

Abstract
Since the prognosis for children with high-risk osteosarcoma (OS) remains suboptimal despite intensive multi-modality therapies, there is a clear and urgent need for the development of targeted therapeutics against these refractory malignancies. Chimeric antigen receptor (CAR) modified T cells can meet this need by utilizing the immune system’s potent cytotoxic mechanisms against tumor specific antigen targets with exquisite specificity. Since OS highly expresses the GD2 antigen, a viable immunotherapeutic target, we sought to assess if CAR modified T cells targeting GD2 could induce cytotoxicity against OS tumor cells. We demonstrated that the GD2 CAR modified T cells were highly efficacious for inducing OS tumor cell death. Interestingly, the OS cells were induced to up-regulate expression of PD-L1 upon interaction with GD2 CAR modified T cells, and the specific interaction induced CAR T cells to overexpress the exhaustion marker PD-1 along with increased CAR T cell apoptosis. To further potentiate CAR T cell killing activity against OS, we demonstrated that suboptimal chemotherapeutic treatment with doxorubicin can synergize with CAR T cells to effectively kill OS tumor cells.
Keywords: GD2, sarcoma, chimeric antigen receptor, PD-L1, PD1, doxorubicin
Introduction
Despite intensive multi-modality therapies employed in the treatment of osteosarcoma (OS) that encompass adjuvant chemotherapy coupled with maximal surgical resection including extremity amputation and/or hemipelvectomy, the prognosis for children with high risk disease remains suboptimal with overall survivals ranging from 20-40% [1-3]. OS requires local control, but remains relatively radio-resistant making surgical resection paramount for reducing primary tumor burden [4]. However, aggressive surgical resection in the up-front setting may have significant morbidity giving credence to neo-adjuvant chemotherapy. This allows post-surgical resections to be more manageable while prognosticating a patient’s risk category based on the percentage of tumor necrosis [5]. Nonetheless, patients with unresectable disease and/or metastatic disease have an ominous overall prognosis. Therapeutic options in this patient population are limited to alternative cytotoxic chemotherapeutic agents and small molecule inhibition, but all with unproven benefit [6-9]. Since the toxicity of systemic standard of care approaches leaves the few survivors of high risk OS with significant morbidity and life-long disabilities, there is a clear and urgent need for the development of targeted therapeutics against these refractory malignancies.
Chimeric antigen receptor (CAR) modified T cells can meet this need by utilizing the immune system’s surveillance capacity and potent cytotoxic mechanisms against tumor cells with exquisite specificity [10]. CAR modified T (CART) cells have already been leveraged in a host of refractory pediatric malignancies with promising therapeutic outcomes but require a tumor specific target to engender efficacious responses [11,12]. Since osteosarcoma highly expresses the GD2 antigen, this can serve as an important target for cellular therapeutics [8]. Meanwhile, GD2 expression has been utilized for immunotherapeutic intervention in neuroblastomas with promising results, and there is data suggesting that GD2 expression in OS may supersede its expression in neuroblastoma [13-15]. Based on these findings, we sought to assess if CAR modified T cells targeting GD2 were a viable therapeutic modality against OS tumor cell lines.
In this study, we established and compared several single chain monoclonal antibody-derived GD2 CARs and demonstrated that OS cells expressing high levels of GD2 (>80%) can be effectively targeted and killed by GD2 CART cells. However, OS cells were induced to up-regulate PD-L1, which in turn promoted apoptotic cell death of GD2 CART cells coupled with overexpression of PD-1. To further potentiate activity against OS, we further demonstrated that suboptimal dose of doxorubicin treatment synergized with CART cells to kill OS cells. To our knowledge, this is the first report demonstrating utility and efficacy of anti-GD2 CART cells against OS that can be enhanced through synergistic application with chemotherapy.
Materials and methods
Cell lines
Osteosarcoma cell lines HOS and U2OS cells were obtained from American Type Culture Collection (ATCC, Manassas VA, USA) and primary OS cells OS156 and OS758 cells were gift of Dr. Lori Rice. OS cells were cultured at 37°C in 5% CO2 atmosphere in Dulbecco’s modified eagle medium (DMEM), supplemented with 10% fetal bovine serum, 1% penicillin and 1% streptomycin (Gibco, Life Technologies, CA, USA).
Antibodies and chemicals
PE conjugated anti-GD2, anti-PD1, and mouse IgG2a antibodies, and PE-Cy7 conjugated mouse IgG1κ antibody were purchased from BD Biosciences, CA, USA. The PE-Cy7 conjugated anti-PD-L1 antibody was purchased from eBiosciences, CA, USA. Carboplatin and ifosfamide were purchased from Sigma-Aldrich (St Louis, MO, USA), and etoposide and doxorubixin were purchased from LC Laboratories (Woburn, MA, USA). All drugs were dissolved in DMSO except for carboplatin, which was dissolved in sterile water. All drugs were adjusted to 10 mM as stock concentration. The drug stocks were aliquoted and kept at -80°C avoid light until use. MTT powder was purchased from Sigma. Stock MTT solution was prepared by dissolving MTT powder in sterile PBS at the concentration of 5 mg/ml. Stock MTT solution was kept the same way as the stock drugs.
