

Abstract
Thousands of unfixed transposable element (TE) insertions segregate in the human population, but little is known about their impact on genome function. Recently, a few studies associated unfixed TE insertions to mRNA levels of adjacent genes, but the biological significance of these associations, their replicability across cell types and the mechanisms by which they may regulate genes remain largely unknown. Here, we performed a TE-expression QTL analysis of 444 lymphoblastoid cell lines (LCL) and 289 induced pluripotent stem cells using a newly developed set of genotypes for 2743 polymorphic TE insertions. We identified 211 and 176 TE-eQTL acting in cis in each respective cell type. Approximately 18% were shared across cell types with strongly correlated effects. Furthermore, analysis of chromatin accessibility QTL in a subset of the LCL suggests that unfixed TEs often modulate the activity of enhancers and other distal regulatory DNA elements, which tend to lose accessibility when a TE inserts within them. We also document a case of an unfixed TE likely influencing gene expression at the post-transcriptional level. Our study points to broad and diverse cis-regulatory effects of unfixed TEs in the human population and underscores their plausible contribution to phenotypic variation.
This article is part of a discussion meeting issue ‘Crossroads between transposons and gene regulation’.
Keywords: unfixed transposable elements, gene expression QTL, chromatin accessibility QTL, human, lymphoblastoid cell line, induced pluripotent stem cell
1. Background
Transposable elements (TEs) are ubiquitous genetic entities that relocate and multiply within genomes. TE sequences occupy a large fraction of the eukaryotic nuclear DNA, including in humans, where they account for more than half of the genetic material [1]. New TE insertions represent an important source of structural genomic variation that can affect both coding and regulatory components of the genome [2–4]. Notably, many TEs deposit cis-regulatory sequences that can modulate flanking gene expression, and sometimes be repurposed for beneficial cellular function [5–7].
While the human genome hosts hundreds of different TE families from various classes, only three retrotransposon families, LINE1 (L1), Alu and SVA, are known to be active and produce de novo insertions, including approximately 100 disease-causing cases thus far documented [8–10]. These three families represent the primary source of unfixed TEs in humans [11,12]. To date, whole-genome sequencing studies have discovered more than 19 000 TE loci segregating in the human population [11–15]. Despite their potential role in shaping human phenotypic variation, including disease susceptibility [16,17], very little is known about the impact of these polymorphic insertions on genome function. Only a handful of recent studies have started to unravel their contribution to gene expression variation [16–19].
The regulation of gene expression is central to cellular function and differentiation in development and physiology, and changes in gene expression are important drivers of phenotypic variation [20]. Steady-state mRNA levels partially govern gene expression in response to inputs integrated by various regulatory sequences acting in cis or in trans [20,21]. Expression quantitative trait loci (eQTL) studies offer a systematic approach to identify such cis-regulatory elements by correlating the genotypes of genomic variants segregating in individuals with mRNA levels of specific genes, which can be measured on a large scale by RNA-sequencing (RNA-seq) [22,23]. Previous eQTL studies have established that virtually every gene in the human genome has its expression affected by at least one genomic variant (typically a single nucleotide polymorphism, SNP) located in cis (usually defined as within a maximum distance of 1 Mb) and generally in non-coding regions [21,24–26]. A recent analysis of 44 different tissues by the GTEx Consortium indicates that such cis-eQTL fall into two broad categories: those shared across most tissues and those apparently acting in a single or a restricted number of similar tissues [21]. Importantly, most eQTL studies thus far have focused on SNPs, yet other types of genomic variants, such as unfixed TE insertions, are common in the human population and likely to have more drastic effects on gene expression [27–30].
