

Abstract
Chronic obstructive pulmonary disease (COPD) is a devastating and common respiratory disease characterized by chronic inflammation and progressive airway remodeling. Ginsenoside Rg1 (GRg1), a major active component of Panax ginseng, has been found to possess beneficial properties against acute lung injury and respiratory diseases. However, the effects of GRg1 on airway remodeling in COPD remain unclear. In this study, we aimed to investigate the potential protective effects of GRg1 on airway remodeling induced by cigarette smoke (CS) and the underlying mechanism. A rat model of COPD was established in which the animals were subjected to CS and GRg1 daily for 12 weeks. Subsequently, we evaluated lung function, inflammatory responses, along with airway remodeling and associated signaling factors. GRg1 treatment was found to improve pulmonary function, reduce airway collagen volume fraction, and markedly reduce the expression of IL-6, TNF-α, α-SMA, and collagen I. Moreover, GRg1 treatment decreased the expression of TGF-β1, TGF-βR1, and phosphorylated-Smad3. In vitro, pretreatment of MRC5 human lung fibroblasts with GRg1 prior to exposure to cigarette smoke extract (CSE) reversed the cell ultrastructure disorder, decreased the expression of IL-6 and TNF-α, and significantly attenuated transdifferentiation of MRC5 cells by suppressing α-SMA and collagen I expression. Additionally, GRg1 suppressed the TGF-β1/Smad3 signaling pathway in CSE-stimulated MRC5 cells, whereas Smad3 over-expression abolished the anti-transdifferentiation effect of GRg1. In conclusion, the results of our study demonstrated that GRg1 improves lung function and protects against CS-induced airway remodeling, in part by down-regulating the TGF-β1/Smad3 signaling pathway.
Keywords: Chronic obstructive pulmonary disease (COPD), ginsenoside Rg1 (GRg1), airway remodeling, cigarette smoke (CS)
Introduction
Chronic obstructive pulmonary disease (COPD) constitutes a primary cause of mortality and morbidity and represents a significant economic burden on the public health system in China [1]. Characterized by progressive and irreversible airflow restriction, COPD is attributed to long-term exposure to noxious gases and particles, most often related to cigarette smoke (CS) [2] and presents with a mixture of chronic inflammation, emphysema, and narrowing of the small airways [3]. It has been demonstrated that the loss of lung function in COPD may result from small airway remodeling, including differentiation of bronchial fibroblasts to myofibroblasts and massive deposition of extracellular matrix (ECM) [4]. The prognosis for patients with COPD has improved in the past 20 years, largely owing to the use of anticholinergics, β2-agonists, methylxanthines, inhaled corticosteroids, and phosphodiesterase-4 inhibitors. However, the efficacy of these therapies in preventing airway remodeling in COPD is still limited, and the number of COPD-related deaths continues to rise. Therefore, it is urgent to explore novel therapeutic strategies for the treatment of this deadly disease.
From the perspective of traditional Chinese medicine, the main cause of COPD is lung Qi deficiency and blood stasis. Panax ginseng, a well-known traditional Chinese medicine, significantly improves the Qi of the lung and exerts protective effects against blood stasis, and has long been widely applied for the management of respiratory diseases [5-7]. Ginsenoside Rg1 (GRg1), a tetracyclic triterpenoid, constitutes one of the bioactive ingredients extracted from Panax ginseng. Several studies indicate that GRg1 possesses anti-oxidative activity [8] and efficiently attenuates fibrosis in the heart, liver, and kidney [9-11]. Moreover, GRg1 can suppress the expression of interleukin (IL)-6 and tumor necrosis factor (TNF)-α, and relieve endoplasmic reticulum stress and the inflammatory response [12,13]. GRg1 has also been demonstrated to down-regulate pulmonary cell apoptosis in lung epithelial cells to protect against acute lung injury and improve lung function [14,15]. However, the role of GRg1 in the airway remodeling of COPD has not been fully elucidated.
Transforming growth factor-β1 (TGF-β1) is recognized as a multifunctional cytokine that affects a wide range of biological processes including cell differentiation, ECM synthesis, tissue repair, and gene transcription [16]. Previous studies have also highlighted TGF-β1 as a strong stimulus that plays a crucial role in pulmonary fibrosis in both humans and animals [17]. Consistent with this, the level of TGF-β1 is elevated in lung tissues from patients with COPD [18] and the canonical TGF-β1/Smad signaling pathway, along with other non-canonical signaling pathways, are reported to be involved in COPD pathogenesis [19,20]. Notably, GRg1 could effectively decrease the TGF-β1 expression in renal fibrosis [21], however, the latent mechanism of GRg1 in pulmonary protection remains to be explored.
Accordingly, in this study we aimed to investigate the potential protective effects of GRg1 on the airway remodeling induced by CS and explore the underlying mechanisms using a rat model of COPD and MRC5 human embryonic lung fibroblasts. The findings of this study may contribute to the identification of potential drug targets and novel therapeutic strategies for treating COPD.
Materials and methods
COPD animal model
The experimental protocol was conducted in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of Tongji University. A total of 30 male Sprague-Dawley (SD, 8 weeks, 200 ± 20 g, five per cage) rats were randomly divided into the following three groups with 10 rats per group: control, COPD, and COPD+GRg1 (20 mg/kg/d). The COPD animal model was established by exposure to the smoke from 15 cigarettes (Daqianmen, 13 mg tar and 1.3 mg nicotine per cigarette, Shanghai Tobacco Company, Shanghai, China) for 40 min, twice daily, with smoke-free intervals of 6 h, for 6 days a week over 12 weeks. GRg1 (Urchem Sinopharm Chemical Reagent Co. Ltd., Shanghai, China) was administered intragastrically 30 min prior to CS exposure. The rats in the control and COPD groups were administrated normal saline (2 ml per rat). At the end of week 12, lung function was evaluated before the rats were sacrificed for sample collection.