Flow cytometry analysis of surface antigens
OS cell lines or primary OS cells were grown to 80-90% confluency and harvested using 2.5-5.0 mM EDTA. The cells were blocked with 10% human-mouse serum (1:1) in FACS buffer, 2% FBS, 0.1% NaN3 in PBS, at 4°C for 30 minutes and stained with anti-GD2 or anti-PD-L1 fluorescence conjugated antibody at 4°C for 30 minutes in the dark. After staining, the cells were washed with FACS buffer and fixed with 1% paraformaldehyde in PBS. GD2 or PD-L1 surface expression were detected by BD LSRII flow cytometer and analyzed with FlowJo (FLOWJO, LLC, BD Biosciences). Percentage of ligand expression and mean fluorescence index (MFI) were determined by subtraction background obtained from isotype control.
Construction of 4th generation GD2 CAR lentiviral vectors
Lentiviral vectors were generated using the NHP/TYF lentiviral vector system as previously described [16,17]. DNA sequences of Anti-GD2 CAR clones hu3F8, c.60C3 and hu14.18 [15,18,19] were human codon-optimized chemically synthesized and cloned into the lentiviral vector pTYF and packaged into lentiviral particles for gene transfer. The hu3F8 and hu14.18 mAbs have been tested in clinical trials and c.60C3 has been reported to be highly tumor-specific [15,18,19]. The monomer GFP wasabi gene is a gift of Dr. Jiwu Wang (Allele Biotech). The final lentiviral constructs were verified by restriction enzyme mapping and DNA sequencing.
All three GD2 CAR sequences were constructed into a 4th generation lentiviral CAR design (4SCAR) as compared with the earlier generations of CAR design as illustrated in Figure 1. The 4SCAR incorporated several intracellular T cell signaling motifs including CD28 trans-membrane and cytoplasmic domain, the co-stimulatory 4-1BB intracellular TRAF binding domain, the CD3ζ chain intracellular domain, and an inducible self-destructive caspase 9 genetic cassette (4SCAR-GD2).
Figure 1.

Illustration of 4SCAR-GD2 lentiviral vector system. The NHP/TYF lentivector system was used for packaging of the 4SCAR-GD2 vectors. The 2nd, 3rd, and 4th generation of CAR lentivectors are illustrated. All three different anti-GD2 scFv genes were cloned into the 4SCAR vector linked to multiple intracellular signaling domains and fused with an inducible caspase 9 gene cassette (iCasp9).
Blood donors, PBMC isolation and T cell activation
Healthy donor blood buffy coats were obtained from Civitan blood center, and osteosarcoma patients’ blood samples were from Shands hospital with approval from the Institutional Review Board (IRB-01) of University of Florida. PBMC were isolated using Ficoll-Paque plus (GE Healthcare). T cells were activated using anti-CD3 and anti-CD28 antibody-conjugated magnetic beads or phytohemagglutinin (PHA). The T cells were maintained in TexMACS (Miltenyi Biotec Inc, San Diego, CA) or AIM-V (Invitrogen, Thermo Fisher Scientific, Waltham, MA) supplemented with interleukin-2, -7 and -15 as previously described [20]. Phenotype analysis of the activated cells were verified to confirm T cell purity. After expansion for two to six days, the T cells were transduced with lentiviral CAR vectors and evaluated killing function.
Osteosarcoma cell killing assay
GFP-positive OS cells were prepared as single cell suspension. The cells were resuspended in DMEM growth medium and seeded into 48-well plate at 3 × 104 cells/well and cultured at 37°C in 5% CO2 for 2 hours. Anti-GD2 CAR-modified T cells were added to the wells of OS cells at various effecter to target (E:T) ratio. The co-cultured cells were monitored for OS cell death either by flow cytometry or under fluorescence microscope (Zeiss Axiovert 25) and photographed.
For flow cytometer analysis of cell death, the cells were harvested using EDTA as described. The harvested cells were washed once with PBS and stained with Annexin V-V450 (BD Biosciences) and propidium iodide (Sigma) for 10 minutes at room temperature. Data were collected by BD LSRII flow cytometer and analyzed with FlowJo. The difference of OS cell death between anti-GD2 CART cells and control-CART cells was analyzed by independent t-test with p value < 0.05 being significant. Percent specific lysis of target cells was calculated based on the following formulation [21]: % specific lysis = (% apoptosis of target cell - % spontaneous cell apoptosis)/(100% - % spontaneous cell apoptosis) × 100.
PD-L1 surface staining of the co-cultured cells
The co-cultured OS cells and CART cells were stained for PD-L1 expression and analyzed using BD LSRII flow cytometer and FlowJo software. Percentage of PD-L1 expression and mean fluorescence index (MFI) were determined by subtraction of background isotype control. The difference of MFI was analyzed by independent T test with p value < 0.05 being significant.
