizing CpG dinucleotides (Ramirez-Carrozzi et al., 2009) in promoter regions. We found that the top 25% target genes (11 genes) regulated synergistically by TA1 AND TA2 had a lower percentage of CG content within ±300 bp from their TSS than the bottom 25% (Figure S7A), supporting a model in which a more stable nucleosome is more restrictive to gene activation. (An alternative model is the co-regulatory function of GC-binding Sp1; Van Essen et al., 2009.) Interestingly, these genes also showed higher κB motif binding z-scores (Figure S7B), possibly leading to longer NF-κB residence times, which may allow both TA domains to be engaged in sequential activation mechanisms. Indeed, upon TNF stimulation, those genes showed higher RNA polymerase II (Pol II) ChIP signals at 0.5 and 3 h (Figure S7C), indicative of higher levels of transcriptional induction.
Examining the types of genes that are regulated redundantly, versus synergistically, by TA1 and TA2, we are struck that many genes that provide for negative regulation of inflammation, such as Il15ra, IL1rl1, Trim47, Slfn2, or RelB fall into the former category. For those genes, TA redundancy means that they constitute an inflammatory core response that includes provisions for inflammatory resolution. In contrast, the latter category includes genes that are key initiators of an inflammatory response, such as TNF, Ccl20, Ccl2, and Cxcl1. For these genes, TA synergy means that their expression is activated only in response to a subset of inflammatory stimuli or when tissue-environmental context provides for the co-stimulatory signal that TA1 requires.
The here-described paradigm of NF-κB RelA-responsive gene expression being either redundantly or synergistically mediated by TA1 and TA2 should prompt a range of further studies that extend past work on the signals and pathways that control TA1 phosphorylation and the molecular mechanisms that determine whether a target gene requires the dual TA1/TA2 functionality. We may imagine that the involvement of differential co-activators, mediator complexes, nucleosome movements, or other complex chromatin interactions or recruitment or release of RNA polymerase may have a role in determining diverse regulatory logics.
More broadly, the present study suggests that the structure-function relationship of key TFs provides an informative probe to dissect the diverse gene regulatory strategies that govern their dozens and hundreds of target genes. Quantitative datasets produced by next-generation sequencing allow a focus on endogenous genes that may be effectively, quantitatively evaluated not only bioinformatically (which evaluates average behavior, assuming commonalities) but also with gene-specific resolution by mathematical models of the gene-expression process to reveal regulatory diversity. Thus, we hope that our study provides an analysis blueprint for future studies; indeed, advances of CRISPR/Cas9 technology that enable the engineering of variants into the endogenous gene locus is powering studies of endogenous gene regulatory circuits at single gene resolution.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexander Hoffmann (ahoffmann@ucla.edu). All plasmids generated in this study are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type and RelA, cRel, p53 gene-deficient C57BL/6 mice were maintained in pathogen-free conditions at the University of California, Los Angeles. The animal protocol for this study were approved by the Institutional Animal Care and Use Committee and by the University of California, Los Angeles Division of Laboratory Animal Medicine. Targeted Neo deleted RelA transactivation domain knock-in mutant mice were generated by ingenious Technology Lab (iTL, https://www.genetargeting.com). Wild-type primary MEFs were generated by crossing wild-type to wild-type mice at 8 weeks of age. p53−/− crel−/− rela−/− primary MEFs were generated by crossing p53−/− crel−/− rela+/ male mouse with p53−/− crel−/− rela+/ female mouse at 8 weeks of age. Wild-type or RelATA/TA primary MEFs were generated by crossing RelATA/+ (heterozygous allele) male mouse with RelATA/+ female mouse at 8 weeks of age.
Primary Cell Culture
Wild-type or mutant primary MEFs were generated from E11.5–13.5 embryos of mixed gender and cultured in DMEM supplemented with 10% bovine calf serum (BCS), 1% penicillin-streptomycin and 1% L-Glutamine. BMDMs were made by isolating 5 × 106 bone marrow cells from mouse femurs of WT or RelATA/TA C57BL/6 mice following red blood cell lysis and seven-day culturing with 30% L929-conditioned medium.
Cell Lines
Platinum-E (Plat-E) retroviral packaging cell line, a modified cell line derived from HEK293T (Morita et al., 2000), were cultured in DMEM with 10% FBS supplemented with 1% penicillin-streptomycin and 1% L-Glutamine, blasticidin (10 μg/mL) and puromycin (1 μg/mL).