Lung function evaluation
All rats were anesthetized with an intraperitoneal injection of 2% pentobarbital (40 mg/kg). The trachea was opened with an inverted T-shaped incision on the upper trachea near the laryngeal part of the pharynx. Each animal was then placed in a forced pulmonary maneuver system connected to an animal pulmonary functionality test machine (AniRes2005, Bestlab, Beijing, China) for measuring resistance inspiration (Ri), resistance expiration (Re), dynamic lung compliance (Cdyn), ratio of forced expiration volume in forced expiratory volume 0.3 s/forced vital capacity (FEV0.3/FVC), and peak expiratory flow (PEF).
Lung histological analysis and immunohistochemistry
The inferior lobes of the right lung were removed, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4 μm thick sections. Hematoxylin and eosin (H&E) staining was performed to evaluate pathological changes of the lung, and Masson Trichrome staining was used to detect airway fibrosis. Some deparaffinized sections were used for immunohistochemical assays to detect alpha-smooth muscle actin (α-SMA) (Abcam, Cambridge, UK) expression. Measurements were performed using Image-Pro Plus 6.0 software.
Enzyme-linked immunosorbent assay (ELISA)
The fractions of IL-6, TNF-α, and TGF-β1 in rat sera were determined using an ELISA Kit (Abcam) according to the manufacturer’s instructions. Plates were read in an ELISA microplate reader (ELx800, BioTek, VT, USA) at 450 nm.
Cigarette smoke extract (CSE) preparation
CSE was prepared according to previously described methods [22], with some modifications. Briefly, one Daqianmen cigarette (Shanghai Tobacco Company), smoked using a peristaltic pump, was bubbled through 20 ml of cell growth medium in 10 min. The solution was adjusted to pH 7.4 and sterilized through a 0.22 μm pore filter. The medium was considered 100% strength CSE and used within 30 min of preparation.
Cell culture and treatment
Human embryonic lung fibroblasts (MRC5 cells) were obtained from the Cell Bank of the China Science Academy (Shanghai, China). Cells were seeded in minimum essential medium (MEM, Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco) and incubated at 37°C in a humidified atmosphere with 5% CO2 and 95% air. The medium was replaced every 2-3 days. To induce airway remodeling, MRC5 cells were first starved for 24 h in serum-free MEM and then exposed to CSE (10%) for 48 h. To explore the role of GRg1 in CSE-induced airway remodeling, MRC5 cells were pretreated with GRg1 (40 μM) for 1 h prior to CSE stimulation. To elucidate whether GRg1 protects MRC5 cells against airway remodeling via the TGF-β1/Smad3 signaling pathway, human Smad3 cDNA was cloned into the pHBLV-CMVIE-ZsGreen vector (Genechem Co., Shanghai, China). Smad3 lentivirus (Lv) packaging and concentration were performed by co-transfecting constructs with two helper plasmids, PSPAX2 and PMD2G, in HEK293T cells with technical help from Genechem Co. MRC5 cells at 50-70% confluence were infected with Smad3-Lv (multiplicity of infection = 20) for 12 h in the presence of 5 mg/ml polybrene (Sigma, St Louis, MO, USA). Infection efficiency was determined by western blot analysis. Subsequently, the cells were subjected to GRg1 treatment and CSE stimulation.
Cytotoxicity assay
A Cell Counting Kit-8 (CCK-8) assay (Dojindo, Kumamoto, Japan) was used to detect the cytotoxicity of GRg1 and CSE toward MRC5 cells. A total of 3×103 cells/well were seeded in 96 well plates and cultured in a 37°C incubator with 5% CO2. Upon reaching 70% confluence, the cells were treated with GRg1 (5, 10, 20, 40, or 80 μM) or CSE (5, 10, 15, or 20%). After 12, 24, 48, or 72 h, 10 μl of CCK-8 was added to the media for 2 h. The CCK-8 assay was repeated at least three times, and the absorbance of all samples was measured at 450 nm using an ELISA reader (MQX200R, BioTek, Winooski, VT, USA).
Transmission electron microscopy
The cells were harvested and fixed with 2% glutaraldehyde and post-fixed in 1% osmium tetroxide. The fixed cells were then dehydrated in ice-cold graded ethanol (30, 50, 70, 80, 95, and 100%). The samples were embedded in Epon 812 (Electron Microscopy Sciences, Hatfield, PA, USA), followed by cutting into ultrathin sections. Each sample was stained with uranyl acetate and lead citrate. Images were acquired on a CM-120 transmission electron microscope (Philips, Holland).
Real-time polymerase chain reaction (PCR) analysis
Total RNA was extracted from rat lung tissues and cultured cells using TRIzol reagent (TaKaRa, Liaoning, China). Equal amounts of total RNA were reverse-transcribed to cDNA according to the manufacturer’s protocols. The primers were synthesized by Sangon Biotech (Shanghai, China). The mRNA levels of α-SMA, TGF-β1, IL-6, TNF-α, and collagen I were quantified using SYBR Green real-time PCR (TaKaRa). Real-time PCR reactions were performed using the ABI7500 real-time PCR System (Applied Biosystems, Foster City, CA, USA). Relative mRNA level was calculated using the 2-ΔΔCt method. GAPDH was used as the endogenous control. The primer sequences are shown in Table 1.
Table 1.
Primer sequence for RT-PCR
| Genes | Forward (5’-3’) | Reverse (3’-5’) |
|---|---|---|
| collagen I* | GGACACTACTGGATCGACCTAAC | CTCACCTGTCTCCATGTTGCA |
| TGF-β1* | ATTCCTGGCGTTACCTTGG | AGCCCTGTATTCCGTCTCCT |
| GAPDH* | GGCACAGTCAAGGCTGAGAATG | ATGGTGGTGAAGACGCCAGTA |
| α-SMA# | CTATGAGGGCTATGCCTTGCC | GCTCAGCAGTAGTAACGAAGGA |
| collagen I# | CCAGGCAGAGATGGTGAAGA | GCAGGTCCTTGGAAACCTTG |
| IL-6# | ATTCGGTACATCCTCGAC | TGATGATTTTCACCAGGC |
| TNF-α# | ACTCTTCTGCCTGCTGCACTTTGG | GTTGACCTTTGTCTGGTAGGAGACGG |
| GAPDH# | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
genes from rat;
genes from human.