In a pioneering study, Wang et al. [18] mapped TE-eQTL in a reference set of 445 EBV (Epstein-Barr virus)-transformed lymphoblasts (lymphoblastoid cell lines, LCL) for which TE insertion genotypes [12] and RNA-seq data [24] had been previously generated as part of the 1000 Genomes Project and GEUVADIS Consortium, respectively. In this dataset, they identified 83 cis-TE-eQTL where the genotype of TE insertions correlated with mRNA levels of adjacent genes [18]. These data, as well as a follow-up study building on these results [17], suggest that unfixed TEs represent a class of structural variants that plays an important role in driving population- and tissue-specific regulatory variation. However, many questions remain unexplored, in particular (i) the size, direction and strength of the discovered associations, (ii) their tissue- or cell type-specificity and (iii) a better understanding of the molecular mechanisms by which unfixed TEs may modulate gene expression.
To begin filling these gaps, we use a newly assembled set of TE genotypes [31], including more than 800 TE insertions not considered in previous studies [17,18,32], in conjunction with reprocessed RNA-seq quantifications to map TE-eQTL in the aforementioned LCL from 444 individuals. We examined the cell type-specificity of these associations by performing another TE-eQTL mapping using 289 induced human pluripotent stem cells (iPSC) from 188 donors available through the HipSci Consortium [24, www.hipsci.org]. To investigate cis-regulatory mechanisms by which unfixed TEs may affect adjacent gene expression, we explored their association with chromatin accessibility QTL (caQTL) mapped for a subset of the LCL. Lastly, we document a case of an unfixed TE insertion likely affecting the expression of a gene involved in lipid metabolism through a post-transcriptional mechanism.
2. Results
(a). Mapping cis-TE-eQTL in LCL and iPSC
After sample filtering based on their expression profiles (see Material and methods), we conducted a search for TE-eQTL in 444 individual LCL and 289 human-induced pluripotent stem cells (iPSC) from 188 donors using the genotypes of unfixed TE insertions. In LCL, the genomic coordinates for these loci (13 986 Alu; 3104 L1 and 844 SVA) were obtained from the previous analysis of 2504 human samples from the 1000 Genomes Project [12]. To improve genotyping quality, we re-analysed all LCL samples available with TypeTE [31]. These recalls enabled us to use 860 TE insertions present in the reference genome (hg19) which have not been interrogated in previous studies [17,18,32], likely owing to the uncertainty of their original genotypes [31]. TypeTE improves the genotype quality for TE by mapping reads against reconstructed ‘presence’ and ‘absence’ alleles of each insertion (see Material and methods). Genotypes predicted by TypeTE are greater than 90% concordant with those obtained by PCR for a large panel of Alu insertions [31]. To call TE genotypes in iPSC, we initially used the genomic alignments generated by the HipSci Consortium (www.hipsci.org) for 326 healthy fibroblast-derived iPSC, predominantly sourced from subjects of British ancestry [33]. TE detection and genotyping in this iPSC dataset were achieved using MELT2 [15] and TypeTE (see Material and methods). We identified 8477 TEs (6931 Alu; 1068 L1 and 478 SVA) with a ‘PASS’ flag following MELT2 analysis. These numbers are in line with those expected based on the results of the 1000 Genome Project [12,15]. After sample filtering (see below), we identified a total of 2743 unfixed TEs segregating at a minimum allele frequency of 5% among 188 and 444 individual donors of iPSC and LCL, respectively (electronic supplementary material, figure S1).
