

Abstract
The limited treatment options and therapeutic failure due to acquired resistance for patients with triple-negative breast cancer (TNBC) represent a significant challenge. Inhibitors against poly (ADP-ribose) polymerase (PARP), olaparib and talazoparib, were recently approved for the treatment of metastatic breast cancer (including TNBC) in patients with germline BRCA1/2 mutations. Despite impressive response rates of ~60%, the prolongation in median progression-free survival with a PARPi is modest, suggesting the emergence of resistance. Several studies have reported that receptor tyrosine kinases (RTKs), such as c-MET (also known as hepatocyte growth factor receptor), are involved in resistance to various anti-neoplastic agents, including PARPi. However, the mechanism by which c-MET contributes to acquired resistance to PARPi in TNBC is not fully understood. In this study, we show that hyperactivated c-Met is detected in TNBC cells with acquired resistance to PARPi, and the combination of talazoparib and crizotinib (a multi-kinase inhibitor that inhibits c-MET) synergistically inhibits proliferation in these cells. Unexpectedly, depleting c-MET had limited effect on talazoparib sensitivity in PARPi-resistant cells. Interestingly, we found evidence of epidermal growth factor receptor (EGFR) hyperactivation and interaction of EGFR/c-Met in these cells. Notably, combining EGFR and PARP inhibitors resulted in greater inhibition of proliferation in c-MET-depleted TNBC cells, and combined c-MET and EGFR inhibition increased sensitivity to talazoparib in TNBC cells with acquired resistance to PARPi. Our findings suggest that combined inhibition of c-MET and EGFR could potentially re-sensitize TNBC to the cytotoxic effects of PARPi.
Keywords: c-MET, EGFR, PARP inhibitor, triple-negative breast cancer
Introduction
Triple-negative breast cancer (TNBC) is defined by the lack of expression of the three major actionable targets in breast cancer, namely, the estrogen receptor (ER), the progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) [1]. Together, this subtype of breast cancer accounts for 10-20% of primary breast cancers [2-5]. In contrast with hormone receptor-positive and/or HER2-positive breast cancers, TNBCs tend to be larger, more likely node-positive and of higher grade [6]. Although TNBCs are more likely to respond to cytotoxic chemotherapy in the neoadjuvant setting compared to other breast cancer subtypes, chemotherapy-resistant TNBC carries a dismal prognosis with a 40-80% risk of disease recurrence in the first 2-3 years following definitive surgical intervention [6-9].
Up to 80% and 35% of breast cancers diagnosed in BRCA1 and BRCA2 mutation carriers, respectively, are TNBCs. In contrast, only 10-30% of breast cancers diagnosed in non-BRCA1/2 mutation carriers are TNBCs [10,11]. On average, 35% of patients with TNBC carry a germline BRCA1 mutation and 8% of patients with TNBC carry a BRCA2 mutation. BRCA1/2 mutations result in defective homologous recombination, leading to accumulation of DNA damage [12], which increases the sensitivity of such tumors to DNA damaging agents, including platinum compounds and poly-ADP-ribose polymerase (PARP) inhibitors [13]. In addition, some BRCA1/2-wild type TNBCs have defective homologous recombination and share the increased sensitivity to DNA damaging agents observed in BRCA1/2-mutant tumors, a phenotype commonly termed BRCAness [13].
Results from two recent randomized phase III clinical trials [14,15] led to the approval of PARP inhibitors (PARPi) olaparib and talazoparib for patients with metastatic or advanced, HER2-negative breast cancer with germline BRCA1/2 mutations, including those with TNBC. The overall response rate to olaparib in the phase III OlympiAD trial in patients with measurable disease was 59.9%, compared to 28.8% in patients receiving standard therapy [14]. Median progression-free survival in patients receiving olaparib was 7.0 months, versus 4.2 months in patients receiving standard therapy [14]. In the phase III EMBRACA study, the response rate to talazoparib was 62.6%, compared to 27.2% in patients receiving standard therapy [15]. The median progression-free survival was 8.6 months in patients receiving talazoparib and 5.6 months in patients receiving standard therapy [15]. Thus, although response rates to PARPi in advanced or metastatic breast cancer are impressive, the 3-month improvement in progression-free survival is modest, suggesting the emergence of resistance to these novel agents.
Several mechanisms of resistance to PARPi have been described. First, restoration of homologous recombination through reversion mutations in BRCA1/2 [16] as well as concurrent mutations in TP53BP1 [17] or PTEN [18] have been shown to contribute to PARPi resistance. Second, increased reliance on alternate means of DNA repair like non-homologous end joining can limit the therapeutic efficacy of PARPi [19]. Third, since PARPi suppress DNA repair at replication forks and promote formation of double-strand breaks [20,21], stabilization of the replication fork can antagonize the anti-tumor effects of PARPi [22-24]. Fourth, reduced PARP expression [25] or binding [26] has been shown to result in PARPi resistance as did increased expression of PARPi efflux pumps [27]. Fifth, cell cycle checkpoint activation has been reported to result in cell cycle delay, giving malignant cells time to repair damaged DNA [28], resulting in resistance to PARP inhibition. Notably, inhibition of cyclin-dependent kinase 12 (CDK12) was found to enhance sensitivity to PARPi [29-32]. Additionally, increased WEE1 expression, which promotes cell cycle arrest and DNA repair, was found to result in PARPi resistance as well [33]. Similarly, CHK1 has been shown to induce cell cycle arrest in response to DNA damage [34] and inhibition of CHK1 can potentiate the anti-neoplastic effects of PARPi [35].
In addition to the above-mentioned mechanisms of resistance to PARPi, the receptor tyrosine kinase (RTK) c-MET has been shown to interact with and phosphorylate PARP1 at the Tyr907 residue, increasing the enzymatic activity of PARP and decreasing its binding to PARPi [36]. In a model of intrinsic resistance to PARPi, the combined inhibition of c-MET and PARP reduces proliferation of TNBC in vitro and in vivo [36,37]. Interestingly, the epidermal growth factor receptor (EGFR), another RTK, has also been shown to interact with c-MET, leading to phosphorylation of PARP1 at the Tyr907 residue, contributing to PARPi-resistance in hepatocellular carcinoma (HCC) [38]. In TNBC, dual targeting of MET and EGFR inhibits tumor growth in a more consistent manner compared to inhibiting either target alone [39] but the effects of c-MET and EGFR crosstalk signaling on PARPi resistance in TNBC remain unknown.
c-MET activity has been shown to enhance intrinsic resistance to PARPi in BRCA1/2 wild type TNBC [36]; however, its role in acquired resistance to PARPi in BRCA1/2-deficient TNBC remains unclear. In addition, because co-expression of RTKs has been shown to contribute to therapeutic resistance [39], the involvement of other RTKs in the setting of acquired resistance to PARPi remains a distinct possibility and an open question.