Western blot
Protein concentrations were determined by the Bradford assay using bovine serum albumin standards. Equal amounts of proteins were separated by 6-10% SDS-PAGE and then transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies overnight at 4°C and then with horseradish peroxidase-conjugated secondary antibodies (1:2000) for 1 h. Antibodies included those against α-SMA (1:400), collagen I (1:200), TGF-β1 (1:500), TGF-βR1 (1:200), phosphorylated-Smad3 (p-Smad3, 1:500), Smad3 (1:1000), and GAPDH (1:1000) (all from Abcam).
Statistical analysis
Values are expressed as the mean ± standard deviation. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test using GraphPad Prism 5.0 software (La Jolla, CA, USA) was performed to analyze the differences between multiple groups. The values of P<0.05 were considered statistically significant.
Results
GRg1 prevents lung function decline in COPD rats
As depicted in Table 2, compared with the control group, CS exposure induced a significant increase in Ri and Re, albeit a decrease in FEV0.3/FVC, Cdyn, and PEF (P<0.05). GRg1 treatment improved the lung function by eliminating the increase in Ri and Re, along with the decrease in FEV0.3/FVC, Cdyn, and PEF, indicating that GRg1 ameliorated airflow restriction and obstructive pulmonary ventilation disorder.
Table 2.
Comparison of lung function in different groups
| Control | COPD | COPD+GRg1 | |
|---|---|---|---|
| Ri (cmH2O•s/ml) | 1.45 ± 0.12 | 2.36 ± 0.18** | 1.92 ± 0.10## |
| Re (cmH2O•s/ml) | 1.94 ± 0.11 | 2.88 ± 0.16** | 2.61 ± 0.14## |
| Cdyn (ml/cmH2O) | 0.18 ± 0.02 | 0.12 ± 0.02** | 0.14 ± 0.01## |
| FEV0.3/FVC (%) | 89.44 ± 3.23 | 72.11 ± 3.58** | 77.61 ± 3.91# |
| PEF (ml/s) | 34.55 ± 3.82 | 23.38 ± 3.87* | 30.08 ± 2.43## |
Notes: resistance inspiration (Ri), resistance expiration (Re), dynamic lung compliance (Cdyn), the ration of forced expiration volume in 0.3 s/forced vital capacity (FEV0.3/FVC) and peak expiratory flow (PEF) were measured by buxco pulmonary function analysis system at the end of week 12. Data were presented as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test.
P<0.05 vs. control;
P<0.01 vs. control.
P<0.05 vs. COPD.
P<0.01 vs. COPD.
GRg1 alleviates inflammatory responses in COPD rats
H&E staining showed that the lung tissue in the COPD group presented severe alveolar wall thickening, inflammatory cell infiltration, pulmonary alveoli consolidation, and emphysema compared with the control group. These impairments were attenuated by treatment with GRg1 (Figure 1A). The ELISA results showed that the serum concentrations of the inflammatory response biomarkers IL-6 and TNF-α significantly increased in the COPD group compared to the control group (P<0.01). Additionally, GRg1 down-regulated the CS-induced increase of IL-6 and TNF-α expression (Figure 1B and 1C).
Figure 1.

GRg1 attenuated the histopathological changes in lung tissues and inflammatory responses in COPD rats. A. After 12 weeks of CS exposure, lung tissue were collected, fixed, dehydrated and embedded in paraffin. 4 μm thickness slices were collected for H&E staining analysis of the pathology changes of lung tissue. N = 6 in each experimental group. Images were taken at ×100 magnification. Scale bar = 100 μm. B, C. Expressions of IL-6 and TNF-α in serum were determined by ELISA. Date were expressed as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. control; #P<0.05 and ##P<0.01 vs. COPD. The experiment was repeated at least three times.
GRg1 alleviates airway fibrosis in COPD rats
To test whether GRg1 modifies fibrosis in the airway, we performed Masson Trichrome staining at the end of 12 weeks. The results showed that the airway collagen fractional area was markedly attenuated in the COPD+GRg1 group compared with the COPD group (Figure 2A and 2B). The content of collagen I was further assessed by real-time PCR. RNA expression of collagen I was also reduced in the COPD+GRg1 group, compared with the COPD group (Figure 2C, P<0.01). Next, we evaluated the effects of GRg1 on myofibroblasts by examining α-SMA expression. Immunohistochemical analysis showed widespread α-SMA expression in the lung tissue of the COPD group compared with the control group whereas GRg1 treatment markedly decreased α-SMA expression (Figure 2D and 2E).
Figure 2.