METHOD DETAILS
Construction of mutant RelA plasmids
The pBABE-puro plasmid vector containing RelA was constructed by ligating the polymerase chain reaction (PCR)-amplified coding region of murine RelA (amino acids 1 to 549) restricted with EcoRI and SalI. Agilent QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) was used for all RelA point mutation and deletion variants. DNA binding mutant (RelADB) within amino-terminal were mutated by a triple substitution at residues Arg35, Tyr36, and Glu39 to Ala (R35A, Y36A, R39A). Mutagenesis of the carboxyl-terminal region for C-termΔ, TADΔ, and TA1Δ were performed by displacing residues Pro326, Ala429, and Ser520, respectively, with a STOP codon sequence. All site-specific mutations were verified by Sanger sequencing analysis and sequencing analyses of the resulting RelA mutants showed that the targeted sites were the only changes in the DNA sequence.
Genetic complementation of MEFs
For transfection, Plat-E cells were plated on 10-cm plates 16-hr the day before transfection at 50% confluent in DMEM supplemented with 10% fetal calf serum, 1% penicillin-streptomycin and 1% L-Glutamine. Cells were transfected using 300 μL of Opti-MEM medium (ThermoFisher Scientific) and polyethylenimine (PEI; 1 μg/μL in 1xPBS pH4.5, Polysciences #23966–2) with 7 mg of retroviral construct DNA, pBABE-puro EV control or RelA expressing constructs (4:1 ratio of PEI (μL):plasmid DNA (μg)). Transfection complex (Opti-MEM medium, PEI reagent, plasmid DNA) were incubated for 20-min at room temperature then added drop-wise to Plat-E cells for 6 hours. Transfected media was replaced with fresh DMEM media containing 10% fetal calf serum, 1% penicillin-streptomycin and 1% L-Glutamine. Cells were further incubated for a total of 48-hr post-transfection and prior to collecting viruses. Viruses-containing supernatant were filtered through a 0.45 mm filter and used to infect p53−/− crel−/− rela−/− MEFs with the addition of 4 μg/mL Polybrene. 48-hr post-infection of cells with viruses the stably transduced cells were selected with 2 mg/mL puromycin for a total of 72 hours. Post selection with puromycin, puromycin-containing medium was removed and cells are passaged twice for recovery prior to being expanded in culture for experiments. pBABE-puro empty vector without the RelA gene fragment was used as a negative control, to maintain a stable retrovirally transduced RelA knockout cell line, meanwhile pBABE-puro containing full-length RelA (RelA-wt) was used as a positive RelA cell line control.
Western Blotting
For whole cell extracts, 3 × 106 cells were lysed with RIPA buffer containing 1mM PMSF and 1mM DTT followed by lysing with 1x SDS-PAGE sample buffer containing 5% β-mercaptoethanol. Protein extracts were separated by SDS-PAGE before transfer to nitro-cellulose membranes and subjected to western blotting. Nuclear and cytoplasmic extracts were performed with standard methods as described previously (Hoffmann et al., 2002; Werner et al., 2005). Western blots were probed with antibodies listed in the Key Resources Table and were visualized using SuperSignal West Femto and West Pico (1:10 mixture) Chemiluminescent reagents to detect chemiluminescence released by HRP-labeled secondary antibodies. The resulting proteins were visualized by ChemiDoc XRS+ Imaging Systems (Bio-Rad) and Image Lab (Bio-Rad). Western blots of resulting proteins were quantified using ImageJ software (NIH, USA).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-RelA | Santa Cruz Biotechnology | sc-372; RRID: AB_632037 |
| Goat polyclonal anti-RelA | Santa Cruz Biotechnology | sc-372G; RRID: AB_632037 |
| Rabbit polyclonal anti-RelA | Santa Cruz Biotechnology | cat# sc-109; RRID:AB_632039 |
| Rabbit polyclonal anti-IκBα | Santa Cruz Biotechnology | cat# sc-371; RRID:AB_2235952 |
| Rabbit polyclonal anti-IκBβ | Santa Cruz Biotechnology | cat# sc-945; RRID:AB_631696 |
| Rabbit anti-p50 serum | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-AB0080 |
| Mouse monoclonal anti-tubulin | Santa Cruz Biotechnology | cat#sc-5286; RRID:AB_628411 |
| Goat polyclonal anti-Actin | Santa Cruz Biotechnology | cat# sc-1615; RRID:AB_630835 |
| Rabbit polyclonal anti-Histone 3 | Abcam | cat# ab1791; RRID:AB_302613 |
| Rabbit monoclonal anti-p84 | Abcam | cat# ab131268; RRID:AB_11156907 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS | Sigma, B5:055 | cat# L2880 |
| Murine TNF | R&D Systems | cat# 410-MT |
| Murine IL-1β | R&D Systems | cat# 401-ML-CF |
| L-Glutamine | Corning | cat# 25–005-CI |