Materials and methods
Chemicals and antibodies
Olaparib and talazoparib were purchased from Selleck Chemical (Houston, TX), Crizotinib was purchased from LC laboratories. Antibodies against MET (#8198), phospho-MET (#3077), EGFR (#4267) and IgG (#2729) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for Lamin B (sc-365962) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody for phospho-EGFR (Ab5650) was purchased from Abcam (Cambridge, UK). The antibody against PARP1 pY907 was obtained from China Medical University (Taichung, Taiwan) [36].
Cell culture
The SUM149 cell line was obtained from Asterand Biosciences (Detroit, MI) and maintained in F12K medium containing 5% FBS, 10 mM HEPES, 1 mg/ml hydrocortisone, 5 µg/ml insulin, 100 units/ml penicillin, and 100 mg/ml streptomycin. Cells were validated by STR DNA fingerprinting using the AmpF_STR identifier kit following the manufacturer’s protocol (Applied Biosystems cat 4322288). The STR profiles were compared to ATCC fingerprints (ATCC.org) and the Cell Line Integrated Molecular Authentication (CLIMA) database version 0.1.200808 (http://bioinformatics.istge.it/clima/) [40]. The PARPi-resistant TNBC cell lines B3 and C12 were developed from SUM149 with 100 nM talazoparib treatment and were cultured as described above for SUM149.
Immunoblotting
Whole cell lysates were prepared in radioimmunoprecipitation (RIPA) buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin) with protease inhibitors (bimake.com) and phosphatase inhibitors (biotool.com). The concentration of protein in whole cell lysates was determined using the Pierce BCA protein assay kit (Fisher PI-23227) according to the manufacturer’s protocol. 10-40 µg of protein from each sample was separated in an 8% Bis-Tris SDS PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Life Technologies). After blocking with 5% bovine serum albumin (BSA), primary antibodies were incubated with the PVDF membranes overnight at 4°C. Membranes were washed in TBST (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.05% Tween-20) and hybridized with appropriate secondary antibodies for 45 min at room temperature and imaged using ECL reagents (Bio-Rad Laboratories).
Immunoprecipitation
Whole cell lysates were prepared in immunoprecipitation buffer (25 mM Tris, pH 7.4; 150 mM NaCl, 0.1% NP-40, 1 mM EDTA, 10% glycerol) and incubated with 1 µg of primary antibody or IgG control antibody overnight at 4°C. Protein G-agarose beads were then added and incubated at 4°C for 1 hr prior to washing and detection by immunoblotting as described in the previous section.
MTT assay
B3 cells (1250 cells/well) and C12 cells (750 cells/well) were seeded in a 96 well plate and incubated overnight. After 24 hr, cells were then treated with varying concentrations of talazoparib and/or crizotinib for 6 days. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added into each well to achieve a final concentration of 0.5 µg/ml before formazan extraction with DMSO. The optical density at 590 nm in each well was measured and then normalized to untreated wells. The Chou-Talalay method [41] was used to calculate the combination index using the Compusyn software (http://www.combosyn.com).
Plasmids and transfection
For knockdown of c-MET, the PARPi-resistant TNBC cell lines, B3 and C12, were transfected with pLKO-shRNA vector (Sigma-Aldrich, St. Louis, MO). shRNA sequences used in generating stable knockdown clones are as follows (5’ to 3’): shRNA1 (3’ UTR): CCGGGTGTGTTGTATGGTCAATAACCTCGAGGTTATTGACCATACAACACAC TTTTTTG; shRNA2: CCGGCCTTCAGAAGGTTGCTGAGTACTCGAGTACTCAGCAACCTTCTGAAG GTTTTTG.
Colony formation assay
B3 (1,500 cells/well) and C12 (1,500 cells/well) were plated into 24-well plates and incubated overnight. After 24 hr, cells were then treated with varying concentrations of talazoparib and/or crizotinib for 10 days. Colonies were fixed and stained with 0.5% crystal violet, washed, dried and imaged. Crystal violet was resolved from colonies using 33% acetic acid and absorbance was measured at 540 nm [36]. The Chou-Talalay method [41] was used to calculate the combination index using the Compusyn software (http://www.combosyn.com).
Tissue specimens
Breast tumor specimens from 19 patients with stage I-III breast cancer and a germline BRCA1/2 mutation were obtained at baseline and at the time of surgery following treatment with talazoparib in the neoadjuvant setting [42]. Reverse phase protein arrays [43] and whole exome sequencing were performed by the proteomics and sequencing core facility at The University of Texas MD Anderson Cancer Center, respectively. The study protocol was reviewed and approved by The University of Texas MD Anderson Cancer Center Institutional Review Board and all patients signed written informed consent (NCT02282345).
Statistical analysis
Student’s t test was used to compare two groups of independent samples. A P value < 0.05 was considered statistically significant.
Results
Phospho-c-MET expression is higher in acquired PARPi-resistant TNBC cells
To determine whether c-MET also plays an important role in acquired resistance to PARPi in TNBC, we examined its expression in PARPi-sensitive SUM149 TNBC cells and two SUM149-derived PARPi-resistant cells (B3 and C12). Both B3 and C12 exhibited significantly higher levels of phospho-c-MET expression compared with the SUM149 PARPi-sensitive parental cells when treated with vehicle control as well as after 24-hr exposure to either olaparib or talazoparib (Figure 1). The total c-MET expression was also higher in those two PARPi-resistant cells compared with the parental cells, albeit to a lesser extent. Therefore, the higher levels of c-MET phosphorylation observed in PARPi-resistant cells suggested that c-MET may play a role in mediating acquired resistance to PARPi.
Figure 1.
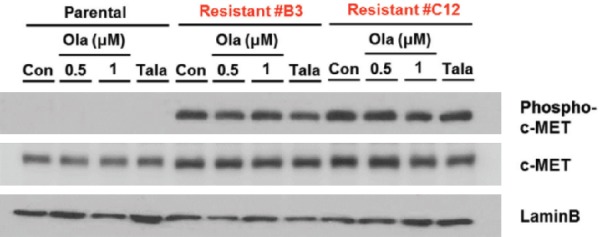
The B3 and C12 PARP inhibitor-resistant TNBC cells exhibit higher levels of phospho-c-MET expression compared with SUM149 PARPi-sensitive parental TNBC cells. The indicated cells were treated with vehicle control (con), olaparib (ola) at a concentration of 0.5 µM or 1.0 µM, or talazoparib (Tala) at a concentration of 25 nM for 24 hr prior to preparation of whole cell lysates. Immunoblotting was used to determine relative levels of phospho-c-MET, total c-MET and lamin B expression.
