

Abstract
Growing evidence have shown that the migration and invasion inhibitory protein (MIIP, also known as IIp45) functions as a tumor suppressor and its expression is downregulated in several types of cancer, yet the function of MIIP in prostate cancer (PCa) and the underlying mechanism of action remains largely unknown. Here we demonstrated that MIIP acts as a suppressor of PCa by inhibiting epithelial-mesenchymal transition (EMT) and cell invasion. Overexpressing MIIP repressed cellular invasion of PC3 and DU145 in vitro, accompanied by a decrease of EMT-inducing factors, and an increase of E-cadherin and KLF17. Moreover, a stable MIIP knockdown in PCa cells promoted the tumor growth or bone osteolytic lesions, when xenografted subcutaneously or via tibia injection. Mechanistically, MIIP represses two onco-miRNAs, miR-181a-5p and miR-181b-5p, thus removing the inhibitory effect of these two miRNAs on their target KLF17, which functions as a negative regulator of EMT by directly suppressing the transcription of SNAIL1/2 and TWIST. Finally, by examining the expression of MIIP, miR-181a/b-5p, KLF17, and E-cadherin in paired cancer samples v.s. adjacent normal tissues from a cohort of human prostate cancer patients, we demonstrated that downregulation of MIIP was well associated with downregulation of KLF17 and E-cadherin, but upregulation of miR-181a/b-5p. The positive correlation between MIIP and KLF17 was also confirmed via immunohistochemical staining of a PCa tissue microarray. Taken together, our findings reveal a novel function of MIIP as an EMT inhibitor in PCa and illustrate the underlying molecular mechanisms, providing new insights into the tumor-suppressor role of MIIP.
Keywords: MIIP, miR-181a/b-5p, KLF17, EMT, prostate cancer
Introduction
Prostate cancer (PCa) is the second most frequently diagnosed cancer and the sixth leading cause of cancer-related death among men worldwide [1]. Although many approaches have been established for prostate cancer treatment, including surgery, hormonal therapy, radiation and chemotherapy, there is currently no curative treatment for advanced, metastatic prostate cancer [2]. It is consequently of great importance to clarify the molecular mechanism(s) of prostate cancer metastasis in order to improve the clinical outcomes for these patients.
Cancer metastasis is a complex, multistep process which includes local invasion and migration of tumor cells, intravasation and survival in vascular circulation, extravasation into distant organs, and final survival as secondary foci [3]. Initiation of metastasis requires a well-known program termed epithelial-mesenchymal transition (EMT), which is characterized by decreased expression of epithelial markers such as E-cadherin and increased expression of mesenchymal markers such as vimentin and N-cadherin, as well as phenotypic changes including loss of cell-cell adhesion, loss of cell polarity, and acquisition of migratory and invasive properties [4]. EMT is fundamental for embryonic development and it is involved in several pathological processes, among which tumor progression has been widely appreciated [4]. In addition to invasion, EMT serves as a central driver of tumor malignancy and confers drug resistance as well as stem-cell-like properties [5,6]. Increasing evidence suggests that EMT is correlated with aggressive behavior and therapy-resistance in prostate cancer [7]. However, the molecular mechanism of EMT regulation in PCa is far from clear.
KLF17 (also known as ZNF393) is a member of the Sp/KLF (specificity protein/Krüppel-like factor) zinc-finger protein family that includes 17 members [8]. Using a forward genetic screen, KLF17 was first identified as a negative regulator of EMT and metastasis in breast cancer [9]. Further studies have demonstrated that KLF17 is downregulated and correlated with tumor progression in various human cancers, including breast cancer, hepatocellular carcinoma, gastric cancer, and lung cancer [8,10-12]. Mechanistically, KLF17 was reported to inhibit the transcription of Id1, which encodes a key metastasis regulator in breast cancer, by directly binding to its promoter region [9]. KLF17 also functions as a cofactor of ERα, p53 or Smad3, thereby interfering with corresponding transcription events [11-13]. As such, KLF17 suppresses tumor growth and metastasis to improve outcomes [11-13]. Interestingly, KLF17 is also regulated by the well-known tumor suppressor p53 and TGF-β/Smad3 signaling through a positive feedback mechanism [11,12]. These data suggest that KLF17 serves as an important EMT regulator and a tumor suppressor that intimately interplays with other tumor-suppressive pathways.
Migration and invasion inhibitory protein (MIIP, also known as IIp45) was first identified as a binding partner of insulin-like growth factor binding protein 2 (IGFBP2) and a negative regulator of cell invasion in glioma [14]. The MIIP gene is located on chromosome 1p36.22; one of the most frequently deleted regions in a broad spectrum of human cancers, including breast and pancreatic cancers, glioma and neuroblastoma [15]. The 43 kDa MIIP protein contains 388 amino acids and is highly hydrophilic. Structurally, MIIP is composed of an RGD motif, several potential serine/threonine/tyrosine phosphorylation sites and a four-helical up-and-down bundle [15]. Due to the lack of MIIP deficient mice, the physiological role of MIIP remains elusive. However, there is accumulating evidence that MIIP is downregulated in several types of cancer [15-17]. When overexpressed, MIIP inhibits tumor initiation, growth and/or metastasis in glioma [18,19], lung cancer [17], endometrial cancer [16] and colorectal cancer [20]. Our most recent study demonstrated that MIIP is downregulated in prostate cancer and its overexpression inhibits tumor growth via interaction with PP1α and negative modulation of AKT signaling, which suggested that MIIP also functions as a tumor suppressor in prostate cancer [21], but its roles and underlying mechanism of action is not fully understood.
In this study, we sought to investigate whether MIIP affects EMT and cell invasion/metastasis in prostate cancer. We demonstrated that MIIP inhibits cell invasion of PC3 and DU145 prostate cancer cell lines in vitro. Moreover, using an ectopic xenograft and intratibial injection model in nude mice, we show that a stable MIIP knockdown in PC3 or DU145 cells promoted tumor growth or bone osteolytic lesions in vivo. Mechanistically, MIIP inhibits EMT and cell invasion by negatively regulating the quantity of miR-181a/b-5p, two onco-miRNAs that directly target KLF17, a critical transcriptional repressor of the EMT. Importantly, in a cohort of human patient samples, the expression of MIIP was downregulated compared to adjacent normal tissues in more than 50% of tumor tissue samples, and at least half of these cases showed a correlated increase of miR-181a/b-5p, reduction of KLF17, and elevation of E-cadherin. Immunohistochemical staining of a PCa tissue microarray further showed a positive correlation between MIIP and KLF17. These data provide new insights into the tumor-suppressor role of MIIP.
Materials and methods
Cell lines, antibodies and reagents
HEK-293T cells and human prostate cancer cell lines PC3, DU145 were all from ATCC, USA, and maintained in RPMI-1640 medium (Life Technologies, USA) supplemented with 10% fetal bovine serum and cultured at 37°C in a humidified atmosphere comprising 5% CO2. Antibodies against MIIP, FLAG and vimentin were from Sigma-Aldrich (St Louis, MO, USA); anti-Snail was from Proteintech (Rosemont, USA); antibodies against E-Cadherin, Slug, Fibronectin and S100A4 were from Cell Signaling Technology (Andover, MA, USA); anti-N-Cadherin was from BD Biosciences (Bedford, MA, USA); antibodies against KLF17, Ki67, GAPDH and Actin were from Abcam (Cambridge, MA, USA). RNAiso reagent, SYBR Green universal master mix and Multiscript RT were purchased from TaKaRa Biotechnology Co., Ltd (Dalian, China). The miScript PCR Starter Kit was purchased from Qiagen (Hilden, Germany). The small interfering RNA (siRNA) for MIIP, miR-181a/b-5p mimics, specific inhibitors and the nonspecific control were purchased from GenePharma (Shanghai, China).
