

Abstract
Recently, the combination of platinum chemotherapy with PD-1/PD-L1 pathway blockades has shown synergistic efficacy in a few clinical trials. However, the exact mechanisms and the optimized sequence of such combinations are not fully clear. In this study, we combined different doses of platinum agents (cisplatin or oxaliplatin) with sequential therapy of PD-1 blockade therapy (anti-PD-1 antibody or anti-PD-L1 antibody) to treat established MC38 murine colon tumors. Although 10 mg/kg platinum (cisplatin or oxaliplatin) showed no significant effect on tumor growth, its combination with sequential anti-PD-1 antibody administration caused complete tumor remission in 80-100% mice. The synergic therapeutic efficacy was found to be associated with more effector and less exhausted CD8 T cell infiltration in the tumor sites. Platinum chemotherapy is generally considered immunosuppressive, with lymphopenia and neutropenia being common side effects. However, our data showed that high-dose (20 mg/kg) platinum treatment induced lymphopenia in MC38 tumor-bearing mice, and low-dose (10 mg/kg) treatment augmented the T cell response with an increased number of peripheral T cells. Notably, increased numbers of PD-1 positive CD8 T cells were found in draining lymph nodes, peripheral blood and tumor tissues three days after 10 mg/kg oxaliplatin treatment, and increased numbers of CD8 T cells and apoptotic tumor cells were detected at the edge of tumor tissues. Further investigation showed that the death of tumor cells induced by platinum compounds promoted T cell activation. Moreover, increased expression of T cell-attracting chemokines (CXCL9, CXCL10 and CCL5) was detected in MC38 cells after platinum treatment. These data indicated that the optimal dose of platinum chemotherapy could trigger T cell activation and recruitment into tumors, and sequential PD-1 blockade could prevent newly arriving T cell from becoming exhausted in tumor sites. These findings highlight the importance of optimizing the dose and timing of platinum chemotherapy combined with PD-1 blockade and provide an indication for the improvement of combined therapies in clinical trials.
Keywords: Platinum chemotherapy, combination therapy, immune checkpoint inhibitor, T cell recruitment
Introduction
“Checkpoint-blocking” antibodies against programmed cell death protein 1 (PD-1) and its ligands have emerged as an increasingly effective treatment modality for many cancers, resulting in remarkable responses [1-3]. Nevertheless, anti-PD-1/PD-L1 antibodies as a single immune therapy are not sufficient to improve clinical outcome in most patients, emphasizing the need to develop combination strategies [2,4,5]. The platinum-based drugs cisplatin (Cis), carboplatin, and oxaliplatin (Oxa) are widely used anticancer drugs that are thought to be immunosuppressive by interfering cell division [6,7]. Recently, the combination of platinum compounds with PD-1/PD-L1 pathway blockade showed synergistic efficacy in some murine tumor models and a few clinical trials [8-13]. However, their exact synergistic mechanism has not yet been elucidated.
In this study, we tested the effect of different doses of Cis and Oxa on peripheral immune cell profiles in mice implanted with murine MC38 colon tumor cells. We found that 10 mg/kg platinum compounds (Cis or Oxa) increased the number of peripheral blood T lymphocytes, whereas high-dose chemotherapy showed conventional lymphopenia. Further investigation showed that a sequential treatment schedule of anti-PD-1 antibody dramatically improved the inhibitory effects of low-dose (10 mg/kg) platinum compounds on tumor growth. Intriguingly, despite the lack of effect of 10 mg/kg platinum compounds alone on tumor eradication, tumor cell death induced by Cis or Oxa could initiate T cell activation and migration to the tumor site, resulting in synergistic antitumor effect with PD-1 monoclonal antibodies.
Materials and methods
Mice
C57BL/6 mice and mice with transgenic T cell receptors specific for H-2Kb OVA257-264 (OT-I) were purchased from the Model Animal Research Center of Nanjing University. All female mice were 6 to 8 weeks old at the beginning of each experiment. All procedures performed in studies involving animals were approved by the Fujian Medical University Institutional Animal Care and Use Committee (IACUC, NO. 2017-033) in accordance with the ethical standards. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Cell lines and antibodies
The murine colorectal cancer cell line MC38 was purchased from the authenticated NIH repository. MC38-OVA cells were generated by stable transfection with chicken egg ovalbumin (OVA). Tumor cells were cultured in DMEM supplemented with 10% fetal calf serum, L-glutamine, nonessential amino acids, sodium pyruvate, and antibiotics (Thermo Fisher Scientific, USA). All tumor cell lines were tested before used and found to be free of Mycoplasma. Antibodies against PD-L1 (10F.9G2), PD-1 (RMP1-30), CD3 (17A2), CD8 (53-6.7), IFN-γ (XMG1.2), CD4 (GK1.5), Foxp3 (FJK-16s) and CD45 (HI30) were obtained from BioLegend, BD Biosciences or Thermo Fisher Scientific. Blocking antibodies against mouse PD-1 (clone G4) and PD-L1 (clone 10B4) were produced in our lab.
Tumor models and treatment
Mice were subcutaneously injected in the right flank with 5×105 MC38 tumor cells. Tumor sizes were measured with digital calipers every 3 days and calculated using the equation (l+w)/2, where l and w refer to the larger and smaller dimensions, respectively, collected at each measurement. When the tumor diameter reached 4-8 mm (at 6-7 days), mice were assigned to homogenous groups of 4-6 mice and intraperitoneally injected with a single dose of Cis or Oxa (Sigma-Aldrich, USA) at different concentrations (0, 10, 20, 40 or 80 mg/kg body weight). For combination treatment, mice were sequentially administered with 250 µg anti-mouse PD-1 or anti-mouse PD-L1 every 4 days for a total of three times, and hamster IgG was used as a control antibody. All the mice that developed tumors reaching a size of 2.0 cm in each dimension were sacrificed in accordance with standards for humane treatment.
Flow cytometry (FCM)
Single-cell suspensions of tumor tissues, spleen and lymph node tissues and blood were prepared on the scheduled days after treatment. Tumor infiltrating lymphocytes (TILs) were isolated from freshly resected tumor tissues using a Gentle MACS mechanical dissociator containing lysis buffer (Miltenyi Biotec, Germany) and enriched in the mouse lymphocyte separation buffer LymphoLyte-M (Cedarlane Laboratories Ltd, Canada) according to the manufacturer’s recommendations. Cells from tissues were blocked with anti-mouse CD16/32 (TruStain fcX™, BioLegend, USA). Single-cell suspensions were stained with antibodies, and then run on a BD FACSVerse™ flow cytometry (BD Biosciences, USA) and analyzed using FlowJo software.