Crizotinib combined with talazoparib synergistically inhibits proliferation of acquired PARPi-resistant TNBC cells
To address whether c-MET inhibition can re-sensitize TNBC cells with acquired resistance to PARPi, we treated B3 and C12 PARPi-resistant cells with varying concentrations of crizotinib, a multi-kinase inhibitor that inhibits c-MET, and talazoparib. Cell killing activities of the inhibitors, either alone or in combination, were determined by the MTT assay and the combination index (CI) calculated using the Chou-Talalay method [41]. As shown in Figure 2A, the CI for crizotinib and talazoparib in B3 and C12 was below 1, indicating synergistic inhibition of cell proliferation. To validate these results, we performed a colony formation assay with results also showing synergistic inhibition of colony formation when cells were treated with the combination of crizotinib and talazoparib (Figure 2B). These findings indicated that combined inhibition of c-MET and PARP synergistically inhibited cell growth of TNBC cells with acquired PARPi resistance.
Figure 2.
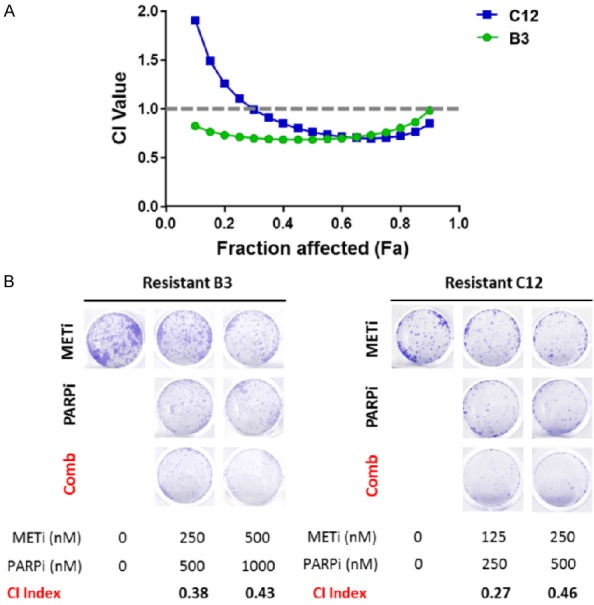
The combination of crizotinib and talazoparib synergistically inhibits cell proliferation of B3 and C12 PARPi-resistant TNBC cells. A. B3 and C12 PARPi-resistant TNBC cells were treated with varying concentrations of crizotinib and talazoparib for 6 days. Following 6 days of treatment, cells were subjected to 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The combination index (CI) was calculated, and synergistic inhibition of cell proliferation was defined as a CI < 1. B. Cells were seeded at low density and treated with varying concentrations of crizotinib and talazoparib for 10 days. Following 10 days of treatment, colonies were stained with 0.5% crystal violet and imaged.
Depletion of c-MET in PARPi-resistant TNBC cells has limited impact on restoring sensitivity to talazoparib
To specifically assess the role of c-MET in acquired resistance to PARPi in TNBC, we depleted c-MET by short hairpin RNA (shRNA) knockdown in B3 and C12 PARPi-resistant cells which express significantly higher levels of phospho-c-MET compared with SUM149 parental cells. Knockdown efficiency of > 50% was achieved in all cases (Figure 3A). We then treated the c-MET-depleted B3 and C12 cells and their c-MET-expressing counterparts with increasing concentrations of talazoparib. Depleting c-MET in C12 modestly increased sensitivity to talazoparib (Figure 3B) whereas depleting c-MET in B3 did not consistently increase sensitivity to talazoparib (Figure 3C). These results suggested that depleting c-MET has limited effect on restoring sensitivity to PARPi in TNBC cells with acquired resistance to PARPi.
Figure 3.
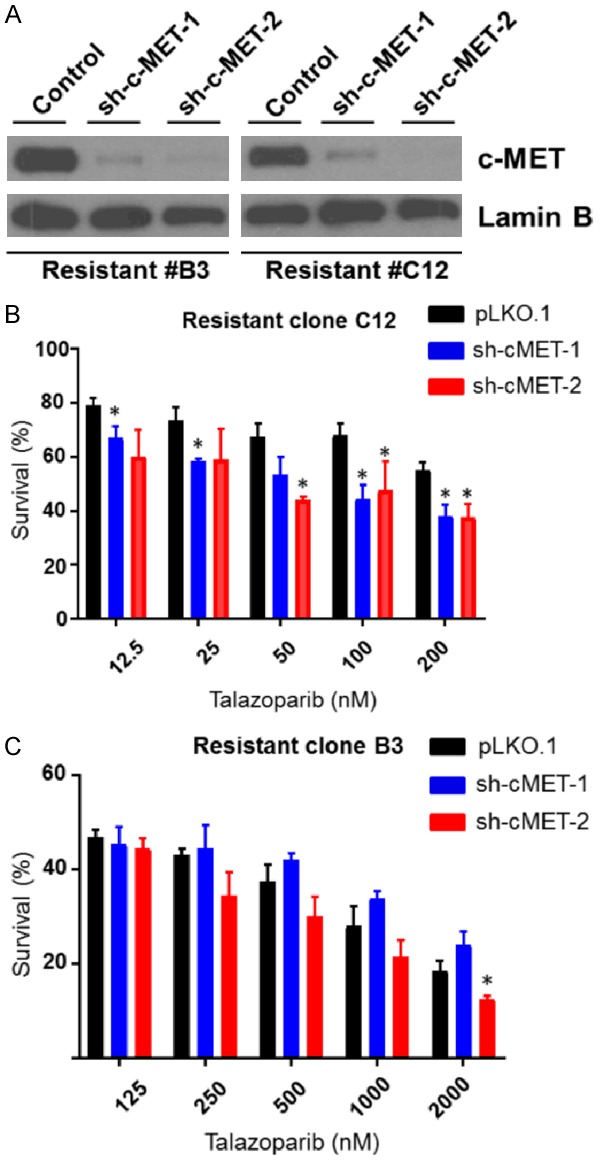
Depleting c-MET results in a limited effect of restoring PARPi sensitivity to talazoparib in the PARPi-resistant TNBC cells. (A) Following lentiviral delivery of two independent short hairpin RNAs (shRNA) targeting c-MET to B3 and C12 PARPi-resistant TNBC cells, c-MET expression levels were reduced by at least 50% relative to cells receiving non-specific shRNA sequences. B3 and C12 cells were infected with control shRNA (control), a shRNA targeting the 3’ UTR of c-MET (sh-c-MET-1), or a shRNA targeting the coding region of c-MET (sh-c-MET-2). Relative expression levels of total c-MET in stable clones were determined by immunoblotting. (B) C12 (750) and (C) B3 (1,500) cells were seeded in each well of a 96-well plate on Day 0. On Day 1, talazoparib was added at varying concentrations to each well (12.5-200 nM). Following 6 days of treatment, cells were subjected MTT, and the percentage of surviving cells in each well was calculated. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test.
