

Abstract
Non-small cell lung cancer (NSCLC) patients with c-MET dysregulation may benefit from c-MET inhibitors therapy as inhibition of c-MET activity has emerged as a therapeutic approach against this disease. Although several c-MET inhibitors have been evaluated in multiple clinical trials in lung cancer, their benefits so far have been modest. Thus, furthering our understanding of the mechanisms contributing to the lack of success of c-MET inhibitors in clinical trials is essential toward the development of rational and effective combination strategies. Here we show that exposure of NCSLC cell lines to c-MET inhibitor tivantinib increases their expression of PD-L1, which in turn causes cells to become more resistant to T-cell killing. Mechanistically, inhibition of c-MET suppresses p-GSK3β, leading to the stabilization of PD-L1 similar to that observed in liver cancer cells. Collectively, our findings suggest a potential crosstalk between c-MET inhibition and immune escape and provide a rationale for the combination therapy of c-MET inhibitors and immune checkpoint blockade in NSCLC.
Keywords: PD-L1, NSCLC, c-MET, GSK3β, combination therapy
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related deaths worldwide, and a large number of NSCLCs, particularly adenocarcinomas, are found to harbor driver genes that promote tumor growth [1]. The receptor tyrosine kinase c-MET (also known as hepatocyte growth factor receptor) is one of the oncogenic drivers whose abnormalities may contribute to the resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) [2]. Because dysregulation of c-MET has been reported to occur in up to 26% of NSCLC after EGFR-TKI treatment [3], c-MET may be an excellent therapeutic target in recurrent NSCLC due to EGFR-TKI resistance. For instance, one of the c-MET inhibitors tivantinib was evaluated in combination with other targeted therapies, such as EGFR TKI erlotinib, in NSCLC [4]. However, compared with patients in the control groups, those who received onartuzumab, a monoclonal antibody against c-MET, plus erlotinib did not show improved clinical outcomes [5]. In advanced non-small cell lung cancer, the combination of c-MET inhibition and other targeted therapies was also unsatisfactory [6]. Moreover, in phase II randomized discontinuation trial of c-MET inhibitor cabozantinib, the interpretation of efficacy outcomes were limited by early termination of the randomized portion of the trial for nine tumor types, including lung cancer [7]. Likewise, although preliminary clinical activity was observed for c-MET inhibitor capmatinib and EGFR TKI gefitinib in combination in a phase Ib/II study (INC280), no significant synergistic effects were reported [3]. The c-MET inhibitor tivantinib also failed to meet its primary endpoint of improving overall survival in phase III NSCLC clinical trial [8]. All of these findings point to the limited benefits of those inhibitors against c-MET activities in the clinic. Although the failure of the combination of c-MET and EGFR inhibitors to overcome EGFR inhibitor resistance may be partially attributed to tumor heterogeneity due to activation of different onco-kinases in different tumor cells within the same lung tumor [9], it is not yet clear why c-MET inhibitors in general failed in clinical trials for NSCLC, which is known to harbor c-MET amplification and/or overexpression.
Programmed death-ligand 1 (PD-L1), also known as B7-H1 or CD274, binds to its receptor, programmed cell death protein-1 (PD-1), which can suppress tumor-infiltrating lymphocytes in tumors. Inhibition of the PD-1/PD-L1 pathway has demonstrated promising results in a number of cancer types [10], including a small population of NSCLC patients [11]. The improved response rate of anti-PD-1/PD-L1 in the clinic is highly encouraging, and preclinical studies investigating various modifications in breast and liver cancer and testing combination therapies are actively ongoing to further enhance its therapeutic efficacy [12,13]. We recently reported that c-MET inhibitors stabilized PD-L1 in liver cancer cells and the combination of capmatinib, a c-MET inhibitor, and anti-PD-1 significantly enhanced the therapeutic efficacy compared with each agent alone in a liver cancer animal models [14]. This prompted us to investigate the effect of c-MET inhibitors on PD-L1 regulation in lung cancer cells, and the potential relationship between c-MET inhibitors and immune escape to develop more effective therapeutic approaches for c-MET dysregulated NSCLC. Our findings provide a partial explanation for the lack of success of c-MET inhibitors in clinical trials for NSCLC and pave the way for the development of more effective combination therapies, such as combining c-MET inhibitors and anti-PD-1/PD-L1 therapy, to treat NSCLC.