Histology and immunohistochemistry
MC38 tumor tissues were fixed and paraffin-embedded following standard procedures. The sections were stained with H&E (Thermo Fisher Scientific, USA) for histologic analysis. For immunohistochemical staining, the sections were immunolabeled with rabbit anti-mouse CD8 antibodies (SP16, 1:100, Thermo Fisher Scientific, USA), followed by the use of the ImmPRESS anti-rabbit Ig Kit (Vector Laboratories, USA), according to the manufacturer’s instructions.
TUNEL staining
TUNEL (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling) staining for tumor tissue was based on the protocol of the TdT-Fragel DNA Fragmentation detection kit (Roche, USA). The tissue sections were imaged with an EVOS FL Auto Cell Imaging System inverted microscope (Thermo Fisher Scientific, USA).
ELISA
MC38-OVA cells were cultivated in phenol red-free medium and then treated with different doses of Cis or Oxa (0, 100, 200 or 400 μM) for 24 hours. The supernatants were collected for HMGB1 detection. HMGB1 levels in culture medium were quantified using an ELISA kit (Chemicon, USA) according to the manufacturer’s instructions.
Immunofluorescent staining
MC38-OVA cells were treated with the indicated concentrations of Cis or Oxa for 4 hours, and then fixed in 4% paraformaldehyde for 20 minutes. Immunofluorescent staining was performed with a primary rabbit anti-calreticulin (CRT) antibody (1:300, Ab2907, Abcam, USA), and followed by staining with an Alexa Fluor 488® goat anti-rabbit IgG antibody (1:500, A-11034, Thermo Fisher Scientific, USA) and DAPI (1:500, Sigma-Aldrich, USA) for 20 seconds. Cells were observed under an inverted microscope using 405 and 488 nm lasers to visualize nuclei and CRT expression on the cell membrane, respectively.
Apoptosis assay
MC38-OVA cells were treated with the indicated concentrations of Cis or Oxa for 24 hours, and then stained with PE-Annexin V and 7-AAD (BD Biosciences, USA) according to the manufacturer’s instructions. The percentages of cell apoptosis were analyzed by FCM. Late apoptosis was defined as Annexin V and 7-AAD double-positivity; early apoptosis was defined as Annexin V positivity with 7-AAD negativity.
T cell proliferation assay
Dendritic cells (DCs) were magnetically isolated from splenocytes using the EasySep™ Mouse Pan-DC Enrichment Kit (StemCell Technologies, Canada). MC38-OVA cells were pretreated with different doses of Cis or Oxa (0, 100, 200 or 400 μM) for 24 hours. OT-I T cells were isolated from the lymph nodes of female OT-I transgenic mice by a negative CD8 T cell isolation kit (Miltenyi Biotec, Germany), and labeled with 3 μM 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) (Sigma-Aldrich, USA). Drug-treated and control MC38-OVA cells were harvested and incubated with DCs for 4 hours, and then DCs were coincubated with CFSE-labeled naïve OT-I T cells for 72 hours. Cell proliferation was analyzed by FCM.
Expression of chemokine genes
MC38-OVA cells were seeded into 12-well plates for 16 hours, and then drugs were added at indicated concentrations. The expression of chemokine genes relative to that of GAPDH was measured by quantitative real-time PCR (qRT-PCR) as previously described [14]. In brief, total RNA was extracted from analyzed specimens and converted to cDNAs using RNeasy Mini Kit and Sensiscript RT Kit (Qiagen, Germany), respectively. qRT-PCR was performed with SYBR Green Master Mix (Applied Biosystems, USA), according to the manufacturer’s instructions. All primers listed in Table S1 were validated according to the protocol. The relative expression of target genes was calculated as using the formula 2-∆∆CT.
Transwell migration assay
OT-I T cells were isolated from the spleen and lymph nodes of OT-I transgenic mice by a negative isolation kit. Non-T cells were irradiated with 40 Gy, and then were pulsed with 3 ng/ml OVA-peptide 257-264 (SIINFEKL) for 1 hour. OT-I CD8 T cells were cocultured with SIINFEKL-pulsed cells in 10% FBS-supplemented medium containing 20 IU/ml IL-2 for 24 hours. The FBS concentrations of media for activated CD8 cells were gradually changed to 5%, 2.5% and 1.25% every 4 hours for serum-starvation treatment. A total of 5×105 activated CD8 cells were added to the upper chamber in 100 μl of 1.25% FBS-supplemented media. The lower chamber contained 600 μl of culture supernatant from MC38-OVA cells after they had been pretreated with Oxa or Cis for 48 hours, which was changed to 5% FBS-supplemented media for another 24 hours. After incubation for 4 hours at 37°C, cells that migrated into the lower chamber were observed under a microscope.
Statistical analysis
Statistical analysis was conducted using Prism 8 (GraphPad, Canada). Numerical data are expressed as the mean ± SEM unless otherwise stated. Statistical differences between groups were compared using Student’s t test or one-way ANOVA with Tukey or Dunnett multiple comparison tests (tumor growth and phenotype comparisons), and individual group comparisons were evaluated by Bonferroni’s multiple comparison test. The log-Rank and Wilcoxon tests were used to analyze the differences in survival time. *P < 0.05; **P < 0.01; ***P < 0.001. The results represent at least three experiments unless otherwise stated.