c-MET cooperates with EGFR to mediate acquired PARPi resistance in TNBC
Although hyperactivated c-MET is observed in PARP-resistant TNBC cells, the modest effect of c-Met depletion on PARPi sensitivity implies that while c-Met may be involved in RTK-driven PARP resistance mechani-sm, additional molecules may work in tandem, contributing to the PARPi-resistant phenotype. c-MET is known to interact with multiple cell surface molecules [44] and form heterodimers with other RTKs to increase their downstream signaling, which in turn drives the malignant phenotype of the tumor [44]. Analysis of the online protein-binding database (https://thebiogrid.org/) identified EGFR as one of the RTK candidates that associates with c-Met. Several drugs targeting EGFR have been approved by the FDA, including gefitinib and erlotinib. Thus, we focused on the EGFR for all subsequent experiments. Based on the importance of EGFR in DNA repair and its clinical impact as a therapeutic target in TNBC [45,46], we evaluated the activity of EGFR in PARPi-resistant and SUM149 PARPi-sensitive cells. As expected, EGFR phosphorylation was highly increased in resistant cells and remained unaffected by PARPi treatment (Figure 4A). Interestingly, the expression of total EGFR and c-Met were not significantly upregulated in PARPi-resistant clones, but their activities (defined by relative levels of phosphorylation) were augmented. On the basis of those results, we hypothesized that enriched heterodimerization of EGFR and c-Met may contribute to acquired resistance to PARPi in TNBC cells through a kinase-dependent mechanism. To this end, we immunoprecipitated c-Met and then tested for interaction with EGFR. Our results revealed enhanced interaction between c-MET and EGFR in C12 compared with SUM149 (Figure 4B). We then immunoprecipitated c-MET in whole cell lysates obtained from B3 and C12 as well as SUM149 under basal conditions followed by treatment with talazoparib. The results showed that interaction between c-MET and EGFR is increased in PARPi-resistant B3 and C12 cells compared with PARPi-sensitive SUM149 parental cells (Figure 4C). Notably, c-MET depletion in the PARPi-resistant cells enhanced the efficacy of combined EGFR and PARP inhibition (Figure 4D and 4E). Next, we examined the effect of combined inhibition of c-MET, EGFR, and PARP on cell proliferation by using the MTT assay. The results showed that combined inhibition of c-MET and EGFR significantly increased the growth inhibitory effect of PARPi in the B3 and C12 PARPi-resistant cells whereas inhibiting either c-MET or EGFR alone did not (Figure 4F and 4G). Collectively, these data suggested that EGFR may cooperate with c-MET in mediating resistance to PARPi through their interaction.
Figure 4.
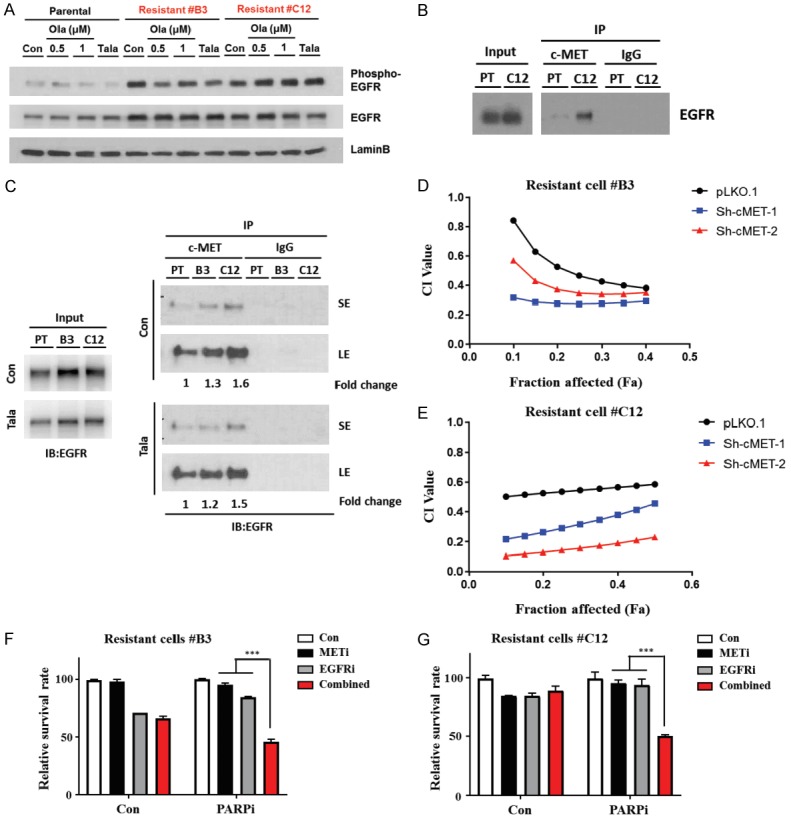
Enhanced interaction between c-MET and EGFR is detected in the PARPi-resistant TNBC cells relative to PARPi-sensitive parental TNBC cells. A. Parental (PT) and resistant (C12) cells were treated with talazoparib (Tala) at a concentration of 25 nM for 24 hr prior to preparation of whole cell lysates. B. Parental (PT) cells and resistant cells (B3 and C12) were treated with vehicle control (Con) or talazoparib (Tala) at a concentration of 25 nM for 24 hr prior to preparation of whole cell lysates. cMET or IgG control antibodies were used to immunoprecipitate the cell lysate. EGFR expression was then analyzed by immunoblotting. C. Cells were treated with vehicle control, olaparib at a concentration of 0.5 µM or 1.0 µM, or talazoparib at a concentration of 25 nM for 24 hr prior to preparation of whole cell lysates. Relative expression levels of phospho- and total-EGFR were determined by immunoblotting. D and E. Cells were seeded in each well of a 96-well plate on Day 0. On Day 1, talazoparib (125-2,000 nM) and gefitinib (31.25-500 nM) were added at varying concentrations to each well. F and G. Cells were seeded in each well of a 24-well plate on Day 0. On Day 1, talazoparib (100 nM), crizontinib (62.5 nM) and gefitinib (62.5 nM) were added at to each well. Following 6 days of treatment, cells were subjected to MTT, and the percentage of surviving cells in each well was calculated. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test.