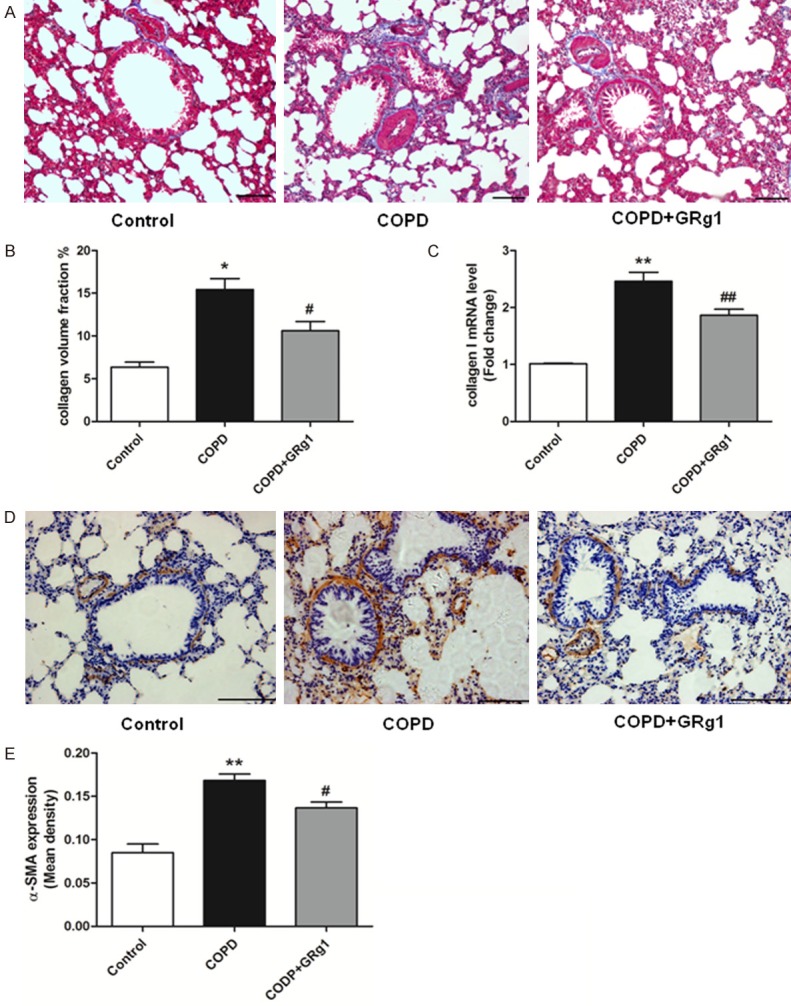
GRg1 attenuated airway fibrosis in COPD rats. A. At the end of week 12, lung sections were prepared, and Masson Trichrome staining analysis was used to assess the fibrotic changes of lung tissue. N = 6 in each experimental group. Images were taken at ×100 magnification. Scale bar = 100 μm. B. Quantitative collagen assay indicated that GRg1 markedly reduced the ratio of area with collagen accumulation in COPD rats. C. The mRNA level of collagen I was measured by real-time PCR. The results expressed as relative mRNA expression (relative to GAPDH mRNA level). CS exposure promoted mRNA expression of collagen I in lung tissue, and could be attenuated by GRg1. D. Lung sections were prepared, and immunohistochemistry was used to assess the expression of α-SMA. Images were taken at ×200 magnification. Scale bar = 100 μm. E. CS increased the expression of α-SMA in lung tissue, and was abated by GRg1. Data were expressed as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. control; #P<0.05 and ##P<0.01 vs. COPD.
GRg1 down-regulates serum TGF-β1 expression and attenuates the TGF-β1/Smad3 signaling pathway in the lung tissue of COPD rats
As the TGF-β1/Smad3 signaling pathway plays a critical role in pulmonary fibrosis, we next examined whether GRg1 could affect this pathway in lung tissues. As shown by western blotting (Figure 3A-D), the protein expression of TGF-β1, TGF-βR1, and p-Smad3 was significantly higher in the COPD group than the control group (P<0.01). Treatment with GRg1 down-regulated the expression of TGF-β1, TGF-βR1, and p-Smad3 (P<0.01). Additionally, GRg1 treatment markedly decreased serum TGF-β1 concentration compared with the COPD group (Figure 3E).
Figure 3.

GRg1 down-regulated TGF-β1 expression in serum and regulated TGF-β1/Smad3 signaling pathway in lung tissue of COPD rats. A-D. At the end of week 12, total proteins were isolated from lung tissue of different groups. Western blot tested the level of TGF-β1, TGF-βR1 and p-Smad3 in lung tissue. CS-induced up-regulation of TGF-β1, TGF-βR1 and p-Smad3 could be reduced by GRg1. E. The concentration of TGF-β1 in serum was determined by ELISA. Date were expressed as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. control; #P<0.05 and ##P<0.01 vs. COPD.
Effects of CSE and GRg1 on MRC5 cell viability
A CCK-8 kit was used to evaluate the changes in cell viability after MRC5 cells were incubated with different concentrations of CSE (0, 5, 10, 15, and 20%) or GRg1 (10, 20, 40, and 80 μM) for 72 h. CSE decreased cell viability in a dose- and time-dependent manner. Following 48 h incubation, 15% and 20% CSE groups displayed striking decreases in cell viability (44.20% and 19.59%, respectively). Notably, 72 h of incubation decreased the cell viability in all CSE groups (>40% viability decline, Figure 4A). We next examined the effect of GRg1 on MRC5 cell viability. Incubation with 80 μM GRg1 for 48 or 72 h down-regulated the cell viability by 14.36% and 19.65%, respectively (Figure 4B). Therefore, 48 h of treatment of 10% CSE and/or 40 μM GRg1 was chosen for the follow-up experiments.
Figure 4.
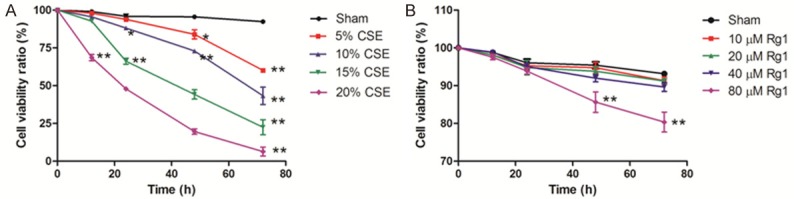
The survival rate of MRC5 cells grown in various concentration of CSE or GRg1 was determined by CCK-8 assay. A. Effect of different concentration of CSE on cell viability. B. Effect of different concentration of GRg1 on cell viability. Data were provided as mean ± SD of three independent experiments. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. corresponding Sham group.
GRg1 decreases the inflammatory response and transdifferentiation of MRC5 cells following CSE exposure
Recent studies have shown that GRg1 exerts a protective effect against lung injury [15,23]. The effects of GRg1 expression on pro-inflammatory cytokines induced by CSE were therefore analyzed. RT-PCR results showed that GRg1 treatment significantly decreased CSE-induced expression of IL-6 and TNF-α (P<0.01, Figure 5A and 5B). Our data also indicated that compared with the CSE group, GRg1 treatment reduced the expression of α-SMA and collagen I both at the mRNA and protein level (P<0.01, Figure 5C-G). Moreover, we evaluated the changes in ultrastructure of MRC5 cells using transmission electron microscopy. When MRC5 cells were treated with CSE, swelling of mitochondrial cristae and deformation and vacuolization in mitochondria were observed. In addition, the cells exhibited obvious distension of the endoplasmic reticulum indicating an increase in synthesis function. These effects were attenuated by GRg1 treatment (Figure 5H).