Results
Optimal dose of platinum chemotherapy in combination with anti-PD-1 antibodies was defined in the treatment of MC38 tumor model
To determine the median lethal doses (LD50) of Cis and Oxa, MC38 tumor-bearing mice were intraperitoneally injected with different doses of Cis or Oxa (0, 10, 20, 40 or 80 mg/kg) on day 6 after tumor implantation. The data showed that high-dose platinum compounds (> 40 mg/kg) caused rapid weight loss (data not shown) and mouse death, and the LD50 values of Cis and Oxa were 17.62 and 28.28 mg/kg, respectively. No therapeutic outcome was found in MC38 tumor-bearing mice treated with different doses of Cis or Oxa (Figure 1A). Nevertheless, the increasing numbers of lymphocytes and the percentages of CD4 and CD8 T cells were observed in the blood 2 days after 10 mg/kg Oxa or Cis administration (Figure 1B), indicating that the low-dose (10 mg/kg) platinum chemotherapy exerted an immune priming effect. Thus, we treated established MC38 tumors with a single dose of platinum compounds on day 6 and three sequential doses of a blocking antibody targeting PD-1 on days 9, 13 and 17 (Figure 1C). The sequential combination of 10 mg/kg Cis (or Oxa) and anti-PD-1 caused almost complete remission of the established MC38 tumor model, with Oxa yielding a more pronounced therapeutic response than Cis. However, there was no synergistic effect on tumor growth in surviving mice that treated with the combination of 5 or 20 mg/kg Cis (or Oxa) and anti-PD-1 antibodies. Anti-PD-1 antibody or Cis (or Oxa) monotherapy showed no significant effect on tumor remission (Figure 1D, 1E).
Figure 1.

The synergistic effect of different doses of platinum compounds combined with anti-PD-1 antibodies in the established MC38 model. (A) The effect and toxicity of different doses of Cis or Oxa in MC38 tumor-bearing mice. C57BL/6 mice were subcutaneously inoculated on day 0 with 5×105 viable MC38 cells. When the mean tumor length reached approximately 4-8 mm (day 6), the mice were randomized by tumor size and intraperitoneally injected with single dose (0, 10, 20, 40 or 80 mg/kg) of platinum compounds (Cis, left panel; Oxa, right panel). The mean tumor lengths and survival rates of mice in each group (n = 5) are shown. Each symbol represents the mean ± SD of 5 mice. (B) Lymphocyte numbers and the frequency of CD4 or CD8 T cells within the lymphocyte population in the peripheral blood was detected by cell number quantification and flow cytometry on day 3 after platinum chemotherapy (n = 5). (C) Treatment schedule of MC38 tumor-bearing mice comparing platinum chemotherapy alone and sequential combination therapy with anti-PD-1 antibodies. MC38 tumor-bearing mice were intraperitoneally administered with Cis or Oxa (0, 5, 10 or 20 mg/kg) on day 6, and then 250 μg of anti-PD-1 (clone G4) or irrelevant hamster IgGs was sequentially injected every four days for a total of three times (days 9, 13 and 17). (D, E). The time-course of tumor growth in different groups. MC38 tumors on the right flank were treated with different doses of Cis (D) or Oxa (E) alone, or drugs combined with anti-PD1 antibodies, as described in (C). Tumors on the right flank were monitored (n = 4-5), and each line represents an individual mouse. Con: isotype antibody control; Cis: cisplatin; Oxa: oxaliplatin; anti-PD-1: anti-PD-1 antibodies. All results shown are representative of at least two independent experiments. Ns, no significant difference; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Platinum chemotherapy in combination with 3-day-delayed sequential PD-L1/PD-1 blockade augmented tumor remission
Correspondingly, low-dose Oxa (10 mg/kg) plus delayed anti-PD-1 antibody administration resulted in a significant increase in survival, with approximately 80% or 100% of the animals experiencing complete tumor remission (Figure 2A). The tumor-free mice from the combination group were observed for 120 days and rechallenged with MC38 tumors, and no tumors developed in these mice (data not shown), suggesting that long-lasting antitumor effects are induced by combined low-dose Oxa and anti-PD-1 antibodies. Similar to the results from the anti-PD-1 antibody, the combination of low-dose Oxa and delayed anti-PD-L1 antibody administration also promoted tumor remission and prolonged mouse survival compared with Oxa or anti-PD-L1 antibody administration alone (Figure 2B). Considering that sequence and timing are important aspects for the optimized therapeutic effect of PD-1 blockade combined with platinum compounds, we compared three treatment strategies, including various combinations of concurrent or sequential treatment with anti-PD-1 antibody and Oxa. The concurrent (d6) and 6-day-delayed anti-PD-1 antibody (d12) combinations slight inhibited tumor progression. However, analysis of tumor growth, tumor-free mice and survival with these combinations showed that they were not significantly different from the respective results with anti-PD-1 treatment. Importantly, the sequential combination of 3-day-delayed anti-PD-1 antibody (d9) and Oxa demonstrated significant inhibition of tumor growth for a longer period of time than the other combinations (Figure 2C). These data demonstrated that 3-day-delayed sequential combination therapy with platinum chemotherapy and anti-PD-1 pathway blockade was superior therapy to concurrent or 6-day-delayed combination therapy in preclinical colon cancer models.
Figure 2.

The combination of platinum chemotherapy and sequential PD-L1/PD-1 blockade significantly promoted MC38 tumor remission. MC38 tumor-bearing mice were established by subcutaneous injection of 5×105 MC38 cells into the right flask of C57BL/6 mice, followed by treatment with platinum compounds alone (Cis or Oxa, 10 mg/kg) on day 6, or with platinum compounds plus antibodies against PD-L1 or PD-1 at different time points (on days 9, 13, and 17). Tumor growth over time (left panel), survival curves for mice (middle panel), and the percentage of tumor-free mice (right panel) were monitored and determined. A. MC38 tumor-bearing mice were treated with Oxa (10 mg/kg) alone, or with a combination of chemotherapy and sequential anti-PD-1 antibodies (clone G4). B. MC38 tumor-bearing mice were treated with Oxa alone or Oxa followed by anti-PD-L1 antibodies (clone 10B4). C. The day of MC38 tumor injection was referred to as day 0. The administration of anti-PD-1 antibodies started on the same day as Oxa treatment (day 6), or 3 days (day 9), or 6 days (day 12) after Oxa treatment. The antibodies were administered every four days for a total of three times. Con: isotype antibody control; Cis: cisplatin; Oxa: oxaliplatin; anti-PD-1: anti-PD-1 antibodies; anti-PD-L1: anti-PD-L1 antibodies. Each symbol represents the mean ± SD of 5-6 mice, and one representative of two independent experiments is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Effective CD8 T cells were essential for the efficacy of the combination therapy
Given the therapeutic efficacy of sequential combinations, we sought to explore the possible synergistic mechanism. Tumor infiltrating lymphocytes (TILs) were isolated from mice treated with different strategies on day 20 (3 days after the last sequential anti-PD-1 dose) and analyzed by flow cytometry. Figure 3A shows that the frequency of CD8 T cells in the combination group (Oxa+anti-PD-1) was significantly increased as compared with that in the untreated group, while there was no difference in intratumoral CD4 T cells between the groups. In addition, the treatment with Oxa or anti-PD-1 alone treatment exerted no effect on the frequency of intratumoral CD8 and CD4 T cells. To evaluate whether combination treatment increased the tumor-reactive CD8 T cell populations, TILs derived from different groups were cocultured with MC38 cell lysate pulsed matured dendritic cells (DCs) followed by intracellular staining of IFN-γ. As expected, combination treatment significantly increased the percentage of CD8+IFN-γ+ cell in response to lysate-pulsed DCs, as compared with no treatment. This increasing response was also observed for mice treated with Oxa monotherapy (Figure 3B). Given the suggestion that combination treatment decreased T cell exhaustion in TILs, we next investigated the PD-1 and TIM-3 expression on the surface of intratumoral T cells. Figure 3C shows that the frequency of both CD4+PD-1+ and CD8+PD-1+ T cells was significantly decreased in combination treatment, as compared with Oxa monotherapy or untreated groups. The frequency of intratumoral CD8 T cells or CD4 T cells expressing TIM-3 was also decreased in the combinational groups (Figure 3D). Unexpectedly, high levels of PD-1 and TIM-3 expression on CD8 T cells were also found in tumor tissues after Oxa monotherapy, indicating that more exhausted CD8 T cells infiltrated the tumor after Oxa treatment than no treatment.