| Penicillin-Streptomycin Solution | Corning | cat# 30002CI |
| β-mercaptoethanol | ThermoFisher | cat# 21985023 |
| Poly(deoxyinosinic-deoxycytidylic) | Sigma-Aldrich | cat# P4929 |
| PEI (polyethylenimine) | Polysciences | cat# #23966–2 |
| Polybrene | Santa Cruz Biotechnology | cat# 134220 |
| Blasticidin | InvioGen | cat# ant-bl |
| Puromycin | InvioGen | cat# ant-pr |
| PMSF (Phenylmethylsulfonyl fluoride) | Sigma-Aldrich | cat# P7626 |
| DTT (Dithiothreitol) | Fisher Scientific | cat# BP172–5 |
| methanol-free Formaldehye solution | ThermoFisher Scientific | cat# 28908 |
| DSG (disuccinimidyl glutarate) | ThermoFisher Scientific | cat# 20593 |
| EDTA-free protease inhibitors | ThermoFisher Scientific | cat# 78439 |
| Proteinase K | Sigma-Aldrich | cat# P2308 |
| PureLink Rnase A | ThermoFisher Scientific | cat# 12091–021 |
| TRIzol | ThermoFisher Scientific | cat# 15596026 |
| Prestained Protein marker | BioPioneer | cat# P-001M |
| Critical Commercial Assays | ||
| Agilent QuikChange II Site-Directed Mutagenesis Kit | Agilent Technologies | cat# 200523 |
| RNeasy Mini Kit | QIAGEN | cat# 74104 |
| Direct-zol RNA Microprep Kit | Zymo Research | cat# R2060 |
| mRNA-seq Library Kit | Kapa Biosystems | cat# KK8421 |
| RNA-seq Library Kit With RiboErase | Kapa Biosystems | cat# KK8483 |
| NEBNEXT Ultra DNA Library Prep Kit for Illumina | New England BioLabs | cat# E7370L |
| dsDNA BR assay Kit | ThermoFisher Scientific | cat# Q32853 |
| dsDNA HS assay Kit | ThermoFisher Scientific | cat# Q32854 |
| AMPure XP beads | Beckman Coulter | cat A63881 |
| Dynabeads Protein G | ThermoFisher Scientific | cat# 1004D |
| SuperSignal West Pico PLUS Chemiluminescent substrate | ThermoFisher Scientific | cat# 34578 |
| SuperSignal West Femto Maximum Sensitivity Substrate | ThermoFisher Scientific | cat# 34095 |
| Deposited Data | ||
| Reference SuperSeries for all sequencing data in this study | NCBI Gene Expression Omnibus | GEO: GSE132540 |
| Experimental Models: Cell Lines | ||
| primary mouse embryonic fibroblasts (MEFs) | This study | N/A |
| Platinum-E (Plat-E) cells | (Morita et al., 2000) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6: All murine embryonic friboblasts | This study | N/A |
| Mouse: C57BL/6: WT and RelATA/TA | This study | N/A |
| Oligonucleotides | ||
| NF-Y EMSA probe (5′-GAIIIIIICCIGAIIGGIIAAA-3′; 5′-ACTTTTAACCAATCAGGAAAAA-3′) | This Lab and this study | N/A |
| κB EMSA probe (5′-GCTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGG-3′) | This Lab and this study | N/A |
| Recombinant DNA | ||
| pBABE-puro vector plasmid DNA | (Morgenstern and Land, 1990) | Morgenstern and Land (Addgene plasmid #1764; RRID:Addgene_1764) |
| Software and Algorithms | ||
| Epoch Spectrophotometer System | Biotek | N/A |
| Qubit 2.0 fluorometer | ThermoFisher Scientific | N/A |
| ImageQuant TL v8.1 | GE Healthcate | N/A |
| Image Lab v6.0 | Bio-Rad | N/A |
| ImageJ v1.50i | NIH, USA | N/A |
| ChemiDoc XRS+ Imaging System | Bio-Rad | N/A |
| Amersham Typhoon Scanner | GE Healthcare | N/A |
| Mathworks MATLAB | MathWorks, Inc. | R2014a |
| ChIPseeker v1.18.0 | (Yu etal., 2015) | N/A |
| deepTools2 | (Ramirez et al., 2016) | N/A |
| HOMER | (Heinz et al., 2010) | N/A |
| Enrichr | (Chen et al., 2013; Kuleshov etal., 2016) | N/A |
| IGV (Integrated Genomics Viewer) | (Robinson et al., 2011) | N/A |
| R(v3.6.1) | N/A | http://www.r-project.org/ |
| Wilcox.test function | R program | N/A |
| mean and standard deviation | Microsoft Excel | N/A |
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out with standard methods as described previously (Hoffmann et al., 2002; Werner et al., 2005). In brief, 2.5 mL total normalized nuclear extracts were incubated for 15-min with 0.01pmol of P32-labeled 38bp double-stranded oligonucleotide containing two consensus κB sites (5′-GCTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGG-3′; or with NF-Y loading control (5′-GATTTTTTCCTGATTGGTTAAA-3′; 5′-ACTTTTAACCAATCAGGAAAAA-3′) in binding buffer [10mM Tris-Cl (pH 7.5), 50mM NaCl, 10% glycerol, 1% NP-40, 1mM EDTA, 0.1mg/mL Poly(deoxyinosinic-deoxycytidylic)], in a final reaction of 6 μL. The reaction mixtures were run on a non-denaturing 5% acrylamide (30:0.8) gel containing 5% glycerol and 1X TGE buffer [24.8mM Tris, 190 mM glycine, 1mM EDTA] at 200 V for 2 hours. The gel was visualized by Amersham Typhoon Scanner (GE Healthcare Life Sciences) and analyzed in ImageQuant TL software, in which the NF-Y loading control gel was used to normalize for loading variability.