Chemotherapeutic drug cytotoxic assay
HOS cells were seeded at 1 × 105 cells in 96 well culture plate for one day before treatment. The working drugs were prepared by diluting the stock drugs in 10% FBS DMEM. Two fold dilution range of 54.0-3.8 µM of carboplatin, 8,000-500 µM of ifosfamide, 2.0-0.125 µM etoposide and 0.25-0.0156 µM doxorubicin were freshly prepared before use. Cell culture media were replaced with the drug containing media at the final volume of 100 µl in triplicate wells. Cells added with 0.01-DMSO v/v and medium alone were included as a solvent control and a blank control, respectively.
After 3-day treatment, cell viability was measured by MTT assay. The stock MTT solution was added to the final concentration of 0.5 mg/ml per well and incubate for 4 hrs. The crystals were dissolved by adding 100 ul of acidified isopropanol and mix until homogeneous. The absorbance was measure using Cary® 50 UV-Vis spectrophotometer plate reader (Agilent Technologies) at 570 nM for test and 640 nM for reference. The cell viability was calculated as [(Drugtest - Drugreference) - (Blanktest - Blankreference)]/[(Controltest - Controlreference) - (Blanktest - Blankreference)] × 100%.
Dose response curve and IC50 were plotted and calculated using GraphPad PRISM® according to nonlinear fit curve analysis.
Cytotoxic assay of chemotherapeutic drugs and anti-GD2 CART cells
HOS cells were pre-treated with chemotherapeutic drugs before co-cultured with anti-GD2 CART cells. The drugs were used at the sub toxic concentration including 2 µM of carboplatin, 240 uM of ifosfamide, 100 µM of etoposide and 10 µM of doxorubicin. After 24 hours of treatment the drugs were removed and the cells were washed twice with sterile PBS. Anti-GD2 CART cells were then added to the drug-treated target cells at the E:T ratio of 1:2 with one half of 10% FBS DMEM and one half of TexMACS media. Twenty four hours after co-culture, cell death was determined by annexin V/PI staining and viability and caspase 3/7 activity multiplex assay.
For the caspase activity, ApoLive-Glo™ Multiplex Assay kit (Promega) was used to determine the cellular viability and caspase 3/7 activity level. Briefly, 10 µl of viability reagent was added to the culture, incubated for 1 hour in dark on 4°C followed by measuring the florescent viability signal. Subsequently, 100 µl of caspase 3/7 reagent was simultaneously added in the same wells, incubated for another 1 hour in dark on 4°C followed by measuring the luminescent caspase 3/7 activity signal. Both fluorescent and luminescent signal were detected using Appliskan® Filter-Based Multimode Microplate Readers (Thermo Scientific). Cellular viability and caspase 3/7 activity were expressed as RFU and RLU, respectively.
Results
Detection of GD2 expression in sarcomas
Two OS cell lines, HOS and U2OS cells, and two OS primary cells, OS156 and OS758, were analyzed for surface expression of GD2 by flow cytometry. GD2 expression was detected 80.1%, 99%, 94.3% and 48% on U2OS, HOS, OS156 and OS758 cells, with MFI of 7,066, 26,496, 72080, and 5212, respectively (Figure 2A-D). Survey of GD2 expression in surgical specimens by immunohistochemical staining, including osteosarcoma, rhabdomyosarcoma, and Ewing’s sarcoma showed that most sarcomas express GD2 (Figure 2E). Therefore, GD2 appears to be a good target antigen for immunotherapy of sarcomas.
Figure 2.
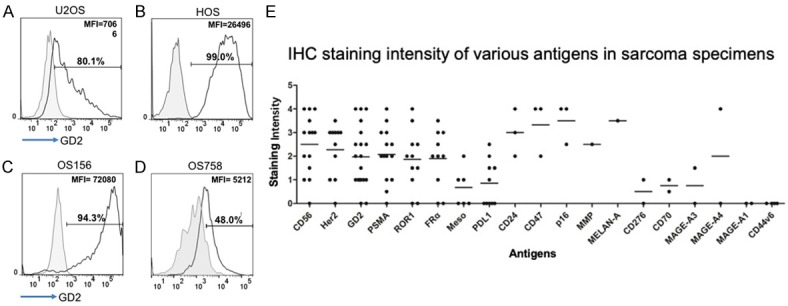
GD2 surface expression in OS cells. Surface expression of GD2 was illustrated by antibody staining and flow cytometry analysis; (A) U2OS (B) HOS (C) OS156 and (D) OS758. The grey area represents isotype control. The percentage and mean fluorescence intensity (MFI) of the positive population are indicated. (E) IHC staining of primary surgical sarcoma tumor sections with antibodies against different antigens. Sarcoma patients’ tumor sections were immunohistochemically stained with different antibodies as indicated and the staining intensities were numerically scored from low to high intensity (0-5) under microscope.