Figure 3.
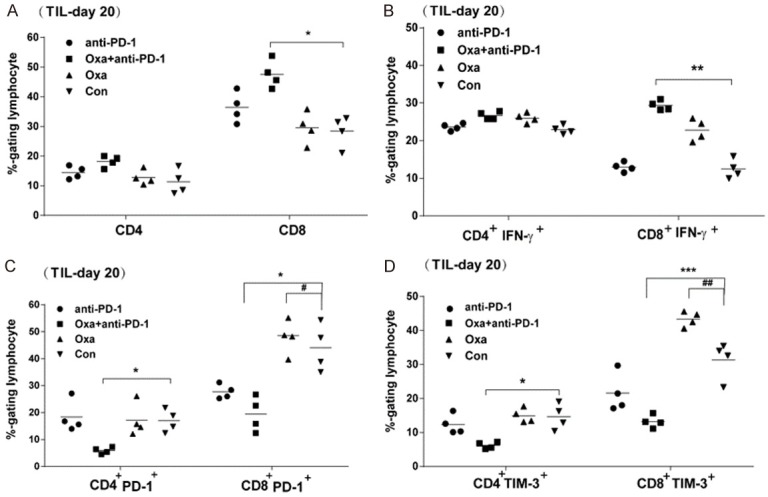
A high frequency of effector CD8 T cells was detected in the tumor site after combination therapy. The MC38 tumor model was established and then treated with different agents as described in Figure 1C. On day 17, mice were treated with the last dose of anti-PD-1 antibody. Three days later (day 20), tumors were removed and digested into single cell suspensions, which were blocked with antibodies against IgG Fc receptor and then subjected to surface and intracellular staining. Summary data pooled from four mice in different treatment groups are shown. A. The frequency of CD4 and CD8 T cells among the lymphocyte population. C, D. The frequency of PD-1- or TIM-3-positive T cells among CD4 and CD8 T cells (gated on the lymphocyte population). B. Tumor-infiltrating lymphocytes were analyzed for cytokine production after stimulation with MC38-lysate-pulsed dendritic cells. The percentages of IFN-γ positive cells in CD4 and CD8 T cells (gated on the lymphocyte population) were analyzed by intracellular staining. TIL: Tumor-infiltrating lymphocytes. Representative data are shown from two independent experiments. Error bars, SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 the combination group compared with the untreated group (Con); #, P < 0.05; ##, P < 0.01; the mice treated with Oxa alone compared with the untreated mice.
Platinum chemotherapy upregulated the expression of PD-1 on CD8 T cells in tumor-draining lymph nodes, blood and the tumor tissues
Next, we investigated whether Oxa treatment (10 mg/kg) was associated with an increase in PD-1 positive T cells in the tumor sites. TILs were analyzed on days 0, 3 and 7 after Oxa treatment, according to the schedule of Oxa treatment and tissue harvest (Figure 4A). The frequency of intratumoral CD8 T cells in the Oxa-treated group was dramatically increased on day 3 and subsequently decreased on day 7; however, no change was found in the untreated group. The frequency of CD4 T cells in the Oxa-treated group remained as similar to that seen in pre-treatment group (Oxa-d0). Curiously, the frequency of PD-1+CD8+ T cells was continuously increased after Oxa treatment (the fourth graph in Figure 4B). Thus, the expression of PD-1 on T cells was examined in the peripheral blood, spleen, draining lymph nodes and TILs on day 3 after Oxa treatment. The data showed that Oxa treatment resulted in a significant upregulation of the frequency of CD8 and CD3 T cells in the peripheral blood, draining lymph nodes and tumor tissues (Figure 4C). The frequency of PD-1+CD8+ T cells from Oxa-treated mice was also increased in the peripheral blood and draining lymph nodes, although at a lower frequency than that in tumor tissues (Figure 4D). In the spleen, however, there were no significant increases in the frequency of CD3, CD8 T and CD8+PD-1+ T cells in Oxa-treated mice compared with untreated mice (Figure 4C and 4D, SP). Additionally, a higher level of PD-L1 (PD-1 ligand) expression was detected on tumor cells (CD45-negative cells) in tumor tissues. There was no difference between Oxa-treated tumors and the untreated tumors (Figure 4E). Consistent with the results from flow cytometry, more tumor-infiltrating CD8 T cells were detected by immunohistological staining in whole tumor sections on day 3 after Oxa treatment than before treatment. Interestingly, the major site of CD8 T cell distribution was the edge of tumor tissues (Figure 4F right panel). To determine the association of tumor-infiltrating CD8 T cells and tumor cell death, TUNEL staining was also performed in whole tumor sections, and the results demonstrated more apoptotic tumor cells were detected in Oxa-treated tumors than the untreated tumor (Figure 4F left panel). Similarly, TUNEL-positive cells were dominantly located at the edge of the tumor in all samples. These data indicated that the tumor cell apoptosis induced by Oxa chemotherapy might promote CD8 T cell infiltration into the tumor site.