In order to identify TE-expression QTL (TE-eQTL), we first quantified the steady-state RNA levels of LCL by applying the program kallisto (v. 0.46.0 [34]) against RefSeq (GRCh37.75) using RNA-seq reads originally generated by Lappalainen et al. [24]. This allowed us to match the RNA-seq quantification data generated for the iPSC by the HipSci Consortium (H. Kilpinen 2019, personal communication; see also Material and methods). We then analysed independently LCL and iPSC datasets to search for correlations between TE genotypes and normalized mRNA levels using QTLtools (v. 1.1 [35]). In LCL, samples of African and European ancestry were kept together to maximize power, as the population was not the main factor structuring gene expression (electronic supplementary material, figure S2; less than 6.4% of the variance). cis-TE-eQTL were searched within a 1 Mb window of associated genes (eGenes). Statistical significance was assessed by performing 10 000 permutations of the gene expression matrix and applying a 5% false discovery rate for multiple testing correction (see Material and methods) [35]. The results showed that two chromosomal regions produced an exceptionally high density of predicted TE-eQTL: one corresponds to the human leucocyte antigen (HLA) locus at 6p21 (chr6: 28 477 797–33 448 354 (hg19), electronic supplementary material, figure S3) and the other corresponds to the 17q21.31 inversion (chr17: 38 100 001–50 200 000 (hg19) electronic supplementary material, figure S3). We suspect both regions to be prone to yield a high rate of false positives in eQTL analyses owing to their complex, highly repetitive nature which makes short read mapping unreliable and hinders both TE insertion mapping/genotyping and RNA-seq quantification [36,37]. Thus, conservatively, we excluded 49 (LCL) and 42 (iPSC) TE-eQTL falling within these two regions from subsequent analysis. After this filtering, we obtained a total of 211 and 176 TE-eQTL in LCL and iPSC, respectively (figure 1 and table 1 and electronic supplementary material, figure S4). Repeating the analysis with increasingly larger, randomly selected subsamples revealed no evidence of saturation regarding the total number of TE-eQTL discovered (electronic supplementary material, figure S5).
Figure 1.

Manhattan plots of TE-eQTL p-value significance [−log10(p-value)] in LCL (up) and iPSC (down). p-values are represented according to chromosomal position of TE-eQTL mapped in LCL (up) and in iPSC (down) TE-eQTL. The grey dashed lines represent the 5% false discovery rate (FDR) cut-off values (LCL: 1.21 × 10−3; iPSC: 2.18 × 10−3). Each p-value is coloured according to the TE family. n.s., not significant. (Online version in colour.)
Table 1.
TE-QTL in LCL and iPSCs. ‘genes 50%’: number of genes expressed in at least 50% of the samples and considered in the analysis. ATAC, assay for transposase-accessible chromatin using sequencing; caTEs, chromatin accessible TE insertions; ca peak, chromatin accessibility peak.
| TE-eQTL |
TE-caQTL |
|||||||
|---|---|---|---|---|---|---|---|---|
| cell type | sample size | genes 50% | eGenes | eTEs (Alu/L1/SVA) | sample size | ATAC peaks | ca peaks | caTEs (Alu/L1/SVA) |
| LCL | 444 | 14 320 | 211 | 157/9/4 | 86 | 277 128 | 656 | 394/28/9 |
| iPSC | 188 | 16 245 | 176 | 129 /14/5 | — | — | — | — |
We compared our results with the TE-eQTL analysis performed by Wang et al. [17] on the same set (+1 sample) of LCL (electronic supplementary material, figure S6). Excluding TE-eQTL mapping in the HLA and 17q21.31 regions, we found that only 20 out of 211 (approx. 10%) TE-eQTL discovered in our study with LCL were previously identified by Wang et al. Conversely, there were 33 TE-eQTL identified by Wang et al. (39%) that were not detected by our LCL analysis (31 if compared with both LCL and iPSC). There are at least four important methodological differences that could account for the discrepancies between the two studies. First and foremost, our analysis considered 860 reference TEs never analysed before, presumably because their genotypes could not be confidently predicted [31]. Second, we used improved genotypes recalled by TypeTE [31], while Wang et al. used the genotypes as originally called by Sudmant et al. [12]. We show in electronic supplementary material, figure S7 that re-processing of the genotypes by TypeTE allows the discovery of new TE-eQTL associations that would have been missed by using the genotypes predicted by Sudmant et al. [12]. Third, we required that the minimum allele frequency of a given TE insertion reach 5% in both LCL and iPSC, so we could compare the results across the two datasets. Wang et al. used the same frequency cut-off but only analysed the LCL dataset. Consequently, some insertions present at greater than 5% frequency in LCL but not in iPSC could not be considered in our analysis, even if they could be identified as TE-eQTL in the LCL dataset alone. For example, an SVA insertion in the B4GALT1 gene previously identified as strong TE-eQTL by Wang et al. [17], which we were able to replicate in a separate analysis of the LCL data (data not shown), could not have been identified in the iPSC data because none of the individuals in this dataset possesses the SVA insertion. Fourth and finally, we reprocessed the RNA-seq data for the LCL dataset with more recent methods matching those applied to the iPSC dataset (see Material and methods). This includes differences in the quantification, normalization and gene sorting which could all have contributed to yielding different results from those of Wang et al. [17].