Functional evaluation of GD2-CARs based on a rapid killing assay
To evaluate CART functions, we used a simplified target cell-killing assay using lentiviral green fluorescence (wasabi GFP) labeled sarcoma cells. CAR-modified T (CART) cells were incubated with GD2-positive green sarcoma cells at different effector/target (E/T) ratios, and at various time points, the cultured cells were analyzed by flow cytometry after staining with annexin V and propidium iodide (PI). Figure 3A illustrates the killing assay; the target cell death is detected by the disappearance of green cells as shown by change in E/T ratios and increased apoptotic and PI-stained dead cells. The cells in Q1 are living cells, Q2 illustrates early apoptotic cells, Q3 illustrates late apoptotic stage, and Q4 illustrates dead cells. Based on this assay, we compared the three GD2 CARs and the results indicated that the hu3F8 and hu14.18 CART cells killed target cells at similar efficiencies but the c.60C3 GD2-CART did not show any target killing effects (Figure 3B). In repetitive studies, the hu3F8 CAR consistently showed the best target killing activities. Therefore, hu3F8 CAR was used in later experiments.
Figure 3.

GD2-CAR T cell killing assay. A. CAR T and target cell coculture and analysis of cytolytic activities by flow cytometry. B. Target cell killing analysis of the three different GD2 CAR T cells, Hu3F8, Hu14.18 and c60C3. The OS cells transduced with a lentiviral wasabi green fluorescence reporter gene were used as target cells, and plated into 96 or 48 wells for 2 hours before adding the different CAR T cells (Jurkat or primary T cells, at E:T ratio 3:1). After co-culture at 37°C, the cells were harvested using 2.5-5 mM EDTA, washed twice with PBS and stained with Annexin V and propidium iodide (PI). The percentage of target cell death was detected by flow cytometry and the % specific lysis was calculated as illustrated.
GD2-CAR-T targeting sarcoma cells
To examine OS cells killing, CART cells were co-cultured with wasabi marked U2OS and HOS reporter cell lines, U2OSw and HOSw. The specific target cell lysis was assessed by flow cytometry. Initial assay was based on Jurkat (JK) T cells transduced with GD2-CAR or control CD19-CAR and cocultured with U2OSw or HOSw to assess the CART killing ability. The result showed that compared with control (non-specific) CD19-CAR-JK cell, the percent specific lysis by GD2 CAR-JK cells was significantly increased against U2OSw (P = 0.0049), and similar trend was found against HOSw (P = 0.078; data not shown). To confirm this, T cells from healthy donors were transduced with GD2 CAR and co-cultured with U2OSw and HOSw in different E/T ratios (4:1, 2:1, 1:1 and 1:2), and the cells were evaluated after 1 day. Compared with control T cells and/or non-specific CAR (CD19-CAR), percent specific lysis of U2OSw was increased when co-cultured with GD2 CART cells but not with non-specific control CD19-CART cells (Figure 4A, 4B). In addition, HOSw cells were also specifically killed by GD2-CART cells but not by the control glypican 3 (GPC3)-CART cells (Figure 4C, 4D). Similar results were also observed with OS patient-derived CART cells (Figure S1).
Figure 4.

GD2-CAR-JK T cell killing of OS cells. Jurkat (JK) T cells were transduced with 4SCAR-GD2 lentivectors to evaluate the GD2 CAR T cell killing activities. A. Representative flow cytometry graphs of short term killing of U2OS cells by GD2 CAR-JK T cells at various E:T ratios. FITC positive target cells (upper panels) were gated and percentage of cell death was determined by Annexin V and PI staining (lower panels). B. Specific lysis of U2OS by GD2 CAR-JK cells vs. CD19 CAR-JK cells; *indicates a significant difference (P = 0.0049). C. Representative flow cytometry graphs of HOSw targeted by GD2 CAR-JK cells at various E:T ratios. FITC positive target cells (upper panels) and Annexin V and/or PI stained cells (lower panels) are shown. D. Target cell lysis of HOSw by GD2 CAR-JK cell vs. control GPC3 CAR-JK cells (P = 0.078).
Next, primary tumor cells, OS156 and OS758, which were OS patient-derived, were tested for GD2-CAR T killing. Lentiviral vector modified OS156w and OS758w green fluorescent cells were used as target cells and examined after 1 day co-culture at E:T ratio 1:1. The results showed that the GD2 CART cells also efficiently killed the primary sarcoma cells (Figure 5A, 5B, OS156w and Figure 5C, 5D, OS758w).
Figure 5.

4SCAR-GD2 T cell targeting patient-derived OS cells. Patient-derived and green fluorescence labeled OS lines, OS156w and OS758w, were cocultured with CAR T cells. Specific lysis was determined 1 day after co-culture at E:T ratio 1:1; (A, B) OS156w and (C, D) OS758w. Additional round of tumors cells were then added to the coculture at an increased target ratio, E:T = 1:3, and the results were recorded under fluorescence microscope on day 3, day 7 and day 12 (E).