Figure 4.

Pretreatment with platinum compounds increased both peripheral blood, draining lymph node and intratumoral PD-1-positive CD8 T cells. A. The MC38 tumor model was established and treated with 10 mg/kg Oxa on day 6. Then, the tissues were harvested, and blood was drawn on the indicated day. B. Tumor-infiltrating lymphocytes (TILs) were isolated on days 6 (d0), 9 (d3), and 13 (d7). The frequencies of CD4 T cells, CD8 T cells and PD-1-positive T cells in tumor sites were analyzed by flow cytometry (n = 4). C. The frequencies of CD4 and CD8 T cells (gated on lymphocytes) in the draining lymph node (DLN), spleen (SP), and peripheral blood (PB) and within the TIL population were analyzed in MC38 tumor-bearing mice on day 3 after Oxa treatment (n = 4). D. The frequencies of CD4+PD-1+ and CD8+PD-1+ T cells (gated on lymphocytes) in TILs, DLN, SP and PB of Oxa-treated and untreated mice. E. The frequencies of CD45-PD-L1+ and CD45+PD-L1+ cells (gated on living cells) in Oxa-treated and untreated tumors. F. Oxa-treated and untreated tumor tissues were harvested for TUNEL staining (left graph) and immunohistochemical staining with an anti-CD8 antibody (right graph) (n = 3). Scale bar, 200, 50 μm. Con: untreated mice; Oxa: Oxa-treated mice; Nor: normal mice. Representative data are shown from at least two experiments conducted using 4 mice per group. Error bars, SEM. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 compared with untreated mice.
Tumor cell apoptosis induced by platinum compounds facilitated cross-priming of antigen-specific T cells in vitro
To further examine the impact of platinum-treated tumor cells on the T cell response, OVA-expressing MC38-OVA cells were prepared. MC38-OVA cells were treated with different concentrations of platinum compounds for 24, 48 and 72 hours to evaluate the cytotoxicity of Cis and Oxa. The half-maximal inhibitory concentrations (IC50) values at 24, 48 and 72 hours for Cis were 357, 162.3 and 125.7 μM, respectively. The IC50 values at 24, 48 and 72 hours for Oxa were 306.3, 123.6 and 94.1 μM respectively. Annexin V and 7-AAD staining showed that Cis (or Oxa) mainly induced late apoptosis 24 hours after treatment with platinum compounds (Figure 5A). To investigate whether cell apoptosis was correlated with the immunogenic cell death, we analyzed the hallmarks of immunogenic cell death in drug-treated MC38-OVA cells. As expected, in response to Cis or Oxa (100, 200 or 400 μM), higher levels of HMGB1 proteins were dose-dependently released into the suspension than those seen in untreated cells (Figure 5B). Calreticulin (CRT) was also transported to the plasma membrane after platinum treatment (Figure 5C). To explore whether immunogenic cell death could facilitate cross-presentation and subsequent priming of antigen-specific T lymphocytes, DCs were incubated with platinum-treated tumor cells before their coculture with naïve OVA-peptide specific T cells (OT-I). The results showed that DCs pulsed with platinum-treated MC38-OVA cells significantly promoted OT-I T cell proliferation. However, OT-I T cells had no proliferative response after coculture with DCs pulsed with nontreated MC38-OVA cells (Figure 5D). Collectively, these findings suggested that Cis and Oxa augmented the cross-presentation and proliferation of antigen-specific T-lymphocytes in vitro.
Figure 5.

Immunogenic tumor cell apoptosis was induced by platinum chemotherapy in vitro. A. MC38-OVA cells were treated with Cis or Oxa (0, 100 or 200 μM) for 24 hours, followed by staining with annexin V-PE and 7-AAD. The percentages of early and late apoptotic cells were determined and the summarized data are pooled in the right panel. B. The level of HMGB1 in the cell medium of platinum-treated and untreated MC38-OVA cells was determined by ELISA. C. MC38-OVA cells were treated with Cis or Oxa for 4 hours, and calreticulin (CRT) exposure on the cell surface was assessed by immunofluorescence staining and imaging under a microscope at a magnification of 200×. D. Platinum-treated and untreated MC38-OVA cells were incubated with DCs. After 4 hours of incubation, DCs were harvested and cocultured with CFSE-labeled naïve OT-I T cells for 72 hours. Proliferation of OT-I T cells was measured by the CFSE spread on flow cytometry and quantified via calculation of the percent of CFSE+ OT-I cells. Error bars, SEM. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Optimal dose platinum chemotherapy promoted the recruitment of effective CD8 T cells into the tumor microenvironment
To gain further insight into the possible role of tumor cell apoptosis in T cell recruitment, the ability of OT-I cell migration was assessed after administration of different MC38-OVA cell culture media. More OT-I cells were attracted to the Cis/Oxa-treated cell culture medium, in which MC38 cells were pretreated with Cis or Oxa for 24 hours, than to control medium (Figure 6A). To characterize chemokines involved in T cell recruitment, we analyzed the expression of several chemokine genes by qRT-PCR in MC38-OVA cells after platinum compound treatment. The expression of CXCL9, CXCL10 and CCL5 was enhanced in Cis- or Oxa-treated tumor cells (50 or 100 μM) compared with control cells (Figure 6B). To evaluate the role of T cell recruitment on the synergistic effect of platinum chemotherapy and subsequent anti-PD-1 antibody, we treated MC38 tumor-bearing mice with a combination therapy of 10 mg/kg Oxa and PD-1 antibody administration in the presence of the S1P receptor inhibitor FTY720, which locks T cells in lymphoid organs (Figure 6C). Importantly, FTY720 mitigated the therapeutic efficacy of the combination therapy (Figure 6D), suggesting that T cell recruitment from lymph nodes dominantly contributed to the synergistic efficacy of Oxa pretreatment and PD-1 antibody administration. Collectively, these observations indicated that optimal doses platinum compounds promoted the release of T cell chemokines from the tumor cells, which could attract more effector T cells to the tumor sites and facilitate subsequent PD-1 blockage.
Figure 6.