Material and method
Antibodies and reagents
The following antibodies were used in Western blotting and immunofluorescence microscopy: PD-L1 (13684; Cell Signaling Technology, Danvers, MA, USA), tubulin (B-5-1-2; Sigma-Aldrich), c-MET (8198; Cell Signaling Technology), and phosphorylated c-MET (3077; Cell Signaling Technology). Tivantinib was purchased from Selleck Chemicals (Houston, TX, USA); Puromycin was obtained from Sigma-Aldrich (St. Louis, MO, USA). Phospho-specific antibodies against phosphorylation of GSK3β at Y56 was generated by EZBiolab (Carmel, IN, USA).
Cell culture, plasmids, and transfection
All cell lines were obtained from the ATCC (Manassas, VA, USA) and independently validated by short tandem repeat DNA fingerprinting using the AmpF_STR Identifiler kit according to the manufacturer’s instructions (Applied Biosystems). The short tandem repeat profiles were compared with and matched to known ATCC fingerprints (ATCC.org) and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/). Tests for mycoplasma contamination were negative. Cells were maintained in Dulbecco’s modified Eagle’s medium/F12 medium or RPMI 1640 medium supplemented with 10% fetal bovine serum. Lung cancer cells were transfected with a pGIPZ shRNA vector (control; Thermo Fisher Scientific, Rockford, IL, USA) or pGIPZ shRNA against c-MET to knockdown its expression. The c-MET shRNA sequences are as follows (5’ to 3’): shRNA1: 5’ CCATCCAGAATGTCATTCT 3’ and shRNA2: 5’ GCATTAAAGCAGCGTATC 3’ (targeting the 3’-untranslated region).
For the generation of stable NSCLC cells using retroviral infection, recombinant retroviruses were produced by co-transfecting HEK 293T cells (Clontech, Palo Alto, CA, USA) with the DVPR plasmid and VSV-G plasmids using Lipofectamine 3000 (Invitrogen). Supernatants containing viruses were harvested 48 hours after transfection, centrifuged to eliminate cell debris, and filtered through 0.22 μm filters. Lung cancer cells at ~70% confluency were cultured in virus-containing medium for 1 day to infect cells. Stable clones of different constructs were subsequently selected and maintained in a culture medium with 2 μg/ml puromycin.
Real-time PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). The first-strand cDNA was prepared using the PrimeScript 1st strand cDNA Synthesis Kit (Takara, Japan) according to the manufacturer’s protocol with 1 µg of total RNA. All RT-PCR reactions were performed in a 10 μl mixture containing 1 × SYBR Green Master Mix (Takara Bio USA, Mountainview, CA, USA), 0.2 µmol/L of each primer, and 1 µl of cDNA template. Primers used are as follows (5’ to 3’): Human PD-L1: 5’ TGGCATTTGCTGAACGCATTT 3’ and 5’ TGCAGCCAGGTCTAATTGTTTT 3’. Human glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 5’ GGAGCGAGATCCCTCCAAAAT 3’ and 5’ GCTGTTGTCATACTTCTCATGG 3’.
Real-time PCR was performed using the Applied Biosystem 7500 system (USA) under the following cycling conditions: (step 1) 95°C for 30 sec, (step 2) 40 cycles of 95°C for 5 sec and 60°C for 34 sec, followed by the melting curve stage. The relative PD-L1 expression level was normalized to that of GAPDH.