Immunoprecipitation
Whole cell extracts (WCE) were lysed with RIPA buffer containing 1mM PMSF and 1mM DTT. Anti-RelA antibody (Santa Cruz Biotechnology, sc-372G) was conjugated to prewashed magnetic protein-G beads (Life Technologies Dynabeads) for 30-min at room temperature followed by addition unstimulated WCE in RIPA buffer containing 1mM PMSF and 1mM DTT and incubation overnight (~16-hr). The beads were then washed thoroughly with 1X TBS-T buffer and IP samples were eluted with 1X SDS-sample buffer containing 5% β-mercaptoethanol. Samples were resolved on SDS-PAGE and western blotting with antibodies specific for anti-RelA, anti-IκBα, anti-IκBβ, and anti-p50.
RNA isolation and sequencing (RNA-seq)
MEF cells at ~80% confluence were serum-starved by overnight incubation in Dulbecco’s Modified Eagle’s Medium supplemented with 0.5% bovine calf serum (BCS), 1% penicillin-streptomycin and 1% L-Glutamine prior to stimulation. For chromatin-associated RNA-seq (caRNA-seq), nascent RNA transcripts were prepared from the chromatin fraction (Tong et al., 2016) using TRIzol according to the manufacturer’s instruction (ThermoFisher Scientific) and purified using Direct-zol RNA Microprep Kit (Zymo Research cat#R2060). RNA libraries were prepared from ribo-depleted using KAPA Stranded RNA-Seq Kit with RiboErase (KAPA Biosystems, cat#KK8483) with 400ng of starting total RNA material. For polyA+RNA-seq analysis, total RNA was isolated with the QIAGEN RNeasy Mini Kit according to the manufacturer’s instructions, quantified using Epoch Spectrophotometer System (Biotek), and purified from 1 μg of starting total RNA material using oligo (dT) magnetic beads before fragmenting at high temperature with divalent cations. Complementary DNA libraries were generated using KAPA Stranded mRNA-Seq Kit (KAPA Biosystems, cat#KK8421), and quantitation was performed using Qubit 2.0 fluorometer using dsDNA BR assay kit #Q32853. Sequencing was performed on Illumina HiSeq 2000 and HiSeq 4000 with single-end 50-bp sequencing, according to manufacturer’s recommendations and prepared by Broad Stem Cell Research Center core facility at the University of California, Los Angeles.
RNA-seq data analysis
Reads were aligned with STAR to Gencode mouse mm10 genome and RefSeq genes and featureCounts were used to obtain aligned raw counts. Only uniquely mapped reads with a mapping quality score of ≥ 20 were kept for further analysis, using samtools. Read counts were normalized for library size and transcript length by conversion to RPKM and gene below the maximum RPKM < 1 were excluded from downstream analysis. DESeq2 was used identify induced genes for subsequent analysis using the following criteria: ≥ 2-fold increase in expression at any one time point over basal with FDR threshold of 0.01. Principal components were calculated with prcomp package and plotted with ggplot. caRNA-seq and polyA+RNA-seq heatmaps were plotted using pheatmap package. HOMER (Heinz et al., 2010) was used to assess for enrichment of de novo and known transcription factor binding motifs for NFκB dependent genes with promoter sequences 1 kB upstream and 0.3 kB downstream of the transcription start site. An in-depth description of this software can be found at homer.ucsd/edu/homer. Gene ontology analysis in biological processes for NFκB target genes were performed using Enrichr (Chen et al., 2013; Kuleshov et al., 2016). The 5 most enriched GO terms were selected for NFκB-dependent genes.
