

Abstract
Programmed cell death 4 (PDCD4) suppresses tumorigenesis, tumor progression, and invasion by inhibiting transcription and translation of oncogenes. However, the role of PDCD4 in lung tumorigenesis is unclear. Sequestosome1/p62 mediates cell proliferation, survival, and death through multiple signaling pathways, including autophagy and cell metabolism. p62/SQSTM1 is transcriptional target of Nrf2 and an important regulator of tumor growth. The aim of this study was to clarify whether and how PDCD4 regulates the p62-Nrf2 pathway, and how this regulation relates to tumorigenesis in human lung cancer cells. We established two stable human lung cancer cell lines, A549 and H460 that each overexpressed PDCD4. We found that PDCD4 overexpression decreased p62 expression levels and inhibited cell proliferation, and also increased the expression levels of cleaved PARP and cleaved caspase 3. Knockdown of p62 markedly increased the apoptotic rate of A549 and H460 cells overexpressing PDCD4. Furthermore levels of the epithelial-mesenchymal transition-related markers Slug, Snail, Twist1 and Vimentin were decreased and expression level of E-cadherin was increased in PDCD4-overexpressing cells. We also found that PDCD4 suppressed transcriptional activation of Nrf2 (an upstream regulator of p62) and increased endogenous levels of Keap1 (a negative regulator of Nrf2). Upregulation of Keap1 induced apoptosis and inhibited cell proliferation by suppressing activity of the p62-Nrf2 pathway in PDCD4-overexpressing cells. As anticipated, results from a mouse xenograft model showed that PDCD4 overexpression in xenografts inhibited cell proliferation and tumorigenesis. Taken together, our results demonstrate that PDCD4 overexpression, which increased Keap1 expression, reduces the levels and activity of the p62-Nrf2 pathway, thereby inhibiting tumorigenesis. Our findings suggest that PDCD4 may be a potential target for lung cancer therapies.
Keywords: PDCD4 (programmed cell death4), p62/Nrf2 signaling, Keap1, apoptosis, epithelial-mesenchymal transition
Introduction
Non-small cell lung cancer (NSCLC) has a poor prognosis. Despite improvements in surgical techniques and chemotherapeutics, NSCLC is the leading cause of cancer deaths in the world [1]. Hence, alternatives to conventional treatment, such as new molecularly targeted therapies, are urgently required.
The tumor suppressor programmed cell death 4 (PDCD4) inhibits cell proliferation, migration, and invasion and promotes apoptosis in tumors [2]. Expression of PDCD4 is downregulated in human cancers and its loss is correlated with tumor progression [3]. Similarly, PDCD4 expression is reduced in primary lung tumors and lung cancer cells [4]. Conversely, PDCD4 overexpression suppresses proliferation of cancer cells and tumors in a xenograft mouse model [5].
Sequestosome1/p62 plays a crucial role in cellular functions including apoptosis, inflammation, cell proliferation, and tumorigenesis [6], and it acts as an oncogene in tumor formation and progression through regulation of NF-κB, Nrf2, and Twist1 [7]. Furthermore, Ras induces p62 expression in lung tumorigenesis [6].
Approximately 30% of human lung cancers acquire mutations in Keap1 and NFE2L2, resulting in the stabilizations of Nrf2. In patients with NSCLC, Nrf2 stabilization is associated with poor overall survival [8]. Additionally, the expression of mutant Keap1 in lung adenocarcinoma tumors correlates with reduced overall survival [9].
Twist1 also is required for oncogene-driven NSCLC [10]. In tumors that are dependent upon the potential number of oncogenic drivers, including tumors with epidermal growth factor receptor (EGFR) mutations, Twist1 suppresses oncogene-induced senescence and apoptosis [11]. PDCD4 interacts directly with Twist1, which reduces cell proliferation through down-regulation of Twist1 target gene Y-box binding protein-1 (YB-1) [12]. p62 stabilizes Twist1 protein to increase cell proliferation and migration in vitro and in mice [13]. Therefore, activation of the p62-Nrf2 pathway promotes cancer progression and worsens prognosis in human lung cancer. Despite these advances, the regulatory and functional role of p62 in lung cancer remains poorly understood. Here, we investigated whether PDCD4 affects p62 expression, and sought to characterize the mechanism by which the p62-Nrf2 signaling pathway regulates lung tumorigenesis. We found that PDCD4 inhibited p62-Nrf2 pathway expression and activation. Downregulation of p62-Nrf2 signaling stimulated apoptosis and suppressed EMT in cells stably overexpressing PDCD4. Moreover, PDCD4 stabilized Keap1 by directly binding to Keap1, which increased Keap1 levels and led to the loss of p62 and Nrf2 expression in lung tumorigenesis.
Materials and method
Cell culture
Human lung cancer cell lines A549 and H460 were stably transfected with pLenti4/V5-DEST (Invitrogen, Thermo Scientific Inc.) encoding human PDCD4, and pooled clones were generated. Lung cancer cells stably expressing PDCD4 were maintained in RPMI medium (Invitrogen, Thermo Scientific Inc.) contained with 5 µg/mL puromycin. Stable cell lines were continuously grown in the media containing the same concentration of puromycin.
Cell proliferation assay
A459 and H460 cells were seeded at 1×104 cells/well in 24-well plates. After culturing for 2 days, the cells were transfected with PDCD4 wild-type or negative vector control. After 48 h, cells were photographed under a light microscope and then cell proliferation was measured using a WST-8 assay kit as per the manufacturer’s instructions (Roche Applied Science, Mannheim, Germany).
NRF2 luciferase reporter assays
Cells (5×104) were transiently co-transfected with 1 µg of antioxidant response element (ARE) reporter plasmid, along with 0.05 µg of pRL-TK (Renilla luciferase) control plasmid, using Lipofectamine 2000 transfection reagent (Invitrogen, Thermo Scientific Inc.). The luciferase activity was measured using the luciferase reporter assay system (Promega) according to the manufacturer’s protocol.
siRNA transfection
p62 and NRF2 siRNAs were synthesized by Bioneer (Seoul, Korea). Cells stably expressing PDCD4 were transfected with p62, NRF2 or non-specific siRNA using TransIT-LT1 siRNA transfection reagent (Mirus bio). Two different target siRNA sequences were used for each gene: 5’-CUUGCAUUAAUUCGGGAUATT-3’ and 5’-GAUGCCCAAUGUGAGAACATT-3’ for NRF2, and 5’-GUGACGAGGAAUUGACAAUTT-3’ and 5’-GGAGUCGGAUAACUGUUCATT-3’ for p62. Forty-eight hours after transfection, cells were harvested for western blot analysis.
Western blot analysis and subcellular extraction
Protein samples were extracted with lysis buffer and total protein was measured using the BCA Protein Assay Kit (Thermo Fisher Scientific, Inc). Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membrane were blocked in PBS containing 5% non-fat milk for 1 h at room temperature and incubated with the following primary antibodies: PDCD4, cleaved caspase-3, cleaved PARP, Nrf2, Twist1, Slug, Snail, Vimentin and antibody from Cell Signaling Technology; p62, Keap1, Lamin B, Flag, E-cadherin, Ki-67 and HO-1 from Santa Cruz Biotechnology; and anti-β-actin from Sigma-Aldrich. Images were detected using Bio-rad chemi-doc imaging system. Densities were measured using NIH ImageJ (Bethesda, MD, USA). Cytoplasmic and nuclear fractions were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Inc.).