Patients’ samples
Freshly dissected prostate tumors and adjacent non-tumor tissues from 21 patients with prostate cancer were obtained from the Department of Urology of Xijing Hospital with informed consent and approval from the Clinical Research Ethics Committee of Xijing Hospital, The Fourth Military Medical University. Tissue microarrays (TMAs) containing 8 cases normal adults samples and 73 cases patients samples [170 points: 30 normal (16 normal prostate tissue plus 14 adjacent normal prostate tissue), 23 Gleason score 3, 71 Gleason score 4, and 46 Gleason score 5] were commercially obtained from Xi’an Alenabio Technology Co., LTD., and the experiments were approved by Research Ethics Committee.
Plasmids construction, lentivirus packaging, cell transfection and infection
The vector pcDNA3.1-MIIP was constructed in a previous study [19]. KLF17 cDNA and corresponding mutant (DNA binding domain deleted variant, ∆DBD) were cloned between the EcoRI and EcoRV sites of the pFLAG-CMV4 vector. Short hairpin RNAs (shRNA) against MIIP were cloned between the EcoRI and AgeI sites of the pLKO.1-TRC lentiviral vector (Carlsbad, CA, USA) and the correctness of the resulting construct confirmed by DNA sequencing. The shRNA oligonucleotides specific for MIIP (MIIP shRNA) are listed in Table 1. Transfection with plasmids DNA or siRNA was performed using Lipofectamine 2000 reagent (Invitrogen; Thermo-Fisher Scientific, USA). Briefly, cells were seeded into multi-well plates or dishes at a density of 60%-70% confluence. Transfection was performed the next day using a final concentration of 20 nM siRNA or 10 µg plasmid DNA/100-mm dish. For virus packaging, HEK-293T cells were transfected with pLKO-Scramble/MIIP-shRNA1/MIIP-shRNA2, together with psPAX2 and pMD2G vectors using Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h, the lentivirus-containing supernatants were collected, filtered (0.45-μm filter; Millipore, Billerica, MA, USA), and added into the culture medium of DU145 or PC3 cells. Stable MIIP-knockdown cell lines were obtained using puromycin (1 μg/mL; Sigma-Aldrich, USA) selection for 2 weeks.
Table 1.
Oligos used in MIIP specific-shRNA vector construction
| MIIP-shRNA1 |
| Forward: CCGGGTGTACTGTTACCGTGTCAACCTCGAGGTTGACACGGTAACAGTACACTTTTTG |
| Reverse: AATTCAAAAAGTGTACTGTTACCGTGTCAACCTCGAGGTTGACACGGTAACAGTACAC |
| MIIP-shRNA2 |
| Forward: CCGGCGTGGAGGAAGACCATGAATGCTCGAGCATTCATGGTCTTCCTCCACGTTTTTG |
| Reverse: AATTCAAAAAGTGTACTGTTACCGTGTCAACCTCGAGGTTGACACGGTAACAGTACAC |
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted using RNAiso reagent (TaKaRa, Dalian, China) according to the manufacturer’s protocol. The first strand cDNA was generated from total RNA (2 μg) with reverse transcriptase for coding region genes or non-coding regions, and used as the template for qRT-PCR analysis. GAPDH cDNA and U6 were used as internal controls. The primers used are listed in Table 2. PCR was performed in a GeneAmp PCR system 2400 Thermal Cycler (Perkin-Elmer, Norwalk CT, USA). The PCR temperature program was composed of 40 cycles comprising 30 s at 95°C, 10 s at 95°C and 30 s at 60°C.
Table 2.
Genes name and primers used in q-PCR
| MIIP: |
| Forward: 5’-ATACCTGGGCTATGACTGGATT-3’ |
| Reverse: 5’-AGTACACGCATTCATGGTCTTC-3’ |
| KLF17: |
| Forward: 5’-GGGGCCACAGTTCAGTATGC-3’ |
| Reverse: 5’-TCTCTGACAGTAAATCATCCGGG-3’ |
| SNAI1 (encoding Snail): |
| Forward: 5’-CACTATGCCGCGCTCTTTC-3’ |
| Reverse: 5’-GGTCGTAGGGCTGCTGGAA-3’ |
| SNAI2 (encoding Slug): |
| Forward: 5’-TGTGACAAGGAATATGTGAGCC-3’ |
| Reverse: 5’-TGAGCCCTCAGATTTGACCTG-3’ |
| TWIST1 (encoding Twist): |
| Forward: 5’-GACGAGCTGGACTCCAAGATGGCA-3’ |
| Reverse: 5’-ATCCTCCAGACCGAGAAGGCGTA-3’ |
| CDH1 (encoding E-cadherin): |
| Forward: 5’-TGCCCAGAAAATGAAAAAGG-3’ |
| Reverse: 5’- GTGTATGTGGCAATGCGTTC-3’ |
| CDH2 (encoding N-cadherin): |
| Forward: 5’-ACAGTGGCCACCTACAAAGG-3’ |
| Reverse: 5’-CCGAGATGGGGTTGATAATG-3’ |
| VIM (encoding Vimenin): |
| Forward: 5’-GACGCCATCAACACCGAGTT-3’ |
| Reverse: 5’-CTTTGTCGTTGGTTAGCTGGT-3’ |
| GAPDH: |
| Forward: 5’-CTGCACCACCAACTGCTTAG-3’ |
| Reverse: 5’-TTCTGGGTGGCAGTGATG-3’ |
| miR-181a-5p: |
| 5’-AACATTCAACGCTGTCGGTGAGT-3’ |
| miR-181b-5p: |
| 5’-AACATTCATTGCTGTCGGTGGGT-3’ |
Western blotting
Total protein was extracted either from cultured cells or tumor samples using RIPA buffer and quantified using a commercial BCA kit (Beyotime Biotechnology, Jiangsu, China). The protein samples were resolved by SDS-PAGE on 8 to 15% polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked and then probed with the indicated primary antibodies and corresponding secondary antibodies, and washed with TBST buffer (PH 8.0), then developed using the enhanced chemiluminescence kit (Tanon, Shanghai, China).
Cell proliferation and invasion assays
For the cell proliferation assay, 96-well plates were seeded with 2000 cells per well in septuple twenty-four hours after transfection. Viability was assessed using the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) to obtain a growth curve of metabolically active cells measured as absorbance at 490 nm.
Cell invasion assays were performed in matrigel-coated transwell chambers (8-μm pore size, BD Pharmingen, USA). Cells in 0.1% FBS medium were seeded at 2 × 104 per upper chamber, and then placed into 24-well tissue culture plates containing 10% FBS medium. After 24 h, invasive cells were stained with Giemsa and analyzed with microscope. The number of invading cells was determined by counting ten high-power fields (× 400) on each membrane and calculated as the mean number of cells per field. The data were presented as the mean ± SD.
All the above experiments were repeated at least three times independently.
Immunofluorescence
5 × 103 PC3 cells were plated on sterile glass coverslips, and pcDNA3.1-MIIP was transfected and incubated for 48 h. Then, the cells were fixed with 4% paraformaldehyde, followed by permeabilization with 0.2% Triton X-100 for 10 min at room temperature. The slips were blocked with 3% BSA for 1 h at room temperature and then incubated with anti-E-Cadherin (Cell Signaling, Andover, MA, USA), anti-N-Cadherin (BD Biosciences, Bedford, MA, USA) and anti-Vimentin (Sigma-Aldrich, St Louis, MO, USA) antibodies at room temperature for 2 h. After washing the slips with PBS, secondary antibody was incubated in the dark for 1 h at room temperature and then washed with PBS; the coverslips were counterstained with DAPI (0.1 mg/mL; Molecular Probes) and imaged with confocal microscope.
Luciferase reporter assay
The DNA fragments of -2000 to +100 bp (upstream 2000 bp from the transcription initiation site; containing KLF17 consensus binding sites), deletion mutant (binding sites deleted) of the human SNAI1, SNAI2 and TWIST promoter region were cloned into the pGL3-Enhancer vector (Promega, USA). HEK-293 cells were co-transfected with human KLF17 cDNA, pGL3 reporter or deletion mutant and Renilla luciferase (0.2 ng; pRL-TK) as a normalizing control. Luciferase activity was determined using Dual-Luciferase Reporter Assay (Promega, USA) 48 h after transfection, according to the manufacturer’s instructions.