Chromatin immunoprecipitation and sequencing (ChIP-seq)
MEF cells (8 × 106 cells per plate) were serum-starved overnight with 0.5% BCS medium, then stimulated with 10ng/mL TNF at indicated time points. For ChIP, cells were double cross-linked with 100mM disuccinimidyl glutarate/PBS solution (DSG, ThermoFisher Scientific #20593) for 30-min followed by 1% methanol-free Formaldehye/PBS solution (ThermoFisher Scientific #28908) for 15-min. Cross-linked cells were quenched with 125mM Glycine for 5 min and washed twicewith cold PBS followed by snap freezing with dry ice. The cell pellets were thawed and lysed in Lysis Buffer 1, on ice. Cells were briefly sonicated with Diagenode Bioruptor 300 sonication system (medium power, 15 s ON/15 s OFF, 2 cycles) in Diagenode 1.5mL TPX microtubes (100 μl-max 300 μl/tube). After centrifugation, cell pellets were lysed in Lysis Buffer 2 and incubated for 10 min at room temperature. Following centrifugation, cell nuclear pellets were lysed in Lysis Buffer 3. Nuclear lysates were sonicated with Diagenode Bioruptor 300 sonication system (low power, 30 s ON/30 s OFF, 12 cycles) in Diagenode 1.5mL TPX microtubes. Sonication was stopped every 4 cycles for incubation on ice for 1 min, gentle inversion and pulse-spin. Sonicated nuclear supernatant containing DNA fragments were consolidated with the same sample into a single 1.5 mL polypropelene tube. The lysates were centrifuged at max speed for 10 min at 4°C. The supernatants were transferred to 2 mL no-stick microtubes (Phenix Research cat# MH-820S) with 3 volumes of Dilution buffer and 5 mg anti-RelA antibody (Santa Cruz Biotechnology, sc-372), incubated overnight at 4°C. Next day, antibody-chromatin complexes were incubated with 30 μL magnetic protein G beads (Life Technologies Dynabeads protein G #1004D) for 5 hours at 4°C. Chromatin-immunoprecipitates were washed 3 times (4 min wash at 4°C) with each of the following buffer in this order: low-salt wash buffer, high-salt wash buffer, LiCl buffer, and TE buffer. ChIP DNA was eluted in elution buffer overnight at 65°C. 1% inputs and ChIP DNA fragments were subjected to reverse cross-linking, RNase A digestion (50 μg), Proteinase K digestion (50 μg), and purification with AMPure XP beads (Beckman Coulter). Input DNA samples were quantified with dsDNA BR assay kit #Q32853, and ChIP DNA samples were quantified with dsDNA HS assay kit (Q32854). Quantitation was performed using Qubit 2.0 fluorometer. ChIP-Seq DNA libraries were prepared from 5ng of Inputs or ChIP DNA using NEBNext Ultra DNA Library Prep Kit for Illumina (New England BioLabs, # E7370L).
ChIP Buffer composition
Lysis Buffer 1: 50mM HEPES-KOH (pH 7.6), 140mM NaCl, 1mM EDTA, 10% Glyercol, 0.5% NP-40, 0.25% Triton X-100 [with freshly added 1X EDTA-free protease inhibitors].
Lysis Buffer 2: 10mM Tris-Cl (pH 8.0), 200mM NaCl, 1mM EDTA, 0.5mM EGTA [with freshly added 1X EDTA-free protease inhibitors].
Lysis Buffer 3: 10mM Tris-Hcl (pH 8.0), 100mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% Na Deoxycholate, 0.5% N-lauroylsarcosine sodium salt, 0.2% SDS [with freshly added 1X EDTA-free protease inhibitors].
Dilution Buffer: 10mM Tris-Cl (pH 8.0), 160mM NaCl, 1mM EDTA, 0.01% SDS, 1.2% Triton X-100 [with freshly added 1X EDTA-free protease inhibitors].
Low Salt Wash Buffer: 50mM HEPES-KOH (pH 7.6), 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS [with freshly added 0.5X EDTA-free protease inhibitors].
High Salt Wash Buffer: 50mM HEPES-KOH (pH 7.6), 500mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS [with freshly added 0.5X EDTA-free protease inhibitors].