Talazoparib-resistant breast tumor specimens exhibit enhanced protein expression of c-MET, total EGFR, and phospho-EGFR
To determine whether tumors from patients with breast cancer harbor enhanced expression of c-MET and EGFR, we identified 19 patients with previously untreated stage I-III breast cancer who received talazoparib in the neoadjuvant setting as part of a clinical trial conducted at The University of Texas MD Anderson Cancer Center (NCT02282345) [42]. Patients enrolled in this study underwent a pretreatment core needle biopsy at baseline and received up to 6 months of treatment with talazoparib in the neoadjuvant setting before undergoing surgery to remove any residual cancer. The amount of residual cancer was quantified using the residual cancer burden (RCB) index [47]. Patients were classified as having talazoparib-sensitive disease if they had a pathologic complete response or minimal residual disease (pCR/RCB-I) and classified as having talazoparib-resistant disease if they had significant residual disease (RCB-II/RCB-III). Of the 19 patients, four had sufficient tissue for paired analysis. Among the four patients with paired tissue samples, two had talazoparib-sensitive disease and the remaining two had talazoparib-resistant disease. Following treatment with talazoparib, tumors from patients with talazoparib-sensitive disease exhibited decreased c-MET (Figure 5A, left), EGFR (Figure 5B, left), and phospho-EGFR (Figure 5C, left) protein expression whereas tumors from patients with talazoparib-resistant disease had increased c-MET (Figure 5A, right), EGFR (Figure 5B, right), and phospho-EGFR protein expression (Figure 5C, right) as detected by RPPA. These results suggested the potential compensatory upregulation of these pathways in talazoparib-resistant breast cancer. To investigate whether any single nucleotide variants (SNVs) or insertions/deletions (indels) in MET or EGFR were present in tumors from this cohort of patients, we performed whole exome sequencing on baseline and surgical specimens obtained from the 19 patients. However, we did not find any SNVs or indels in either MET or EGFR, suggesting that changes in MET or EGFR expression in these tumors are not mediated by mutations in the respective genes.
Figure 5.
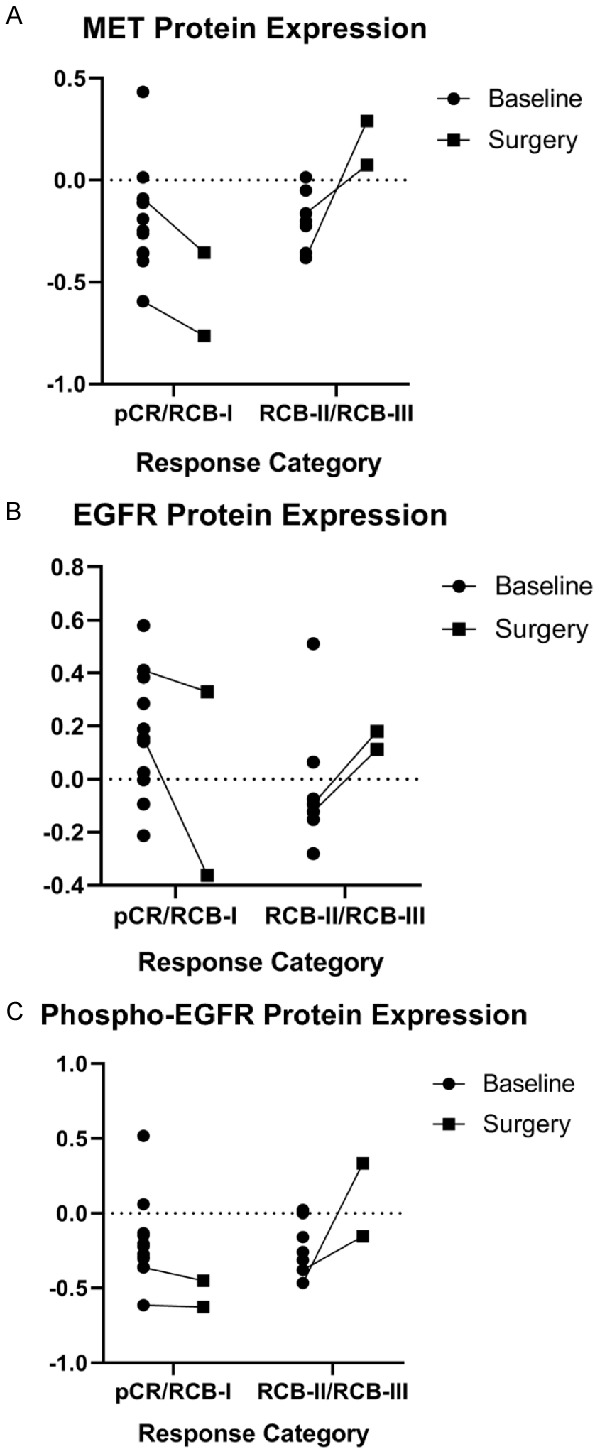
MET, EGFR and phospho-EGFR protein expression increased in patients with talazoparib-resistant breast cancer following treatment with single-agent talazoparib in the neoadjuvant setting. Patients with stage I-III breast cancer with a germline BRCA1/2 mutation were treated with talazoparib in the neoadjuvant setting prior to undergoing definitive surgery. Patients underwent a baseline core needle biopsy prior to initiating therapy. (A) MET (B) EGFR and (C) phospho-EGFR protein levels in pre-treatment biopsy specimens and surgical specimens obtained at the end of treatment were measured by reverse phase protein arrays. Patients were classified as having talazoparib-sensitive disease if they had a pathologic complete response or minimal residual disease (pCR/RCB-I) and classified as having talazoparib-resistant disease if they had significant residual disease (RCB-II/RCB-III).