Detection of apoptotic cells by flow cytometry
PDCD4-expressing and control cells were harvested, washed twice with pre-chilled PBS, and resuspended in 100 µL of 1× binding buffer. The cells were then stained with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) using the Annexin V-FITC & PI Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA).
Immunoprecipitation
A549 and H460 cells were lysed in IP lysis buffer (Thermo Fisher Scientific, Inc.). Lysates were precleared using magnetic beads; 10% of the supernatant was saved as the input sample, and the remainder was used for IP. Magnetic beads were incubated with PDCD4 antibody or normal rabbit IgG (Cell Signaling Technology) at 4°C overnight. The beads were washed three times with wash buffer and eluted from the beads with elution buffer and used to western blot. For the reciprocal IP experiment, PDCD4-overexpressing A549 cells were transfected with FLAG-tagged Keap1 for 24 h, and immunoprecipitation was performed with anti-FLAG antibody.
Caspase-3 assay
PDCD4-overexpressing and control cells were seeded at 1×105 cells/well in 6-well plates. After 24 h, the cells were collected, centrifuged, and lysed on ice for 10 min in 50 μL of lysis buffer, and incubated with DEVD-AFC substrate and reaction buffer at 37°C for 1.5 h. Caspase-3 activity was detected using a colorimetric caspase-3 assay kit (ab39401, Abcam). Each experiment was performed in duplicate.
Immunofluorescence staining
PDCD4-overexpressing cells grown on 4-well chamber slides were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Cells were incubated with rabbit polyclonal anti-PDCD4 antibody (1:100 dilution; Cell Signaling Technology) and mouse polyclonal anti-Keap1 antibody (1:100 dilution; Santa Cruz Biotechnology). After three washes with phosphate-buffered saline, the slides were incubated for 2 h with FITC-conjugated goat anti-rabbit IgG (to detect anti-PDCD4) and RITC-conjugated goat anti-mouse IgG (to detect anti-Keap1) (Molecular Probes). The cells were washed twice with phosphate-buffered saline, and nuclear DNA was stained with DAPI. Fluorescence images were obtained on an Olympus confocal microscope.
Reverse transcription-quantitiative polymerase chain reaction
Total RNA was extracted with TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) in A549 and H460 cells. The SuperScript IV Reverse transcriptase kit (Thermo Fisher Scientific, Inc.) was used to synthesize cDNA. The mRNA expression levels of PDCD4, p62, Keap1, Nrf2 and HO-1 were assessed by SYBR Green incorporation on a Roche LightCycler real-time PCR system (Roche Diagnositcs, Indianapolis, IN, USA), with the specific primers: human PDCD4: forward, 5’-AGGCCGAGGTGGGCGGATCATTTG-3’, reverse, 5’-GCCACCATGCCTGGCTACT-3’; SQSTM1 (p62): forward, 5’-CGGGAGTCCGCAGTCTTA-3’, reverse, 5’-GCTTGAGGGTCTTG-3’; KEAP1: forward, 5’-TGTTTCAGCATCAACACTGGCACG-3’, reverse, 5’-TGCATGACAACTAAAGCACCGCAC-3’; NFE2L2 (NRF-2): forward, 5’-ATCCAGACAGACACCAGTGGATC-3’, reverse, 5’-GGCAGTGAAGACTGAACTTTCA-3’; HMOX1 (HO-1): forward, 5’-TGCTCAACATCCAGCTCTTTG-3’, reverse, 5’-GCAGAATCTTGCACTTTGTTGCT-3’. Relative transcript levels were calculated using the 2-ΔΔCq method, with GAPDH used for normalization. Results from three independent experiments are expressed as fold change relative to the corresponding values in the control cell line (defined as 1.0). Statistical significance was determined by Student’s t-test analysis: *, P<0.05; **, P<0.01.
Xenograft study
Male nude mice (5 weeks old) were purchased from Central Lab Animal (Seoul, Korea) and housed in a pathogen-free room. All animal studies were approved by the Institutional Review Board Committee at Daegu Catholic University Medical Center, which conforms to the US National Institutes of Health guidelines for care and use of laboratory animals. Cells were washed and resuspended in a 50% mixture of Matrigel (BD Bioscience) in culture medium at a final concentration of 1.0×107 cells/mL. A 0.1 mL volume of the cell suspension (1×106 cells) was injected subcutaneously into the left hind legs of ten mice. The mice were checked for tumor growth every other day, and weighed every week. Tumor tissues from representative mice in each group were sectioned, embedded in paraffin, and processed for hematoxylin and eosin (H&E) staining.
Immunohistochemistry
Tumor tissue section were deparaffinized in xylene and rehydrated with graded alcohol, then washed and incubated in 3% hydrogen peroxide for 15 minutes to quench endogenous peroxidase activity. After washing in phosphate-buffered saline (PBS), the tissue sections were incubated with 3% bovine serum albumin (BSA) in PBS for 1 hour at room temperature to block the unspecific binding sites. Primary antibodies were applied on tissue sections overnight at 4°C. The following day, the tissue sections were washed and incubated with secondary horseradish peroxide-conjugated antibodies for 2 hours at room temperature. Antigen-antibody complexes were detected with the streptavidin-peroxidase method with diaminobenxidine using Vectastain elite ABC kit (Vector Laboratories, Inc.). After washing, tumor tissue sections were stained with hematoxylin for 1 min at room temperature and washed with xylene. Cover slips were mounted using Permount (Thermo Fisher Scientific, Inc.), and the slides were reviewed using a light microscope (Olympus Corporation).
Statistical analyses
Data are represented as mean ± SD. Statistical analysis was performed using Student’s t-test. All study represents at least three independent replications. The statistical significance of differences (*, P<0.05 and **, P<0.01) was indicated in figures.
Results
PDCD4 inhibits p62 expression in lung cancer cells
The NF-κB pathway is activated by increased p62 expression, and PDCD4 inhibits NF-κB transactivation in cancer cells. To investigate whether PDCD4 also regulates p62 expression, we transfected A549, H1299 and H460 human lung cancer cells with wild-type PDCD4 and then performed real-time PCR and western blot assays. Wild-type PDCD4 significantly decreased p62 protein and mRNA expression in A549 and H460 cells, but only slightly decreased p62 mRNA expression in H1299 cells (Figure 1A, 1B). For further studies, we generated A549 and H460 cell lines stably overexpressing PDCD4. We then investigated whether the effect of p62 downregulation on cell proliferation is dependent on PDCD4 overexpression, by using A549 and H460 cell lines expressing inducible wild-type PDCD4. Wild-type PDCD4 induced a morphology typical of an apoptotic cell compared to the non-transfected and vector transfected cells (Figure 1C). Moreover, proliferation of A549 and H460 cells (measured using the WST-8 assay) was significantly decreased after wild-type PDCD4 transfection, but remained unchanged when vector control was expressed (Figure 1D). These results indicate that wild-type PDCD4 expression suppresses expression of p62 and induces cell death in lung cancer cells. In contrast to findings in other cancer cells, PDCD4 overexpression did not decrease p62 protein expression (Figure S1). These findings indicate that overexpression of PDCD4 specifically inhibits p62 gene and protein expression in lung cancer cells.