The 3’-UTR of KLF17 that was predicted to interact with miR-181a/b-5p was amplified from human genomic DNA and cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, USA). The wild-type and mutant inserts were sequenced to verify the mutations. HEK-293 cells were harvested 48 h after co-transfection of miRNA with the reporter vector and assessed using the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s protocol.
In vivo tumor xenograft study
All procedures involving animals were approved by, and in accordance with, the ethical standards of the Institutional Animal Care and Use Committee of The Fourth Military Medical University. Four-to six-week-old athymic mice were injected with prostate cancer cells which were infected with either the MIIP shRNA or the scramble control shRNA (shRNA1, shRNA2 or Scramble; in DU145 or PC3) at bilateral axillae. Tumor growth was monitored by measuring the tumor size using a vernier caliper every 5 days for a 1-month period and calculating tumor volume using the standard formula: tumor volume (mm3) = width2 (mm2) × length (mm) × 0.5. The mice were sacrificed 30 days after cell injection, the tumors were dissected and weights were recorded. Portions of tumors were formalin-fixed, paraffin-embedded, sectioned and mounted on slides for immunostaining.
Intratibial injection, bioluminescence imaging and X-ray examination
Two groups of 7 male athymic mice each were used and anesthetized with inhaled 3% isoflurane. Luciferase-expressing PC3-Scramble and PC3-MIIP shRNA cells were injected into the right tibia medullary cavity of the two groups at a concentration of 5 × 105/20 μL of 10 μL PBS and 10 μL Matrigel (BD, USA). A 21-G syringe was used to drill a hole though the tuberosity of tibia and cells were injected though 29-G insulin syringe. Injection was done very slowly to prevent cells entering the soft tissue. No incision was made.
For bioluminescence imaging and analysis. Mice were anesthetized with 3% isoflurane every 7 days monitor the tumor status. D-Luciferin (Xenogen) was injected at 150 mg/kg (body weight). Five minutes later, bioluminescent images were acquired with an IVIS imaging system (Xenogen). Analysis was performed by using LivingImage software (Xenogen) by measuring the photon flux within a region of interest drawn around the bioluminescence signals. Blank regions of interest were also measured for each scan and deducted from each tumor photon flux to normalize.
Mice were euthanized when they showed signs of morbidity, and the right legs were harvested, osteolytic lesions were identified on radiographs. The semiquantitation scoring method was formulated as: 0 = no lesions, 1 = minor changes, 2 = small lesions, 3 = significant lesions (minor peripheral margin breaks, 1-10% of bone surface disrupted), 4 = significant lesions (major peripheral margin breaks. >10% of bone surface disrupted).
Immunohistochemistry
Xenograft tumor sections were dewaxed and rehydrated in a series of graded alcohol solutions and then subjected to antigen retrieval by high pressure cooking for 2 min in 0.01 M sodium citrate buffer, pH 6.0. A rabbit antibody against human Ki67 (Abcam, Cambridge, MA, USA) was used at a dilution of 1:50. Staining specificity was confirmed by substitution with normal rabbit IgG as a negative control. Antibody binding was visualized by a horseradish peroxidase-conjugated secondary antibody system using 3,3’-diaminobenzidine (Sigma, St Louis, MO, USA) as substrate.
For TMA immunohistochemistry staining, the primary antibodies anti-MIIP (1:200) and anti-KLF17 (1:200) were used for staining. The results were obtained by digital slice scanner (3DHISTECH, Hungary). Protein expression was analyzed by immunoreactivity scored. The total score = cell score × color score. The cell score standard used the number of cells with positive staining (≤5%: 0, 6-25%: 1, 25-50%: 2, 51-75%: 3, ≥75%: 4). The color score standard used the staining intensity (colorless: 0, mild: 1, moderate: 2, strong: 3).
Statistical analysis
Data were plotted with GraphPad Prism v5.0 and Excel, and presented as the means ± SD from three independent experiments. Statistical analysis was performed using the SPSS14.0 software package for Windows (IBM Corp., USA) using Student’s t-test for independent groups. Pearson’s correlation tests were used to evaluate the pairwise expression correlation between MIIP and KLF17 in PCa tissues. Statistical significance was based on a threshold value of P≤0.05.
Results
MIIP inhibits EMT factors and cell invasion of prostate cancer cell lines by KLF17
Our previous study have shown that MIIP functions as a tumor suppressor in prostate cancers via inhibiting cell proliferation [21], here we sought to investigate the potential role of MIIP in prostate cancer invasion. Upon overexpressing or silencing MIIP in DU145 and PC3 prostate cancer cell lines, the transwell invasion assay were performed. As shown in Figure 1A, MIIP overexpression significantly reduced, whereas MIIP knockdown increased the number of invasive cells in both cell lines, suggesting that MIIP inhibits cell invasion. It has been well documented that EMT is the primary driving force of cell invasion and cancer metastasis [5,6], so we wondered if MIIP is involved in the regulation of EMT-related factors. We examined epithelial and mesenchymal markers and well-known transcriptional factors that regulate the EMT process. Strikingly, MIIP overexpression in PC3 and DU145 cells increased the protein levels of the epithelial marker E-cadherin while decreased the mesenchymal markers N-cadherin, vimentin and EMT transcriptional factors, such as Slug and Snail, whereas MIIP silence had the opposite effect (Figure 1B). Consistently, q-PCR and immunofluorescence assay in MIIP-overexpressing or silencing DU145 cells showed a similar result (Figure 1C, 1D). In addition, through gene set enrichment analysis (GSEA) of the RNA-seq data of PCa cell line C4-2, which were previously conducted with stable MIIP-overexpression C4-2 v.s. the control cells, we identified that epithelial-to-mesenchyma-transition as one of gene sets that are strongly associated with MIIP expression (Figure S1). These data corroborated that MIIP represses EMT, accounting for the inhibition of PCa cell invasion.
Figure 1.

MIIP inhibits EMT factors and cell invasion of prostate cancer cells by KLF17. A. MIIP-encoding plasmid, MIIP siRNAs or their corresponding controls were transiently transfected into the prostate cancer cell lines PC3 and DU145, and invasive ability of the cells was evaluated by a transwell invasion assay. B. Western blot analysis was performed to examine MIIP, KLF17, Snail, Slug, E-cadherin, N-cadherin, and vimentin in MIIP overexpressing and silencing PCa cell lines, respectively. C. Total RNA was isolated from DU145 cells transfected with MIIP siRNAs and relative mRNA levels of MIIP, KLF17, SNAI1, SNAI2, TWIST, CDH1, CDH2, and VIM were measured by quantitative real-time PCR. D. MIIP-encoding plasmid and empty vector were transiently transfected into DU145. EMT markers were examined by imunofluorescence assay. (Scale bar, 20 μm (× 80 objective magnification)). E. DU145 cells were transfected with MIIP-encoding plasmid, together with or without KLF17-specific siRNAs. Expression of MIIP, KLF17 and E-cadherin were examined by Western blot analysis. F. PC3 and DU145 cells were transfected with MIIP-encoding plasmid, together with or without KLF17-specific siRNAs. Cell invasive ability was evaluated by a transwell invasion assay. All data are shown as mean ± SD. *P<0.05, **P<0.01, ***P<0.001.
Having examined the differential expression of genes in MIIP-overexpressing versus control cells, we noticed that Krueppel-like factor 17 (KLF17), a newly identified regulator of the EMT [9], was upregulated significantly by MIIP overexpression (data not shown). To validate this, we examined KLF17 alteration upon MIIP manipulation. Obviously, KLF17 was upregulated in both PC3 and DU145 cells when MIIP was overexpressed, while downregulated in MIIP knocked down cells (Figure 1B, 1C). Next, to determine whether KLF17 participates in and is required for MIIP-mediated regulation of invasion and EMT-related factors, MIIP-overexpressing DU145 cells were further transfected with KLF17-specific siRNAs to examine EMT markers and cell invasion. Notably, the MIIP-caused increase of E-cadherin was markedly abrogated by three KLF17-specific siRNAs (Figure 1E). Moreover, KLF17 silencing significantly reversed the inhibitory effect of MIIP on cell invasion (Figure 1F). Taken together, these results suggest MIIP functions as a negative regulator of EMT and cell invasion by KLF17 in prostate cancer.