To see if the GD2-CART cells could kill target tumors for extended period, after all tumors cells were killed in the co-culture, more tumor cells were added to the GD2-CART cells to set up a second round of killing, at an increased target ratio, E/T = 1:3. Evident under fluorescent microscope, the results showed that GD2 CART cells, but not control T cells or non-specific CD19-CART cells, were able to kill the target sarcoma cells (HOSw) continuously at an increased tumor ratio (Figure 5E).
Upregulation of PD-L1 and PD1 expression on OS cells and GD2-CART cells, respectively, after CART and target interaction
PD-L1 is a ligand of PD1 related to immune checkpoint inhibition of T cell functions [22]. To see if CART could affect PD-L1 expression in OS cells, we analyzed PD-L1 on OS cells by flow cytometry. While all three sarcoma cells, HOS, U2OS, and OS156 expressed high levels of GD2, the expression of PD-L1 was high on HOS and U2OS but not on the primary OS156 (Figure 6A). After incubation with GD2 CART versus control CD19 CART cells, flow cytometry showed that the percentage of PD-L1 expression was not changed (HOSw and U2OSs, Figure 6B), but the quantitative levels of PD-L1 expression on cell surface as determined by mean fluorescence index (MFI) revealed significant increase upon interaction with GD2 CART cells but not with the control CART cells (Figure 6C, P < 0.05 for both HOS and U2OS).
Figure 6.
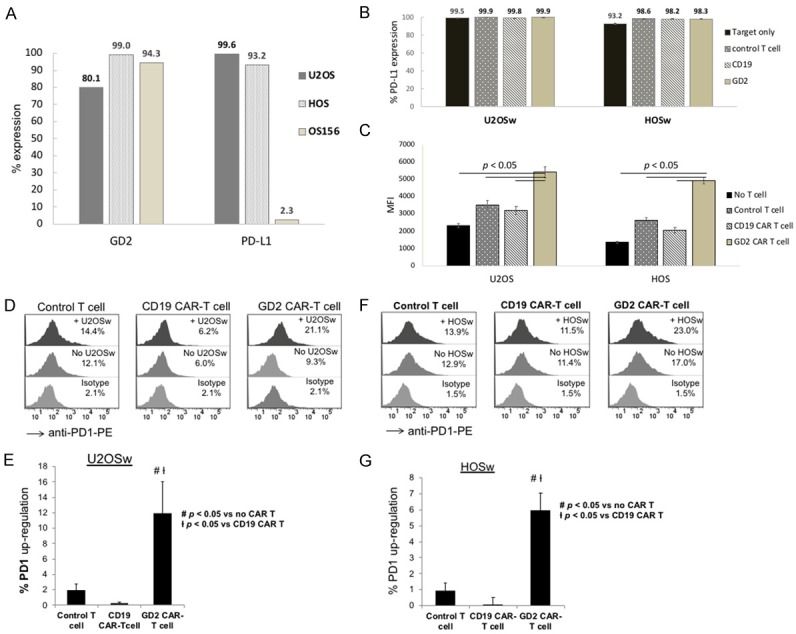
Upregulation of PD-L1 and PD1 on OS cells and CAR T cells, respectively. (A) Analysis of PD-L1 expression on OS cell lines. The percentage of PD-L1 expression was plotted for U2OS, HOS and OS156 cells after surface staining and flow cytometry analysis. (B, C) Analysis of PD-L1 expression levels on OS cells after CAR T interaction. OS cell lines, U2OSw and HOSw were incubated with different CAR T cells as indicated, and after 24 hr, PD-L1 expression was analyzed by flow cytometry, and percentage (B) and MFI (C) of surface PD-L1 expression were plotted. The levels of PD-L1 expression were significantly higher for the two OS cells incubated with the 4SCAR-GD2 T cells (P < 0.05). (D-G) Analysis of PD1 expression on CAR T cells. Control T cells and CAR T cells were incubated with U2OSw cells, and 24 hr later, stained with anti-PD1 antibody and analyzed by flow cytometry. Flow cytometry histograms of T cells stained with isotype control antibody, or anti-PD1 antibody cocultured with or without U2OSw or HOSw cells are shown (D and F). The percentage of PD1 upregulation after subtraction of background PD1 expression was determined and illustrated by bar graphs (E and G), and significant difference (P < 0.05) was established between GD2 CAR T vs. control T and CD19 CAR T cocultures.
To see if target interaction up-regulated PD-1 in CART cells, we examined PD-1 expression on GD2 CART cells upon target cell engagement. The result showed that GD2 CART cells, but not control T or the non-specific CD19-CART cells, displayed significantly increase in PD-1 expression upon interaction with U2OSw (Figure 6D, 6E) and HOSw target cells (Figure 6F, 6G).
Synergistic effect of anti-GD2 CART cells with doxorubicin
Chemotherapy is known to have immune modulatory effects besides killing cancer cells. The chemotherapy drugs for OS include platinum-based carboplatin, doxorubicin, etoposide and ifosfamide. To see if metronomic application of chemotherapy drugs can modulate CART response, we investigated possible synergistic effect of these drugs at sub-toxic levels with CART cells.
