

Abstract
Glycosylation plays an important role in the genesis of various cancers. The inhibition of glycosylation disturbs the protein folding machinery, causing the accumulation of unfolded proteins in the cell endoplasmic reticulum (ER) and inducing ER stress. Tunicamycin (TM) is an inhibitor of glycosylation that has shown marked antitumor activity. In this study, we investigated the effect of TM on the tumorigenesis of head and neck cancer cells. The effects of TM on cell proliferation, colony formation and tumorsphere formation in vitro and tumorigenicity in vivo were investigated in head and neck cancer cells. ER stress was determined by the evaluation of PERK, PDI, IRE1-α, BIP, Ero1-Lα and calnexin expression using western blotting and immunofluorescence. We found that TM inhibited colony formation and tumorsphere formation of head and neck cancer cells in vitro and suppressed tumor growth in vivo. After incubation with TM, the expression of the cancer stem cell markers CD44 and Bmi-1 was reduced, and the expression of the ER stress markers BIP, Ero1-Lα and calnexin was elevated. Moreover, the EGFR signaling pathway was inhibited, and nonglycosylated EGFR degradation was accelerated with TM treatment. Our results suggest that inhibition of glycosylation by TM may be a novel treatment strategy for use with HNSCC patients.
Keywords: Head and neck squamous cell carcinoma, tunicamycin, glycosylation, ER stress, EGFR, CD44
Introduction
Head and neck cancer (HNC) is the sixth most common cancer in the world, squamous cell carcinoma accounts for over 90% of HNCs [1]. Despite continuous improvement in traditional treatments (surgery, radiotherapy and chemotherapy) over the past three decades, the 5-year survival rate for HNC patients has been unsatisfactory [2]. Thus, the discovery of novel treatment strategies and the application of novel chemotherapy agents are important and urgent for improving the outcomes of HNC.
Tunicamycin, initially identified as a natural antibiotic, inhibits N-linked glycosylation by blocking the UDP-HexNAc:polyprenol-P HexNAc-1-P family of enzymes in the human enzyme GlcNAc phosphotransferase (GPT) [3,4]. As a major posttranslational modification, glycosylation plays an important role in the folding, stability, subcellular localization and biological functions of glycoproteins. Aberrant glycosylation is recognized as a hallmark of cancer and is significantly correlated with the development, progression, metastasis and chemoresistance of tumors [5-13]. Thus, TM has been identified as a potential anticancer therapeutic. It has been reported that TM can sensitize cancer cells to chemotherapy, reverse chemoresistance and promote apoptosis in many kinds of cancers [14-17]. Furthermore, inhibition of N-linked glycosylation by TM eventually causes the accumulation of unfolded proteins in the endoplasmic reticulum (ER) lumen and potentially triggers ER stress. The ER is the widest intracellular organelle, spanning from the nuclear envelope to the cell membrane, and is involved to several different activities, including calcium storage, detoxification of chemical compounds, and lipid synthesis. It is also the site of protein folding and posttranslational modification of proteins. To overcome imbalanced ER protein-folding capacity, cells have evolved an evolutionarily conserved signal transduction pathway called the unfolded protein response (UPR) that establishes ER homeostasis [18,19]. This UPR mechanism has been elucidated through the activation of three different pathways that operate in parallel to each other upon induction by the ER transmembrane proteins inositol-requiring enzyme 1a (IRE1a), PRKR-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6) [19,20]. Essentially, ER stress facilitates the activation of these signaling pathways, leading to the upregulation of the molecular chaperones involved in protein folding and degradation and the maintenance of normal cellular function by halting protein translation and synthesis [21]. It has been reported that TM-induced ER stress leads cells towards death via apoptosis [22-24]. However, few studies have focused on the effects of TM on HNSCC.
Since cancer stem cells (CSCs) are capable of self-renewal and proliferation, they are thought to initiate tumorigenesis and tumor recurrence after treatment. CD44, a CSC marker expressed in HNSCC, plays an important role in tumorigenesis. CD44 is a cell-surface glycoprotein that functions as a receptor for hyaluronic acid (HA) and is involved in cell acquisition of stemness and ability to become a CSC in HNSCC [25,26]. However, the molecular mechanisms underlying glycosylation and stemness in HNSCC are obscure. Here, we report that TM treatment triggers ER stress in head neck squamous cell carcinoma (HNSCC) cells and inhibits stem cell-like markers. Furthermore, we found that TM inhibited tumorigenesis both in vitro and in vivo. Then, we discovered that the EGFR signaling pathway was inhibited by the inhibition of glycosylation by TM and that the degradation of nonglycosylated EGFR was accelerated. Therefore, novel drugs that specifically inhibit the glycosylation of tumor cells is expected to improve the overall efficacy of clinical treatment of HNSCC and deserves further development.