A stable MIIP-knockdown promoted tumor progression and bone metastatic lesion in vivo
Consistent with our most recent study [21], MIIP inhibits the proliferation of prostate cancer cells in vitro (Figure S2A, S2B). To gain deeper insights into MIIP’s role in prostate cancer, stable MIIP-knockdown cell lines derived from DU145 and PC3 cells were established. Notably, the two stable MIIP-knockdown cell lines (MIIP shRNA1, MIIP shRNA2) which had markedly reduced levels of MIIP (Figure 2A), exhibited more mesenchymal morphology and grew in a more dispersed fashion than the scramble-RNA control cells (Figure 2B). The results of western blotting and real-time PCR showed that the stable knockdown of MIIP resulted in similar alterations of the levels of KLF17, EMT markers and SNAI1, as had been observed after transient knockdown (Figure 2A, 2C). Whereafter, we injected these stable cells into the bilateral axillae of nude mice to monitor and compare their growth in vivo. The tumors derived from stable MIIP-knockdown DU145 and PC3 cells grew faster than the corresponding scramble-RNA control tumors (Figure 2D). Accordingly, the average tumor weight from each MIIP-knockdown group was significantly heavier than that of the controls (Figure 2E). Tumor samples were then subjected to western blot analysis of MIIP and KLF17 expression (Figure 2F), and immunostaining of Ki67 (Figure 2G). Clearly, the tumors derived from the stable MIIP-knockdown cells revealed lower MIIP and KLF17 expression and higher Ki67 expression in comparison to the control group. Taken together, this xenograft model provides further evidence that MIIP-knockdown promotes a more mesenchymal phenotype and accelerates prostate tumor progression.
Figure 2.

A stable MIIP-knockdown promoted tumor progression in vivo. (A-C) Two stable MIIP-knockdown (MIIP shRNA1 or MIIP shRNA2) or scramble control cells of DU145 and PC3 were generated. levels of MIIP, KLF17 and EMT markers were examined in protein level by Western blot (A), cell morphology is shown (B, × 10 objective magnification) and mRNA level of MIIP, KLF17 and EMT markers were determined by quantitative real-time PCR (C). (D, E) Stable MIIP-knockdown or scramble cells were injected into nude mice at bilateral axillae. Tumor growth was monitored (D) and the tumors were removed and weighed at the end of the experiment (E). (F) Protein extracted from xenograft tumor tissues were subjected to Western blot analysis for MIIP and KLF17. (G) Tumor tissue sections were subjected to immunostaining for Ki67 (brown), (× 20 objective magnification). All data are shown as mean ± SD.*P<0.05, **P<0.01.
Because bone metastasis is the major outcome for advanced PCa, we further utilized orthotopic intra-tibial implantation model to assess the effect of MIIP on bone metastatic growth of PCa in vivo. To this end, luciferase-expressing, scramble or MIIP-shRNA1 (hereafter referred to as MIIP-shRNA) PC3 cells were generated and inoculated into the tibia of the nude mice to monitor progression of bone metastatic tumors and osteolytic lesions. As shown in Figure 3A and 3B, the MIIP-silenced PC3 cells displayed greater tumor burden in bone compared with the PC3-Scramble control group by bioluminescence imaging (BLI). In addition, X-ray examination of the tibias of injected mice at the end of the experiment demonstrated that MIIP knocked-down cells exhibited larger osteolytic area of tumors than scramble control did (Figure 3C and 3D). Collectively, these in vivo findings demonstrate that knockdown of MIIP promotes the tumor burden and osteolytic lesions of PCa in bone.
Figure 3.

MIIP-knockdown promoted tumor bone metastatic lesion in vivo. A. Representative BLIs signal of bone metastasis of a mouse inoculated with luciferase-expressing, PC3-Scramble or PC3-MIIP shRNA cells via intra-tibial injection. B. Quantification of the BLI signaling in the Scramble (n=7) and MIIP-shRNA (n=7) groups at 21 and 35 day respectively. *P<0.05. C. Representative radiographic images of bone metastases in the indicated mice at 42 day (arrows indicate osteolytic lesions). D. The quantification of radiographic score in tumor-bearing mice inoculated via tibia with Scramble or MIIP shRNA cells. *P<0.05.
miRNA-181a/b-5p is suppressed by MIIP, and directly targets the 3’UTR of KLF17 in prostate cancer
Next, we explored how MIIP promotes KLF17 expression. Since MIIP dramatically influenced KLF17 mRNA level, we examined whether MIIP promotes its transcription by luciferase reporter assay. The promoter region of human KLF17 was cloned upstream of a luciferase gene in pGL3-basic reporter vector and transfected together with pcDNA3.1-MIIP into HEK293 cells. MIIP expression did not change the luciferase signal from the reporter (Figure 4A), suggesting that MIIP is not involved in the regulation of KLF17 at the transcriptional level directly. We then asked if MIIP participates in the regulation of KLF17 through microRNAs, thus we screened the overlapping candidate microRNAs predicted to target KLF17 based on three miRNA databases (miRBase, TargetScan, miRWalk) (Figure 4B). To determine whether these miRNAs were regulated by MIIP, qRT-PCR analysis in MIIP-overexpressing and -silencing DU145 cell lines was conducted. As a result, miR-181a/b-5p were identified according to their consistent alterations caused by MIIP overexpression and knockdown (Figure 4C). Moreover, miR-181a/b-5p has been documented as onco-miRs in many types of cancers [22-24], and their expression were both positively correlated with prostate cancer progression and metastasis according to dbDEMC 2.0 (database of Differentially Expressed MiRNAs in human Cancers, www.picb.ac.cn/dbDEMC/) database (Figure S3). Importantly, ectopic expression of either miR-181a-5p or miR-181b-5p mimics downregulated KLF17 expression at mRNA levels, while the antisense oligonucleotide (AS) had the opposite effect (Figure 4D).
Figure 4.
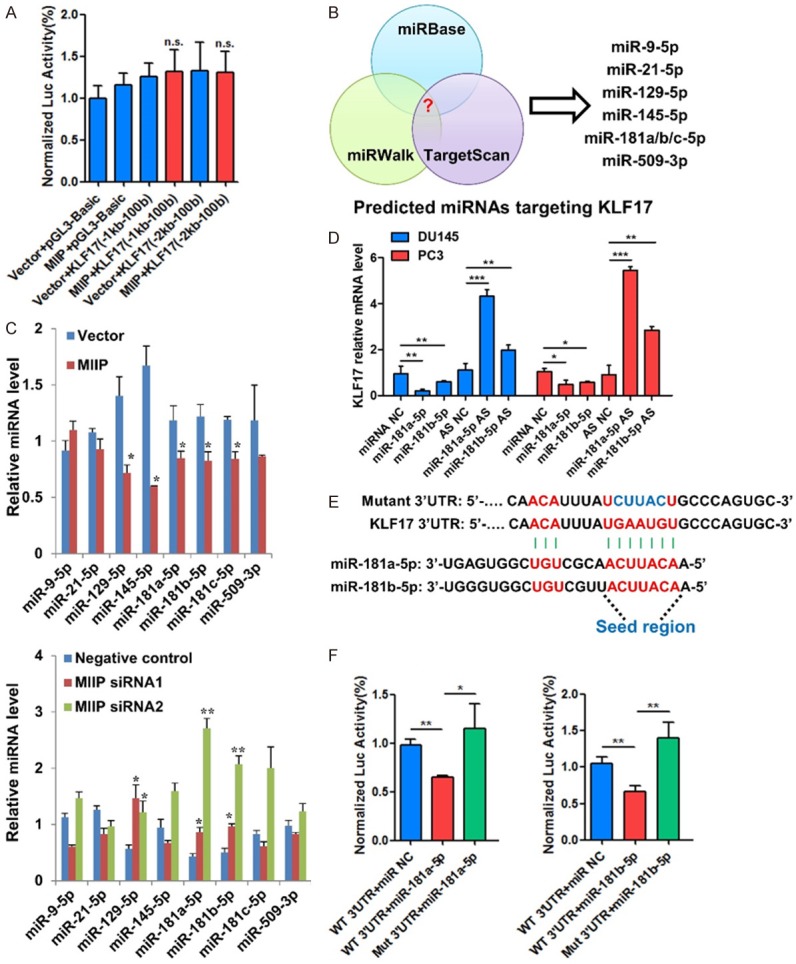
miRNA-181a/b-5p is suppressed by MIIP, and directly targets the 3’UTR of KLF17 in prostate cancer. A. Relative luciferase activity of the reporter constructs harboring KLF17 promoter with different length upon co-transfection with empty vector or MIIP expressing plasmid. B. Schematic diagram of screening of miRNAs that target KLF17. Potential miRNAs were predicted by integrating the results of three algorithms (Targetscan, microRNA and miRanda). C. Candidate miRNAs regulated by MIIP were identified with qPCR in MIIP overexpression and knockdown PCa cells. D. KLF17 mRNA levels in DU145 and PC3 cells transfected with miR-181a/b-5p mimics or corresponding antisense oligonucleotide (AS) were examined by qPCR. E. Predicted miR-181a/b-5p binding sites on the 3’UTR of KLF17 and the mutant 3’UTR. F. Relative luciferase activity of the reporter constructs harboring WT or mutated 3’UTR of KLF17 upon co-transfection with miR-NC or miR-181a/b-5p. All data are shown as mean ± SD. *P<0.05, **P<0.01.