Western blot analysis
Cells were harvested, washed, and lysed in radioimmunoprecipitation buffer (10 mM Tris-HCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 140 mM NaCl, and 1 × protease inhibitor). Sample buffer was then added to the cell lysates and proteins were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Germany). The membranes were blocked in Tris-buffered saline containing 0.2% Tween 20 and 5% fat-free dry milk and incubated first with primary antibodies and then with horseradish peroxidase-conjugated secondary antibodies. Specific proteins were visualized with enhanced chemiluminescence detection reagent according to the manufacturer’s instructions (Pierce Biotechnology, Waltham, MA, USA). Image acquisition and band intensity quantitation was performed using an Odyssey infrared imaging system.
Flow cytometric analysis
To detect cell surface PD-L1 expression, single NSCLC cells were resuspended in phosphate-buffered saline (PBS) and stained with APC anti-human PD-L1 (329707, BioLegend, US) antibody as previously described using standard protocols for flow cytometry [14]. An isotype IgG antibody was used as a negative control. Stained cells were evaluated using a BD FACSCanto II cytometer and data analyzed using the FlowJo software.
T-cell-mediated tumor cell killing assay
T cells were activated by a CD3 antibody (100 ng/ml) and interleukin-2 (10 ng/ml). After 4 days of tumor cell and T cell co-culturing in 24-well plates, the wells were washed with PBS twice to remove the T cells, and the surviving tumor cells were fixed and stained with a crystal violet solution. The dried plates were then scanned and quantified.
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 15 min, and then blocked with 5% BSA for 1 hour. After incubation with primary antibodies overnight at 4°C, cells were then incubated with a secondary antibody tagged with fluorescein isothiocyanate (Life Technologies, Thermo Fisher Scientific) for 1 hour at room temperature followed by staining nuclei with DAPI in the mounting reagent (Invitrogen). Confocal fluorescence images were captured using a Zeiss LSM 710 laser microscope.
Statistical analysis
Statistical analyses were performed using the SPSS software program (v20.0, IBM, Chicago, IL). ANOVA was used to evaluate the differences between groups. P values < 0.05 were considered statistically significant.
Results
c-MET inhibition enhances PD-L1 expression in NSCLC cells
The failure of c-MET inhibitor tivantinib in phase III NSCLC clinical trials and the recent preclinical study on the c-MET inhibitors and PD-L1 relationship prompted us to ask whether c-MET inhibitors regulate PD-L1 expression in NSCLC cells. To this end, we first validated c-MET inhibitor-mediated upregulation of PD-L1 in NSCLC cell lines, including human NSCLC cell lines H1975 and H1993 by Western blot analysis (Figure 1A and 1B). To determine whether tivantinib-mediated PD-L1 upregulation is c-MET dependent, we used two independent short hairpin RNAs (shRNAs) to knockdown c-MET expression in NSCLC cells. As shown in Figure 1C and 1D, knocking down c-MET in H1975 and H1993 induced PD-L1 expression. Flow cytometric analysis further validated the above results in which c-MET knockdown enhanced the expression of cell-surface PD-L1 in H1975 and H1993 cells similar that of the positive control, IFNγ (Figure 1E and 1F). To corroborate the above findings, we treated H1975 and H1993 with increasing concentrations of tivantinib and for different time periods. Our findings indicated that the PD-L1 expression increased in a dose- and time-dependent manner (Figure 2A-D). Likewise, the expression of PDL1 on the cell surface was also upregulated (Figure 2E, 2F). Together, these results indicated that inactivation of c-MET inhibitor upregulates PD-L1 expression in NSCLC cells.
Figure 1.