Materials and methods
Cell cultures and reagents
HN4 cells, from an HNSCC-derived cell line, were kindly provided by the University of Maryland School of Dentistry. The CAL27 cell line was purchased from the American Type Culture Collection (ATCC). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin-streptomycin. All cells were maintained in a humidified atmosphere of 5% CO2 at 37°C. Antibodies against GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against p-EGFR, p-AKT, p-Erk, p-STAT3, EGFR, AKT, Erk and STAT3 were purchased from Cell Signaling Technology, Inc. An ER stress sample kit, including antibodies against PERK, PDI, IRE1-α, BIP, Ero1-Lα and calnexin, was purchased from Cell Signaling Technology, Inc. HRP-conjugated secondary antibodies were obtained from eBioscience (San Diego, CA).
Cell viability assay
Cell viability was measured with Cell Counting Kit-8 (DOJINDO, Kumamoto, Japan) according to the manufacturer’s instructions. Cells were seeded in 96-well plates at a density of 2×103 cells/well, in triplicate, and incubated overnight. We then treated the cells with the appropriate drugs. After treatment, the original medium was replaced with a mixture of 10 μl of CCK-8 reagent and 100 μl of fresh medium. The cells were incubated for another 4 hours at 37°C. Finally, the absorbance of each well was measured at 450 nm by a microplate reader (Varioskan Flash, Thermo Scientific). Every experiment was performed in triplicate.
Western blotting
The experimental protocol was performed as described previously [27]. Cells were lysed with RIPA lysis buffer (Cell Signaling Technology). Cell lysates were separated by SDS-PAGE in a 10% acrylamide gel and transferred onto a nitrocellulose membrane for immunoblot analysis.
Immunofluorescence
The experimental protocol was performed as described previously [27]. Cultured cells were rinsed three times with PBS and fixed with 3.7% formaldehyde and then permeabilized with 0.1% Triton X-100. After being blocked in 1% BSA for 1 hour, the cells were incubated with the primary antibody in a moist, 4°C chamber overnight, washed and then incubated with Alexa Fluor 488 donkey anti-rabbit IgG antibody (Invitrogen, Grand Island, NY) in the dark for 1 hour at room temperature. After being mounting onto a slide, the washed cells (with PBS containing 0.02% Tween 20) were stained with aqueous mounting medium containing 0.5 mg/ml 40-6-diamidino-2-phenylindole and examined with a fluorescence microscope.
Tumorsphere formation
A sphere formation analysis was performed as described elsewhere [27]. Briefly, HN4 and CAL27 cells were cultivated in serum-free Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with or without tunicamycin treatment in a low-adhesion plate. After two weeks, all spheres were poured through a 70 μm filter membrane. Only spheres greater than 70 μm in diameter were counted. The sphere formation efficiency was calculated.
In vivo experiment
This study was approved by the ethics committee of our institution. Nude mice were transplanted with tumors established with CAL27 and HN4 cells at 1×106 cells/point. The experimental cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) and treated with tunicamycin for 48 hours, and the controls were treated with normal saline. Then, the cells were injected into the mice subcutaneously. Four weeks after the injection, the mice were sacrificed, and tumor tissues were excised. The animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978).
Colony formation assay
A total of 1×103 cells were plated in 60 mm culture dishes and incubated with TM or DMSO for 2 weeks to allow for colony formation. The colonies were fixed by treatment with 70% ethanol for 1 hour, stained with 0.1% Coomassie Brilliant Blue R-250 (Thermo, USA) for 2 hours and washed with PBS. Colonies with more than 50 cells were counted under a dissecting microscope. The data from the colony formation assays were calculated as the means (± SD) from 3 independent experiments, each performed in triplicate.
Statistical analysis
All assays were in triplicate test. The data were expressed as mean ± standard deviation (SD). Student’s t test or Wilcoxon test were used for comparison between groups. P<0.05 was considered statistically significant.