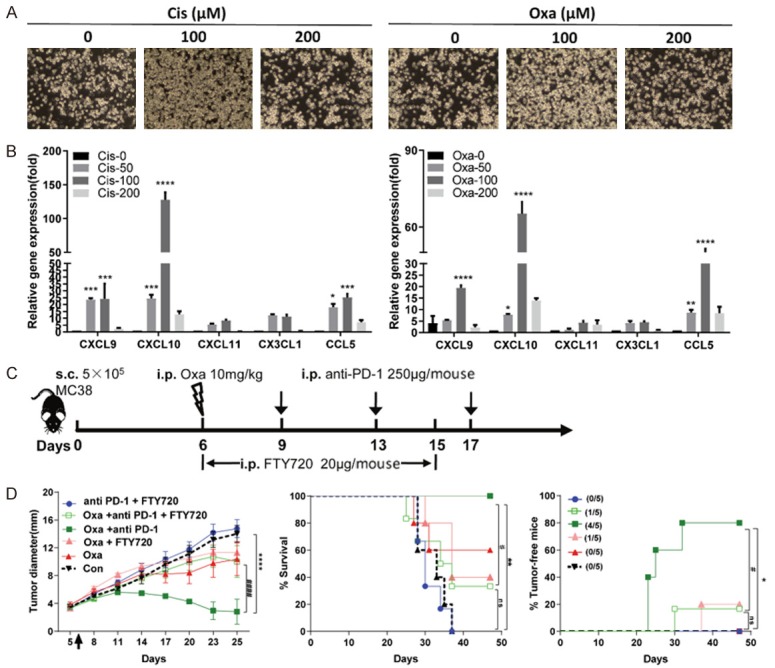
CD8 T cell recruitment into the tumor site contributed to the synergistic efficacy. A. MC38-OVA cells were pretreated with Cis or Oxa for 24 hours, and the medium was changed to 5% FBS for incubation for another 24 hours. Migration assays were performed with culture supernatant, and the cell number of activated OT-I cells migrating to the lower chamber was determined with imaging under a microscope at a magnification of 100×. B. MC38-OVA cells were treated with increasing doses of Cis or Oxa for 24 hours, and chemokine mRNA expression was determined by qRT-PCR and calculated as using the formula 2-∆∆CT, with the GAPDH gene used as an endogenous control. Error bars, SEM. C. Treatment schedule of MC38 tumor-bearing mice comparing FTY720 blockade and vehicle control. MC38 cells were inoculated into C57BL/6 mice (n = 5) on day 0, and then intraperitoneally treated with Oxa (day 6) and/or anti-PD-1 (days 9, 13 and 17). FTY720 (20 μg/mouse) or vehicle control was intraperitoneally administered from day 6 to day 15. D. Tumor growth curve (left panel), survival curve (middle panel) and the percentage of tumor-free mice (right panel) were monitored and analyzed. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 the combination group (Oxa plus anti-PD-1 antibodies) compared with the untreated group (Con); #, P < 0.05; ####, P < 0.0001; the combination group (Oxa plus anti-PD-1 antibodies) compared with the group treated with the combination of drugs (Oxa plus anti-PD-1 antibodies) and FTY720.
Discussion
The programmed death 1 (PD-1) pathway is a negative feedback system that inhibits antitumor T cell responses and drives tumor immune escape in cancer. Blockade of this pathway with antibodies against PD-1 or its ligands has elicited durable antitumor responses and long-term remissions in patients with many different types of solid tumors [15]. Nevertheless, clinical trials have demonstrated that PD-L1 or PD-1 antagonistic antibodies only elicited responses in a subset of cancer patients (15%-25%), speficically in tumors with favorable mutational landscapes, PD-1 ligand (PD-L1) expression and pre-existing tumor-infiltrating CD8 T cells [2,16,17]. Recent clinical trials have focused on the combination of immunotherapy and chemotherapy, offering a promising approach to augment the effect of current immunotherapies targeting the PD-L1/PD-1 pathway [10-13]. Nonetheless, how to optimally combine chemotherapy with antibodies targeting PD-L1/PD-1 currently being developed remains a major problem in cancer research.
Conventional and new platinum-based compounds (cisplatin, carboplatin, oxaliplatin, satraplatin and picoplatin) have become the backbone of combination chemotherapy for a wide range of solid tumors [18]. Increasing evidence demonstrates that chemotherapeutic compounds are less effective in immunodeficient hosts than in immunocompetent hosts [19], indicating that the antitumor effects of platinum drugs occur partially through modulation of the immune system. However, lymphopenia is frequent in advanced cancer progression on or after platinum-based chemotherapy, predicting the toxicity of chemotherapy [20]. Our results raise the question of what optimal biologic dose of chemotherapy regimens is associated with maximum antitumor activity and minimum immunosuppression. To address this question, we tested the effect of different doses of Cis and Oxa on established murine colon tumor model. The LD50 of Cis and Oxa was 17.62 mg/kg and 28.28 mg/kg respectively. There was no difference in therapeutic outcomes for different doses of Cis or Oxa in established MC38 colon cancers. However, low-dose Cis or Oxa (10 mg/kg) increased peripheral CD4 and CD8 T cell numbers after 2 days of treatment, while high-dose drugs (> 20 mg/kg) dramatically decreased lymphocyte numbers. Overall, our findings demonstrated that low-dose Cis and Oxa could trigger an immune response despite the lack of an identified role for them in tumor growth control.
The immunotherapeutic efficacy of PD-1 pathway blockade depends on ongoing immune responses, especially those in the tumor microenvironment [21,22]. Our findings suggested that optimal dose platinum chemotherapy (10 mg/kg Cis or Oxa) could trigger an immune response. Thus, we tested the synergetic effect of anti-PD-1 antibody combined with platinum chemotherapy in the immunogenic MC38 tumor model. Notably, complete tumor remission was observed in almost all mice when low-dose platinum compounds (10 mg/kg Cis or Oxa) were combinated with 3 days-delayed anti-PD-1 antibodies, and the injection of Oxa plus subsequent anti-PD-L1 antibodies showed the similar results. However, the anti-PD-1 or anti-PD-L1 antibody monotherapy showed no significant effect on tumor remission. Combination therapy has been reported to only enhance the survival rate but not induce tumor eradication in CT26 mouse tumor models, in which anti-PD-1 antibodies were concomitantly injected with Oxa [8]. In MCA205 tumor-bearing mice, Oxa injection was initiated early (on day 0, the day of tumor inoculation), followed by anti-PD-1 antibodies (on day 8), and then complete tumor remission was observed in 9 out of 10 tumor-bearing mice [23]. These findings indicated that different therapeutic efficacies might be due to the timing and sequence of chemotherapy-immunotherapy combinations. To address this question, a treatment schedule of concurrent (on day 6) and 6-day-delayed anti-PD-1 antibody (on day 12) combination with Oxa was tested in MC38 tumor-bearing mice, and then complete tumor remission was only observed in half of mice. Our findings demonstrated that the sequential treatment has the combined benefit of both optimizing the timing of PD-1/PD-L1 inhibitors and potentially minimizing the possible toxicity of platinum compounds. Considering the fact that the majority of patients enrolled in clinical trials of anti-PD-1 therapy are heavily pretreated with chemotherapy, our findings suggested that low-dose chemotherapy might be beneficial for the patients who sequentially received PD-1/PD-L1 pathway blocking antibodies during the optimal treatment regimens.