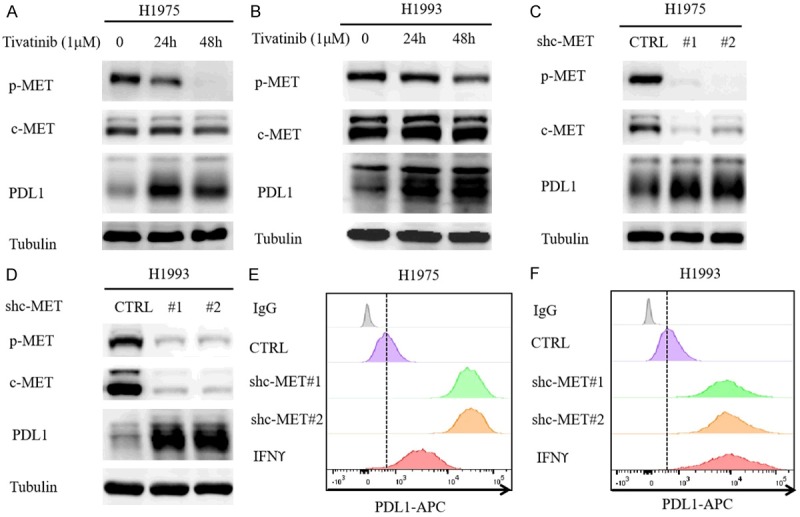
c-MET inhibitor upregulates PD-L1 expression in NSCLC cells. A and B. Western blot analysis of PD-L1 levels in NSCLC cell lines H1975 and H1993 treated with c-MET inhibitor tivantinib (1 μM). C and D. Western blot analysis of PD-L1 levels in H1975 and H1993 shc-MET cells. E and F. Flow cytometric analysis of cell-surface PD-L1 in H1975 and H1993 shc-MET cells.
Figure 2.
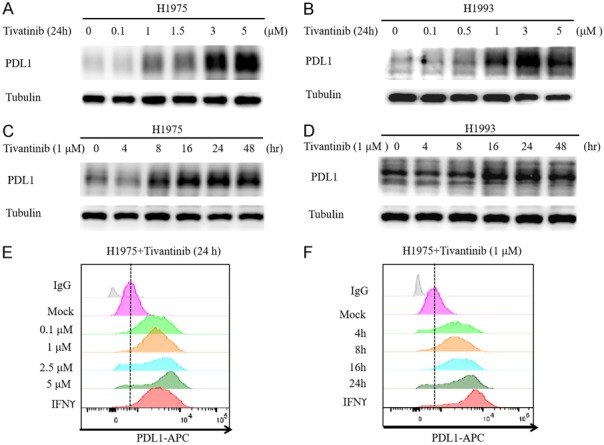
c-MET inhibitor induces PD-L1 expression in NSCLC cells in dose and time-dependent manner. A and B. Western blot analysis of whole cell lysates from H1993 and H1975 treated with the indicated concentrations of c-MET inhibitor tivantinib for 24 hours. C and D. Western blot analysis of whole cell lysates from H1993 and H1975 treated with c-MET inhibitor tivantinib (1 μM) for the indicated time. E. H1975 cells were treated with the indicated concentration of tivantinib for 24 hours followed by flow cytometric analysis of cell surface PD-L1 levels. F. H1975 cells were treated with tivantinib (1 μM) for the indicated time followed by flow cytometric analysis of cell surface PD-L1 levels.
c-MET inhibition drives PD-L1 expression by suppressing GSK3β
Next, we investigated the mechanisms by which c-MET inhibitor increases PD-L1 expression in NSCLC cells and asked whether this occurs via transcriptional or post-transcriptional regulation. To this end, we first examined PD-L1 mRNA levels in H1975 and H1993 cells treated with or without tivantinib. Compared with the untreated cells, tivantinib had no effects on PD-L1 mRNA expression (Figure 3A and 3B) in H1975 and H1993 cells, implying that the regulation is not at the transcriptional level. Pulse-chase analysis using cycloheximide indicated that knocking down c-MET increased the PD-L1 protein half-life in H1975 and H1993 cells (Figure 3C and 3D). Previously, we reported that glycogen synthase kinase 3 beta (GSK3β) downregulates PD-L1 protein stability [13], and c-MET can phosphorylate and activate GSK3β at Y56, which reduced expression of PDL1 by liver cancer cells [14]. To determine whether c-MET-mediated PD-L1 upregulation is GSK3β dependent in NSCLC cells, we treated the H1975 and H1993 with c-MET inhibitor tivantinib and evaluated the levels of p-GSK3βY56. As shown in Figure 3G and 3H, p-GSK3βY56 was abrogated whereas PD-L1 increased under tivantinib treatment. These results suggested that c-MET-mediated PD-L1 downregulation may occur via GSK3β in NSCLC cells similar to that previously reported in liver cancer cells [14].