Results
TM triggers ER stress in HNSCC cells and inhibits the proliferation of HNSCC cells
To explore the effects of TM on HNSCC cells, we identified the dose-response curves of the treated HN4 and CAL27 cells. CAL27 and HN4 cell exposure to TM resulted in a dose-dependent inhibition of cell viability (Figure 1). To further explore the effects of TM on HNSCC cells, the HN4 and CAL27 cells were treated with TM (2 µg/ml) for 24 hours, and the ER stress levels were monitored. ER stress was assessed by western blot analysis of PERK, PDI, IRE1α, BIP, Ero1-Lα and calnexin protein expression. The results showed that PDI, IRE1α, BIP, Ero1-Lα and calnexin were all upregulated in the TM-treated cells, but not PERK, which was slightly downregulated in the TM-treated cells (Figure 2A). Immunofluorescence examination of BIP in CAL27 cells also revealed upregulated expression with TM treatment (Figure 2B). As the expression of all the proteins was considered comprehensively, we inferred that TM induced ER stress in the HNSCC cell lines.
Figure 1.
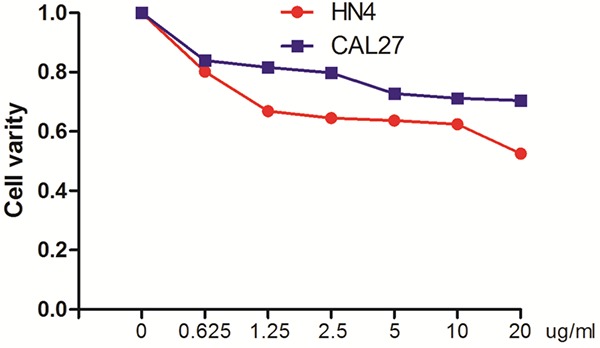
Tunicamycin exerts cytotoxic effects in HNSCC cells. Cell survival after TM treatment for 24 hours as assayed by CCK-8. The values in the untreated group serve as the baseline, representing 100% survival.
Figure 2.
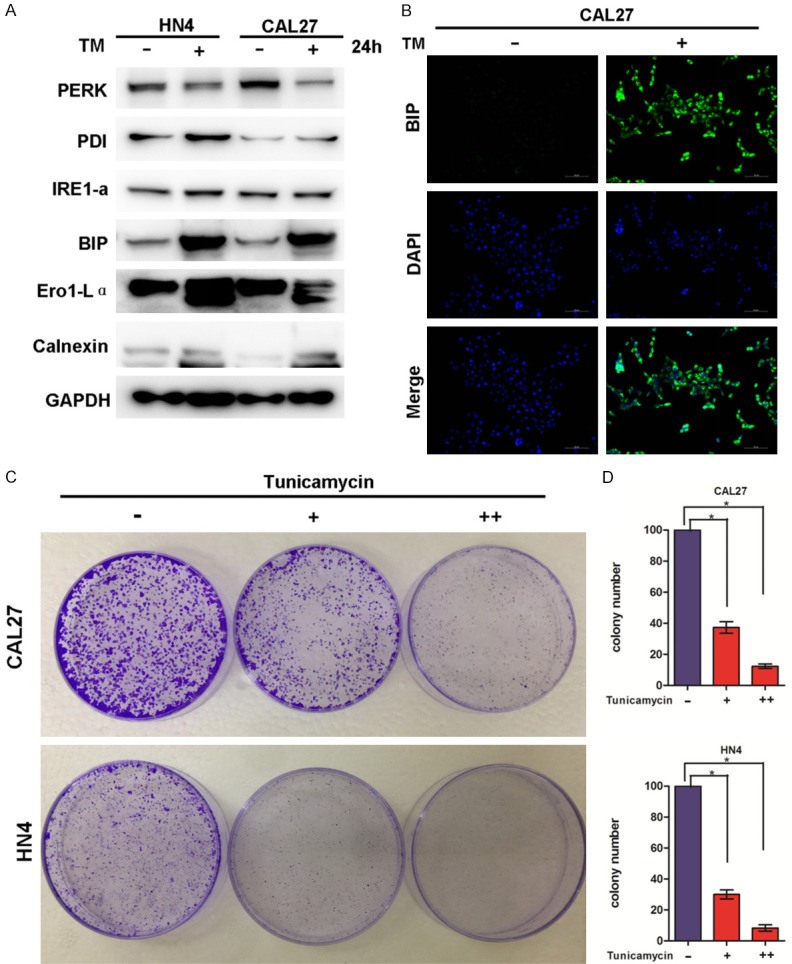
TM triggers ER stress and inhibits the proliferation of HNSCC cells. A. HN4 and CAL27 cells were treated with TM (2 µg/ml) for 24 hours, and the protein levels of ER stress markers PERK, PDI, IRE1-α, BIP, Ero1-Lα and calnexin was analyzed by western blotting. B. Immunofluorescence staining for BIP in CAL27 cells treated with or without 2 ng/mL TM for 24 hours. Scale bar = 50 μm. C. Representative image of colony formation in HN4 and CAL27 cells treated with TM (0, 1 µg/ml, 2 µg/ml). D. Measurement of colony formation (mean ± SD from 3 separate experiments) in CAL27 and HN4 cells, *P<0.05.