Figure 1.
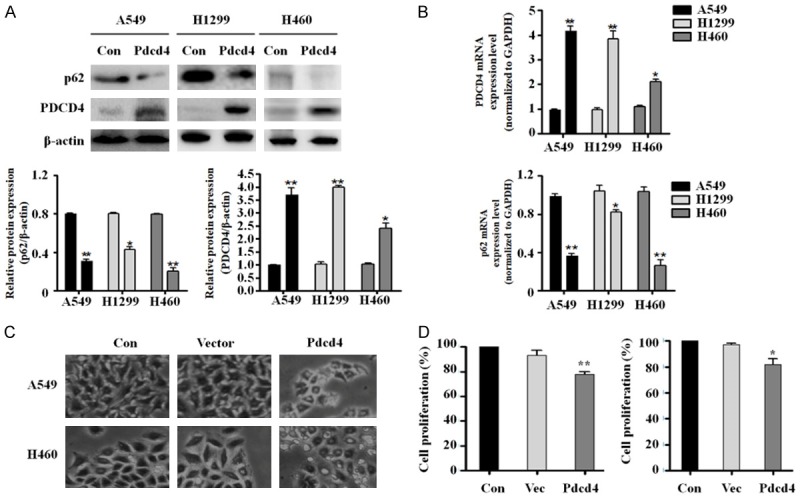
PDCD4 inhibits p62 expression and suppressed cell proliferation in human lung cancer cells. Three lung cancer cell lines were transiently transfected with PDCD4 wild-type and analyzed by qRT-PCR and western blot. A. PDCD4 and p62 protein expression levels. B. PDCD4 and p62 mRNA levels. C. Phase-contrast images of the control (left), vector control (middle) and PDCD4 overexpression cells (right) at 48 h after PDCD4 wild-type treatment. D. PDCD4 wild-type transfected into A549 (left panel) and H460 cells (right panel) and after 48 h, cell growth were analyzed by WST-8 assay.
Inhibition of p62 promotes apoptosis and suppress EMT in PDCD4-overexpressing lung cancer cells
To determine whether PDCD4-mediated inhibition of p62 can also regulate autophagy, we measured autophagy-related protein expression by western blot in A549 and H460 cells stably expressing PDCD4. As shown in Figure S2, overexpression of PDCD4 did not affect the expression of autophagy-related proteins in several cancer cell lines. This result indicates that PDCD4 does not affect autophagy in lung cancer cells, even though p62 expression is suppressed.
PDCD4 overexpression facilitates apoptosis in cancer cells, we hypothesized that PDCD4-mediated suppression of p62 might activate apoptosis. To test this idea, we first examined the expression of pro-apoptotic proteins using western blot analysis in PDCD4-overexpressing A549 and H460 cells. Cleaved caspase-3 and cleaved PARP expression levels were significantly elevated in PDCD4-overexpressing cells relative to vector controls (Figure 2A). We confirmed the effect of p62 silencing on apoptosis by annexin V/FITC staining. Flow cytometry revealed that the proportion of apoptotic cells was significantly higher in PDCD4-overexpressing cells transfected with p62 siRNA than in non-transfected control cells (Figure 2B). We also measured caspase-3 activation as an indicator of apoptosis induction. Caspase-3 activity was significantly higher in PDCD4-overexpressing cells transfected with p62 siRNA than in non-transfected control cells (Figure 2C). Based on these results, we explored the possibility that p62 overexpression inhibits apoptosis in PDCD4-overexpressing cells. For this purpose, we expressed HA-tagged p62 plasmid, and then performed western blot and cleaved caspase-3 immunofluorescence assays. Overexpression p62 decreased the expression levels of cleaved caspase-3 and cleaved PARP (Figure 2D). Caspase-3 activity also was significantly reduced in A549 and H460 cells by overexpression of p62 (Figure 2E). Furthermore, immunofluorescence assays revealed that the level of cleaved caspase-3 was lower in p62-overexpressing cells than in p62-knockdown cells (Figure 2F and 2G).
Figure 2.

p62 expression regulates apoptotic cell death in PDCD4-overexpressing cells. A. Expression levels of cleaved caspase-3, cleaved PARP, and β-actin were measured by immunoblotting in two independent PDCD4-overexpressing cell lines. B. Annexin V-FITC and PI assays were used to evaluate apoptosis in PDCD4-overexpressing cells (left panel). The quantification of the apoptotic rates was analyzed by flow cytometry. C. PDCD4-overexpressing cells were transiently transfected with control and siRNA p62, and analyzed for caspase-3 activity using a colorimetric assay kit. D. PDCD4-overexpressing cells were transfected with plasmid encoding HA-tagged p62, and cell lysates were immunoblotted with antibodies against cleaved caspase-3, cleaved PARP, and β-actin. E. PDCD4-overexpressing cells were transiently transfected with plasmid encoding HA-tagged p62 for 24 h, and then analyzed for caspase-3 activity using a colorimetric assay kit. Mean ± SD shown for three independent experiments, and statistical significance was calculated by Student’s t-test (*, P<0.05; **, P<0.01). F. PDCD4 overexpressing A549 cells were co-transfected with plasmid encoding HA-tagged p62 and siRNA targeting p62, and then immunostained with anti-cleaved caspase-3 antibody (green) and DAPI (blue). Stained cells were visualized on an Olympus confocal microscope. Scale bar; 50 µm. G. Quantification of the cleaved caspase-3-positive cells. Error bars represent s.d. *P<0.05 from Student’s t-test. H. PDCD4-overexpressing cells were transfected with plasmid encoding HA-tagged p62, and cell lysates were immunoblotted with antibodies against Vimentin, Slug, Twist1, E-cadherin and β-actin. I. PDCD4-overexpressing cells were transfected with siRNA targeting p62, and cell lysates were immunoblotted with antibodies against Vimentin, Slug, Twist1, E-cadherin and β-actin.
We also explored the possibility that p62 overexpression induces EMT in PDCD4-overexpressing cells. For this purpose, we expressed a HA-tagged p62 plasmid in PDCD4-overexpressing cells, and then performed western blot analysis of EMT marker proteins. Overexpression of p62 increased the expression levels of Slug, Vimentin and Twist1. Expression of E-cadherin was significantly higher in A549 and H460 cells overexpressing PDCD4 but not in cells overexpressing p62 (Figure 2H). Moreover, we also confirmed the effect of p62 silencing on EMT in PDCD4-overexpressing cells. Expression of Slug, Vimentin and Twist1 was lower while E-cadherin expression was higher in PDCD4-overexpressing cells transfected with siRNA p62 than in non-transfected control cells (Figure 2I). Together, these results show that PDCD4-dependent inhibition of p62 promotes apoptosis and suppresses EMT in lung cancer cells.