Further investigation showed that the combination treatment (Oxa+anti-PD-1) significantly increased the frequency of CD8 T cells in the tumor site and the ability of IFN-γ production compared with other treatments (Oxa or anti-PD-1 monotherapy mice or no treatment). Additionally, our data showed that PD-1 expression on intratumoral CD8 T cells was sustainably increased following Oxa treatment, and an increase in the PD-1 positive CD8 T cell population was also detected in draining lymph nodes and peripheral blood. These results implied that low-dose platinum compounds, compared with high-dose platinum compounds, could induce more activated CD8 T cells to expand in the lymph nodes and then migrate into tumor sites.
Increasing evidence from several studies emphasized the importance of immunogenic cell death contributing to the effects of cancer treatment in different tumor models [24-29]. In our study, when the tumor cells were induced to undergo apoptosis by Cis or Oxa, we also noted a significant increase in HMGB1 levels in the supernatant and CRT expression on the surface of tumor cells. In many cases, cell apoptosis is a physiological process that is frequently viewed as immunologically silent (no immune response) [30,31]. Here, we showed that tumor cell apoptosis induced by platinum-based compounds was capable of initiating an immune response in vivo. More infiltrating CD8 T cells were detected at the edge of the tumor tissue 3 days after Oxa or Cis treatment than on day 0. This response correlated with the increased numbers of apoptotic tumor cells seen in the tumor site of platinum-treated mice compared with untreated mice. The in vitro results re-vealed that optimal dose platinum compounds (100 μM Cis or Oxa) induced cell apoptosis and simultaneously upregulated T cell-attracting chemokine expression in MC38-OVA tumor cells, including CXCL9, CXCL10 and CCL5. The migration assay further confirmed that the media from 100 μM platinum-treated tumor cells promoted T cell migration. The in vivo data showed that FTY720, which locks lymphocytes in the lymph node, impaired the therapeutic efficacy of Oxa combined with PD-1 antibody administration. Together, our findings suggested that the immunogenic cell death induced by platinum compounds initiated T cell proliferation in the lymph node and T cell infiltration into the tumor site.
In summary, our work describes a previously uncharacterized mechanism by which the PD-1 blockade enhances platinum-based chemotherapy: Stressed and dying cancer cells induced by optimal dose of platinum compounds could release immunogenic signals. These signals are perceived by dendritic cells and trigger T cell activation in the lymph node. Stressed cancer cells also released chemokines that attracted activated T cell to the edge of the tumor site. PD-L1 expression on the tumor cells impairs newly arrived T cell function by engaging with it receptors (PD-1) on the surface of effector T cells. PD-L1/PD-1 pathway blockade reversed the impairment and boosted the T cell activation initiated by platinum chemotherapy (Figure 7). These findings could provide insight into clinical study design for chemotherapy in combination with PD-L1/PD-1 pathway blockade. Chemotherapeutic modalities, including compounds, doses, and timing, are important aspects of study design.
Figure 7.
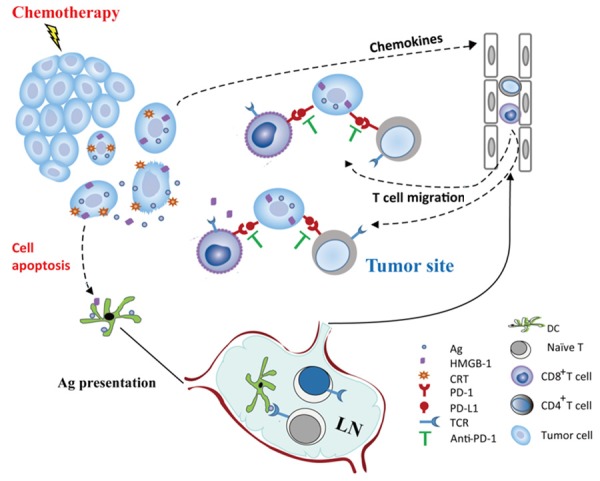
The potential synergistic mechanism of optimal dose platinum chemotherapy and PD-1 antibodies in cancer prevention. Platinum chemotherapeutic agents induced tumor cell apoptosis, and the stressed tumor cells released immunogenic signals (such as CRT and HMGB1). These signals allowed dendritic cells, the essential antigen presenting cells of the innate immune system, to take up portions of the stressed tumor cells and then presented tumor antigen to cognate T cells. Tumor antigen specific T cells with PD-1 expression are activated in lymph nodes and recruited to the tumor site. Unfortunately, in the tumor microenvironment, the PD-L1 molecule, which is expressed on the surface of tumor cells, engages with PD-1 on recruited T lymphocytes and blocks T cell function. Effective blockade of the PD-1/PD-L1 pathway interferes with the immune evasion of tumor cells and allows the immune system to control residual tumor cells.