To further determine whether miR-181a/b-5p directly targets the 3’UTR of KLF17, a luciferase reporter assay was performed. The binding sites for miR-181a/b-5p present in the 3’UTR of KLF17 were shown and corresponding mutant sequences were designed (Figure 4E). The reporter assay showed that miR-181a/b-5p significantly repressed the luciferase expression of reporter vectors harboring wild-type but not mutant 3’UTRs of KLF17 (Figure 4F). Overall, these data suggested that miR-181a/b-5p serve as the mediators for downregulation of KLF17 by MIIP, through directly targeting the 3’UTR of KLF17 and causes its downregulation.
KLF17 inhibits the transcription of EMT factors
KLF17 belongs to KLF family, which comprises transcription factors with diverse functions [25]. KLF family members can function as either activators or repressors, depending on the promoters they bind to and the cellular proteins they interact with [26]. It has been shown that KLF17 can bind to the DNA consensus sequence (5’-CACCC-3’) of its target genes to either transactivate or repress gene transcription [25]. Snail, Slug, Twist and Zeb1 are critical transcriptional factors for EMT, and we found several potential binding sites for KLF17 in the promoter region of SNAI1, SNAI2 and TWIST (Figure 5A), but no binding sites in ZEB1 on this region. To address whether MIIP directly regulates the transcription of these genes, we cloned the promoter region (-2000~100 bp) of human SNAI1, SNAI2 and TWIST upstream of a luciferase gene in reporter plasmids pGL3-basic (Figure 5A), and transfected each of them together with the pFLAG-CMV4-KLF17 into HEK293 cells. The result of luciferase reporter assay showed that KLF17 expression suppressed the luciferase signal driven by the SNAI1, SNAI2 or TWIST promoter (Figure 5B), nevertheless, deletion of the potential KLF17 binding sites (Mut promoter) relieved the transcriptional suppression caused by KLF17 (Figure 5B). However, KLF17 with DNA binding domain deleted variant (∆DBD) failed to restrain the luciferase signal (Figure 5B). Additionally, for ZEB1, with no potential binding sites of KLF17 on the established promoter region (-2000~100 bp), the luciferase signal had no changes even in the presence of wild type KLF17 (Figure 5B). Hence, KLF17 can directly suppress SNAI1, SNAI2 and TWIST transcription and requires CACCC binding sites.
Figure 5.
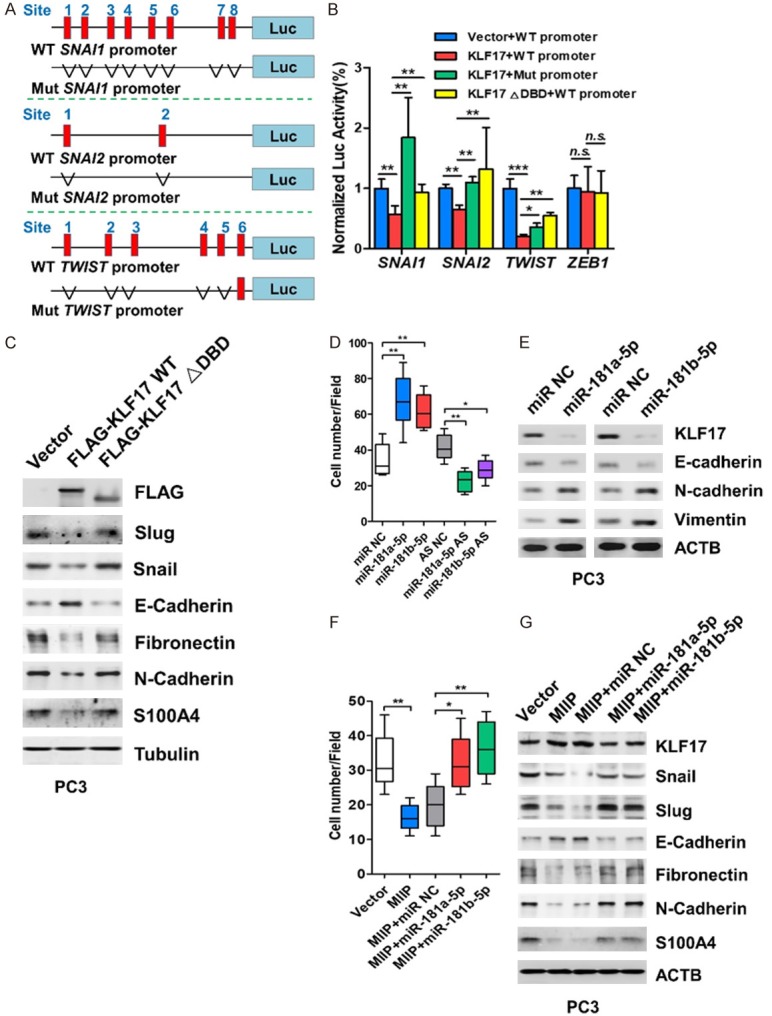
KLF17 inhibits the transcription of EMT factors and MIIP exerts its invasion-inhibitory role through repressing miRNA-181a/b-5p. (A) Luciferase (Luc) reporter constructs contain the SNAI1, SNAI2 and TWIST promoter respectively with potential KLF17 binding sites upstream of a luciferase gene, or the promoter with the deletion of potential binding sites. Red indicates the potential binding sites (SNAI1: site 1, -1502 to -1498, site 2, -1442 to-1438, site 3, -1298 to -1294, site 4, -1231 to -1227, site 5, -962 to -958, site 6, -859 to -855, site 7, -73 to -69, site 8, -63 to -59); (SNAI2: site 1, -1675 to -1671, site 2, -1803 to -1799); (TWIST: site 1, -1588 to -1584, site 2, -1333 to -1329, site 3, -1139 to -1135, site 4, -487 to -483, site 5, -472 to -468, site 6, -402 to -398). (B) Relative luciferase activity of the reporter constructs harboring WT or mutated (5’-CACCC-3’ deletion) promoter upon co-transfection with empty vector or KLF17 expressing plasmid. (C) PC3 cell were transfected with WT or mutant KLF17 constructs and subjected to Western blot analysis for FLAG, Snail, Slug, E-cadherin, N-cadherin, Fibronectin and S100A4. (D, E) miR-181a/b-5p, miRNA antisense oligonucleotide (AS), or their corresponding controls were transiently transfected into PC3 cells. Cell invasion were evaluated by transwell invasion assay (D), and KLF17 and EMT biomarkers were analyzed by Western blot (E). (F, G) MIIP-encoding plasmid or miR-181a/b-5p were transiently transfected into PC3 cells, and cell invasion was evaluated by transwell invasion assay (F). KLF17 EMT factors and biomarkers were analyzed by Western blot (G). All data are shown as mean ± SD. *P<0.05, **P<0.01, ***P<0.001.