Pdcd4 inhibits p62 expressions by suppressing of Nrf2 activation
p62 is a downstream target of Nrf2, and non-canonical pathways for Nrf2 activation involve competitive inhibition of the Keap1-Nrf2 interaction through activation of p62 [14]. The results described above imply PDCD4 plays a role in inhibition of Nrf2 activation. Hence, we investigated whether PDCD4 inhibits endogenous expression levels of Nrf2 and its transcriptional activity. To this end, we transfected PDCD4-overexpressing and vector control cells with an ARE-luciferase reporter plasmid. Nrf2 transactivation was significantly inhibited in PDCD4-overexpressing cells, but not in control cells (Figure 3A). Nrf2 is upstream regulator of p62; thus, accumulation of Nrf2 expression in the nucleus should be associated with elevated p62 expression levels. To investigate whether PDCD4 directly regulates Nrf2 to modulate p62 expression, we analyzed Nrf2 protein expression in cell lysates of A549 and H460 cells overexpressing PDCD4. Endogenous levels of Nrf2 were significantly lower in PDCD4-overexpressing cells than in vector control cells. The expression of Keap1 protein (a repressor of Nrf2) was upregulated in PDCD4-overexpressing cells (Figure 3B).
Figure 3.
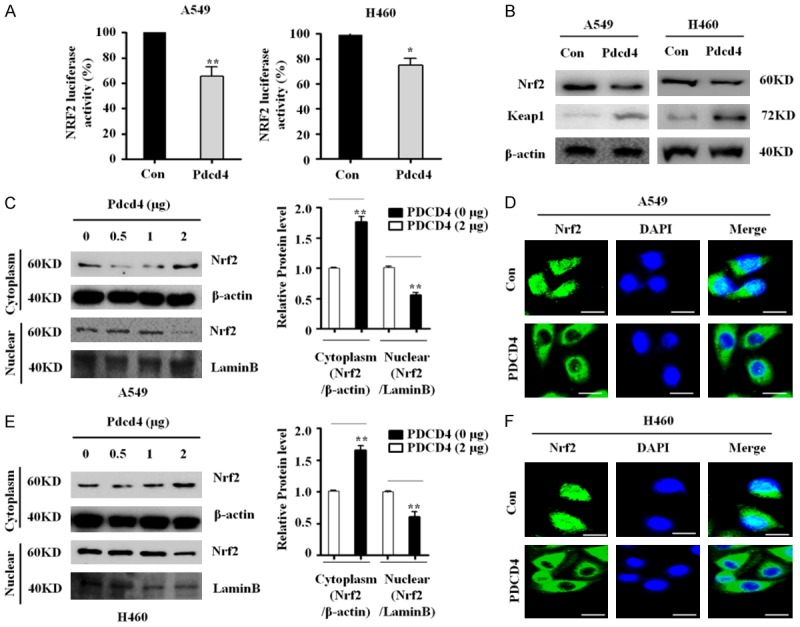
PDCD4 inhibits Nrf2 activity and nuclear localization. A. Western blots for Nrf2 and Keap1 in PDCD4-overexpressing cells. B. Two lung cancer cell lines (A549 and H460) stably expressing PDCD4 were co-transfected with pRL-TK Renilla and ARE (NRF2-luciferase) plasmids, and subjected to luciferase reporter activity assays. Mean ± SD shown for three independent experiments, and statistical significance was calculated by Student’s t-test (*, P<0.05; **, P<0.01). C. Subcellular localization of Nrf2 in A549 cells. A549 cells were transfected with the indicated amounts of PDCD4 WT plasmid for 24 h and then Nrf2 levels in the cytosolic and nuclear fractions of indicated cells were determined by western blotting. Lamin B was used as a nuclear protein marker, and β-actin as a loading control. D. PDCD4-overexpressing cell lines (A549) were immunostained with anti-Nrf2 antibody (green) and counterstained with DAPI (blue). E. Subcellular localization of Nrf2 in H460 cells. H460 cells were transfected with the indicated amounts of PDCD4 WT plasmid for 24 h and then Nrf2 levels in the cytosolic and nuclear fractions of indicated cells were determined by western blotting. Lamin B was used as a nuclear protein marker, and β-actin as a loading control. Scale bar; 20 µm. F. Two PDCD4-overexpressing cell lines (H460) were immunostained with anti-Nrf2 antibody (green) and counterstained with DAPI (blue).
Nuclear translocation of Nrf2 is essential for activation of the Nrf2 signaling pathway, and nuclear accumulation of Nrf2 is required for transcriptional activation of p62 [15]. To investigate whether PDCD4 overexpression mediated nuclear translocation of Nrf2, we monitored the subcellular localization of Nrf2 protein by western blot analysis. As shown in Figure 3C and 3E, PDCD4 expression substantially decreased the level of Nrf2 protein in the nuclear fraction in both lung cancer cell lines. Confocal microscopy confirmed that PDCD4 overexpression inhibited nuclear translocation of Nrf2. Thus, PDCD4 regulates Nrf2 transcriptional activity by inhibiting Nrf2 nuclear localization translocation (Figure 3D, 3F), implying that Nrf2 inhibition could suppress p62 by driving overexpression of PDCD4. Together, these results indicate that PDCD4 suppresses the p62-Nrf2 signaling pathway in lung cancer.
Downregulation of the Nrf2/p62 signaling pathway involves a direct interaction of PDCD4 with Keap1
Keap1 is a substrate adaptor protein that recruits the CUL3 ubiquitin ligase to Nrf2, targeting it for proteasomal degradation [16]. To determine whether Keap1 levels must be increased in order for Nrf2 inhibition to lead to suppression of p62, we measured Keap1 and Nrf2 protein levels by western blotting. Based on our findings that PDCD4 significantly upregulates Keap1 and inhibits Nrf2 activation, we tested the possibility that PDCD4 directly interacts with Keap1. Therefore, we immunoprecipitated whole-cell lysates from PDCD4-overexpressing and vector control cells with PDCD4 antibody or control rabbit sera, and then performed immunoblotting with Keap1 and PDCD4 antibodies. Endogenous Keap1 co-precipitated with PDCD4 in PDCD4-overexpressing cells, but not in control immunoprecipitations. Furthermore, immunoprecipitation with a PDCD4 antibody followed by immunoblotting for p62 or Nrf2 revealed no interaction between PDCD4 and p62 or PDCD4 and Nrf2 (Figure 4A). We also performed a reciprocal experiment in which lysates of PDCD4-overexpressing cells transfected with FLAG-Keap1 were immunoprecipitated with FLAG antibody; the results confirmed that FLAG-Keap1 physically interacted with PDCD4 (Figure 4B). To further confirm the PDCD4-Keap1 interaction in the cytoplasm, we used a multicolor confocal immunofluorescence microscope to ascertain the subcellular localizations of PDCD4 and Keap1 in PDCD4-overexpressing A549 cells. The immunofluorescence signal for PDCD4 (green) was distributed in both the cytoplasm and nucleus, whereas the signal for Keap1 (red) was mainly localized to the cytoplasm (Figure 4C). Colocalization of PDCD4 and Keap1 (yellow) was detected almost exclusively in the cytoplasm, whereas little or no colocalization signal was observed in the nucleus (blue). These results suggest that PDCD4 directly and specifically interacts with Keap1, leading to reduced expression of Nrf2 and p62 in A549 cells. We next sought to confirm that Keap1 promotes apoptosis in PDCD4-overexpressing cells. We transiently transfected PDCD4-overexpressing cells with a Flag-Keap1 plasmid and measured the expression levels of apoptotic marker proteins such as cleaved caspase-3 and cleaved PARP by western blot analysis. Overexpression Keap1 increased the protein levels of apoptotic marker proteins such as cleaved caspase-3 and PARP, while it decreased Nrf2 and p62 protein expression (Figure 4D). Moreover, Keap1 overexpression significantly suppressed Nrf2 transcriptional activity in PDCD4-overexpressing A549 and H460 cells (Figure 4E). These data indicate that inhibition of p62-Nrf2 activation through the PDCD4-Keap1 interaction results in suppression of downstream signaling events such as cell proliferation and induces apoptosis.