Moreover, colony formation assays were also performed on the HN4 and CAL27 cells treated with different concentrations of TM for 2 weeks. TM treatment inhibited colony formation in a dose-dependent manner, as shown in Figure 2C and 2D, confirming the inhibitory effect of TM on the proliferation of HNSCC cells.
TM suppresses the CSC characteristics of HNSCC cells
Since cancer stem cells (CSCs) are capable of self-renewal and proliferation, they are thought to initiate tumorigenesis and promote tumor recurrence after treatment. To determine the effects of TM on the CSC characteristics of HNSCC cells, we performed a tumorsphere assay with the HN4 and CAL27 cells. We found that tumorsphere formation was significantly suppressed in the cells from both lines treated with TM (Figure 3A and 3B). Next, we examined the CSC markers CD44, Bmi-1 and Oct4 in HNSCC cells by western blot analysis; CD44 expression was decreased in the HN4 and CAL27 cells treated with TM for 48 hours (Figure 3C). For the TM-treated cells, western blots developed with an anti-CD44 antibody had a band lower than 90 kDa, indicating the glycosylation of CD44, which is localized to the apical domain of plasma membranes, was inhibited by TM. These results showed that the inhibitory effect of TM on tumorsphere formation was partly the result of inhibition of CD44 glycosylation. In addition, we also determined the expression of Bmi-1 and Oct4, specifically finding that the expression of Bmi-1 was inhibited.
Figure 3.
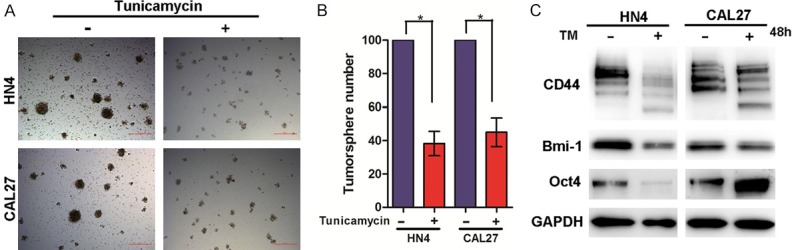
TM suppresses the CSC characteristics of HNSCC cells. A. Representative image of tumorsphere formation in HN4 and CAL27 cells treated with or without TM (2 µg/ml) for 7 days. Scale bar =100 μm. B. Measurement of tumorsphere formation (mean ± SD from 3 separate experiments) in CAL27 and HN4 cells, *P<0.05. C. Western blot analysis of CD44, Bmi-1 and Oct-4 protein levels of HN4 and CAL27 cells with or without of TM (2 µg/ml) treatment for 48 hours.
TM suppresses the EGFR signaling pathway and facilitates the translocation of EGFR from cell membrane to cytosol
The EGFR signaling pathway plays a critical role in DNA damage repair and HNSCC cell proliferation, aggression and apoptosis. To investigate the molecular mechanism involved in TM-inhibited proliferation and tumorigenesis, we examined the EGFR signaling pathway in TM-treated and untreated HN4 and CAL27 cells. The results showed that EGFR activation was inhibited in both the TM-treated HN4 and CAL27 cells (Figure 4A). In addition, the downstream pathways of EGFR, including the AKT, STAT3 and Erk pathways, were all inhibited in cells treated with TM (Figure 4A).
Figure 4.

TM suppresses the EGFR signaling pathway and facilitates the translocation of EGFR. A. Western blot analysis of p-EGFR, EGFR, p-AKT, AKT, p-STAT3, STAT3, p-Erk and Erk protein levels of HN4 and CAL27 cells with or without of TM (2 µg/ml) treatment for 48 hours. B. Image of immunofluorescence staining for EGFR expression in HN4 cells treated with or without TM (2 µg/ml) for 48 hours with nuclei stained with DAPI (blue). Scale bar = 50 μm.