Figure 5.

GRg1 lessened inflammatory responses and transdifferentiation of MRC5 cells after CSE exposure. MRC5 cells were subjected to 10% CSE and treated with GRg1 (40 μM) for 48 h. A-D. Real-time PCR analysis indicated that CSE significantly increased the IL-6, TNF-α, α-SMA and collagen I mRNA expression. In the GRg1 treatment group, the mRNA levels of IL-6, TNF-α, α-SMA and collagen I were decreased significantly. E-G. Western blot analysis showed that the protein expression of α-SMA and collagen I was up-regulated by CSE, and such effects were alleviated by GRg1 treatment. H. Ultrastructure of MRC5 cells were determined by transmission electron microscopy. GRg1 protected the normal ultrastructural morphology of mitochondria (white arrows) and endoplasmic reticulum (black arrows). Images were taken at ×33000 magnification. Scale bar = 500 nm. Data were expressed as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. Sham group; #P<0.05 and ##P<0.01 vs. CSE group.
GRg1 attenuates MRC5 cell transdifferentiation by suppressing the TGF-β1/Smad3 signaling pathway
Next, we tested whether the TGF-β1/Smad3 signaling pathway was involved in the suppression of transdifferentiation in MRC5 cells. We found that the expression of TGF-β1, TGF-βR1, and p-Smad3 was upregulated in MRC5 cells exposed to CSE. GRg1 treatment significantly reduced TGF-β1, TGF-βR1, and p-Smad3 protein levels compared with the CSE group (P<0.05, Figure 6A-D). Together with the in vivo results, our data indicated that GRg1 relieved CSE-induced transdifferentiation of MRC5 cells, which may be related to inhibition of the TGF-β1/Smad3 signaling pathway. To further clarify the result, we first up-regulated the expression of Smad3 in MRC5 cells (Figure 6E and 6F) by lentivirus vector transfection. We determined that Smad3 over-expression enhanced the protein levels of α-SMA and collagen I, demonstrating that Smad3 over-expression blocked the anti-transdifferentiation of GRg1 in MRC5 cells exposed to CSE (Figure 6G-I). However, Smad3 over-expression did not significantly affect IL-6 and TNF-α expression in MRC5 cells treated with GRg1 and CSE (Figure 6J and 6K).
Figure 6.

GRg1 attenuated MRC5 cells transdifferentiation through suppressing the TGF-β1/Smad3 signaling pathway. A-D. Western blot analysis demonstrated that TGF-β1, TGF-βR1 and p-Smad3 were increased in MRC5 cells exposed to CSE, and could be inhibited by GRg1. E, F. MRC5 cells were infected with Smad3-lentivirus. Western blot assay was performed to confirm the transfection efficiency. G-I. MRC5 cells were subjected to Smad3 over-expression, followed by CSE and GRg1 treatment. Western blot analysis demonstrated that over-expression of Smad3 abated the protective effect of GRg1 in MRC5 cells by increasing α-SMA and collagen I level in MRC5 cells. J, K. The concentrations of IL-6 and TNF-α in MRC cells were determined by real-time PCR. Smad3 over-expression did not alter the expression of IL-6 and TNF-α. Data were expressed as mean ± SD. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc test. *P<0.05 and **P<0.01 vs. Sham group; #P<0.05 and ##P<0.01 vs. CSE group. ΔP<0.05 and ΔΔP<0.01 vs. CSE+GRg1 group.
Discussion
Airway remodeling in COPD is a complex pathophysiological process that involves inflammation, fibrosis, oxidative stress, and other pathological processes, which eventually lead to irreversible and progressive airflow limitation. Panax ginseng represents a traditional Chinese herb widely used to treat a range of respiratory diseases including COPD [24]. In the present study, we investigated the protective effect of one of the main active compounds of Panax ginseng, GRg1, on airway remodeling and the underlying mechanisms using in vivo and in vitro systems. We demonstrated that 1) GRg1 suppressed airway remodeling in a rat model of COPD and improved lung function; 2) GRg1 attenuated the transdifferentiation and collagen secretion of MRC5 cells; and 3) GRg1 exerted its anti-airway remodeling property by inhibiting the TGF-β1/Smad3 signaling pathway.
Ginseng has been found to effectively and safely improve the pulmonary function and exercise capacity in patients with COPD [25,26]. In our COPD animal model, we found that GRg1 prevented the disruption of Cydn, FEV0.3/FVC, and PEF, and suppressed the deterioration of RI. Our data suggested that GRg1 effectively abolished pulmonary dysfunction during CS exposure. Small airways of less than 2-3 mm in diameter constitute the main site of airway obstruction in COPD [27]. Consistent with the pulmonary function results, GRg1 treatment improved the small airway obliteration and reduced alveolar cavity, along with decreasing the disruption and loss of epithelial cilia and airway wall fibrosis. Thus, GRg1 appeared to block airway remodeling during COPD development.
Chronic lung inflammation induced by inhalation of CS or other noxious particles plays an important role in the airway remodeling of COPD and is related to disease progression and mortality [28,29]. The effects of ginseng and ginseng-derived ingredients against this phenomenon have been extensively explored [30]. Ginseng extracts could strongly inhibit the activation of NLRP3 and AIM2 inflammasomes, resulting in the suppression of pyroptosome formation and inflammatory cytokines in vivo and in vitro [31]. Fan and colleagues further revealed that GRg1 protected against neurodegeneration by ameliorating the over-expression of pro-inflammatory cytokines including IL-1β, IFN-γ, and TNF-α in chronic stress models [32]. Moreover, GRg1 alleviated sepsis-induced lung injury by decreasing IL-6 and TNF-α levels [13]. These findings are consistent with the results of the current study. In vivo, GRg1 treatment markedly decreased inflammatory cell infiltration in lung tissues and reduced the plasma levels of the inflammatory cytokines IL-6 and TNF-α in COPD rats. GRg1 treatment also decreased the expression of these cytokines in CSE-induced MRC5 cells. In addition, recent studies have revealed the involvement of the mitochondria and the endoplasmic reticulum in inflammatory signaling [33,34]. Notably, we observed that GRg1 suppressed mitochondrial deformation and swelling, along with endoplasmic reticulum distention in vitro, indicating that GRg1 could attenuate the pro-inflammatory responses induced by CSE.