Figure 4.
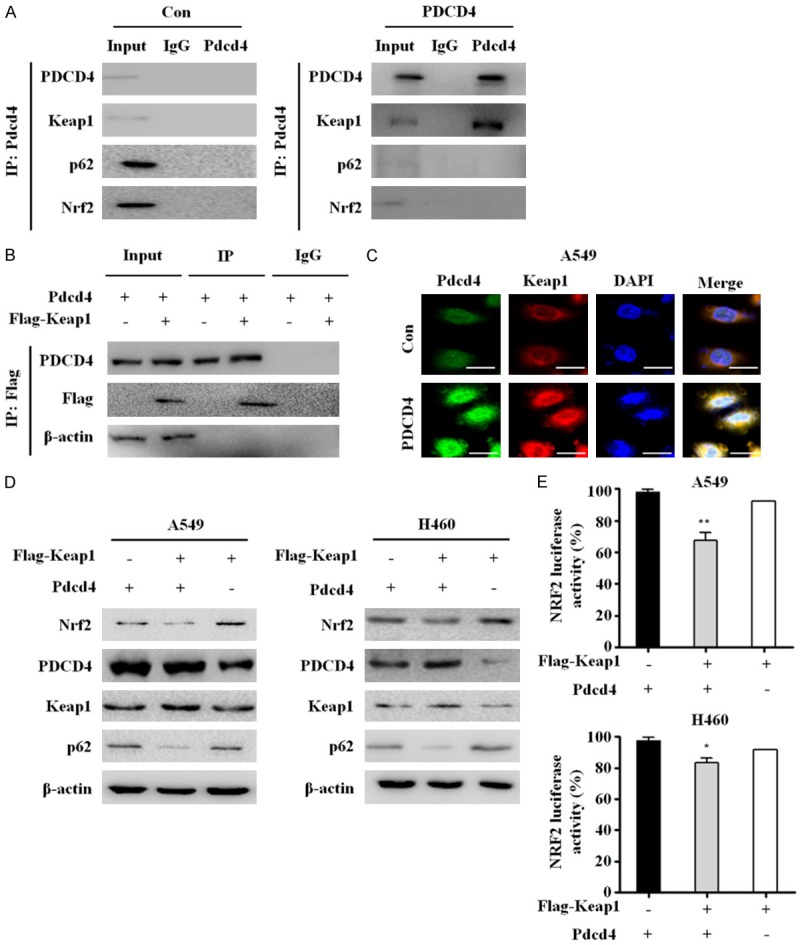
Interaction PDCD4 with Keap1 inhibits Nrf2-p62 signaling activation in lung cancer. A. Total protein extracts from control-A549 cells and PDCD4-overexpressing cells (A549) were immunoprecipitated with anti-PDCD4 or anti-IgG antibodies. PDCD4, Keap1, p62, and Nrf2 were detected by western blot after IP with anti-PDCD4. B. PDCD4-overexpressing A549 cells were transfected with plasmid encoding FLAG-tagged Keap1 and immunoprecipitated with anti-Keap1 antibody; western blots were performed to detect PDCD4 and Keap1 proteins. C. PDCD4-overexpressing A549 cells were immunostained with anti-PDCD4 (green), anti-Keap1 (red), and DAPI (blue). Stained cells were visualized on an Olympus confocal microscope. Scale bar; 10 µm. D. A549 and H460 cells were transiently co-transfected with plasmids encoding FLAG-tagged Keap1 and PDCD4 WT for 24 h, and then analyzed by western blotting with antibodies against Nrf2, PDCD4, Keap1, p62. E. Two lung cancer cell lines (A549 and H460) were co-transfected with FLAG-tagged Keap1 and PDCD4 WT, pRL-TK Renilla and ARE (NRF2-luciferase) plasmids, and subjected to luciferase reporter activity assays. Mean ± SD shown for three independent experiments, and statistical significance was calculated by Student’s t-test (*, P<0.05; **, P<0.01).
Inhibition of Nrf2 suppresses cancer cell growth and activates apoptosis in PDCD4-overexpressing cells
Activation of Nrf2 confers several advantages on cancer cells, including protection from apoptosis, promotion of cell growth, and resistance to chemotherapy [17]. We performed quantitative RT-PCR analyses to examine the expression levels of Nrf2 target genes including NQQ, MRL and HO-1 in PDCD4-overexpressing and vector control A549 and H460 cells. PDCD4 overexpression decreased HO-1 mRNA expression by about 20% relative to control cells, but had no effect on NQQ and MRL mRNA levels (Figure 5A). In addition, the changes in protein levels mirrored those of the corresponding mRNAs (Figure 5B).
Figure 5.
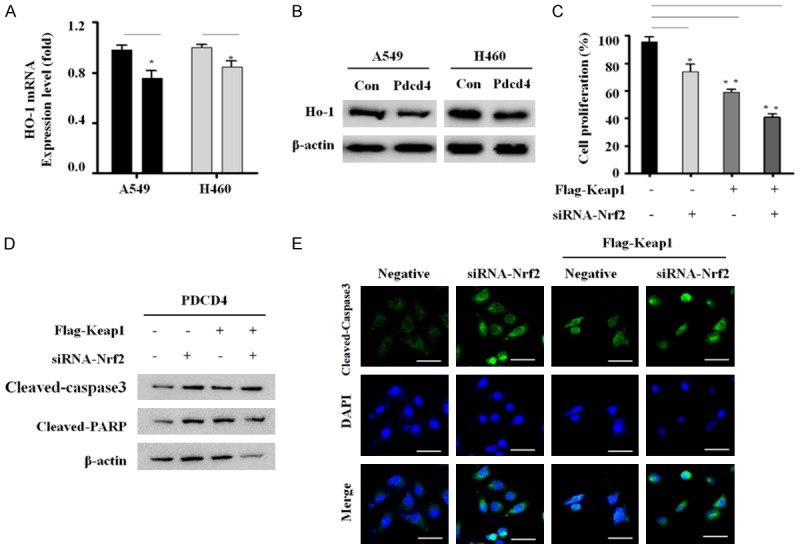
Inhibition of Nrf2 signaling activates apoptosis in PDCD4/Keap1 overexpressing cells. A. HO-1 mRNA levels were analyzed by qRT-PCR in two PDCD4-overexpressing cell lines. Expression levels were normalized against GAPDH mRNA. Results were derived from three independent experiments, and are plotted as fold change relative to the corresponding values in control cells (defined as 1.0). B. HO-1 protein level was determined by western blot analysis. C. PDCD4-overexpressing A549 cells were co-transfected with FLAG-tagged Keap1 and siRNA targeting Nrf2 for 24 h, and then cell proliferation was detected using a WST-8 assay. Mean ± SD shown for three independent experiments, and statistical significance was calculated by Student’s t-test (*, P<0.05; **, P<0.01). D. PDCD4-overexpressing A549 cells were transiently co-transfected with FLAG-tagged Keap1 and siRNA targeting Nrf2 for 24 h, and then analyzed by western blotting with antibodies against cleaved-caspase-3 and cleaved-PARP. E. PDCD4-overexpressing A549 cells were transfected with FLAG-tagged Keap1 and siRNA targeting Nrf2 for 24 h, and then immunostained with anti-cleaved caspase-3 (green) and counterstained with DAPI (blue). The stained cells were visualized on an Olympus confocal microscope. Scale bar; 50 µm.