(b). TE-eQTL are enriched near and within genes
For both cell types, the vast majority of the TE-eQTL (92.4% in LCL and 83% in iPSC) have the implicated TE (eTE) located within 250 kb of the gene body (from transcription start site (TSS) to the end of the 3′-UTR, figure 2a,b). To further examine whether the distribution of eTEs relative to eGenes departs from that of all unfixed TEs found within 1 Mb of a quantified gene (figure 2c, ‘TE/gene < 1 Mb’), we compared the location of the two TE categories (eTEs and all TEs within 1 Mb) with randomly sampled genomic positions (see Material and methods). The results (figure 2c) indicate that eTEs are enriched within introns (1000 permutations of the eTE position, mean enrichment ±1 (s.d.)) and within a 10 kb window upstream and downstream of eGenes, but depleted in intergenic regions, coding exons and UTRs. By contrast, when all TEs present in the same 1 Mb window are considered, we observe that they are generally depleted in exons, UTRs and within 10 kb upstream and downstream regions of genes, but their densities in introns and intergenic regions do not depart from random expectations (figure 2c). We conclude that eTEs are more closely associated with eGenes than other TEs, but remain generally excluded from exons and UTRs, presumably because insertions in these compartments tend to be strongly deleterious and rapidly removed from the population.
Figure 2.

TE-eQTL distribution. (a,b) Distribution of the distances between eTE and eGene for LCL (left) and iPSC (right). Grey shading corresponds to the distribution for a matched number of SNP-eQTL. (c) TE enrichments in different genomic compartments: expected values are estimated by selecting a random set of genomic intervals equal to the number of each type of variant. Error bars indicate ±1 s.d. dist. TE/gene < 1 MB: unfixed TE located within 1 Mb of a gene; eTE: TE associated to a given TE-eQTL. up10: region spanning 10 kb upstream of gene transcription start site (TSS); down10: region spanning 10 kb downstream of gene termination site. UTR5: 5′-UTR; UTR3: 3′-UTR. intergenic: genomic region distant by more than 10 kb upstream from TSS or more than 10 kb downstream from 3′-UTR. (Online version in colour.)
(c). Effect size and TE-eQTL significance
Next, we focused on the effect size and p-value distributions of TE-eQTL as a first step to assess their potential biological relevance. The effect size captures the magnitude of changes in gene expression between the three possible TE genotypes (0/0: no TE, 0/1: heterozygous TE insertion, 1/1: homozygous TE insertion) as the slope of the linear regression. The p-value associated with each TE-eQTL reflects the strength of this correlation [38].
We found little to no correlation between the distance of eTEs to eGenes and the effect size of the TE-eQTL (electronic supplementary material, figure S8, Spearman correlation test, r = −0.02, p > 0.05 for LCL; r = −0.20, p < 0.01 for iPSC). Similarly, there was no strong correlation between the distance of eTEs to eGenes and the strength (p-value) of the eQTL (electronic supplementary material, figure S9, Spearman correlation test, LCL: r = −0.11, p > 0.05; iPSC: r = −0.16, p = 0.039). However, as expected, effect sizes and significance (−log10(p-values)) were positively correlated (electronic supplementary material, figure S10, Spearman correlation test, LCL: r = 0.80, p < 0.01; iPSC: r = 0.73, p < 0.01), which means that there is generally stronger statistical support for TE-eQTL candidates associated with the largest change in gene expression between genotypes.