LiCl Buffer: 20mM Tris-HCl (pH 8.0), 250mM LiCl, 1mM EDTA, 0.5% Na Deoxycholate, 0.5% NP-40
TE Buffer: 10mM Tris-HCl (pH 8.0), 1mM EDTA
Elution Buffer: 10mM Tris-HCl (pH 8.0), 1mM EDTA, 1% SDS
ChIP-seq data analysis
Sequencing reads were aligned to the mouse mm10 genome and RefSeq genes using Bowtie 2 (Langmead and Salzberg, 2012) and filtered using Samtools. Uniquely mapped reads were used to identify peaks for each sample individually with MACS2 version 2.1.0 using default settings except for FDR of 0.01. Differential RelA binding events were plotted as heatmap using plotHeatmap from deepTools2 (Ramírez et al., 2016). ChIP-seq peak annotation for genomic locations were plotted as pie chart using ChIPseeker (Yu et al., 2015). For mapping of κB elements to NFκB target genes, any TNF-inducible RelA ChIP-seq peaks within ± 10kb from gene TSS were annotated to κB elements (unique half and full κB DNA sequences were annotated within the mm10 genome) and were mapped to each RelA ChIP-seq peak centered at 500bp window (±250bp from each peak of κB sequence) on every gene using Bedtools intersect. Full-length κB elements were defined by the consensus sequence 5′GGRNNNNNYCC-3′ for unique 9, 10, or 11bp DNA sequence; where R is a A or G, Y is a T or C, and N is any nucleotide. κB half sites were defined by the consensus sequence 5′GGR or YCC-3′ DNA sequence. κB motifs were extracted from UCSC mm10 genome using Biostrings and BSgenome packages. Mapping of κB motifs and RelA ChIP-seq peaks were visualized by ggplot2 geom_segment. Scatterplots and boxplots of RelA ChIP-seq data were visualized by ggplot. Wilcox.test function in R was used to find the statistical significance of reduced versus non-reduced RelA peaks (Mann-Whitney U-test). Integrative Genomics Viewer (IGV) was used to acquire RelA ChIP-seq tracks for example genes (Robinson et al., 2011).
GC content analysis
GC sequence information was obtained from UCSC annotation for mouse mm10 genome using getSeq package in R with Granges objects. GC sequences are analyzed for ± 300 bp from each gene TSS. Percentage of GC were plotted as bar graphs using ggplot2 (v3.2.1) with Tidyverse. Wilcox.test function in R (Mann-Whitney U-test) was used to find the statistical significance of GC content.
Computational Modeling
An ODE model was constructed representing the rate of change of mRNA for each gene.
kact is the mRNA synthesis rate that depends on the activation strength of RelA. KD is the dissociation constant which quantifies the nuclear abundance of RelA activity that achieves half-maximal gene induction. kdeg is the degradation rate of the mRNA that relates to the half-life with the identity t1/2 = ln(2)/ kdeg. nhill is the Hill coefficient that describes ultra-sensitivity in gene activation, potentially the result of the cooperative effect of multiple κB sites, or multiple-sequential mechanistic steps that are not explicitly represented in this model (e.g., NFκB binding, recruitment of PIC components, promoter opening, initiation, RNA Pol II CTD phosphorylation, etc. RelA is a time-dependent variable obtained by quantifying RelA DNA-binding activity from EMSA and linearly interpolating between discrete time points. Basal mRNA for a given parameter set was obtained by analytically solving the ODE with RelA fixed at the basal value:
Induced time-course response was obtained using ode23s in MATLAB (The MathWorks, Inc.). The distance between experimental and modeled mRNA time courses was quantified using a combination of Pearson correlation and MSE to take into account both dynamics and absolute values (dist = (1 – Pearson) + MSE). As KD, nhill, kdeg are assumed to be gene-specific properties, unaffected by TAD mutations these were first fit to WT (RelA-wt) data using particle swarm optimization (PSO) to minimize the distance between modeled mRNA time-course and measured RNA-seq (50 particles, terminating at either 100 epochs or distance below 0.001). As the RNA-seq time-course did not capture a decrease in mRNA levels for late-induced genes, kdeg could not be fit but was assumed to be equivalent to an 8 hour half-life. Following the fit to the WT data KD, nhill, kdeg were fixed for data from mutant cells and a new kact was obtained for each TAD mutation using the same optimization approach.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using Excel or R (version 3.6.1). Where applicable, statistical significance are reported in the Figures and corresponding Figure Legends. Western blotting were quantified using ImageJ software and data are presented as mean + SD. All comparisons in sequencing data were analyzed using Wilcox.test function in R (Mann-Whitney U-test).
DATA AND CODE AVAILABILITY
All sequencing data presented in this publication have been deposited to the NCBI Gene Expression Omnibus (GEO) and accessible under SuperSeries accession number GSE132540, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132540. All quantified RelA DNA-binding activity as RelA concentration for the model and all values for model parameters for each 3 replicate model runs are presented in Table S5.
Supplementary Material
Highlights.