HO-1 activation promotes lung cancer progression and metastasis, leading to cancer cell growth, whereas low expression of HO-1 promotes apoptosis by regulating the transcription factors Nrf2, NF-κB, and AP-1 [18]. This suggests that PDCD4 suppresses HO-1 expression by inhibiting Nrf2 activation, and that inhibition of Nrf2 signaling may be a consequence of PDCD4-mediated effects on cell growth and apoptosis. To determine this, we performed cell proliferation assays. PDCD4 significantly suppressed proliferation in Keap1-overexpressing/Nrf2-knockdown cells compared with Nrf2 knockdown or Keap1 overexpression individually (Figure 5C). We also sought to determine whether inhibition of cell proliferation is associated with induction of apoptosis in PDCD4-overexpressing cells. We measured the expression levels of cleaved caspase-3 and PARP in these cells by western blotting. Expression levels of the cleaved caspase-3 and PARP were increased in Keap1-overexpressing/Nrf2-knockdown cells (Figure 5D). Induction of apoptosis in Keap1-overexpressing/Nrf2-knockdown cells was confirmed by cleaved caspase-3 immunofluorescence assay (Figure 5E). These results indicate that inhibition of Nrf2 attenuates apoptosis of lung cancer cells via mechanisms that are mediated by PDCD4 overexpression.
PDCD4 inhibits lung tumorigenesis in vivo
To explore the inhibitory effects of PDCD4 on tumorigenesis in vivo, we injected PDCD4-overexpressing cells subcutaneously into the flanks of nude mice. Tumors derived from the PDCD4-overexpressing cells formed earlier (Figure 6A and 6B), and grew slower than those derived from control cells. Tumor cell proliferation was also decreased by PDCD4 overexpression (Figure 6C) and average tumor weight was significantly lower in the PDCD4 group than in the control group (Figure 6D). Hematoxylin/eosin staining confirmed that the control tumors were larger than those derived from PDCD4-overexpressing cells (Figure 6G). Thus, PDCD4 inhibits tumor formation in vivo.
Figure 6.
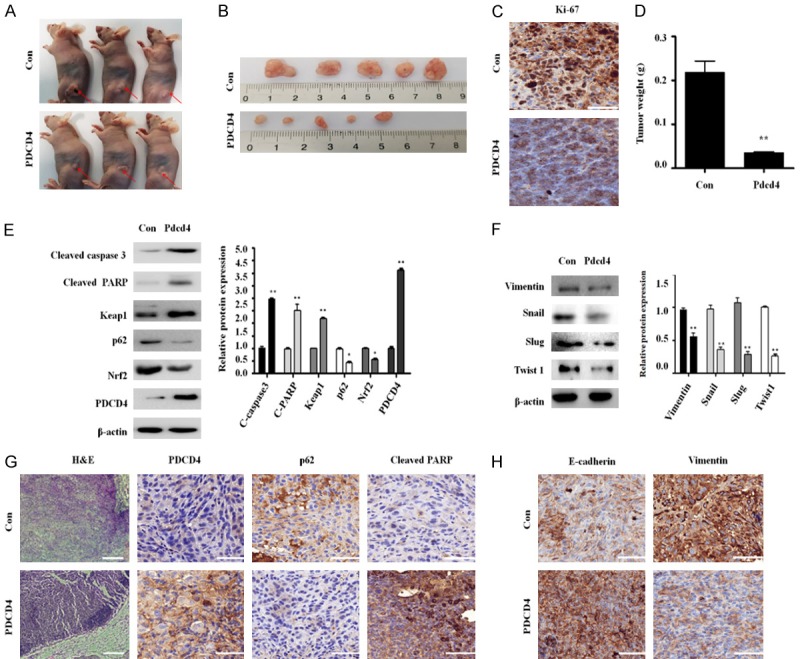
Effect of PDCD4 overexpression in tumors derived from lung cancer cells. A and B. For xenograft studies, 1×106 PDCD4-overexpressing cells in a final volume of 100 µL were injected subcutaneously in the left flanks of mice. Representative tumors are shown. C. Immunohistochemical analysis of Ki-67 in tumor tissues. Dark brown color indicates the Ki-67 expression. Scale bar; 50 µm. D. Tumor weight, measured after the injection of PDCD4-overexpressing or control A549 cells into nude mice. E. Tumor tissue homogenates were subjected to western blot analysis with antibodies against cleaved-caspase-3, cleaved-PARP, Nrf2, PDCD4, Keap1 and p62. F. Tumor tissue homogenates were subjected to western blot analysis with antibodies against Vimentin, Snail, Slug and Twist1. G. Immunohistochemical analysis of PDCD4, p62 and cleaved PARP in tumor tissues. Dark brown color indicates the PDCD4, p62 and cleaved PARP expression. Scale bar; 50 µm. H. Immunohistochemical analysis of E-cadherin and Vimentin in tumor tissues. Dark brown color indicates the E-cadherin and Vimentin expression. Scale bar; 50 µm.
Furthermore, the protein expression of apoptotic-related genes such as cleaved PARP and cleaved caspase 3 were higher in the PDCD4 group than in the control group. Expression of p62 and Nrf2 was also significantly lower in the PDCD4 group than in the control group (Figure 6E). Immunohistochemistry results also showed that cleaved caspase-3 levels were significantly higher and p62 expression levels significantly lower in the PDCD4 group (Figure 6G). Moreover, the protein expression of EMT-related genes such as Vimentin, Snail, Slug and Twist1 were lower in the PDCD4 group (Figure 6F). Immunohistochemistry results also showed that PDCD4 inhibited Vimentin, whereas E-cadherin expression was increased (Figure 6H). These results indicate that PDCD4 suppresses lung tumorigenesis in vivo by inhibiting p62-Nrf2 signaling.
Discussion
p62 plays an important role in tumorigenesis by regulating multiple cellular signal pathways, including autophagy, apoptosis, and cell survival [19]. Accumulating evidence indicates that excess p62 expression may have an oncogenic role in human cancers [20,21]. High expression of p62 has been detected in human lung cancer and in lymph node metastasis in adenocarcinoma patients [22]. These data suggest that the pathway involving p62 may enhance the metastatic potential of lung cancer cells. Furthermore, excess p62 expression may be involved in the activation of various oncogenic signaling pathways, including the NF-κB, Twist1, NRF2 or mTOR pathways [23,24].