IC50s of the different drugs were first determined based on MTT viability assay. The IC50s of carboplatin, ifosfamide, etoposide and doxorubicin on osteosarcoma were determinted to be 10.39 µM, 1,202 µM, 0.51 µM and 0.102 µM, respectively (Figure 7). We chose sub-toxic doses including 2 µM of carboplatin, 240 µM of ifosfamide, 100 nM of etoposide and 10 nM of doxorubicin for the combination treatment also to minimize possible drug cytotoxic effect on immune cells. After pre-treatment with the different drugs, tumor cells were co-cultured with GD2 CART cells at E/T ratio of 2/1 to observe the drug-CART synergistic effect. The results after annexin V and PI staining showed that only treatment with doxorubicin significantly increased the percentage of tumor cell killing by GD2 CART cells (Figure 8A, 8B), which was confirmed by significantly decreased target cell viability and increased caspase 3/7 activities after doxorubicin pretreatment (Figure 8C). We then analyzed PD-L1 expression after sub-toxic chemotherapy on the target cells, which were shown to express high level of PD-L1. The result demonstrated that treatment with low dose of doxorubicin significantly decreased the intensity of PD-L1 on the tumor cells (Figure 8D, 8E).
Figure 7.

IC50 determination of chemotherapeutic drugs on HOS cells. HOS cells were treated with different concentrations of carboplatin, ifosfamide, etoposide, and doxorubicin and cell viability was determined by MTT assay. IC50 of each drug was determined and shown in the graphs.
Figure 8.

Sub-toxic chemo-conditioning synergizes 4SCAR-GD2 T cell killing activity. HOSw cells were treated for one day with sub-toxic doses of the following: 2 µM of carboplatin, 240 µM of ifosfamide, 100 nM of etoposide and 10 nM of doxorubicin, and the drug was removed, and GD2 CART cells were then added at E:T ratio of 2:1. Target cell death was determined by annexin V and PI staining (A), and cell death percentage (B) and cell viability and caspase 3/7 activities (C) were plotted, and significant difference was established (**, P < 0.05). After low dose (10 nM) of doxorubicin treatment, the expression of PD-L1, shown by % (D) and MFI (E) by flow cytometry, was significantly decreased on the tumor cells (#, *P < 0.05).
Discussion
Patients with residual and refractory sarcoma may benefit from targeted therapies to eliminate metastatic resistant OS cells and metastatic propagation. Adoptive CART cell therapy represents a novel targeted therapeutic modality to exquisitely eliminate resistant neoplastic cells with unparalleled specificity and provide a novel approach to subvert relapsed/refractory sarcoma [12,23]. Here we demonstrated that T cells expressing a 4th generation GD2 CAR with a safety design (4SCAR-GD2) effectively killed the tumor cells and, in combination with sub-toxic level of doxorubicin, displayed a synergistic target killing effect.
GD2 represents a promising immunotherapeutic target not only for osteosarcoma, but also for several other sarcomas including Ewing’s sarcoma, rhabdomyosaromca and synovial sarcoma, all of which have been shown to express the GD2 antigen [24,25]. Sarcomas are difficult to eradicate without appropriate local control, but CART cells may assist in making surgical resections more manageable for tumors with limited responsiveness to chemotherapy. CART cells may also hold promise for eradicating micro-metastatic disease through the induction of long-lasting immunologic memory.
Importantly, GD2 has been identified as an efficacious therapeutic target in neuroblastoma where a chimeric antibody CH14.18 targeting GD2 has demonstrated activity in patients with high-risk disease [15,26]. While this chimeric antibody appears to be safe and efficacious, it is administered in the context of GM-CSF and high dose IL-2 in order to induce antibody dependent cytotoxic killing, but these agents are not easily tolerated [27]. Moreover, antibodies induce passive immunity without demonstrable immunological memory whereas CAR modified T cells can act directly on tumor cells independent of the endogenous immune system and requirement for antigen presentation, while engendering long term recall responses [28]. We have established several GD2 CARs based on different anti-GD2 scFvs and illustrated that both CH14.18 scFv- and 3F8 scFv-derived CART cells can effectively kill sarcoma cells. There have been attempts to generate anti-GD2 CAR modified T cells against neuroblastomas; a recent phase I study corroborated the safety of first generation anti-GD2 CART cells despite limited progression free survival [13]. While first generation anti-GD2 CART cells appear to be safe, later generation CARs embedded with additional co-stimulatory domains to improve their activation and persistence against cognate antigens, may be more efficacious. However, enhanced efficacy from later generation CART cells may impose increased toxicity from systemic cytokine release and increased risk of targeting normal cells expressing GD2. Consequently, novel CAR designs embedded with suicide genes may prevent acute toxicities and mitigate against long-term sequela in patients.