Airway fibrosis also plays a pivotal role during lung function decline and airway remodeling in COPD [35]. During CS or CSE exposure, numerous inflammatory cytokines and pro-fibrogenic factors are synthesized and secreted [36,37]. In response to these stimuli, the intrapulmonary fibroblasts transform into myofibroblasts, which secrete elevated levels of collagen and serve as potent effector cells of airway fibrosis [38,39]. GRg1 was reported to reduce myocardial fibrosis and ameliorate cardiac remodeling in acute myocardial infarction in a rat model [40]. Similarly, the present study indicated that GRg1 treatment significantly down-regulated collagen I expression and fibrotic friction in the lung tissues of COPD rats. GRg1 also inhibited MRC5 cell activation and differentiation by reducing α-SMA and collagen I expression. These results indicated that GRg1 exerts a protective effect against airway fibrosis.
We further investigated the mechanism by which GRg1 inhibits airway remodeling. TGF-β1 is considered as a “master switch” that is tightly involved in pulmonary fibrosis [41]. Studies showed that activating the TGF-β1/Smad3 signaling pathway induces fibroblasts to transdifferentiate into myofibroblasts, and secrete large quantities of ECM [42]. Therefore, targeting the TGF-β1/Smad3 signaling pathway might constitute a promising therapeutic approach for COPD. Recent studies have demonstrated that GRg1 can suppress the activity of the TGF-β1/Smad signaling cascade. Specifically, it was observed that GRg1 could block the TGF-β1/Smad signaling pathway in HCS-T6 rat hepatic stellate cells, thereby decreasing the collagen level and ameliorating hepatic fibrosis [43]. GRg1 has also been reported to protect against renal fibrosis by suppressing TGF-β1 and Smad2/3 expression [44]. Consistent with these findings, we observed that GRg1 could reduce the content of TGF-β1 and reverse the activation of TGF-βR1 and p-Smad3 in COPD rats and CSE-induced MRC5 cells, leading to the down-regulation of α-SMA and collagen I expression. Furthermore, over-expression of Smad3 increased α-SMA and collagen I expression which abolished the anti-transdifferentiation effect of GRg1. Therefore, we conclude that GRg1 attenuates airway remodeling at least in part through the suppression of the TGF-β1/Smad3 signaling pathway.
A limitation of our study is that we only examined the TGF-β1/Smad3 signaling pathway. In addition to this pathway, other signaling pathways may also be activated by CS, such as the Wnt/β-catenin, PI3K/AKT, or Nrf2/DAMP signaling pathways [45-47], which are all involved in airway remodeling. It is unclear whether other signaling pathways participate in the GRg1-mediated protection against airway remodeling. We observed that over-expression of Smad3 did not affect IL-6 or TNF-α levels in CSE-stimulated MRC5 cells, indicating that GRg1 may regulate inflammatory responses through non-Smad3 dependent signaling pathways. In addition, although our study demonstrated that GRg1 improved CSE-induced mitochondria injury, we did not determine whether mitophagy or mitochondrial dynamics mediate the protective effect of GRg1 in airway remodeling. Further studies are warranted to elucidate the mechanisms underlying the GRg1-mediated benefits on pulmonary function in COPD.
In conclusion, the results of the present study demonstrated that the administration of GRg1 has notable benefits on pulmonary function and inhibits the progression of airway remodeling in COPD by down-regulating inflammatory responses and airway fibrosis. Our data also revealed that GRg1 exerts its anti-fibrotic effects primarily by suppressing the TGF-β1/Smad3 signaling pathway. These findings provide insights into the molecular mechanisms of GRg1 function and its potential as a novel therapeutic agent for COPD.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant number: 81903420), The Health Industry Project of Pudong Health Bureau of Shanghai (Grant number: PW2013E-1), Shanghai Three-year plan on promoting TCM development (Grant number: ZY (2018-2020)-FWTX-2007), and Clinical Peak Discipline of TCM/Clinical Plateau Discipline of TCM (Grant number: PDZY-2018-0603).
Disclosure of conflict of interest
None.
References
- 1.Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, Li X, Wang L, Wang L, Liu Y, Liu J, Zhang M, Qi J, Yu S, Afshin A, Gakidou E, Glenn S, Krish VS, Miller-Petrie MK, Mountjoy-Venning WC, Mullany EC, Redford SB, Liu H, Naghavi M, Hay SI, Wang L, Murray CJL, Liang X. Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;394:1145–1158. doi: 10.1016/S0140-6736(19)30427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Criner GJ, Frith P, Halpin DMG, Han M, Lopez Varela MV, Martinez F, Montes de Oca M, Papi A, Pavord ID, Roche N, Sin DD, Stockley R, Vestbo J, Wedzicha JA, Vogelmeier C. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019:53. doi: 10.1183/13993003.00164-2019. [DOI] [PubMed] [Google Scholar]
- 3.Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389:1931–1940. doi: 10.1016/S0140-6736(17)31222-9. [DOI] [PubMed] [Google Scholar]
- 4.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa-Ochiai K, Kawasaki K. Panax ginseng for frailty-related disorders: a review. Front Nutr. 2018;5:140. doi: 10.3389/fnut.2018.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Chen Y, Ren W, Zhu CD, Li CY, Zhou Q, Ji HY. Effect of Qi benefiting blood activating method on plasma fibrinogen and D-dimer in patients with acute exacerbation of chronic obstructive pulmonary disease. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2015;35:537–540. [PubMed] [Google Scholar]