Phosphorylation of PARP1 and other DNA repair proteins interacting with c-MET and EGFR may be potential mechanisms underlying PARPi acquired resistance in TNBC cells
Previously, in a model of intrinsic resistance to PARPi in TNBC, oxidative stress was shown to increase c-MET phosphorylation, which in turn phosphorylates PARP1 at the Tyr907 residue (PARP1 pTyr907 or pY907) [36], enhancing the enzymatic activity of PARP1 and reducing PARPi binding [36], leading to PARPi resistance. Although another study also reported oxidative damage-mediated increase in PARP1 pY907 expression [38], inhibiting c-MET activity with crizotinib did not consistently reduce PARP1 pY907 expression levels in HCC cell lines due to EGFR-mediated PARP1 phosphorylation [38]. Thus, because we observed increased interaction between c-MET and EGFR in B3 and C12 c-MET-overexpressing PARPi-resistant cells relative to SUM149 PARPi-sensitive parental cells, we hypothesized that c-MET and EGFR cooperate to augment resistance to PARPi through increased phosphorylation of PARP1 at the Tyr907 residue. However, while PARP1 pY907 levels were significantly higher in C12 compared to SUM149, they were not significantly higher in B3 (Figure 6). These data suggested that in addition to increased PARP1 pY907 expression, there are other potential mechanisms contributing to acquired resistance to PARPi in B3 cells.
Figure 6.
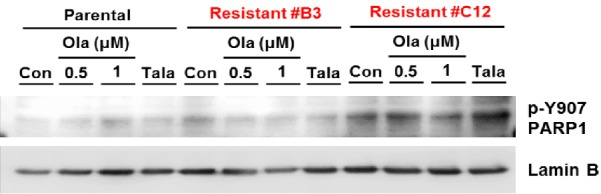
PARPi treatment increases p-Y907 PARP1 expression in C12 but not B3 resistant cells. PARPi-sensitive (SUM149) and PARPi-resistant TNBC cells (B3 and C12) were treated with vehicle control (Con), olaparib (Ola) at a concentration of 0.5 µM or 1.0 µM, or talazoparib (Tala) at a concentration of 25 nM for 24 hr prior to preparation of whole cell lysates. Cell lysates were then subjected to Western blotting with the indicated antibodies.
To explore other potential mechanisms of resistance that could be mediated by c-MET and EGFR, we utilized BioGRID (https://thebiogrid.org) and identified 91 and 1254 proteins known to interact with c-MET and EGFR, respectively. Of these, 33 were found to interact with both c-MET and EGFR (Table 1). Functional annotation of these 33 proteins using The Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) led to the identification of 6 proteins involved in DNA damage repair (Table 2). Among them, FBXO6 is an F box protein that is known to promote replication stress-induced Chk1 degradation. Low levels of FBXO6 has been associated with impairment of Chk1 degradation, leading to replication fork stabilization and activation of DNA repair responses, resulting in resistance to PARPi through an increased Chk1-mediated DNA damage repair [48]. STUB1 encodes an E3 ubiquitin-protein ligase, CHIP, which has been shown to ubiquitylate and degrade base excision repair proteins that are not part of the DNA repair complex, resulting in greater efficiency of base excision repair [49], which may in turn lead to resistance to PARPi. SFN encodes the protein 14-3-3 sigma which increases non-homologous end joining and upregulates PARP1 expression, enhancing DNA repair [50] and potentially contributing to PARPi resistance. CDH1 binds to and activates the anaphase promoting complex or cyclosome (APC/C) which degrades Plk1 in response to DNA damage and prevents cell cycle progression past G2 in cells with DNA damage [51]. Additionally, depleting CDH1 impairs Chk1 phosphorylation which may also reduce a cell’s ability to repair damaged DNA [51]. Thus, since CDH1 contributes maintaining genomic integrity in cells, its activation may limit sensitivity to PARPi. TP53 plays a crucial role in maintaining genomic stability [52], and it is plausible that EGFR and/or c-MET may, through its interaction with p53, promote its ability to prevent catastrophic DNA damage, inducing enhancing PARPi resistance. PTEN was recently demonstrated to promote repair of double strand DNA breaks via homologous recombination and loss of PTEN improved sensitivity to PARPi [53]. Thus, whether EGFR and/or c-MET promotes PARPi resistance by enhancing PTEN activity remains an open question. Together, the DNA repair proteins identified to interact both c-MET and EGFR may also contribute acquired TNBC resistant cells in tandem with increased PARP1 phosphorylation.
Table 1.
List of proteins known to interact with both c-MET and EGFR
| List of binding proteins of c-MET and EGFR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CBL | CBLC | CD44 | CDH1 | CRK | CTNNB1 | DCN | EPHA2 | ERBB3 | FBXO6 | GAB1 |
| GRB2 | HGS | HSP90AA1 | HSP90AB1 | HSPA4 | INPPL1 | ITGB1 | LRIG1 | MAP2K3 | MAP2K5 | NF2 |
| PTEN | RAF1 | SFN | SH3KBP1 | SHC1 | SOCS1 | SOS1 | SRC | STAT3 | STUB1 | TP53 |
Table 2.
List of proteins known to interact with both c-MET and EGFR that are involved in DNA damage repair
| Protein/Gene | Function |
|---|---|
| FBXO6 | DNA repair |
| STUB1 | DNA repair |
| SFN | Apoptotic response to DNA damage |
| CDH1 | DNA repair |
| TP53 | DNA damage response |
| PTEN | DNA damage repair |
Discussion
PARPi are the first targeted agents to receive FDA approval for the treatment of TNBC in the setting of germline BRCA1/2 mutations. However, despite having over double the response rates compared with standard therapy [14,15], the modest prolongation of median progression-free survival [14,15] suggests the emergence of resistance shortly after an initial response. We previously described the role of the RTK, c-MET, in a model of intrinsic resistance to PARPi using in vitro and in vivo models recapitulating BRCA1/2 wild-type TNBC [36]. However, whether c-MET plays a role in acquired resistance to PARPi in TNBCs is not clear.