We also noticed that EGFR expression was suppressed by TM treatment and that EGFR migration in cells treated with TM was slow on an SDS-PAGE gel (Figure 4A). Because TM strongly interferes with the N-linked glycosylation of the proteins in the ER and thus potently induces ER stress, we speculated that the slowly migrating band represented nonglycosylated EGFR. Moreover, evidence from the immunofluorescence assay revealed that EGFR was located on the plasma membrane of the untreated cells. EGFR in the TM-treated HN4 cells was determined (Figure 4B) and suggested that deglycosylation of EGFR by TM treatment is involved in promoting the translocation of EGFR to the cytosol. Taken together, the evidence indicated that TM treatment leads to dysregulation of EGFR signaling in HNSCC cells.
TM induces EGFR degradation by inhibiting its glycosylation
We noticed that the EGFR band in the western blot of the TM-treated cells was lower than the parallel band in the blot of the untreated cells (Figure 4A). As EGFR is a glycosylated transmembrane protein, we attempted to clarify whether the inhibition of glycosylation by TM resulted in the suppression of EGFR expression. To determine the regulatory mechanism, we examined the protein levels of EGFR in cells treated with or without TM in the absence or presence of the protein translation inhibitor cycloheximide (CHX) (Figure 5A and 5B). The western blots of the TM-treated cells showed two bands for EGFR representing glycosylated and nonglycosylated forms. The nonglycosylated EGFR showed accelerated degradation, while the glycosylated EGFR showed sustained stability (Figure 5C and 5D). To determine the involvement of the 26S proteasome machinery, we subsequently treated the TM-treated cells with the proteasome inhibitor MG132. We found that only the nonglycosylated EGFR exhibited additional ubiquitination in the presence of MG132 (Figure 5E), suggesting that nonglycosylated EGFR undergoes relatively fast degradation. The results suggest that the TM treatment suppressed the glycosylation of EGFR and promoted EGFR degradation.
Figure 5.
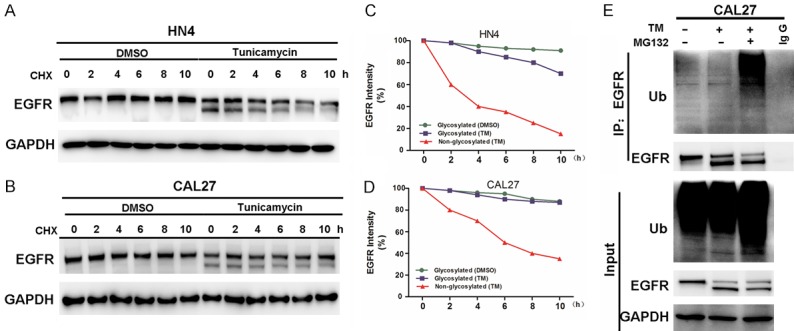
TM promotes degradation of nonglycosylated EGFR. A, B. HN4 and CAL27 cells were treated with or without TM for 2 hours and then incubated with cycloheximide (CHX; 10 μM) for an extended period of time. The levels of EGFR were determined by western blot analysis. C, D. Graphic representation of the densitometry results of EGFR after cycloheximide (CHX) treatment (circle, glycosylated EGFR with DMSO; square, glycosylated EGFR with TM; triangle, nonglycosylated EGFR with TM). E. Inhibition of EGFR glycosylation enhances ubiquitination. CAL27 cells treated with TM and/or MG132 were subjected to EGFR immunoprecipitation (IP) and western blot analyses with anti-ubiquitin.
TM suppresses tumorigenesis of HNSCC cells in vivo
Because TM inhibited CSC characteristics and the EGFR signaling pathway of the HNSCC cells, we determined whether TM suppresses tumorigenesis in vivo. HN4 and CAL27 cells with or without TM treatment were used to establish transplantation tumors in nude mouse. The results showed that untreated cells formed more bulky tumors with greater weight than were established by the TM-treated cells (Figure 6A and 6B). Thus, TM treatment significantly suppressed HNSCC tumorigenesis in vivo.
Figure 6.

TM suppresses tumorigenesis of HNSCC cells in vivo. A, B. CAL27 and HN4 cells treated with or without TM were injected into nude mice. Tumor growth was monitored every 3 days; tumor weight was recorded. Data are represented as the mean ± SEM from five mice. *P<0.05.