- 7.Min J, Mao B, Jiang HL, Fan T, Zhou W. [Effects of “reinforcing Qi and activating blood” on stable chronic obstructive pulmonary disease (Qi deficiency and blood stasis syndrome] . Sichuan Da Xue Xue Bao Yi Xue Ban. 2014;45:601–605. [PubMed] [Google Scholar]
- 8.Li J, Cai D, Yao X, Zhang Y, Chen L, Jing P, Wang L, Wang Y. Protective effect of ginsenoside Rg1 on hematopoietic stem/progenitor cells through attenuating oxidative stress and the Wnt/beta-Catenin signaling pathway in a mouse model of d-Galactose-induced aging. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17060849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang YJ, Zhang XL, Li MH, Iqbal J, Bourantas CV, Li JJ, Su XY, Muramatsu T, Tian NL, Chen SL. The ginsenoside Rg1 prevents transverse aortic constriction-induced left ventricular hypertrophy and cardiac dysfunction by inhibiting fibrosis and enhancing angiogenesis. J Cardiovasc Pharmacol. 2013;62:50–57. doi: 10.1097/FJC.0b013e31828f8d45. [DOI] [PubMed] [Google Scholar]
- 10.Li JP, Gao Y, Chu SF, Zhang Z, Xia CY, Mou Z, Song XY, He WB, Guo XF, Chen NH. Nrf2 pathway activation contributes to anti-fibrosis effects of ginsenoside Rg1 in a rat model of alcohol- and CCl4-induced hepatic fibrosis. Acta Pharmacol Sin. 2014;35:1031–1044. doi: 10.1038/aps.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie XS, Liu HC, Wang FP, Zhang CL, Zuo C, Deng Y, Fan JM. Ginsenoside Rg1 modulation on thrombospondin-1 and vascular endothelial growth factor expression in early renal fibrogenesis in unilateral obstruction. Phytother Res. 2010;24:1581–1587. doi: 10.1002/ptr.3190. [DOI] [PubMed] [Google Scholar]
- 12.Fan X, Zhang C, Niu S, Fan B, Gu D, Jiang K, Li R, Li S. Ginsenoside Rg1 attenuates hepatic insulin resistance induced by high-fat and high-sugar by inhibiting inflammation. Eur J Pharmacol. 2019;854:247–255. doi: 10.1016/j.ejphar.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 13.Wang QL, Yang L, Peng Y, Gao M, Yang MS, Xing W, Xiao XZ. Ginsenoside Rg1 regulates SIRT1 to ameliorate sepsis-induced lung inflammation and injury via inhibiting endoplasmic reticulum stress and inflammation. Mediators Inflamm. 2019;2019:6453296. doi: 10.1155/2019/6453296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji Q, Sun Z, Yang Z, Zhang W, Ren Y, Chen W, Yao M, Nie S. Protective effect of ginsenoside Rg1 on LPS-induced apoptosis of lung epithelial cells. Mol Immunol. 2018 doi: 10.1016/j.molimm.2018.11.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Bao S, Zou Y, Wang B, Li Y, Zhu J, Luo Y, Li J. Ginsenoside Rg1 improves lipopolysaccharide-induced acute lung injury by inhibiting inflammatory responses and modulating infiltration of M2 macrophages. Int Immunopharmacol. 2015;28:429–434. doi: 10.1016/j.intimp.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Aschner Y, Downey GP. Transforming growth factor-beta: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol. 2016;54:647–655. doi: 10.1165/rcmb.2015-0391TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, Jenkins G, Gauldie J, Ask K, Kolb M. Stretch-induced activation of transforming growth factor-beta1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 18.de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, van Krieken JH. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:1951–1957. doi: 10.1164/ajrccm.158.6.9803053. [DOI] [PubMed] [Google Scholar]
- 19.Mahmood MQ, Reid D, Ward C, Muller HK, Knight DA, Sohal SS, Walters EH. Transforming growth factor (TGF) beta1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology. 2017;22:133–140. doi: 10.1111/resp.12882. [DOI] [PubMed] [Google Scholar]
- 20.Asano K, Shikama Y, Shoji N, Hirano K, Suzaki H, Nakajima H. Tiotropium bromide inhibits TGF-beta-induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways in vitro. Int J Chron Obstruct Pulmon Dis. 2010;5:277–286. doi: 10.2147/copd.s11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li SS, He AL, Deng ZY, Liu QF. Ginsenoside-Rg1 protects against renal fibrosis by regulating the Klotho/TGF-beta1/Smad signaling pathway in rats with obstructive nephropathy. Biol Pharm Bull. 2018;41:585–591. doi: 10.1248/bpb.b17-00934. [DOI] [PubMed] [Google Scholar]
- 22.Song L, Guan XJ, Chen X, Cui ZL, Han FF, Guo XJ, Xu WG. Mesenchymal stem cells reduce cigarette smoke-induced inflammation and airflow obstruction in rats via TGF-beta1 signaling. Copd. 2014;11:582–590. doi: 10.3109/15412555.2014.898032. [DOI] [PubMed] [Google Scholar]
- 23.Ye Y, Shan Y, Bao C, Hu Y, Wang L. Ginsenoside Rg1 protects against hind-limb ischemia reperfusion induced lung injury via NF-kappaB/COX-2 signaling pathway. Int Immunopharmacol. 2018;60:96–103. doi: 10.1016/j.intimp.2018.04.040. [DOI] [PubMed] [Google Scholar]
- 24.Shergis JL, Di YM, Zhang AL, Vlahos R, Helliwell R, Ye JM, Xue CC. Therapeutic potential of panax ginseng and ginsenosides in the treatment of chronic obstructive pulmonary disease. Complement Ther Med. 2014;22:944–953. doi: 10.1016/j.ctim.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Gross D, Shenkman Z, Bleiberg B, Dayan M, Gittelson M, Efrat R. Ginseng improves pulmonary functions and exercise capacity in patients with COPD. Monaldi Arch Chest Dis. 2002;57:242–246. [PubMed] [Google Scholar]
- 26.An X, Zhang AL, Yang AW, Lin L, Wu D, Guo X, Shergis JL, Thien FC, Worsnop CJ, Xue CC. Oral ginseng formulae for stable chronic obstructive pulmonary disease: a systematic review. Respir Med. 2011;105:165–176. doi: 10.1016/j.rmed.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Sohal SS, Ward C, Danial W, Wood-Baker R, Walters EH. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev Respir Med. 2013;7:275–288. doi: 10.1586/ers.13.26. [DOI] [PubMed] [Google Scholar]