Figure 3.
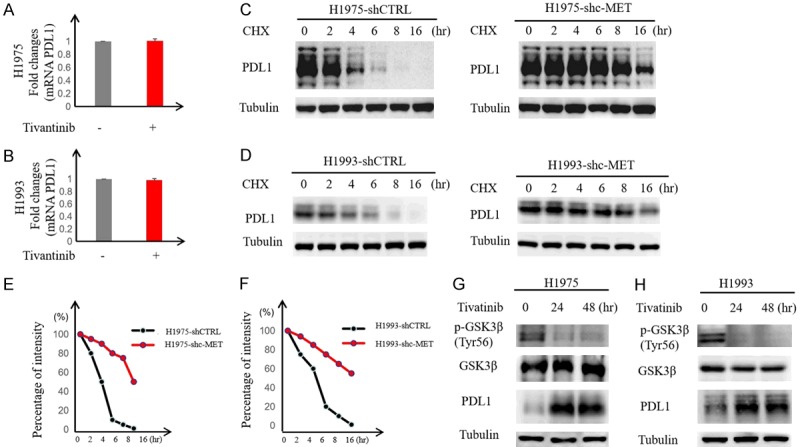
c-MET inhibition enhances PD-L1 expression by suppressing GSK3β. A and B. H1975 and H1993 cells were treated with tivantinib (1 μM) for 24 hours followed by RT-PCR to measure PD-L1 mRNA levels. Relative fold ratio of PD-L1 mRNA levels is shown. C and D. H1975 and H1993 shc-MET cells were treated with 20 mM cycloheximide (CHX) at the indicated intervals and subjected to Western blot analysis to evaluate PD-L1 expression levels. E and F. Quantification of PD-L1 half-life in the indicated groups. G and H. H1975 and H1993 cells were treated with tivantinib followed by Western blot analysis with the indicated antibodies. Tubulin served as loading control.
c-MET inhibitor blocks T-cell-mediated cell killing by stabilizing PD-L1
Given that binding of PD-L1 to PD-1 suppresses T-cell activation and that c-MET knockdown upregulates PD-L1 in NSCLC cells, we asked whether c-MET inhibition enables NSCLC cells to escape T-cell-mediated cancer cell killing. To this end, H1975 and H1993 cells were co-incubated with activated T cells in an in vitro co-culture assay. Indeed, knocking down c-MET blocked T-cell killing of H1975 and H1993 cells (Figure 4A-D). Those findings are consistent with the observed increase in PD-L1 expression under tivantinib treatment by confocal microscopy (Figure 4E and 4F). Together, these results suggested that c-MET inhibition stabilizes PD-L1 expression and allows NSCLC cells to escape from T-cell killing.
Figure 4.

c-MET inhibitor blocks T-cell-mediated cell killing by stabilizing PD-L1. (A and B) H1975 and H1993 shc-MET cells were co-cultured with or without activated T cells for 4 days followed by crystal violet staining to evaluate the cancer cell survival. Relative fold change in survival cell intensity is shown. *P < 0.05. (C and D) Quantitation of panel (A and B). (E and F) H1975 and H1993 cells were treated with or without tivantinib (1 μM) for 24 hours and then subjected to immunofluorescence assay to evaluate PD-L1 expression and localization.