NF-κB target gene control is defined by transcriptomics, epigenomics, and math modeling
NF-κB target genes require RelA transactivation domains TA1/TA2 in AND-or-OR logics
For inflammatory initiators, constitutive TA2 boosts TA1’s PTM-regulated activity
For inflammatory resolvers, TA1/TA2 redundancy renders context-independent control
ACKNOWLEDGMENTS
We thank S. Basak, M. Asagiri, J.V. Almaden, and J.D. Vargas for critical discussions. K.A.N. acknowledges Ning Wang for help in HOMER motif analysis, Diane Lefaudeux for help with RNA-seq and ChIP-seq data analyses, and Yi Liu for help with BMDM isolation from RelATA/TA mutant mice. This study was supported by National Institute of Health grants GM117134 and AI127864 to A.H. and a University of California, Los Angeles (UCLA) Warsaw Fellowship to K.A.N.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.108.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bae JS, Jang MK, Hong S, An WG, Choi YH, Kim HD, and Cheong J (2003). Phosphorylation of NF-κ B by calmodulin-dependent kinase IV activates anti-apoptotic gene expression. Biochem. Biophys. Res. Commun 305, 1094–1098. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, and Baltimore D (1989). A 65-kD subunit of active NF-κB is required for inhibition of NF-κB by I κB. Genes Dev 3, 1689–1698. [DOI] [PubMed] [Google Scholar]
- Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, Stein B, and Greene WC (1992). The 65-kDa subunit of human NF-κ B functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc. Natl. Acad. Sci. USA 89, 1875–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Indukuri H, Liu T, Liao SL, Tian B, Brasier AR, Garofalo RP, and Casola A (2010). IKKε modulates RSV-induced NF-κB-dependent gene transcription. Virology 408, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S, Behar M, and Hoffmann A (2012). Lessons from mathematically modeling the NF-κB pathway. Immunol. Rev 246, 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TA, Schooley K, Dower SK, Hagen H, and Virca GD (1997). Activation of nuclear transcription factor NF-κB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J. Biol. Chem 272, 32606–32612. [DOI] [PubMed] [Google Scholar]
- Blair WS, Bogerd HP, Madore SJ, and Cullen BR (1994). Mutational analysis of the transcription activation domain of RelA: identification of a highly synergistic minimal acidic activation module. Mol. Cell. Biol 14, 7226–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohuslav J, Chen LF, Kwon H, Mu Y, and Greene WC (2004). p53 induces NF-κκB activation by an κIκB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J. Biol. Chem 279, 26115–26125. [DOI] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, and Kracht M (2004a). Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem 279, 55633–55643. [DOI] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Frank R, Livingstone M, Resch K, and Kracht M (2004b). Phosphorylation of serine 468 by GSK-3b negatively regulates basal p65 NF-κB activity. J. Biol. Chem 279, 49571–49574. [DOI] [PubMed] [Google Scholar]
- Chen FE, Huang DB, Chen YQ, and Ghosh G (1998a). Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 391, 410–413. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Ghosh S, and Ghosh G (1998b). A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol 5, 67–73. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Sengchanthalangsy LL, Hackett A, and Ghosh G (2000). NF-κB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure 8, 419–428. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Johnson TL, and Hoffmann A (2008). Epigenetic control: slow and global, nimble and local. Genes Dev 22, 1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, Chang M, Ponomarenko JV, Sun SC, Benedict CA, et al. (2011). The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci. Signal 4, ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Taylor B, Ourthiague DR, and Hoffmann A (2015). Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci. Signal 8, ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, and Hoffmann A (2017). Iterative modeling reveals evidence of sequential transcriptional control mechanisms. Cell Syst 4, 330–343.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian F, Smith EL, and Carmody RJ (2016). The regulation of NF-κB subunits by phosphorylation. Cells 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Moreno E, and Johnston SA (1998). Alterations in the GAL4 DNA-binding domain can affect transcriptional activation independent of DNA binding. J. Biol. Chem 273, 13776–13780. [DOI] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zhong H, Hayden MS, and Ghosh S (2008). Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev 22, 1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, and Schmitz ML (2009). Phosphorylation of NF-κB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep 10, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller D, Russo MP, Sartor RB, and Jobin C (2002). IKKβ and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-κ B activation in both primary and intestinal epithelial cell lines. J. Biol. Chem 277, 38168–38178. [DOI] [PubMed] [Google Scholar]
- Hao S, and Baltimore D (2009). The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol 10, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2008). Shared principles in NF-κB signaling. Cell 132, 344–362. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrainer K, Racchumi G, and Anrather J (2013). Site-specific phosphorylation of the p65 protein subunit mediates selective gene expression by differential NF-κB and RNA polymerase II promoter recruitment. J. Biol. Chem 288, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, and Baltimore D (2006). Circuitry of nuclear factor κB signaling. Immunol. Rev 210, 171–186. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, and Baltimore D (2002). The IκBNF-κB signaling module: temporal control and selective gene activation. Science 298, 1241–1245. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, and Baltimore D (2003). Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J 22, 5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, and Ghosh G (2006). Transcriptional regulation via the NF-κB signaling module. Oncogene 25, 6706–6716. [DOI] [PubMed] [Google Scholar]