CART cell therapy can be highly effective in targeting antigen-specific tumor cells but could also harm normal tissue expressing these antigens [29]. Anti-CD19 CAR modified T cells have resulted in extended B cell deficiency in responders rendering these patients dependent on IVIG replacement [30]. Therefore, as this technology is further leveraged towards a plethora of refractory malignancies, it will be requisite to engineer a control switch into CART cells to prevent systemic toxicity to normal tissue [31,32]. Here we have prioritized the development of an iCasp9 suicide gene-incorporated 4th generation CAR (4SCAR-GD2) to target the GD2 antigen in OS. Our results demonstrated high target killing effect of the 4SCAR-GD2 against OS.
Although effective in OS recognition and killing, the GD2 CART cells may also face potential tumor resistance. We have observed that increased tumor cells to effector CAR T cell ratios diminished the tumor killing efficacy and longevity of the CART cells. Through surface staining, we discovered that OS cells expressed high level of PD-L1, which was further up-regulated when in contact with GD2 CART cells (Figure 6C). Furthermore, CART cells showed PD-1 up-regulation and increased apoptosis upon incubation with OS cells (Figure 6E, 6G). Such tumor/effector T cell interplay may substantially diminish CART cell killing efficacy. We have further examined the possibility of sensitizing tumor cells with optimized chemotherapeutics in combination with the 4SCAR-GD2 therapy (Figure 7). Upon screening of several existing drugs, we showed that doxorubicin, but not carboplatin, Ifosfamide, and etoposide, could specifically sensitize OS to GD2 CART killing, which induced decreased expression of PD-L1 on the target cells (Figure 8).
Recently, checkpoint blockade strategies have been shown to induce anti-tumor activity in clinical trials [33-35]. Blockade of exhaustion markers along PD-1/PD-L1 axis has been shown to precipitate immunologic recognition and rejection of neoplastic cells, in combination with CART cell therapy [36]. Since CART cells often require lymphodepletion strategies to enhance their in vivo viability, concomitant blockade of PD-1 may obviate the need for cytotoxic lymphodepletion in PD-L1 expressing cancers such as OS. Further safety and efficacy studies are required to examine the relationship of checkpoint blockade with concomitant CART cells with and without lymphodepletion.
In summary, the current outcomes for many children with high-risk sarcomas including OS are unacceptable necessitating the development of more targeted therapeutics. Anti-GD2 expressing CART cells provide a novel targeted approach to eliminate residual disease with consummate specificity. We have utilized this technology to engineer newer generation CART cells that harnesses the vital co-stimulatory domains imperative for T cell activation while embedding the safety features of suicide gene control. These CART cells have demonstrable cytotoxicity against GD2 expressing OS cells that can be further synergized with sub-toxic chemotherapy. These results warrant further validation in relevant animal models and human trials. Thus, as immunotherapy using CART cells becomes further entrenched in the forefront against refractory malignancies, OS provides a new and compulsory niche to further test the feasibility, safety and potential efficacy of this promising new technology.
Acknowledgements
This study was supported by grants from research funds of UF Foundation (P0021205), Science and Technology Planning Technical Research Project of Shenzhen (JCYJ20170413154349187, JCYJ20170817172416991, JCYJ20170817172541842, and KQTD20140630143254906) and UESTC (SQ2016YFSF090567).
Disclosure of conflict of interest
LJC serves as President of Shenzhen Geno-Immune Institute and may benefit financially as a result of the outcomes of LJC’s research or work reported in this publication. The authors declare no competing financial interests.
Supporting Information
References
- 1.Ferrari S, Briccoli A, Mercuri M, Bertoni F, Picci P, Tienghi A, Del Prever AB, Fagioli F, Comandone A, Bacci G. Postrelapse survival in osteosarcoma of the extremities: prognostic factors for long-term survival. J. Clin. Oncol. 2003;21:710–715. doi: 10.1200/JCO.2003.03.141. [DOI] [PubMed] [Google Scholar]
- 2.Boye K, Del Prever AB, Eriksson M, Saeter G, Tienghi A, Lindholm P, Fagioli F, Skjeldal S, Ferrari S, Hall KS. High-dose chemotherapy with stem cell rescue in the primary treatment of metastatic and pelvic osteosarcoma: final results of the ISG/SSG II study. Pediatr Blood Cancer. 2014;61:840–845. doi: 10.1002/pbc.24868. [DOI] [PubMed] [Google Scholar]
- 3.Aksnes LH, Hall KS, Folleraas G, Stenwig AE, Bjerkehagen B, Taksdal I, Winderen M, Bruland OS, Saeter G. Management of high-grade bone sarcomas over two decades: the Norwegian Radium Hospital experience. Acta Oncol. 2006;45:38–46. doi: 10.1080/02841860500466624. [DOI] [PubMed] [Google Scholar]