To further confirm such regulation between KLF17 and EMT factors, we transfected pFLAG-CMV4-KLF17 into PC3 cells. The results of Western blotting clearly showed that overexpression of KLF17 led to obvious decrease of Snail and Slug in protein levels, which was accompanied by alterations in E-cadherin, N-cadherin, Fibronectin and S100A4 (Figure 5C), whereas overexpression of KLF17 ∆DBD variant was unable to cause such alterations (Figure 5C). Together, these data imply that KLF17 is able to suppress the transcription of EMT factors and EMT.
MIIP exerts its invasion-inhibitory role through repressing miRNA-181a/b-5p
To determine the functions of miR-181a/b-5p in prostate cancer, PC3 cells were transfected with miR-181a/b-5p mimics or AS and cell invasion and proliferation were assessed. The results revealed that miR-181a/b-5p mimics upregulated miR-181a/b-5p levels (Figure S4A) and enhanced cell invasion and proliferation whereas miR-181a/b-5p AS had the opposite effect (Figures 5D and S4B). Meanwhile, the expression of KLF17 and E-cadherin was remarkably decreased but N-cadherin and vimentin were increased following miR-181a/b-5p overexpression (Figure 5E). These data suggest that miR-181a/b-5p promote PCa cell proliferation, invasion and EMT.
We then asked whether miR-181a/b-5p is required for MIIP-mediated inhibition of cell invasion and EMT. So miR-181a/b-5p were further overexpressed in MIIP-overexpressing PC3 cells. We found that MIIP-caused repression of cell invasion and proliferation were abrogated by miR-181a/b-5p co-expression (Figures 5F and S4C). In addition, miR-181a/b-5p reversed the MIIP-induced upregulation of KLF17 and E-cadherin and downregulation of Snail, Slug, N-cadherin, and fibronectin (Figures 5G and S5). Thus, MIIP’s inhibitory effect on cell invasion relies on downregulating oncogenic miR-181a/b-5p. Taken together, these data suggested that MIIP exerts its tumor suppressor role in PCa cell invasion and EMT via the miR-181a/b-5p/KLF17 axis.
The expression of MIIP is downregulated in prostate cancer tissues and positively correlated with the expression of KLF17
To determine the clinical significance of MIIP in PCa, we investigated MIIP protein levels in a series of paired samples, comparing tumors and adjacent normal tissues from prostate cancer patients. Downregulation of the MIIP protein levels was detected in more than half (11/21, Sample#1-8, 10-12) of the tumor samples when compared to the corresponding normal tissues (Figures 6A and S6). Furthermore, among these 11 pairs of tissues, the level of miR-181a-5p and miR-181b-5p was dramatically enhanced in most of tumor samples compared to normal samples (Figure 6B), implying that there was an inverse correlation between MIIP and miR-181a/b-5p in prostate cancer patients. Importantly, in 7 pairs of tissues in which MIIP was clearly downregulated in the cancer, the expression of both KLF17 and E-cadherin were also reduced (Figure 6A). Additionally, we analyzed the expression of MIIP and KLF17 in tissue microarrays (TMAs) containing 73 cases PCa patients [170 points: 30 normal (16 normal prostate tissue plus 14 adjacent normal prostate tissue), 23 Gleason score 3, 71 Gleason score 4, and 46 Gleason score 5] by immunohistochemistry (IHC). The data showed that the expression of MIIP decreased significantly along with the increase of PCa Gleason score (Figure 6C), which is in consistent with our recent study [21]. KLF17 expression was also reduced as the Gleason score increased (Figures 6C and S7). Of note, there was a significant correlation between MIIP and KLF17 expression in PCa tissues (Figure 6D). These data clearly demonstrate the clinical relevance of the MIIP/miR-181a/b-5p/KLF17/EMT regulatory axis in prostate cancer. Taken together, these results demonstrated that MIIP inhibits EMT and cell invasion in prostate cancer through miR-181a/b-5p-KLF17 axis, and loss of MIIP may contribute to PCa EMT and metastasis (Figure 7).
Figure 6.
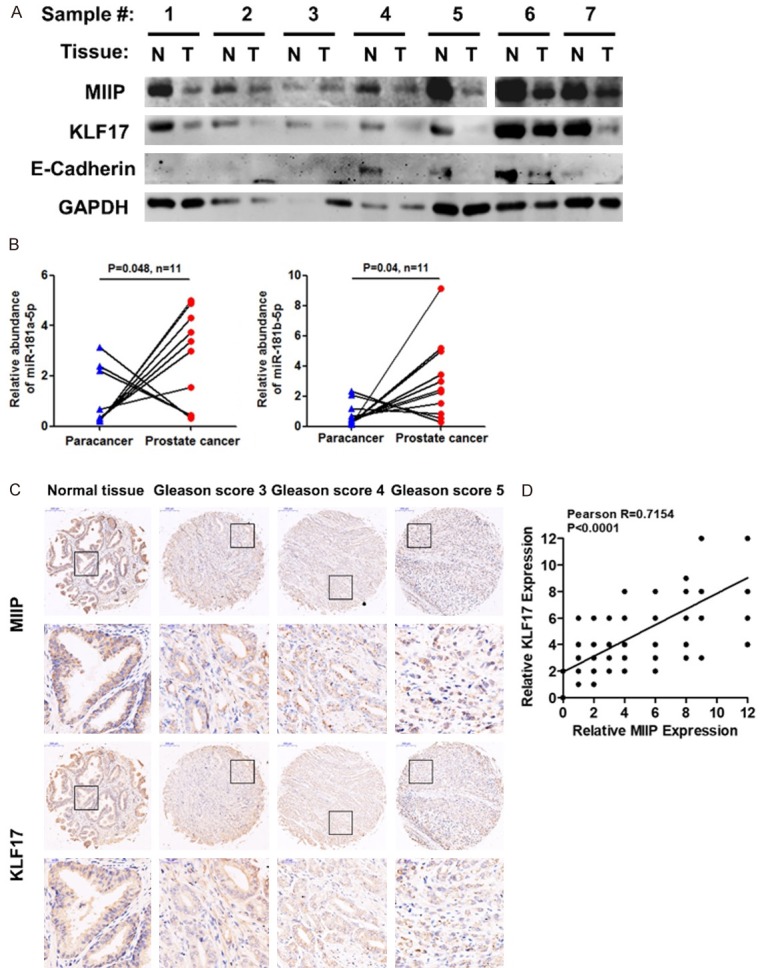
The expression of MIIP is downregulated in prostate cancer tissues and positively correlated with the expression of KLF17. A. MIIP, KLF17 and E-cadherin protein levels were detected by Western blot in 7 paired prostate cancer samples from human patients; tumor and adjacent normal tissue. N, normal tissue; T, tumor tissue. B. miR-181a/b-5p expression was enhanced in clinic prostate cancer samples (n = 11) compared with paired adjacent normal tissues; two-tailed Student’s t-test. C, D. MIIP and KLF17 expression was evaluated by immunohistochemical staining on two independent TMAs composed of normal and adjacent normal prostate tissues, and PCa tissues of Gleason score 3, 4 and 5 grades. D. The correlation between MIIP and KLF17 expression level in normal, adjacent normal prostate tissues, and PCa tissues, Pearson R = 0.7154. All data are shown as mean ± SD.
Figure 7.

Schematic diagram depicting the regulatory mode of MIIP in prostate cancer EMT via miR-181a/b-5p inhibition and KLF17 acceleration.