To assess the contribution of eTEs to eQTLs—relative to linked SNPs—we performed regional conditional eQTL analysis using SNPs directly located within 1 Mb of each detected eGene (see Material and methods). Thirty-eight TE-eQTL in LCL (18%) and 76 (43%) in iPSC had the eTE ranked as the ‘top-variant’ following the procedure implemented in QTLtools (electronic supplementary material, data S1). While the high level of linkage disequilibrium in humans generally obscures the identification of causal variants in QTL and association studies [39], these results strengthen a substantial amount of TE-eQTL candidates as being causal variants. Still, this analysis suggests caution must be used when interpreting the biological impact of TE-eQTL relative to other linked variants.
(d). TE-eQTL contribution of different TE families
We then examined the relative contribution of different TE families (Alu, L1, SVA) to TE-eQTL. Alu-eTEs—which constitute the bulk of all TEs examined here (89% of the TEs analysed, 92 and 89% of the eTEs, respectively, in LCL and iPSC)—appear equally distributed between positive and negative correlations with eGene expression regardless of the genomic compartments or cell type examined (figure 3). This suggests that Alu-eTEs by themselves are not a predictor of the TE-eQTL direction and may equally contribute to the up- or downregulation of adjacent genes. While the limited numbers of L1 and SVA eTEs do not allow us to infer robust patterns, we note interesting trends in the direction of their effects on gene expression in the two cell types (figure 3). Notably, all SVA eTEs identified in LCL (n = 4) were involved in positive correlations (i.e. the insertion is associated with increased gene transcript levels) and intronic L1 were generally involved in negative correlation in LCL (3/4) but in positive correlation in iPSC (7/8) (figure 3). While larger samples are needed to more rigorously test such patterns, they might reflect cell type-specific mechanisms by which these TE families influence adjacent gene expression (see Discussion).
Figure 3.

TE-eQTL effect sizes. Direction of the effect sizes for LCL (a) and iPSC (b) relative to RefSeq gene model positions (compartments as defined in legend of figure 2). (Online version in colour.)
(e). Conservation of TE-eQTL across cell types
Fifty-seven (18%) of the total number of TE-eQTL were detected in both LCL and iPSC. When applying a Bonferroni correction to the initial p-values (threshold = initial p-value threshold at 5% false discovery rate (FDR)/2), 48 of these shared TE-eQTL remain significant, accounting for 22.7 and 27.2% of the TE-eQTL identified in LCL and iPSC, respectively (figure 4a). Conversely, cell type-specific TE-eQTL accounted for more than half of the TE-eQTL identified in each cell type: 52.1% (110/211) in LCL and 56.8% (100/176) in iPSC, after applying Bonferroni correction. The effect size of the 48 shared TE-eQTL were strongly correlated across the two cell types (figure 4b, inverse hyperbolic sine transformation, r = 0.87, p < 0.01). Nearly all (46/48, 95.8%) of the shared TE-eQTL also shared the direction of their effect on gene expression (figure 4b). We observed that average transcript levels of shared eGenes were also highly correlated across the two cell types (figure 4c, Pearson product-moment correlation, r = 0.87, p < 0.01). More generally, the fold change expression of cell type-specific and shared eGenes is not significantly different (LCL-specific vs shared or iPSC-specific vs shared: Tukey HSD, p > 0.05) (figure 4d, ANOVA: F = 0.168; p = 0.682 (cell type-specific versus shared), F = 0.005, p = 0.942 (interaction ‘cell type’בspecific versus shared’)). However, the statistical significance (p-value) of shared TE-eQTL was stronger than that of cell type-specific TE-eQTL (figure 4e, ANOVA, F = 4.563, p = 0.0334). In other words, TE-eQTL shared between cell types appear statistically more robust than cell type-specific TE-eQTL, and this distinction cannot be merely explained by differences in basal eGene expression levels.
Figure 4.