PDCD4 is a novel, multifunctional tumor suppressor that inhibits cell growth, tumor invasion, metastasis, and induces apoptosis [2]. PDCD4 also inhibits carcinogenesis and tumor progression by inhibiting translation of oncogenes [25]. In addition, loss or reduction of PDCD4 expression is observed in multiple types of human tumor cell lines and primary tumors such as lung tumor [4,26]. Hence, we determined the function of PDCD4 in lung tumorigenesis. We showed that in NSCLC cells, p62 activity and expression are decreased by PDCD4 overexpression. Overexpression PDCD4 inhibited lung cancer cell proliferation and induced apoptosis. We found that p62 overexpression decreases apoptosis in PDCD4-overexpressing cells, whereas knockdown of p62 induces apoptosis, suggesting p62 downregulation promotes cell death through induction of apoptosis. Our results indicate that the loss of p62 was related to apoptosis through PDCD4 overexpression, suggesting that PDCD4 might have a contributing role in the inhibition of lung tumorigenesis.
Epithelial-mesenchymal transition (EMT) is an important process that acts as an indispensable driver for invasion and metastasis of a variety of cancer cells [27]. Many transcription factors, such those belonging to the Twist, Snail and ZEB families drive EMT [28,29]. Indeed, TWIST1 and Slug help drive NSCLC tumorigenesis, metastasis, and drug resistance [30,31]. Several studies have found that loss of E-cadherin expression is associated with poor prognosis in lung cancer [32]. High expression levels of EMT-associated markers are found in most lung cancer specimens, especially in squamous cell carcinoma [33]. Our results showing that overexpression p62 increased the expression levels of Slug, Vimentin and Twist1 is in agreement with a recent study showing that that overexpression p62 stabilizes Twist1, and induces tumor cell proliferation and progression. We observed that PDCD4 reduced the expression levels of EMT markers such as Slug, Vimentin and Twist1. Knockdown of p62 also reduced these markers, indeed to a greater extent than the reduction seen with PDCD4 alone. Our data suggests that PDCD4 reduces p62 expression and that this reduction in p62 expression inhibits EMT and facilitates apoptosis.
p62 can modulate the activity of Nrf2, a transcription factor that is constitutively elevated in lung, breast, and head and neck cancers [34,35]. Indeed Nrf2 is important for A549 lung cancer cell proliferation [36]. Constitutively high levels of Nrf2 also protect cancer cells against ionizing radiation and confer radio-resistance in NSCLC cells [37]. Our results showed that PDCD4 inhibits Nrf2 expression in A549 and H460 cells. Therefore, inhibition of the p62-Nrf2 signaling pathway and inactivation of p62-Nrf2-dependent target genes by PDCD4 overexpression could represent a new therapeutic approach against NSCLC.
When autophagy is impaired, p62 accumulates in the cytosol and binds to Keap1, a negative regulator of Nrf2, thereby activating Nrf2 and promoting cancer cell survival [38]. Our finding that PDCD4 increased Keap1 protein expression by directly binding to Keap1 in the cytoplasm suggests that PDCD4 competitively inhibits the Keap1-p62 interaction by decreasing the endogenous levels of p62. Furthermore, Nrf2 regulates expression of SQSTM1 (which encodes p62), leading to a positive feedback loop in the p62-Keap1-Nrf2 axis [39,40]. Our results showed that PDCD4 inhibits Nrf2 transactivation by inhibiting Nrf2 nuclear translocation, demonstrating that PDCD4 suppresses Nrf2 activation by inhibiting binding of p62 to Keap1 in cytoplasm.
Nrf2 translocates into the nucleus, where it binds to antioxidant response element (ARE) target genes, leading to upregulation of GCLM, NQO1, and HMOX1 [41]. Consequently, PDCD4 overexpression specifically suppresses HO-1 mRNA and protein expression but does not affect GCLM and NQQ1 expression. In particular, HO-1 plays important roles in carcinogenesis and tumor progression, and its overexpression promotes proliferation and survival in cancer cells [42]. These findings indicate that suppression of Nrf2 and HO-1 are important in mediating the potential ant-tumorigenic effects seen in PDCD4-overexpressing cells.
Abnormalities in the Keap1-Nrf2 pathways can occur in lung cancer [43]. For example, elevated Nrf2 levels and Keap1 dysfunction have been identified, and are associated with tumor progression and resistance to chemotherapeutic drugs [44]. In this study, we observed that Keap1 overexpression decreased cell proliferation in Nrf2 knockdown/PDCD4-overexpressing cells. Moreover, the combination of Keap1 overexpression and Nrf2 knockdown significantly increased the expression of cleaved caspase-3 and cleaved PARP proteins, suggesting that Keap1 overexpression and knockdown of Nrf2 suppressed proliferation and led to increased apoptosis in PDCD4-overexpressing cells. Our results also showed that PDCD4 inhibits cancer growth and induces apoptosis by inhibiting the expression of p62 and Nrf2 while inducing Keap1 expression in vivo. Together, our results suggest that down-regulation of the Nrf2-p62 signaling accelerates apoptosis and inhibits EMT marker expression in lung cancer cells by activating Keap1 through PDCD4 overexpression (Figure 7).
Figure 7.
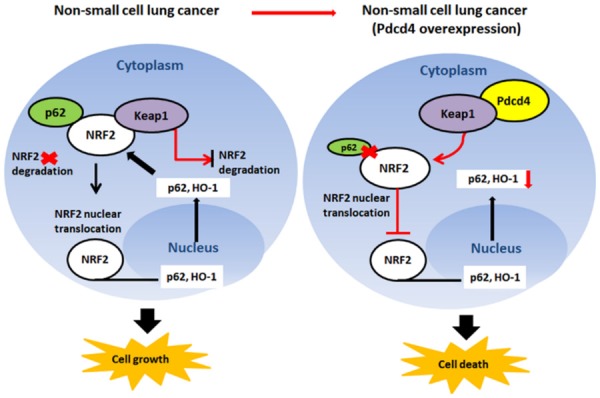
Schematic diagram of the mechanisms of PDCD4 induced cell death through suppression of p62/Nrf2 signaling in NSCLC cells. In this diagram, PDCD4 activates Keap1 by directly binding to PDCD4, leading to p62 and Nrf2 inhibition; Inhibition of p62/Nrf2 pathway promotes cell death in NSCLC cells.