Discussion
Recent studies reported a regulatory mechanism underlying the ineffectiveness of c-MET inhibitor in HCC treatment through PD-L1 upregulation [14]. However, it is not clear whether and how c-MET directly regulates PD-L1 expression in NSCLC. It has been proposed that PD-L1 may be regulated by multiple factors and activated in a complex process in different types of cancer cells [15]. Therefore, understanding the role of c-MET in immunotherapy in lung cancer may identify additional combination therapies to increase therapeutic efficacy. Here, we revealed a mechanism to explain the adverse reactions of c-MET inhibitors in clinical trials for lung cancer. Similar to HCC, c-MET inhibition reduced p-GSK3βY56, leading to the stabilization of PD-L1, which enabled tumor cells to evade immune surveillance. The addition of immunotherapy to c-MET inhibitors has the potential to eliminate cancer cell immunosuppressive activity of c-MET inhibitors to NSCLC patients. Recently, Glodde et al. reported that anti-PD-1 immunotherapy consistently led to c-MET-positive neutrophil recruitment in the tumors and regional lymph nodes and that c-MET inhibition could enhance the efficacy of anti-PD-1 immunotherapy [16]. Moreover, Zhang et al. demonstrated that the cyclin D-CDK4 axis upregulates PD-L1 stability through the ubiquitin E3 ligase Cullin 3SPOP, which provides a complementary molecular rationale for combining CDK4/6 inhibitor treatment with anti-PD-1/PD-L1 immunotherapy to enhance tumor regression [17]. These findings lend additional support to our findings that anti-PD-1 therapy combined with c-MET inhibitors can overcome the expression of PD-L1 induced by c-MET inhibitors in lung cancer. Posttranslational modifications of PD-L1 that stabilize its expression have emerged as important regulatory mechanisms that modulate immunosuppression in patients with cancer [18-20]. In this study, we found that c-MET inhibition also stabilizes PD-L1 in NSCLC, providing a rationale to combine c-MET inhibitor and anti-PD-1/-PD-L1 therapy to treat lung cancer.
In summary, our findings that inhibition of c-MET activity stabilizes PD-L1 and allows tumor cells to escape from T-cell killing partially explain the failure of c-MET inhibitors NSCLC in clinical trials. Together with the prior studies, our findings suggested that the combination of anti-PD-L1 with c-MET inhibitors may be an effective therapeutic strategy against NSCLC worthwhile to explore in the future.
Acknowledgements
This work was funded in part by the following: National Institutes of Health (CCSG CA016672); Cancer Prevention & Research Institutes of Texas (RP160710); Breast Cancer Research Foundation (BCRF-17-069); The University of Texas MD Anderson-China Medical University and Sister Institution Fund; Heilongjiang Provincial Innovation funding (2017LCZX85 to X.S.); China Scholarship Council (to X.S.); Swallow Fund of Tumor Hospital, Harbin Medical University (JJQN2014-03 to X.S.); and Ting Tsung and Wei Fong Chao Research Fund (to M.-C.H).