- 28.Brightling C, Greening N. Airway inflammation in COPD-progress to precision medicine. Eur Respir J. 2019;54:1900651. doi: 10.1183/13993003.00651-2019. [DOI] [PubMed] [Google Scholar]
- 29.Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, Calverley P, Coxson H, Crim C, Edwards LD, Lomas DA, Duvoix A, MacNee W, Rennard S, Silverman E, Vestbo J, Wouters E, Agusti A. Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1065–1072. doi: 10.1164/rccm.201110-1792OC. [DOI] [PubMed] [Google Scholar]
- 30.Hofseth LJ, Wargovich MJ. Inflammation, cancer, and targets of ginseng. J Nutr. 2007;137:183S–185S. doi: 10.1093/jn/137.1.183S. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Ahn H, Han BC, Lee SH, Cho YW, Kim CH, Hong EJ, An BS, Jeung EB, Lee GS. Korean red ginseng extracts inhibit NLRP3 and AIM2 inflammasome activation. Immunol Lett. 2014;158:143–150. doi: 10.1016/j.imlet.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 32.Fan C, Song Q, Wang P, Li Y, Yang M, Yu SY. Neuroprotective effects of ginsenoside-Rg1 against depression-like behaviors via suppressing glial activation, synaptic deficits, and neuronal apoptosis in rats. Front Immunol. 2018;9:2889. doi: 10.3389/fimmu.2018.02889. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Martinon F. Inflammation initiated by stressed organelles. Joint Bone Spine. 2018;85:423–428. doi: 10.1016/j.jbspin.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Mohanty A, Tiwari-Pandey R, Pandey NR. Mitochondria: the indispensable players in innate immunity and guardians of the inflammatory response. J Cell Commun Signal. 2019;13:303–318. doi: 10.1007/s12079-019-00507-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray LA. Commonalities between the pro-fibrotic mechanisms in COPD and IPF. Pulm Pharmacol Ther. 2012;25:276–280. doi: 10.1016/j.pupt.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Ko JW, Jeong SH, Kwon HJ, Shin NR, Seo YS, Kim JC, Shin IS, Kim JS. Preventive effect of garlic oil and its organosulfur component diallyl-disulfide on cigarette smoke-induced airway inflammation in mice. Nutrients. 2018;10 doi: 10.3390/nu10111659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current concepts in pathogenesis, diagnosis, and management of smoking-related interstitial lung diseases. Chest. 2018;154:394–408. doi: 10.1016/j.chest.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 38.Rehan VK, Wang Y, Sugano S, Romero S, Chen X, Santos J, Khazanchi A, Torday JS. Mechanism of nicotine-induced pulmonary fibroblast transdifferentiation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L667–676. doi: 10.1152/ajplung.00358.2004. [DOI] [PubMed] [Google Scholar]
- 39.Milara J, Serrano A, Peiro T, Artigues E, Gavalda A, Miralpeix M, Morcillo EJ, Cortijo J. Aclidinium inhibits cigarette smoke-induced lung fibroblast-to-myofibroblast transition. Eur Respir J. 2013;41:1264–1274. doi: 10.1183/09031936.00017712. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Wang L, Dong Z, Wang S, Qi L, Cho K, Zhang Z, Li N, Hu Y, Jiang B. Cardioprotection of salvianolic acid B and ginsenoside Rg1 combination on subacute myocardial infarction and the underlying mechanism. Phytomedicine. 2019;57:255–261. doi: 10.1016/j.phymed.2018.12.040. [DOI] [PubMed] [Google Scholar]
- 41.Jin J, Togo S, Kadoya K, Tulafu M, Namba Y, Iwai M, Watanabe J, Nagahama K, Okabe T, Hidayat M, Kodama Y, Kitamura H, Ogura T, Kitamura N, Ikeo K, Sasaki S, Tominaga S, Takahashi K. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-beta1. Respir Res. 2019;20:119. doi: 10.1186/s12931-019-1093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chellini F, Tani A, Vallone L, Nosi D, Pavan P, Bambi F, Zecchi Orlandini S, Sassoli C. Platelet-rich plasma prevents in vitro transforming growth factor-beta1-induced fibroblast to myofibroblast transition: involvement of vascular endothelial growth factor (VEGF)-A/VEGF receptor-1-mediated signaling (dagger) Cells. 2018;7:142. doi: 10.3390/cells7090142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei X, Chen Y, Huang W. Ginsenoside Rg1 ameliorates liver fibrosis via suppressing epithelial to mesenchymal transition and reactive oxygen species production in vitro and in vivo. Biofactors. 2018;44:327–335. doi: 10.1002/biof.1432. [DOI] [PubMed] [Google Scholar]
- 44.Du N, Xu Z, Gao M, Liu P, Sun B, Cao X. Combination of ginsenoside Rg1 and astragaloside IV reduces oxidative stress and inhibits TGF-beta1/Smads signaling cascade on renal fibrosis in rats with diabetic nephropathy. Drug Des Devel Ther. 2018;12:3517–3524. doi: 10.2147/DDDT.S171286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M, Novellino E, Brusselle GG, Wagner DE, Yildirim AO, Konigshoff M. Reduced frizzled receptor 4 expression prevents WNT/beta-catenin-driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196:172–185. doi: 10.1164/rccm.201605-0904OC. [DOI] [PubMed] [Google Scholar]
- 46.Pirozzi F, Ren K, Murabito A, Ghigo A. PI3K signaling in chronic obstructive pulmonary disease: mechanisms, targets, and therapy. Curr Med Chem. 2018;26:2791–2800. doi: 10.2174/0929867325666180320120054. [DOI] [PubMed] [Google Scholar]
- 47.Lee H, Lee J, Hong SH, Rahman I, Yang SR. Inhibition of RAGE attenuates cigarette smoke-induced lung epithelial cell damage via RAGE-mediated Nrf2/DAMP signaling. Front Pharmacol. 2018;9:684. doi: 10.3389/fphar.2018.00684. [DOI] [PMC free article] [PubMed] [Google Scholar]