In this study, we demonstrated that phospho-c-MET levels were higher in PARPi-resistant TNBC cells than in their PARPi-sensitive counterparts. Notably, this difference in phospho-c-MET expression between PARPi-resistant and PARPi-sensitive TNBC cells was present under basal conditions as well as following treatment with olaparib or talazoparib. In contrast, oxidative stress is required to induce phospho-c-MET expression in TNBC cells which were intrinsically resistant to PARPi [36]. However, although the combination of crizotinib and talazoparib resulted in synergistic inhibition of proliferation of PARPi-resistant TNBC cells, depleting c-MET did not consistently restore sensitivity to talazoparib. Following our previous work showing that EGFR interacts with c-MET to enhance resistance to PARPi in HCC [38], as well as the existence of a crosstalk between c-MET and EGFR in TNBC [39], we systematically evaluated the existence of c-MET-interacting RTKs in the PARPi-resistant TNBC cells and identified EGFR as a c-MET-interacting protein. Notably, the observed interaction between EGFR and c-MET remained relatively consistent under both basal conditions and following treatment with talazoparib in the C12 PARPi-resistant cells but appeared to be diminished following treatment with talazoparib in the B3 PARPi-resistant cells. The levels of phospho-c-MET and phospho-EGFR expression in B3 were also diminished following treatment with talazoparib, potentially explaining the reduced interaction between c-MET and EGFR, a finding consistent with earlier data from our laboratory demonstrating that phosphorylation of c-MET and EGFR promotes their interaction [38]. Of note, resistance to PARPi in HCC appears to be enhanced by phosphorylation of PARP1 by EGFR/MET heterodimers following oxidative DNA damage [38]. In addition, inhibitors of EGFR have been reported to inhibit its nuclear translocation [54] which may be necessary for EGFR/MET-mediated phosphorylation of PARP1, a nuclear protein. Given the role of PARPi in the DNA damage response, we identified six c-Met/EGFR-interacting proteins which can regulate DNA repair pathway and may contribute to PARPi resistance resulting from the interaction of c-MET and EGFR. In addition to these proteins, we also found enhanced PARP1 Y907 phosphorylation in C12 PARPi-resistant cells, consistent with our earlier findings in HCC [36]. Together, these data suggested that the interaction between EGFR and c-MET contributes to acquired resistance to PARPi in TNBC and that combined inhibition of EGFR, c-MET, and PARP may overcome therapeutic resistance and inhibit malignant proliferation of TNBC.
The identification of EGFR as an interacting protein of c-MET in PARPi-resistant TNBC cells suggest that dual inhibition of c-MET and EGFR may be required to restore sensitivity to PARPi in the setting of RTK-mediated resistance. In addition, since phospho-c-MET and phospho-EGFR levels were significantly elevated in PARPi-resistant TNBC cell lines compared to PARPi-sensitive TNBC cells, phospho-c-MET and phospho-EGFR could serve as potential biomarkers to identify patients likely to benefit from this combinatorial treatment strategy following development of resistance to PARPi.
Acknowledgements
This work was supported in part by the following: National Institutes of Health (CCSG CA016672, CA211615, and CA201777); Breast Cancer Research Foundation (BCRF-17-069); National Breast Cancer Foundation, Inc.; MD Anderson/China Medical University Sister Institution fund; Patel Memorial Breast Cancer Endowment Fund; Allison and Brian Grove Endowed Fellowship for Breast Medical Oncology, Susan Papizan Dolan Fellowship in Breast Oncology, and 2018 Gianni Bonadonna Breast Cancer Research Fellowship to C.Y.; Taiwan’s New Partnership Program For The Connection To The Top Labs In The World (Dragon Gate Program; 107-2911-I-006-519) to Y.-Y.C.; and National Institutes of Health T32 Training Grant in Cancer Biology (5T32CA186892) to M.-K.C. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the sponsors.
Disclosure of conflict of interest
Dr. Litton has received grant and/or research support from Novartis, Medivation/Pfizer, Genentech, GSK, EMD-Serono, Astra-Zeneca, Medimmune, Zenith and Jounce; served in Speaker’s Bureau for MedLearning, Physician’s Education Resource, Prime Oncology, Medscape, and Clinical Care Options; is an advisory committee, review panel, and/or board member of Astra-Zeneca, Pfizer (all uncompensated), NCCN, ASCO, NIH PDQ.
References
- 1.Yam C, Mani SA, Moulder SL. Targeting the molecular subtypes of triple negative breast cancer: understanding the diversity to progress the field. Oncologist. 2017;22:1086–1093. doi: 10.1634/theoncologist.2017-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K, Mitchell EP. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National cancer institute’s surveillance, epidemiology, and end results database. Cancer. 2007;110:876–884. doi: 10.1002/cncr.22836. [DOI] [PubMed] [Google Scholar]
- 3.Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7:4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer registry. Cancer. 2007;109:1721–1728. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 5.Thike AA, Cheok PY, Jara-Lazaro AR, Tan B, Tan P, Tan PH. Triple-negative breast cancer: clinicopathological characteristics and relationship with basal-like breast cancer. Mod Pathol. 2010;23:123–133. doi: 10.1038/modpathol.2009.145. [DOI] [PubMed] [Google Scholar]
- 6.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 7.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 8.Haffty BG, Yang Q, Reiss M, Kearney T, Higgins SA, Weidhaas J, Harris L, Hait W, Toppmeyer D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006;24:5652–5657. doi: 10.1200/JCO.2006.06.5664. [DOI] [PubMed] [Google Scholar]
- 9.Symmans WF, Wei C, Gould R, Yu X, Zhang Y, Liu M, Walls A, Bousamra A, Ramineni M, Sinn B, Hunt K, Buchholz TA, Valero V, Buzdar AU, Yang W, Brewster AM, Moulder S, Pusztai L, Hatzis C, Hortobagyi GN. Long-term prognostic risk after neoadjuvant chemotherapy associated with residual cancer burden and breast cancer subtype. J. Clin. Oncol. 2017;35:1049–1060. doi: 10.1200/JCO.2015.63.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peshkin BN, Alabek ML, Isaacs C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis. 2010;32:25–33. doi: 10.3233/BD-2010-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Wu J, Zhang Z, Tang Y, Li X, Liu S, Cao S, Li X. Association between BRCA status and triple-negative breast cancer: a meta-analysis. Front Pharmacol. 2018;9:909. doi: 10.3389/fphar.2018.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389:2430–2442. doi: 10.1016/S0140-6736(16)32454-0. [DOI] [PubMed] [Google Scholar]
- 13.Belli C, Duso BA, Ferraro E, Curigliano G. Homologous recombination deficiency in triple negative breast cancer. Breast. 2019;45:15–21. doi: 10.1016/j.breast.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 15.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roche H, Im YH, Quek RGW, Markova D, Tudor IC, Hannah AL, Eiermann W, Blum JL. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–763. doi: 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, Lee JS, Hennessy B, Hanash S, Mills GB, Lin SY. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCann KE. Poly-ADP-ribosyl-polymerase inhibitor resistance mechanisms and their therapeutic implications. Curr Opin Obstet Gynecol. 2019;31:12–17. doi: 10.1097/GCO.0000000000000517. [DOI] [PubMed] [Google Scholar]