Discussion
Cancer development is characterized by uncontrolled growth and proliferation of transformed cells, resulting in a compact mass of cells a tumor environment characterized by oxygen and glucose shortage, at least in solid tumors, two conditions that are considered to be canonical and well-characterized ER stress stimuli. A large number of studies have demonstrated the tight relationship between ER stress and cancer hallmarks, such as angiogenesis, and cell invasion, proliferation and survival. Although ER stress is initially activated as a cytoprotective mechanism, excess or prolonged ER stress can result in apoptosis [22-24]. Therefore, it seems that TM has potential as an anticancer therapeutic, as demonstrated in recent studies. For example, cotreatment with TM and Adriamycin dramatically decreased the viability of gastric cancer cells, especially multidrug-resistant cells, by triggering ER stress-associated apoptosis [28]. Our results revealed that, in the HNSCC cells examined, TM induced ER stress, especially the most represented ER-resident chaperone BIP/GRP78 (78-kDa glucose-regulated protein), and repressed the proliferation of tumor cells.
To clarify whether TM inhibits HNSCC tumorigenesis, we used HN4 and CAL27 HNSCC cells for in vitro and in vivo experiments. Tumorigenesis of cancer cells is highly correlated with proliferation capacity and cancer stem cell characteristics. CD44 is a transmembrane glycoprotein that includes an extracellular region that interacts with growth factors and hyaluronan (HA), as well as a cytoplasmic moiety that is capable of interacting with cytoskeletal components. CD44 has been shown to be a cancer stem marker for several kinds of cancers, such as breast cancer, head and neck cancer, and ovarian cancer. Previous studies have shown that inhibition of CD44 blocks tumor growth, invasion and metastasis [29,30]. In this study, we show that TM has a direct effect on HNSCC cell proliferation partially through the inhibition of CD44. Furthermore, we found that TM not only inhibited glycosylation of CD44, showing a serial band of approximately 70 kDa or lower in the western blot but also downregulated the expression of Bmi-1, another potential CSC marker.
In HNSCC, EGFR is recruited by CD44 to form the CD44-EGFR complex; then, the downstream signaling pathways are activated [31]. Activation of EGFR leads to a phosphorylation cascade mediated via tyrosine kinases that function downstream through the PI3K/AKT, MAPK/ERK, and Jak/STAT pathways and promote cell proliferation, invasion, metastasis and other tumor progression behaviors. EGFR has been found to be highly N-glycosylated, and there are 11 N-glycosylation sites in the extracellular domain [32]. Previous studies have reported the importance of N-glycosylation on the functional properties of EGFR, including its dimerization [33], endocytosis [34], cell surface expression [35,36], ligand binding [37], and interaction with membranes [38,39]. It has been shown that the conformational stability of EGFR is influenced partly by N-linked glycosylation [40]. Deglycosylation may weaken EGFR functions. Research has reported that, in the presence of TM, an immature EGFR protein of 130-135 kDa is synthesized that apparently does not reach the cell surface and does not acquire the capacity to bind EGF [41]. It has also been reported that RPN2-mediated glycosylation of EGFR regulates colorectal cancer cell proliferation by affecting the G1/S transition [42]. In this study, we found that EGFR is glycosylated in HNSCC cells and that TM inhibited EGFR expression by regulating its glycosylation to weaken its stability (Figure 5). It was reported that TM inhibited the proliferation and migration of HCC cells by attenuating the activation of ERK1/2 [28]. Our study showed that TM inhibited the expression of pAKT/AKT, pERK/ERK, and pSTAT3/STAT3, indicating the inhibition of the overall EGFR pathway. The results also demonstrated that glycosylation enhanced the stability of EGFR. Therefore, we speculated that TM inhibits HNSCC cell proliferation and expression of CSC characteristics possibly through regulating the glycosylation of CD44 and EGFR, have a further impact on downstream signaling pathways. However, EGFR signaling pathway mediation by CD44 needs to be confirmed in the future.
Studies have reported that TM inhibits proliferation and induces apoptosis in hepatocellular carcinoma cells, breast cancer cells and colon cancer cells [28,43,44]. However, it remains unclear whether TM suppresses HNSCC tumorigenesis in vivo. Our results provide positive evidence that TM inhibits HNSCC transplantation tumors in vivo, indicating the likelihood that TM can be used as an antitumor therapeutic and that glycosylation may be a target of novel antitumor drugs.
In summary, our study suggests that the glycosylation inhibitor TM attenuates HNSCC tumorigenesis in a CD44- and EGFR-dependent manner.
Acknowledgements
This work was supported by the grant of National Nature Science Foundation of China 81802696 (to Shuli Liu), grant of Shanghai Natural Science Foundation of China 17ZR1416300 (to Yang Wang).
Disclosure of conflict of interest
None.