TE-eQTL sharing between cell type. (a) Venn diagram representing the intersection of the total (in parentheses) and significant (in bold) number of eQTL discovered in LCL and iPSC. (b) Relationship between the effect size of shared eQTL between LCL (x axis) and iPSC (y axis). (c) Relationship between average gene expression in LCL and iPSC for shared and cell type-specific eGenes. TPM, transcripts per million. (d) Comparison of gene expression fold change between cell type-specific and shared eGenes; red dots indicate eGenes with more than mean fold change of ±3 s.d. (e) Comparison of p-value distributions between cell type-specific and shared TE-eQTL. Significance: ANOVA, *p < 0.05. (Online version in colour.)
Next, we examined whether the genes associated with TE-eQTL (eGenes) were enriched for particular biological functions, states or processes. We performed an over-representation analysis using iPSC-specific, LCL-specific and shared eGenes, restricting each gene set to genes actually expressed in the relevant cell type or, for shared eGenes, to genes expressed in both cell types (table 1 and see Material and methods). We compared the candidate eGenes with the gene ontology (GO) ‘Biological Process’ gene sets, as well as KEGG, PANTHER and REACTOME pathways. No enrichment for any gene sets was detected among the LCL-specific, iPSC-specific or shared eGenes.
In summary, we found that more than 20% of TE-eQTL in one cell type overlap those identified in the other and the statistical significance of shared TE-eQTL is stronger than that of cell type-specific TE-eQTL. Additionally, we found no obvious association of cell type-specific TE-eQTL with genes involved in specific pathways or ontologies.
(f). Chromatin accessibility TE-QTL
To shed light on the mechanisms by which unfixed TEs may regulate gene expression, we leveraged ATAC-seq data available for a subset of 86 LCL from the GBR (Great Britain) population [40] to perform a caQTL analysis. Using the TE genotypes predicted for these cell lines, the analysis yielded a total of 656 significant TE-caQTL (FDR 5%, with 10 000 permutations of the ATAC peaks quantification values), involving 431 distinct TE insertions (caTEs) (figure 5a). Nearly 90% (585/656) of the predicted associations between a caTE and an ATAC peak occur within 250 kb (figure 5b), a distance compatible with a direct effect of the TEs on the accessibility of the regulatory DNA through chromatin looping or spreading [41,42]. We also observe that the distance between caTEs and their associated ATAC peaks follows a distribution that is slightly broader (F-test, F = 0.85623, p = 0.0472) than the one obtained with a matching number of SNP-caQTL, but is indistinguishable from the distance distribution between eGenes and eTEs mapped in the LCL (F-test, F = 0.91358, p = 0.4061).
Figure 5.

Chromatin accessibility QTL in LCL (TE-caQTL). (a) Venn diagram representing the overlap between eTEs, caTEs and caTEs directly mapping in the associated ATAC peak. (b) Distribution of the TE-caQTL size effect. Families of caTEs inserted directly within the associated ATAC peak are coloured according to the key. (c) Distribution of the distance between caTE and their associated ATAC peaks. (d) Relationship between eQTL and caQTL effect sizes for 78 TEs involved in the two QTL. Blue dots are caTEs inserted in the associated ATAC peak. (e) Enrichments of eTEs and (f) effect size of TE-eQTL in different regulatory regions according to the ChroMM + Segway integrated track generated for one of the LCL (GM12878). Error bars indicate ±1 s.d. T: transcribed region; R: repressed region; PR: promoter (includes transcription start site); ENH: enhancer; CTCF: CTCF binding site. (Online version in colour.)
Interestingly, 78 caTE-QTL (18%) were also detected as involved in at least one eTE-QTL in the LCL (figure 5a), a much greater overlap than expected by chance if the two were independent (χ-squared test, χ-squared = 131.61, p < 2.2 × 10−16). Such overlap raises the possibility that some of these eTEs affect gene expression by modulating chromatin accessibility at nearby cis-regulatory elements. Consistent with this model, the 78 TEs involved in both caQTL and eQTL display a significant correlation of their effects on each type of QTL (figure 5d, Pearson's product-moment correlation test, r = 0.47, p < 0.01). In other words, TE insertions associated with decreased chromatin accessibility tend to be associated with decreased gene expression, while those associated with increased accessibility are generally associated with increased gene expression.