- 20.Sugimura K, Takebayashi S, Taguchi H, Takeda S, Okumura K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J Cell Biol. 2008;183:1203–1212. doi: 10.1083/jcb.200806068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, Petermann E, Lakin ND. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun. 2018;9:746. doi: 10.1038/s41467-018-03159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhat KP, Cortez D. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol. 2018;25:446–453. doi: 10.1038/s41594-018-0075-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, D’Andrea AD. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–1378. doi: 10.1038/ncb3626. [DOI] [PubMed] [Google Scholar]
- 25.Makvandi M, Pantel A, Schwartz L, Schubert E, Xu K, Hsieh CJ, Hou C, Kim H, Weng CC, Winters H, Doot R, Farwell MD, Pryma DA, Greenberg RA, Mankoff DA, Simpkins F, Mach RH, Lin LL. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J Clin Invest. 2018;128:2116–2126. doi: 10.1172/JCI97992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, Frankum J, Ranes M, Pemberton HN, Rafiq R, Fenwick K, Swain A, Guettler S, Lee JM, Swisher EM, Stoynov S, Yusa K, Ashworth A, Lord CJ. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849. doi: 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 29.Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, Chen L, Kozarewa I, Assiotis I, Fenwick K, Natrajan R, Lord CJ, Ashworth A. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, Lazaro JB, Kochupurakkal B, Sun H, Unitt C, Moreau LA, Sarosiek KA, Scaltriti M, Juric D, Baselga J, Richardson AL, Rodig SJ, D’Andrea AD, Balmana J, Johnson N, Geyer M, Serra V, Lim E, Shapiro GI. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17:2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alagpulinsa DA, Ayyadevara S, Yaccoby S, Shmookler Reis RJ. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther. 2016;15:241–250. doi: 10.1158/1535-7163.MCT-15-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, Jordan CT, Venkataraman S, Vibhakar R, Porter CC. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16:2058–2068. doi: 10.1158/1535-7163.MCT-16-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tapia-Alveal C, Calonge TM, O’Connell MJ. Regulation of chk1. Cell Div. 2009;4:8. doi: 10.1186/1747-1028-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Booth L, Roberts J, Poklepovic A, Dent P. The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to kill carcinoma cells. Cancer Biol Ther. 2018;19:786–796. doi: 10.1080/15384047.2018.1472189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GRt, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han Y, Chen MK, Wang HL, Hsu JL, Li CW, Chu YY, Liu CX, Nie L, Chan LC, Yam C, Wang SC, He GJ, Hortobagyi GN, Tan XD, Hung MC. Synergism of PARP inhibitor fluzoparib (HS10160) and MET inhibitor HS10241 in breast and ovarian cancer cells. Am J Cancer Res. 2019;9:608–618. [PMC free article] [PubMed] [Google Scholar]
- 38.Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK, Zhao X, Chu YY, Qiu Y, Qin L, Yamaguchi H, Hung MC. EGFR and c-MET cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019;79:819–829. doi: 10.1158/0008-5472.CAN-18-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linklater ES, Tovar EA, Essenburg CJ, Turner L, Madaj Z, Winn ME, Melnik MK, Korkaya H, Maroun CR, Christensen JG, Steensma MR, Boerner JL, Graveel CR. Targeting MET and EGFR crosstalk signaling in triple-negative breast cancers. Oncotarget. 2016;7:69903–69915. doi: 10.18632/oncotarget.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell Line Data Base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Res. 2009;37:D925–932. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 42.Litton JK, Scoggins M, Ramirez DL, Murthy RK, Whitman GJ, Hess KR, Adrada BE, Moulder SL, Barcenas CH, Valero V, Gomez JS, Mittendorf EA, Thompson A, Helgason T, Mills GB, Piwnica-Worms H, Arun BK. A feasibility study of neoadjuvant talazoparib for operable breast cancer patients with a germline BRCA mutation demonstrates marked activity. NPJ Breast Cancer. 2017;3:49. doi: 10.1038/s41523-017-0052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yam C, Xu X, Davies MA, Gimotty PA, Morrissette JJD, Tetzlaff MT, Wani KM, Liu S, Deng W, Buckley M, Zhao J, Amaravadi RK, Haas NB, Kudchadkar RR, Pavlick AC, Sosman JA, Tawbi H, Walker L, Schuchter LM, Karakousis GC, Gangadhar TC. A multicenter phase I study evaluating dual PI3K and BRAF inhibition with PX-866 and vemurafenib in patients with advanced BRAF V600-mutant solid tumors. Clin Cancer Res. 2018;24:22–32. doi: 10.1158/1078-0432.CCR-17-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viticchie G, Muller PAJ. c-Met and other cell surface molecules: interaction, activation and functional consequences. Biomedicines. 2015;3:46–70. doi: 10.3390/biomedicines3010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa R, Shah AN, Santa-Maria CA, Cruz MR, Mahalingam D, Carneiro BA, Chae YK, Cristofanilli M, Gradishar WJ, Giles FJ. Targeting epidermal growth factor receptor in triple negative breast cancer: new discoveries and practical insights for drug development. Cancer Treat Rev. 2017;53:111–119. doi: 10.1016/j.ctrv.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Lee HJ, Lan L, Peng G, Chang WC, Hsu MC, Wang YN, Cheng CC, Wei L, Nakajima S, Chang SS, Liao HW, Chen CH, Lavin M, Ang KK, Lin SY, Hung MC. Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response. Cell Res. 2015;25:225–236. doi: 10.1038/cr.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Assad L, Poniecka A, Hennessy B, Green M, Buzdar AU, Singletary SE, Hortobagyi GN, Pusztai L. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J. Clin. Oncol. 2007;25:4414–4422. doi: 10.1200/JCO.2007.10.6823. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell. 2009;35:442–453. doi: 10.1016/j.molcel.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parsons JL, Tait PS, Finch D, Dianova II, Allinson SL, Dianov GL. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29:477–487. doi: 10.1016/j.molcel.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Li Z, Dong Z, Beebe J, Yang K, Fu L, Zhang JT. 14-3-3 sigma contributes to radioresistance by regulating DNA repair and cell cycle via PARP1 and CHK2. Mol Cancer Res. 2017;15:418–428. doi: 10.1158/1541-7786.MCR-16-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mansour WY, Tennstedt P, Volquardsen J, Oing C, Kluth M, Hube-Magg C, Borgmann K, Simon R, Petersen C, Dikomey E, Rothkamm K. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci Rep. 2018;8:3947. doi: 10.1038/s41598-018-22289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim HP, Yoon YK, Kim JW, Han SW, Hur HS, Park J, Lee JH, Oh DY, Im SA, Bang YJ, Kim TY. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS One. 2009;4:e5933. doi: 10.1371/journal.pone.0005933. [DOI] [PMC free article] [PubMed] [Google Scholar]