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 3.Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, Ahn MJ, Vansteenkiste JF, Su WC, Felip E, Chia V, Glaser S, Pultar P, Zhao S, Peng B, Akimov M, Tan DSW. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 2018;36:3101–3109. doi: 10.1200/JCO.2018.77.7326. [DOI] [PubMed] [Google Scholar]
- 4.Scagliotti GV, Von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, Hirsh V, Shepherd FA, Sequist LV, Sandler A, Ross JS, Wang Q, von Roemeling R, Shuster D, Schwartz B. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 2015;33:2667–74. doi: 10.1200/JCO.2014.60.7317. [DOI] [PubMed] [Google Scholar]
- 5.Spigel DR, Edelman MJ, O’Byrne KJ, Paz-Ares L, Mocci S, Shames DS, Smith D, Yu W, Paton VE, Mok T. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J. Clin. Oncol. 2017;35:412–420. doi: 10.1200/JCO.2016.69.2160. [DOI] [PubMed] [Google Scholar]
- 6.Jänne PA, Shaw AT, Camidge DR, Giaccone G, Shreeve SM, Tang Y, Goldberg Z, Martini JF, Xu H, James LP, Solomon BJ. Combined Pan-HER and ALK/ROS1/MET inhibition with dacomitinib and crizotinib in advanced non-small cell lung cancer: results of a phase I study. J Thorac Oncol. 2016;11:737–747. doi: 10.1016/j.jtho.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 7.Schöffski P, Gordon M, Smith DC, Kurzrock R, Daud A, Vogelzang NJ, Lee Y, Scheffold C, Shapiro GI. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur J Cancer. 2017;86:296–304. doi: 10.1016/j.ejca.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Yoshioka H, Azuma K, Yamamoto N, Takahashi T, Nishio M, Katakami N, Ahn M, Hirashima T, Maemondo M, Kim SW, Kurosaki M, Akinaga S, Park K, Tsai CM, Tamura T, Mitsudomi T, Nakagawa K. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study) Ann Oncol. 2015;26:2066–2072. doi: 10.1093/annonc/mdv288. [DOI] [PubMed] [Google Scholar]
- 9.Lee PC, Fang YF, Yamaguchi H, Wang WJ, Chen TC, Hong X, Ke B, Xia W, Wei Y, Zha Z, Wang Y, Kuo HP, Wang CW, Tu CY, Chen CH, Huang WC, Chiang SF, Nie L, Hou J, Chen CT, Huo L, Yang WH, Deng R, Nakai K, Hsu YH, Chang SS, Chiu TJ, Tang J, Zhang R, Wang L, Fang B, Chen T, Wong KK, Hsu JL, Hung MC. Targeting PKCδ as a therapeutic strategy against heterogeneous mechanisms of EGFR inhibitor resistance in EGFR-Mutant lung cancer. Cancer Cell. 2018;34:954–969. doi: 10.1016/j.ccell.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, Zou W, Chen DS, Yi J, Sandler A, Rittmeyer A POPLAR Study Group. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 12.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, Chang SS, Lee HH, Chou CK, Liu YL, Yeh HC, Perillo EP, Dunn AK, Kuo CW, Khoo KH, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Huang TH, Sahin AA, Hortobagyi GN, Yoo SS, Hung MC. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201. e110. doi: 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Ding Q, Wang Y, Yao J, Lee CC, Wu HJ, Sahin AA, Allison JP, Yu D, Hortobagyi GN, Hung MC. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Li CW, Li X, Ding Q, Guo L, Liu S, Liu C, Lai CC, Hsu JM, Dong Q, Xia W, Hsu JL, Yamaguchi H, Du Y, Lai YJ, Sun X, Koller PB, Ye Q, Hung MC. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PD-L1. Gastroenterology. 2019;156:1849–1861. doi: 10.1053/j.gastro.2019.01.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balan M, Mier y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290:8110–20. doi: 10.1074/jbc.M114.612689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O’Donnell JS, Szczepanski S, Brandes M, Eickhoff S, Das I, Shridhar N, Hinze D, Rogava M, van der Sluis TC, Ruotsalainen JJ, Gaffal E, Landsberg J, Ludwig KU, Wilhelm C, Riek-Burchardt M, Müller AJ, Gebhardt C, Scolyer RA, Long GV, Janzen V, Teng MWL, Kastenmüller W, Mazzone M, Smyth MJ, Tüting T, Hölzel M. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity. 2017;47:789–802. e789. doi: 10.1016/j.immuni.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, Guo J, Huang YH, Fan C, Ren S, Sun Y, Freeman GJ, Sicinski P, Wei W. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–86. doi: 10.1038/s41422-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, Wang HL, Yang WH, Yen EY, Chang WC, Zha Z, Lim SO, Lai YJ, Liu C, Liu J, Dong Q, Yang Y, Sun L, Wei Y, Nie L, Hsu JL, Li H, Ye Q, Hassan MM, Amin HM, Kaseb AO, Lin X, Wang SC, Hung MC. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. 2019;129:3324–3338. doi: 10.1172/JCI126022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018;78:6349–6353. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]