- 4.Zuch D, Giang AH, Shapovalov Y, Schwarz E, Rosier R, O’Keefe R, Eliseev RA. Targeting radioresistant osteosarcoma cells with parthenolide. J Cell Biochem. 2012;113:1282–1291. doi: 10.1002/jcb.24002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47:2431–2445. doi: 10.1016/j.ejca.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 6.Baldini N, Scotlandi K, Barbanti-Brodano G, Manara MC, Maurici D, Bacci G, Bertoni F, Picci P, Sottili S, Campanacci M, et al. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med. 1995;333:1380–1385. doi: 10.1056/NEJM199511233332103. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz CL, Gorlick R, Teot L, Krailo M, Chen Z, Goorin A, Grier HE, Bernstein ML, Meyers P Children’s Oncology Group. Multiple drug resistance in osteogenic sarcoma: INT0133 from the Children’s Oncology Group. J. Clin. Oncol. 2007;25:2057–2062. doi: 10.1200/JCO.2006.07.7776. [DOI] [PubMed] [Google Scholar]
- 8.Picci P, Sangiorgi L, Rougraff BT, Neff JR, Casadei R, Campanacci M. Relationship of chemotherapy-induced necrosis and surgical margins to local recurrence in osteosarcoma. J. Clin. Oncol. 1994;12:2699–2705. doi: 10.1200/JCO.1994.12.12.2699. [DOI] [PubMed] [Google Scholar]
- 9.Lagmay JP, Krailo MD, Dang H, Kim A, Hawkins DS, Beaty O 3rd, Widemann BC, Zwerdling T, Bomgaars L, Langevin AM, Grier HE, Weigel B, Blaney SM, Gorlick R, Janeway KA. Outcome of patients with recurrent osteosarcoma enrolled in seven phase II trials through children’s cancer group, pediatric oncology group, and children’s oncology group: learning rrom the past to move forward. J. Clin. Oncol. 2016;34:3031–3038. doi: 10.1200/JCO.2015.65.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–2635. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, Gill J, Gorlick R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer. 2014;120:548–554. doi: 10.1002/cncr.28461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, Gillies SD, Cohn SL, Maris JM, Sondel PM Children’s Oncology Group. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang LJ, Liu X, He J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Therapy. 2005;12:1133–1144. doi: 10.1038/sj.gt.3302509. [DOI] [PubMed] [Google Scholar]
- 17.Wang B, He J, Liu C, Chang LJ. An effective cancer vaccine modality: lentiviral modification of dendritic cells expressing multiple cancer-specific antigens. Vaccine. 2006;24:3477–3489. doi: 10.1016/j.vaccine.2006.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014;588:288–297. doi: 10.1016/j.febslet.2013.11.030. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez-Rueda N, Leprieur S, Clemenceau B, Supiot S, Sebille-Rivain V, Faivre-Chauvet A, Davodeau F, Paris F, Barbet J, Aubry J, Birkle S. Binding activities and antitumor properties of a new mouse/human chimeric antibody specific for GD2 ganglioside antigen. Clin Cancer Res. 2007;13:5613s–5620s. doi: 10.1158/1078-0432.CCR-07-1057. [DOI] [PubMed] [Google Scholar]
- 20.Okada S, Han S, Patel ES, Yang LJ, Chang LJ. STAT3 signaling contributes to the high effector activities of interleukin-15-derived dendritic cells. Immunol Cell Biol. 2015;93:461–471. doi: 10.1038/icb.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godal R, Keilholz U, Uharek L, Letsch A, Asemissen AM, Busse A, Na IK, Thiel E, Scheibenbogen C. Lymphomas are sensitive to perforin-dependent cytotoxic pathways despite expression of PI-9 and overexpression of bcl-2. Blood. 2006;107:3205–3211. doi: 10.1182/blood-2005-07-2880. [DOI] [PubMed] [Google Scholar]
- 22.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA, June CH, Kalos M. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castel V, Segura V, Canete A. Treatment of high-risk neuroblastoma with anti-GD2 antibodies. Clin Transl Oncol. 2010;12:788–793. doi: 10.1007/s12094-010-0600-y. [DOI] [PubMed] [Google Scholar]
- 25.Navid F, Santana VM, Barfield RC. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr Cancer Drug Targets. 2010;10:200–209. doi: 10.2174/156800910791054167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, Gan J, Hutson P, Seo S, Kim K, Goldberg J, Hank JA, Billups CA, Wu J, Furman WL, McGregor LM, Otto M, Gillies SD, Handgretinger R, Santana VM. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J. Clin. Oncol. 2014;32:1445–52. doi: 10.1200/JCO.2013.50.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013;73:1547–1558. doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozlowska A, Mackiewicz J, Mackiewicz A. Therapeutic gene modified cell based cancer vaccines. Gene. 2013;525:200–207. doi: 10.1016/j.gene.2013.03.056. [DOI] [PubMed] [Google Scholar]
- 29.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, Levine BL, Bushman FD, June CH. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC, Riddell SR, Press OW. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D, Panageas KS, Ritter G, Sznol M, Halaban R, Jungbluth AA, Allison JP, Old LJ, Wolchok JD, Gnjatic S. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011;108:16723–16728. doi: 10.1073/pnas.1110814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.