Discussion
Although there has been some debate on the contribution of the EMT to metastatic colonization and outgrowth, EMT has recently been identified as a crucial driver of cancer malignancy and progression [5,6]. EMT is a plastic and dynamic process during which cells exhibit a spectrum of phenotypic states encompassing fully epithelial, intermediate and fully mesenchymal characteristics [5,6]. The activation of EMT relies on the expression and activation of a group of EMT-inducing transcription factors -Zeb1/Zeb2, Snail/Slug, Twist etc., which control epithelial and mesenchymal genes [27]. Such a core EMT transcriptional program is further subjected to multiple layers of modulation, including transcriptional, epigenetic and post-transcriptional regulation [6]. Not surprisingly, oncogenes and tumor suppressors are closely implicated at almost all levels of EMT regulation. Here we for the first time demonstrate that MIIP, a newly identified tumor suppressor, functions as a critical modulator of the EMT in prostate cancer, thus inhibiting prostate cancer malignancy in vitro and in vivo.
Through the overexpression and knock-down of MIIP levels in prostate cancer cells, we clearly demonstrated that MIIP enhances the expression of the epithelial marker E-cadherin, but inhibits the mesenchymal marker N-cadherin and EMT-inducing transcriptional factors (Snail, Slug and Twist). As a result, MIIP inhibits invasion of prostate cancer cells, which is consistent with its role in other types of cancer [14,16,18,20]. MIIP was previously reported to reduce invasion by interacting and antagonizing IGFBP-2-stimulated signaling in glioma [14]. MIIP also binds to HDAC6 and decreases its stability, leading to increased acetylation of α-tubulin and thus reduced cell migration [18]. Most recently, MIIP was reported to block the Rac1 signaling pathway by competitively binding to Rac1’s downstream effector PAK1, thereby inhibiting the formation of lamellipodia and migration of endometrial cancer cells [16]. Here, we present data which imply that MIIP at least partially depends on KLF17 to inhibit EMT-related factors and prostate cancer cell invasion. KLF17 levels were dramatically altered upon MIIP manipulation, and KLF17 silencing to a large extent abrogated the effect of MIIP on EMT and cell invasion. Moreover, miR-181a/b-5p are two critical node molecules between MIIP and KLF17. Ectopic expression of these miRs rescued the effect of MIIP on alterations of KLF17 and EMT-related factors and inhibition of cell invasion. Therefore, MIIP functions as a tumor suppressor in prostate cancer via the downregulation of the oncogenic miR-181a/b-5p, leading to KLF17 elevation, alterations of EMT-related factors and cell invasion inhibition (Figure 7). Together with the published literature, this study corroborates that MIIP utilizes different molecular targets and mechanisms to function as a universal migration and invasion inhibitory protein.
Increasing numbers of studies have indicated that miR-181a/b-5p are deregulated in diverse types of cancers and promote tumor progression by targeting crucial molecules [28-31]. For example, miR-181a-5p promotes cell proliferation, invasion and induces EMT by activating the MAPK signaling pathway by directly inhibiting the expression of RASSF6 in gastric cancer [32]. However, the function of miR-181a/b-5p in prostate cancers has, to our best knowledge, not been reported so far. Our data reveal for the first time that miR-181a/b-5p serves as oncomiRs, accelerating EMT and cell invasion in PCa. In addition, although there is a report showing that KLF17 acts as an EMT inducer via direct activation of TWIST1 in endometrioid endometrial cancer [33], the majority of evidence suggests that KLF17 functions as a tumor suppressor and EMT inhibitor [8]. KLF17 can exert its anti-EMT effect by repressing the transcription of Id1, an inducer of EMT and metastasis in breast cancer [9]. KLF17 also physically binds to the promoters of EMT target genes such as vimentin, fibronectin and ZO-1, and interestingly, p53 enhances such association by interacting with KLF17 [11]. Notably, KLF17 can be transcriptionally regulated by p53 and the TGF-beta/Smad3 pathway [11,12]. KLF17 is also crucial for and enhances TGF-beta/Smad signaling in human cancers [12]. Here we demonstrate that KLF17 directly inhibit the transcription of EMT key regulators via CACCC sequence within their promoters in prostate cancer for the first time. Moreover, KLF17 is controlled by MIIP via miRNAs and also serves as an important mediator of MIIP’s role in cell invasion in prostate cancer. Notably, the clinical relevance of the proposed MIIP-miR181-5a/b-5p-KLF17-EMT axis was further confirmed in more than half of prostate cancer tissues we tested. Our data, together with other studies [11,12], suggests a complex interplay and reciprocal regulation across tumor suppressors. However, the mechanism through which MIIP regulates miR-181a/b-5p, and targets of these miRNAs other than KLF17, as well as other downstream effectors of KLF17 that may play roles in EMT and invasion, remains to be investigated.
In addition to the inhibition of migration and invasion, MIIP is implicated in other cellular behaviors. Through direct interaction with Cdc20 and by blocking APC/CCdc20-mediated degradation of cyclinB1 and securin, MIIP delays the mitotic transition and thus tumor initiation/progression of glioma and colon cancer [19,20]. MIIP also associates with and modulates the activity of topoisomerase II, so that MIIP haploinsufficiency alone can induce chromosomal instability and tumor progression in colorectal cancer [20]. By accelerating EGFR turnover, MIIP inhibits cell proliferation in non-small cell lung cancer [17]. Most recently, MIIP was shown to inhibit tumorigenesis of pancreatic cancer via a reciprocal repression between MIIP and HIF-1α [34]. Our most recent study further confirmed MIIP’s inhibitory effect on cell proliferation via interaction with PP1α and negative modulation of AKT signaling in prostate cancer. Thus, MIIP likely taps into multi-faceted mechanisms to play a general inhibitory role in cell proliferation and invasion in various cancer types, including prostate cancer. However, the physiological role of MIIP and its upstream regulation still remain largely unknown and deserve further study.
In summary, we have demonstrated that the tumor-suppressor role of MIIP in prostate cancer proceeds through its regulation of the EMT. MIIP inhibits invasion of prostate cancer cell lines PC3 and DU145. Mechanistically, MIIP enhanced the expression of epithelial markers while inhibiting mesenchymal markers and EMT-inducing transcriptional factors. This was at least partially due to the inhibition of miR-181a/b-5p, which in turn resulted in the upregulation of KLF17. Xenograft and bone metastasis modeling in nude mice demonstrated a stable MIIP-knockdown in prostate cancer cell markedly increased tumor growth and progression. Importantly, the expression of MIIP was shown to be downregulated in more than 50% of prostate tumor samples from a cohort of human patients and was associated with reduction of KLF17 expression observed in these samples. Collectively, our data illustrate a novel MIIP-miR-181a/b-5p-KLF17-EMT axis in prostate cancer and expand the knowledge surrounding the tumor-suppressor role of MIIP.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81572504, 81872421, 81502405, 81672542, 81572815 and 81421003), the State Key Laboratory of Cancer Biology (CBSKL2014Z09, CBSKL2014Z02, CBSKL2017Z12 and CBSKL2017Z09), Natural Science Basic Research Program of Shaanxi Province (2019JM-315).
The procedures of this study were approved by the Institutional Review Board of The Fourth Military Medical University. The archival samples were collected after signed informed consent obtainment from the Department of Urology, Xijing Hospital, Xi’an, Shaanxi.
Disclosure of conflict of interest
None.