In this study, we demonstrated that PDCD4 inhibit lung tumorigenesis through induction apoptosis and suppression EMT by inhibiting p62-Nrf2 signaling in lung cancer cells. These findings indicated that PDCD4 could represent a novel target for NSCLC therapeutics, which would act by suppressing activation of p62-Nrf2 signaling. On the basis of our findings, we consider PDCD4 to be a candidate biomarker involved in the regulation of the p62-Nrf2 signaling pathway with potential applications in the prevention and treatment of lung cancer.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2020R1A2B5B01002648) and also supported by the National Research Foundation of Korea funded (NRF-2017R1A6A3A11033639).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5:288–300. doi: 10.21037/tlcr.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH. Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene. 2004;23:8135–8145. doi: 10.1038/sj.onc.1207983. [DOI] [PubMed] [Google Scholar]
- 3.Wei NA, Liu SS, Leung TH, Tam KF, Liao XY, Cheung AN, Chan KK, Ngan HY. Loss of Programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Mol Cancer. 2009;8:70. doi: 10.1186/1476-4598-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Knosel T, Kristiansen G, Pietas A, Garber ME, Matsuhashi S, Ozaki I, Petersen I. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol. 2003;200:640–646. doi: 10.1002/path.1378. [DOI] [PubMed] [Google Scholar]
- 5.Gaur AB, Holbeck SL, Colburn NH, Israel MA. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro Oncol. 2011;13:580–590. doi: 10.1093/neuonc/nor033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christian F, Krause E, Houslay MD, Baillie GS. PKA phosphorylation of p62/SQSTM1 regulates PB1 domain interaction partner binding. Biochim Biophys Acta. 2014;1843:2765–2774. doi: 10.1016/j.bbamcr.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 7.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Barrera-Rodríguez R. Importance of the Keap1-Nrf2 pathway in NSCLC: is it a possible biomarker? Biomed Rep. 2018;9:375–382. doi: 10.3892/br.2018.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadal E, Palmero R, Munoz-Pinedo C. Mutations in the antioxidant KEAP1/NRF2 pathway define an aggressive subset of NSCLC resistant to conventional treatments. J Thorac Oncol. 2019;14:1881–1883. doi: 10.1016/j.jtho.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Tran PT, Shroff EH, Burns TF, Thiyagarajan S, Das ST, Zabuawala T, Chen J, Cho YJ, Luong R, Tamayo P, Salih T, Aziz K, Adam SJ, Vicent S, Nielsen CH, Withofs N, Sweet-Cordero A, Gambhir SS, Rudin CM, Felsher DW. Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genet. 2012;8:e1002650. doi: 10.1371/journal.pgen.1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burns TF, Dobromilskaya I, Murphy SC, Gajula RP, Thiyagarajan S, Chatley SN, Aziz K, Cho YJ, Tran PT, Rudin CM. Inhibition of TWIST1 leads to activation of oncogene-induced senescence in oncogene-driven non-small cell lung cancer. Mol Cancer Res. 2013;11:329–338. doi: 10.1158/1541-7786.MCR-12-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiota M, Izumi H, Tanimoto A, Takahashi M, Miyamoto N, Kashiwagi E, Kidani A, Hirano G, Masubuchi D, Fukunaka Y, Yasuniwa Y, Naito S, Nishizawa S, Sasaguri Y, Kohno K. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res. 2009;69:3148–3156. doi: 10.1158/0008-5472.CAN-08-2334. [DOI] [PubMed] [Google Scholar]
- 13.Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, Thorburn A, He YY. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111:9241–9246. doi: 10.1073/pnas.1322913111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niture SK, Kaspar JW, Shen J, Jaiswal AK. Nrf2 signaling and cell survival. Toxicol Appl Pharmacol. 2010;244:37–42. doi: 10.1016/j.taap.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furfaro AL, Traverso N, Domenicotti C, Piras S, Moretta L, Marinari UM, Pronzato MA, Nitti M. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxid Med Cell Longev. 2016;2016:1958174. doi: 10.1155/2016/1958174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su J, Liu F, Xia M, Xu Y, Li X, Kang J, Li Y, Sun L. p62 participates in the inhibition of NF-kappaB signaling and apoptosis induced by sulfasalazine in human glioma U251 cells. Oncol Rep. 2015;34:235–243. doi: 10.3892/or.2015.3944. [DOI] [PubMed] [Google Scholar]
- 21.Iwadate R, Inoue J, Tsuda H, Takano M, Furuya K, Hirasawa A, Aoki D, Inazawa J. High expression of SQSTM1/p62 protein is associated with poor prognosis in epithelial ovarian cancer. Acta Histochem Cytochem. 2014;47:295–301. doi: 10.1267/ahc.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langer R, Neppl C, Keller MD, Schmid RA, Tschan MP, Berezowska S. Expression analysis of autophagy related markers LC3B, p62 and HMGB1 indicate an autophagy-independent negative prognostic impact of high p62 expression in pulmonary squamous cell carcinomas. Cancers (Basel) 2018;10 doi: 10.3390/cancers10090281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65:6034–6041. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- 26.Gao F, Zhang P, Zhou C, Li J, Wang Q, Zhu F, Ma C, Sun W, Zhang L. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol Rep. 2007;17:123–128. [PubMed] [Google Scholar]
- 27.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–134. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 28.Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol. 2017;11:755–769. doi: 10.1002/1878-0261.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng DH, Ungewiss C, Tong P, Byers LA, Wang J, Canales JR, Villalobos PA, Uraoka N, Mino B, Behrens C, Wistuba II, Han RI, Wanna CA, Fahrenholtz M, Grande-Allen KJ, Creighton CJ, Gibbons DL. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene. 2017;36:1925–1938. doi: 10.1038/onc.2016.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih JY, Yang PC. The EMT regulator slug and lung carcinogenesis. Carcinogenesis. 2011;32:1299–1304. doi: 10.1093/carcin/bgr110. [DOI] [PubMed] [Google Scholar]
- 31.Pallier K, Cessot A, Cote JF, Just PA, Cazes A, Fabre E, Danel C, Riquet M, Devouassoux-Shisheboran M, Ansieau S, Puisieux A, Laurent-Puig P, Blons H. TWIST1 a new determinant of epithelial to mesenchymal transition in EGFR mutated lung adenocarcinoma. PLoS One. 2012;7:e29954. doi: 10.1371/journal.pone.0029954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myong NH. Reduced expression of E-cadherin in human non-small cell lung carcinoma. Cancer Res Treat. 2004;36:56–61. doi: 10.4143/crt.2004.36.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X, Brown KC, Bekele BN, Moran C, Wistuba II. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Pathol. 2009;22:668–678. doi: 10.1038/modpathol.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, Jaeschke H, Ding WX. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625. doi: 10.1016/j.jhep.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 36.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 37.Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal. 2010;13:1627–1637. doi: 10.1089/ars.2010.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 39.Kageyama S, Saito T, Obata M, Koide RH, Ichimura Y, Komatsu M. Negative regulation of the Keap1-Nrf2 pathway by a p62/Sqstm1 splicing variant. Mol Cell Biol. 2018;38 doi: 10.1128/MCB.00642-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah SZA, Zhao D, Hussain T, Sabir N, Mangi MH, Yang L. p62-Keap1-NRF2-ARE pathway: a contentious player for selective targeting of autophagy, oxidative stress and mitochondrial dysfunction in prion diseases. Front Mol Neurosci. 2018;11:310. doi: 10.3389/fnmol.2018.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong L, Xie J, Song C, Liu J, Zheng J, Liu C, Zhang X, Li P, Wang F. The activation of Nrf2 and its downstream regulated genes mediates the antioxidative activities of xueshuan xinmaining tablet in human umbilical vein endothelial cells. Evid Based Complement Alternat Med. 2015;2015:187265. doi: 10.1155/2015/187265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nitti M, Piras S, Marinari UM, Moretta L, Pronzato MA, Furfaro AL. HO-1 induction in cancer progression: a matter of cell adaptation. Antioxidants (Basel) 2017;6 doi: 10.3390/antiox6020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reisman SA, Csanaky IL, Aleksunes LM, Klaassen CD. Altered disposition of acetaminophen in Nrf2-null and Keap1-knockdown mice. Toxicol Sci. 2009;109:31–40. doi: 10.1093/toxsci/kfp047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.