- Keegan L, Gill G, and Ptashne M (1986). Separation of DNA binding from the transcription-activating function of a eukaryotic regulatory protein. Science 231, 699–704. [DOI] [PubMed] [Google Scholar]
- Kovesdi I, Reichel R, and Nevins JR (1986). Identification of a cellular transcription factor involved in E1A transactivation. Cell 45, 219–228. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq L, Raiola L, Chabot PR, Cyr N, Arseneault G, Legault P, and Omichinski JG (2017). Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-κB and transcription regulatory factors. Nucleic Acids Res 45, 5564–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, Repaka A, Mayo MW, and Burstein E (2009). GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev 23, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Kracht M, and Schmitz ML (2014). The cytokine-induced conformational switch of nuclear factor κB p65 is mediated by p65 phosphorylation. Biochem. J 457, 401–413. [DOI] [PubMed] [Google Scholar]
- Mitchell S, Vargas J, and Hoffmann A (2016). Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med 8, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PA, Ruben SM, and Rosen CA (1993). Conservation of transcriptional activation functions of the NF-κB p50 and p65 subunits in mammalian cells and Saccharomyces cerevisiae. Mol. Cell. Biol 13, 1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R, Sobotzik JM, Schultz C, and Schmitz ML (2010). Specification of the NF-κB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKKε. Nucleic Acids Res 38, 6029–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, and Kitamura T (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, and Ghosh G (2013). Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol 11, e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulero MC, Wang VYF, Huxford T, and Ghosh G (2019). Genome reading by the NF-κB transcription factors. Nucleic Acids Res 47, 9967–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyqvist I, Andersson E, and Dogan J (2019). Role of conformational entropy in molecular recognition by TAZ1 of CBP. J. Phys. Chem. B 123, 2882–2888. [DOI] [PubMed] [Google Scholar]
- O’Shea JM, and Perkins ND (2008). Regulation of the RelA (p65) transactivation domain. Biochem. Soc. Trans 36, 603–608. [DOI] [PubMed] [Google Scholar]
- Ourthiague DR, Birnbaum H, Ortenlöf N, Vargas JD, Wollman R, and Hoffmann A (2015). Limited specificity of IRF3 and ISGF3 in the transcriptional innate-immune response to double-stranded RNA. J. Leukoc. Biol 98, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, and Black DL (2013). Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA 19, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, and Smale ST (2009). A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatel H, Di Valentin E, Gloire G, Dequiedt F, Piette J, and Habraken Y (2012). Phosphorylation of p65(RelA) on Ser(547) by ATM represses NF-κB-dependent transcription of specific genes after genotoxic stress. PLoS ONE 7, e38246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, and Saiki I (2003). Tumor necrosis factor-a-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem 278, 36916–36923. [DOI] [PubMed] [Google Scholar]
- Savaryn JP, Skinner OS, Fornelli L, Fellers RT, Compton PD, Terhune SS, Abecassis MM, and Kelleher NL (2016). Targeted analysis of recombinant NFκB (RelA/p65) by denaturing and native top down mass spectrometry. J. Proteomics 134, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, and Baeuerle PA (1991). The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J 10, 3805–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, dos Santos Silva MA, Altmann H, Czisch M, Holak TA, and Baeuerle PA (1994). Structural and functional analysis of the NF-κB p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an α-helical conformation. J. Biol. Chem 269, 25613–25620. [PubMed] [Google Scholar]
- Schwabe RF, and Sakurai H (2005). IKKβ phosphorylates p65 at S468 in transactivaton domain 2. FASEB J 19, 1758–1760. [DOI] [PubMed] [Google Scholar]
- Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, and Bulyk ML (2011). Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol 13, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starczynowski DT, Reynolds JG, and Gilmore TD (2005). Mutations of tumor necrosis factor α-responsive serine residues within the C-terminal transactivation domain of human transcription factor REL enhance its in vitro transforming ability. Oncogene 24, 7355–7368. [DOI] [PubMed] [Google Scholar]
- Todaro GJ, and Green H (1963). Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol 17, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, and Smale ST (2016). A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell 165, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Essen D, Engist B, Natoli G, and Saccani S (2009). Two modes of transcriptional activation at native promoters by NF-κB p65. PLoS Biol 7, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, and Baldwin AS Jr. (2000). Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem 275, 32592–32597. [DOI] [PubMed] [Google Scholar]
- Werner SL, Barken D, and Hoffmann A (2005). Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 309, 1857–1861. [DOI] [PubMed] [Google Scholar]
- Yoboua F, Martel A, Duval A, Mukawera E, and Grandvaux N (2010). Respiratory syncytial virus-mediated NF-κB p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKKβ. J. Virol 84, 7267–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, and Ghosh S (1998). Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, and Ghosh S (2002). The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol. Cell 9, 625–636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data presented in this publication have been deposited to the NCBI Gene Expression Omnibus (GEO) and accessible under SuperSeries accession number GSE132540, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132540. All quantified RelA DNA-binding activity as RelA concentration for the model and all values for model parameters for each 3 replicate model runs are presented in Table S5.