Abbreviations
- PCa
prostate cancer
- MIIP
The migration and invasion inhibitor protein
- EMT
epithelial-mesenchymal transition
- KLF17
Krueppel-like factor 17
- NC
nonspecific control siRNA
- AS
antisense oligonucleotide
Supporting Information
References
- 1.Zhou CK, Check DP, Lortet-Tieulent J, Laversanne M, Jemal A, Ferlay J, Bray F, Cook MB, Devesa SS. Prostate cancer incidence in 43 populations worldwide: an analysis of time trends overall and by age group. Int J Cancer. 2016;138:1388–1400. doi: 10.1002/ijc.29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T, Zattoni F, Mottet N. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65:467–479. doi: 10.1016/j.eururo.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Nakazawa M, Kyprianou N. Epithelial-mesenchymal-transition regulators in prostate cancer: androgens and beyond. J Steroid Biochem Mol Biol. 2017;166:84–90. doi: 10.1016/j.jsbmb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Zhou S, Tang X, Tang F. Kruppel-like factor 17, a novel tumor suppressor: its low expression is involved in cancer metastasis. Tumour Biol. 2016;37:1505–1513. doi: 10.1007/s13277-015-4588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, Coukos G, Zhang L, Huang Q. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nat Cell Biol. 2009;11:1297–1304. doi: 10.1038/ncb1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu FY, Deng YL, Li Y, Zeng D, Zhou ZZ, Tian DA, Liu M. Down-regulated KLF17 expression is associated with tumor invasion and poor prognosis in hepatocellular carcinoma. Med Oncol. 2013;30:425. doi: 10.1007/s12032-012-0425-3. [DOI] [PubMed] [Google Scholar]
- 11.Ali A, Bhatti MZ, Shah AS, Duong HQ, Alkreathy HM, Mohammad SF, Khan RA, Ahmad A. Tumor-suppressive p53 signaling empowers metastatic inhibitor KLF17-dependent transcription to overcome tumorigenesis in non-small cell lung cancer. J Biol Chem. 2015;290:21336–21351. doi: 10.1074/jbc.M114.635730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ali A, Zhang P, Liangfang Y, Wenshe S, Wang H, Lin X, Dai Y, Feng XH, Moses R, Wang D, Li X, Xiao J. KLF17 empowers TGF-beta/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015;6:e1681. doi: 10.1038/cddis.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ali A, Ielciu I, Alkreathy HM, Khan AA. KLF17 attenuates estrogen receptor alpha-mediated signaling by impeding ERalpha function on chromatin and determines response to endocrine therapy. Biochim Biophys Acta. 2016;1859:883–895. doi: 10.1016/j.bbagrm.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Song SW, Fuller GN, Khan A, Kong S, Shen W, Taylor E, Ramdas L, Lang FF, Zhang W. IIp45, an insulin-like growth factor binding protein 2 (IGFBP-2) binding protein, antagonizes IGFBP-2 stimulation of glioma cell invasion. Proc Natl Acad Sci U S A. 2003;100:13970–13975. doi: 10.1073/pnas.2332186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Wen J, Zhang W. MIIP, a cytoskeleton regulator that blocks cell migration and invasion, delays mitosis, and suppresses tumorogenesis. Curr Protein Pept Sci. 2011;12:68–73. doi: 10.2174/138920311795659434. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Hu L, Ji P, Teng F, Tian W, Liu Y, Cogdell D, Liu J, Sood AK, Broaddus R, Xue F, Zhang W. MIIP remodels Rac1-mediated cytoskeleton structure in suppression of endometrial cancer metastasis. J Hematol Oncol. 2016;9:112. doi: 10.1186/s13045-016-0342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen J, Fu J, Ling Y, Zhang W. MIIP accelerates epidermal growth factor receptor protein turnover and attenuates proliferation in non-small cell lung cancer. Oncotarget. 2016;7:9118–9134. doi: 10.18632/oncotarget.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Song SW, Sun J, Bruner JM, Fuller GN, Zhang W. IIp45 inhibits cell migration through inhibition of HDAC6. J Biol Chem. 2010;285:3554–3560. doi: 10.1074/jbc.M109.063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji P, Smith SM, Wang Y, Jiang R, Song SW, Li B, Sawaya R, Bruner JM, Kuang J, Yu H, Fuller GN, Zhang W. Inhibition of gliomagenesis and attenuation of mitotic transition by MIIP. Oncogene. 2010;29:3501–3508. doi: 10.1038/onc.2010.114. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Ji P, Chen T, Zhou X, Yang D, Guo Y, Liu Y, Hu L, Xia D, Multani AS, Shmulevich I, Kucherlapati R, Kopetz S, Sood AK, Hamilton SR, Sun B, Zhang W. MIIP haploinsufficiency induces chromosomal instability and promotes tumour progression in colorectal cancer. J Pathol. 2017;241:67–79. doi: 10.1002/path.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan G, Ru Y, Yan F, Xiong X, Hu W, Pan T, Sun J, Zhang C, Wang Q, Li X. MIIP inhibits the growth of prostate cancer via interaction with PP1alpha and negative modulation of AKT signaling. Cell Commun Signal. 2019;17:44. doi: 10.1186/s12964-019-0355-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mi Y, Zhang D, Jiang W, Weng J, Zhou C, Huang K, Tang H, Yu Y, Liu X, Cui W, Zhang M, Sun X, Zhou Z, Peng Z, Zhao S, Wen Y. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer Lett. 2017;389:11–22. doi: 10.1016/j.canlet.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 23.Liu Z, Sun F, Hong Y, Liu Y, Fen M, Yin K, Ge X, Wang F, Chen X, Guan W. MEG2 is regulated by miR-181a-5p and functions as a tumour suppressor gene to suppress the proliferation and migration of gastric cancer cells. Mol Cancer. 2017;16:133. doi: 10.1186/s12943-017-0695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian F, Shen Y, Chen Z, Li R, Lu J, Ge Q. Aberrant miR-181b-5p and miR-486-5p expression in serum and tissue of non-small cell lung cancer. Gene. 2016;591:338–343. doi: 10.1016/j.gene.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 25.van Vliet J, Crofts LA, Quinlan KG, Czolij R, Perkins AC, Crossley M. Human KLF17 is a new member of the Sp/KLF family of transcription factors. Genomics. 2006;87:474–482. doi: 10.1016/j.ygeno.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Lomberk G, Urrutia R. The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem J. 2005;392:1–11. doi: 10.1042/BJ20051234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Sun F, Hong Y, Liu Y, Fen M, Yin K, Ge X, Wang F, Chen X, Guan W. MEG2 is regulated by miR-181a-5p and functions as a tumour suppressor gene to suppress the proliferation and migration of gastric cancer cells. Molecular Cancer. 2017;16:133. doi: 10.1186/s12943-017-0695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyu X, Li J, Yun X, Huang R, Deng X, Wang Y, Chen Y, Xiao G. miR-181a-5p, an inducer of Wnt-signaling, facilitates cell proliferation in acute lymphoblastic leukemia. Oncol Rep. 2017;37:1469–1476. doi: 10.3892/or.2017.5425. [DOI] [PubMed] [Google Scholar]
- 30.Tian F, Shen Y, Chen Z, Li R, Lu J, Ge Q. Aberrant miR-181b-5p and miR-486-5p expression in serum and tissue of non-small cell lung cancer. Gene. 2016;591:338–343. doi: 10.1016/j.gene.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Liu K, Xie F, Gao A, Zhang R, Zhang L, Xiao Z, Hu Q, Huang W, Huang Q, Lin B, Zhu J, Wang H, Que J, Lan X. SOX2 regulates multiple malignant processes of breast cancer development through the SOX2/miR-181a-5p, miR-30e-5p/TUSC3 axis. Mol Cancer. 2017;16:62. doi: 10.1186/s12943-017-0632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi Y, Zhang D, Jiang W, Weng J, Zhou C, Huang K, Tang H, Yu Y, Liu X, Cui W, Zhang M, Sun X, Zhou Z, Peng Z, Zhao S, Wen Y. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer Lett. 2017;389:11–22. doi: 10.1016/j.canlet.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 33.Dong P, Kaneuchi M, Xiong Y, Cao L, Cai M, Liu X, Guo SW, Ju J, Jia N, Konno Y, Watari H, Hosaka M, Sudo S, Sakuragi N. Identification of KLF17 as a novel epithelial to mesenchymal transition inducer via direct activation of TWIST1 in endometrioid endometrial cancer. Carcinogenesis. 2014;35:760–768. doi: 10.1093/carcin/bgt369. [DOI] [PubMed] [Google Scholar]
- 34.Niu Y, Jin Y, Deng SC, Deng SJ, Zhu S, Liu Y, Li X, He C, Liu ML, Zeng Z, Chen HY, Zhong JX, Ye Z, Wang CY, Zhao G. MiRNA-646-mediated reciprocal repression between HIF-1alpha and MIIP contributes to tumorigenesis of pancreatic cancer. Oncogene. 2018;37:1743–1758. doi: 10.1038/s41388-017-0082-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.