Acknowledgements
Authors thank Professor Lieping Chen and Professor Liqun Luo for the kind gift of blocking antibodies. The members of Fujian Medical University Public Technology Service Center and Laboratory Animal Center are also acknowledged. This study was supported by National Natural Science Foundation of China (No. 81702828), Key Research Program of Fujian Provincial Heath and Education Joint Committee (No. WKJ2016-2-31), Education and Scientific Research Foundation for Young and Middle-aged teachers in Fujian Province (No. JAT160210), and National Collaboration Center in Immuno-Oncology (No. 2016sysbz02).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt EV. Developing combination strategies using PD-1 checkpoint inhibitors to treat cancer. Semin Immunopathol. 2019;41:21–30. doi: 10.1007/s00281-018-0714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, Gough MJ, Urba WJ, Fox BA. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res. 2017;23:6165–6177. doi: 10.1158/1078-0432.CCR-16-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerard B, Bleiberg H, Van Daele D, Gil T, Hendlisz A, Di Leo A, Fernez B, Brienza S. Oxaliplatin combined to 5-fluorouracil and folinic acid: an effective therapy in patients with advanced colorectal cancer. Anticancer Drugs. 1998;9:301–305. doi: 10.1097/00001813-199804000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Leonardi V, Savio G, Laudani A, Blasi L, Agostara B. New approaches to breast cancer: oxaliplatin combined with 5-fluorouracil and folinic acid in pretreated advanced breast cancer patients: preliminary reports. Ann N Y Acad Sci. 2002;963:91–97. [PubMed] [Google Scholar]
- 8.Dosset M, Vargas TR, Lagrange A, Boidot R, Vegran F, Roussey A, Chalmin F, Dondaine L, Paul C, Lauret Marie-Joseph E, Martin F, Ryffel B, Borg C, Adotevi O, Ghiringhelli F, Apetoh L. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. 2018;7:e1433981. doi: 10.1080/2162402X.2018.1433981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo R, Firat E, Gaedicke S, Guffart E, Watanabe T, Niedermann G. Cisplatin facilitates radiation-induced abscopal effects in conjunction with PD-1 checkpoint blockade through CXCR3/CXCL10-mediated T-cell recruitment. Clin Cancer Res. 2019;25:7243–7255. doi: 10.1158/1078-0432.CCR-19-1344. [DOI] [PubMed] [Google Scholar]
- 10.Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, Cheng SY, Bischoff HG, Peled N, Grossi F, Jennens RR, Reck M, Hui R, Garon EB, Boyer M, Rubio-Viqueira B, Novello S, Kurata T, Gray JE, Vida J, Wei Z, Yang J, Raftopoulos H, Pietanza MC, Garassino MC KEYNOTE-189 Investigators. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 11.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Jalal SI, Panwalkar A, Yang JC, Gubens M, Sequist LV, Awad MM, Fiore J, Ge Y, Raftopoulos H, Gandhi L KEYNOTE-021 investigators. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–1508. doi: 10.1016/S1470-2045(16)30498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, Rodriguez-Cid J, Wilson J, Sugawara S, Kato T, Lee KH, Cheng Y, Novello S, Halmos B, Li X, Lubiniecki GM, Piperdi B, Kowalski DM. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379:2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 13.Rizvi NA, Hellmann MD, Brahmer JR, Juergens RA, Borghaei H, Gettinger S, Chow LQ, Gerber DE, Laurie SA, Goldman JW, Shepherd FA, Chen AC, Shen Y, Nathan FE, Harbison CT, Antonia S. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non-small-cell lung cancer. J. Clin. Oncol. 2016;34:2969–2979. doi: 10.1200/JCO.2016.66.9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang QY, Wu LQ, Zhang T, Han YF, Lin X. Autophagy-mediated HMGB1 release promotes gastric cancer cell survival via RAGE activation of extracellular signal-regulated kinases 1/2. Oncol Rep. 2015;33:1630–1638. doi: 10.3892/or.2015.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, Liniker E, Kong B, Cooper A, Howle JR, Saw RPM, Jakrot V, Lo S, Thompson JF, Carlino MS, Kefford RF, Long GV, Scolyer RA. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin Cancer Res. 2017;23:5024–5033. doi: 10.1158/1078-0432.CCR-16-0698. [DOI] [PubMed] [Google Scholar]
- 18.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 19.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 20.Ray-Coquard I, Cropet C, Van Glabbeke M, Sebban C, Le Cesne A, Judson I, Tredan O, Verweij J, Biron P, Labidi I, Guastalla JP, Bachelot T, Perol D, Chabaud S, Hogendoorn PC, Cassier P, Dufresne A, Blay JY European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009;69:5383–5391. doi: 10.1158/0008-5472.CAN-08-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA, Hinrichs CS. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356:200–205. doi: 10.1126/science.aak9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levesque S, Le Naour J, Pietrocola F, Paillet J, Kremer M, Castoldi F, Baracco EE, Wang Y, Vacchelli E, Stoll G, Jolly A, De La Grange P, Zitvogel L, Kroemer G, Pol JG. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology. 2019;8:e1657375. doi: 10.1080/2162402X.2019.1657375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marciscano AE, Ghasemzadeh A, Nirschl TR, Theodros D, Kochel CM, Francica BJ, Muroyama Y, Anders RA, Sharabi AB, Velarde E, Mao W, Chaudhary KR, Chaimowitz MG, Wong J, Selby MJ, Thudium KB, Korman AJ, Ulmert D, Thorek DLJ, DeWeese TL, Drake CG. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin Cancer Res. 2018;24:5058–5071. doi: 10.1158/1078-0432.CCR-17-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, Krishnan S, Allison JP, Sharma P, Hwu P, Komaki R, Overwijk WW, Gomez DR, Chang JY, Hahn SM, Cortez MA, Welsh JW. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017;77:839–850. doi: 10.1158/0008-5472.CAN-15-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks ED, Chang JY. Time to abandon single-site irradiation for inducing abscopal effects. Nat Rev Clin Oncol. 2019;16:123–135. doi: 10.1038/s41571-018-0119-7. [DOI] [PubMed] [Google Scholar]
- 27.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 28.Park SJ, Ye W, Xiao R, Silvin C, Padget M, Hodge JW, Van Waes C, Schmitt NC. Cisplatin and oxaliplatin induce similar immunogenic changes in preclinical models of head and neck cancer. Oral Oncol. 2019;95:127–135. doi: 10.1016/j.oraloncology.2019.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jessup JM, Kabbout M, Korokhov N, Joun A, Tollefson AE, Wold WSM, Mattoo AR. Adenovirus and oxaliplatin cooperate as agnostic sensitizers for immunogenic cell death in colorectal carcinoma. Hum Vaccin Immunother. 2019:1–9. doi: 10.1080/21645515.2019.1665960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zitvogel L, Kroemer G. The immune response against dying tumor cells: avoid disaster, achieve cure. Cell Death Differ. 2008;15:1–2. doi: 10.1038/sj.cdd.4402267. [DOI] [PubMed] [Google Scholar]
- 31.Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.