References
- 1.Mignogna MD, Fedele S, Lo Russo L. The World Cancer Report and the burden of oral cancer. Eur J Cancer Prev. 2004;13:139–42. doi: 10.1097/00008469-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890–900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 3.Duksin D, Bornstein P. Impaired conversion of procollagen to collagen by fibroblasts and bone treated with tunicamycin, an inhibitor of protein glycosylation. J Biol Chem. 1977;252:955–62. [PubMed] [Google Scholar]
- 4.Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc: dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18:2186–92. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- 5.Very N, Lefebvre T, El Yazidi-Belkoura I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget. 2017;9:1380–1402. doi: 10.18632/oncotarget.22377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Legler K, Rosprim R, Karius T, Eylmann K, Rossberg M, Wirtz RM, Muller V, Witzel I, Schmalfeldt B, Milde-Langosch K, Oliveira-Ferrer L. Reduced mannosidase MAN1A1 expression leads to aberrant N-glycosylation and impaired survival in breast cancer. Br J Cancer. 2018;118:847–856. doi: 10.1038/bjc.2017.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones RB, Dorsett KA, Hjelmeland AB, Bellis SL. The ST6Gal-I sialyltransferase protects tumor cells against hypoxia by enhancing HIF-1alpha signaling. J Biol Chem. 2018;293:5659–5667. doi: 10.1074/jbc.RA117.001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui J, Huang W, Wu B, Jin J, Jing L, Shi WP, Liu ZY, Yuan L, Luo D, Li L, Chen ZN, Jiang JL. N-glycosylation by N-acetylglucosaminyltransferase V enhances the interaction of CD147/basigin with integrin beta1 and promotes HCC metastasis. J Pathol. 2018;245:41–52. doi: 10.1002/path.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty A, Dorsett KA, Trummell HQ, Yang ES, Oliver PG, Bonner JA, Buchsbaum DJ, Bellis SL. ST6Gal-I sialyltransferase promotes chemoresistance in pancreatic ductal adenocarcinoma by abrogating gemcitabine-mediated DNA damage. J Biol Chem. 2018;293:984–994. doi: 10.1074/jbc.M117.808584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao TD, Fernández-Tejada A, Axelrod A, Rosales N, Yan X, Thapi S, Wang A, Park KJ, Nemieboka B, Xiang J, Lewis JS, Olvera N, Levine DA, Danishefsky SJ, Spriggs DR. Antibodies against specific MUC16 glycosylation sites inhibit ovarian cancer growth. ACS Chem Biol. 2017;12:2085–2096. doi: 10.1021/acschembio.7b00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D, Davalos V, McDermott M, Nayak S, Darvishian F, Castillo M, Ueberheide B, Osman I, Fenyo D, Mahal LK, Hernando E. A systems biology approach identifies FUT8 as a driver of melanoma metastasis. Cancer Cell. 2017;31:804–819. e7. doi: 10.1016/j.ccell.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yen HY, Liu YC, Chen NY, Tsai CF, Wang YT, Chen YJ, Hsu TL, Yang PC, Wong CH. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc Natl Acad Sci U S A. 2015;112:6955–60. doi: 10.1073/pnas.1507329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 14.Jung YH, Lim EJ, Heo J, Kwon TK, Kim YH. Tunicamycin sensitizes human prostate cells to TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of cIAP2. Int J Oncol. 2012;40:1941–8. doi: 10.3892/ijo.2012.1402. [DOI] [PubMed] [Google Scholar]
- 15.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68:3803–9. doi: 10.1158/0008-5472.CAN-07-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de-Freitas-Junior JC, Bastos LG, Freire-Neto CA, Rocher BD, Abdelhay ES, Morgado-Díaz JA. N-glycan biosynthesis inhibitors induce in vitro anticancer activity in colorectal cancer cells. J Cell Biochem. 2012;113:2957–66. doi: 10.1002/jcb.24173. [DOI] [PubMed] [Google Scholar]
- 17.Hou H, Sun H, Lu P, Ge C, Zhang L, Li H, Zhao F, Tian H, Zhang L, Chen T, Yao M, Li J. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther. 2013;12:2874–84. doi: 10.1158/1535-7163.MCT-13-0201. [DOI] [PubMed] [Google Scholar]
- 18.van den Berg B, Wain R, Dobson CM, Ellis RJ. Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell. EMBO J. 2000;19:3870–5. doi: 10.1093/emboj/19.15.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777–93. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- 21.Shenkman M, Tolchinsky S, Lederkremer GZ. ER stress induces alternative nonproteasomal degradation of ER proteins but not of cytosolic ones. Cell Stress Chaperones. 2007;12:373–83. doi: 10.1379/CSC-281.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 23.Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–181. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 24.Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer. 2016;2:252–262. doi: 10.1016/j.trecan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Bourguignon LY. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol Cancer. 2014;13:52. doi: 10.1186/1476-4598-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourguignon LY. Hyaluronan-CD44 interaction promotes microRNA signaling and RhoGTPase activation leading to tumor progression. Small GTPases. 2012;3:53–9. doi: 10.4161/sgtp.19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Ye D, Guo W, Yu W, He Y, Hu J, Wang Y, Zhang L, Liao Y, Song H, Zhong S, Xu D, Yin H, Sun B, Wang X, Liu J, Wu Y, Zhou BP, Zhang Z, Deng J. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6:6887–901. doi: 10.18632/oncotarget.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, Chen S, Liu H, Zhang Z, Ni Z, Chen J, Yang Z, Nie Y, Fan D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J Exp Clin Cancer Res. 2018;37:272. doi: 10.1186/s13046-018-0935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou H, Ge C, Sun H, Li H, Li J, Tian H. Tunicamycin inhibits cell proliferation and migration in hepatocellular carcinoma through suppression of CD44s and the ERK1/2 pathway. Cancer Sci. 2018;109:1088–1100. doi: 10.1111/cas.13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weidle UH, Maisel D, Klostermann S, Weiss EH, Schmitt M. Differential splicing generates new transmembrane receptor and extracellular matrix-related targets for antibody-based therapy of cancer. Cancer Genomics Proteomics. 2011;8:211–26. [PubMed] [Google Scholar]
- 31.Wang SJ, Bourguignon LY. Role of hyaluronan-mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am J Pathol. 2011;178:956–63. doi: 10.1016/j.ajpath.2010.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, Lee J, Yarden Y, Libermann TA, Schlessinger J, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Natur. 1984;309:418–25. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 33.Wang XQ, Sun P, O’Gorman M, Tai T, Paller AS. Epidermal growth factor receptor glycosylation is required for ganglioside GM3 binding and GM3-mediated suppression [correction of suppresion] of activation. Glycobiology. 2001;11:515–22. doi: 10.1093/glycob/11.7.515. [DOI] [PubMed] [Google Scholar]
- 34.Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–57. doi: 10.1016/j.sbi.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–83. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 36.Coskun Ü, Grzybek M, Drechsel D, Simons K. Regulation of human EGF receptor by lipids. Proc Natl Acad Sci U S A. 2011;108:9044–8. doi: 10.1073/pnas.1105666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lax I, Bellot F, Howk R, Ullrich A, Givol D, Schlessinger J. Functional analysis of the ligand binding site of EGF-receptor utilizing chimeric chicken/human receptor molecules. EMBO J. 1989;8:421–7. doi: 10.1002/j.1460-2075.1989.tb03393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakash A, Janosi L, Doxastakis M. Self-association of models of transmembrane domains of ErbB receptors in a lipid bilayer. Biophys J. 2010;99:3657–65. doi: 10.1016/j.bpj.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azimzadeh Irani M, Kannan S, Verma C. Role of N-glycosylation in EGFR ectodomain ligand binding. Proteins. 2017;85:1529–1549. doi: 10.1002/prot.25314. [DOI] [PubMed] [Google Scholar]
- 40.Taylor ES, Pol-Fachin L, Lins RD, Lower SK. Conformational stability of the epidermal growth factor (EGF) receptor as influenced by glycosylation, dimerization and EGF hormone binding. Proteins. 2017;85:561–570. doi: 10.1002/prot.25220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soderquist Am, Carpenter G. Glycosylation of the epidermal growth factor receptor in A-431 cells. The contribution of carbohydrate to receptor function. J Biol Chem. 1984;259:12586–94. [PubMed] [Google Scholar]
- 42.Li H, Al-Japairai K, Tao Y, Xiang Z. RPN2 promotes colorectal cancer cell proliferation through modulating the glycosylation status of EGFR. Oncotarget. 2017;8:72633–72651. doi: 10.18632/oncotarget.20005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nami B, Donmez H, Kocak N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24- phenotypebreast cancer stem cells. Exp Toxicol Pathol. 2016;68:419–26. doi: 10.1016/j.etp.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Guo X, Meng Y, Sheng X, Guan Y, Zhang F, Han Z, Kang Y, Tai G, Zhou Y, Cheng H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs. 2017;28:66–74. doi: 10.1097/CAD.0000000000000431. [DOI] [PubMed] [Google Scholar]