A particularly interesting subset of TE-caQTL are 27 instances where the position of the TE overlaps with its associated ATAC peak (dark blue, figure 5a), which suggests that the TE is directly responsible for the modulation of chromatin accessibility at its insertion site. Strikingly, 25 out of 27 such caTEs were associated with reduced ATAC peak size (i.e. negative effect size, figure 5c,d). While the sample size is small, this trend suggests that the insertion of a TE within a cis-regulatory element generally lowers its accessibility, which in turn could lead to a repressive effect on adjacent gene transcription. Indeed, out of the five caTEs within peaks that were also associated with eQTL, four were associated with reduced eGene expression (figure 5d). Together these data support a model whereby the insertion of TEs within cis-regulatory elements reduces their chromatin accessibility, which could lead to the downregulation of their target genes.
An evocative example is an AluYb8 element inserted within the third intron of the MAP3K13 gene (figure 6). The presence of the TE correlates with reduced chromatin accessibility at three ATAC peaks surrounding the gene, including one predicted as a transcribed enhancer in LCL directly overlapping with the TE. The same TE insertion was also associated with reduced MAP3K13 expression in our TE-eQTL analysis (figure 6). Because MAP3K13 is known to be a positive regulator of the proto-oncogenic transcription factor c-Myc [43], it is tempting to speculate that the AluYb8-containing allele might confer an anti-tumourigenic effect by attenuating MAP3K13 expression (see Discussion).
Figure 6.

Unfixed AluYb8 insertion correlates with chromatin accessibility and gene expression of MAP3K13. The insertion of an AluYb8 (red diamond) in a putative enhancer within the second intron of the MAP3K13 gene is associated with both reduction in MAP3K13 mRNA levels (1) and reduced chromatin accessibility at three ATAC peaks (A, B and C). The figure reproduces the H3K27Ac track from ENCODE for the LCL GM12878. Genome segmentation according to figure key from the combined ChromHMM + Segway ENCODE track for GM12878. TSS, transcription start site; CTCF, CTCF binding site; TPM, transcripts per million. (Online version in colour.)
(g). Post-transcriptional effects
To identify plausible cases of post-transcriptional effects of unfixed TEs on gene expression, we focused on nine TEs included in our original dataset located within the 3′-UTR of genes. We found that only one was a significant TE-eQTL: an AluYa5 element (ALU-NREF_1825) inserted within the 3′-UTR of the major transcript for HSD17B12. This transcript encodes a hydroxysteroid 17-beta dehydrogenase involved in long-chain fatty-acid elongation and its expression level has been associated with cancer prognosis in humans and fertility in mice [44–46] (figure 7). This was one of the strongest TE-eQTL mapped in both LCL and iPSC (electronic supplementary material, data S1). In both cell types, mRNA levels of HSD17B12 were negatively correlated with the presence of the Alu insertion (figure 7a). Regional eQTL analysis including TEs, SNPs and indels (see Material and methods) revealed a strong linkage block, with markers reaching the highest p-values within the 3′-UTR of HSD17B12 (figure 7b). Conditional analysis of these significant markers (including SNPs, indels and TEs) ranked ALU-NREF_1825, 1/107 (top variant) and 74/346 in iPSC and LCL, respectively. Together, these data strongly implicate this Alu insertion in modulating the expression of HSD17B12.
Figure 7.

Unfixed AluYa5 insertion within the 3′-UTR of HSD17B12 is associated with transcript downregulation. (a) Boxplots comparing the expression of the gene HSD17B12 (RNA-seq) according to the TE genotype (the TE insertion is represented by a red diamond) in LCL (left) and iPSC (right). TPM, transcripts per million. (b) Regional analysis of unfixed TE and other variants (SNPs) associated with the expression levels of HSD17B12. Top: LCL, bottom: iPSC (Alu is top variant). (Online version in colour.)
To investigate whether the Alu insertion within the 3′-UTR could affect protein expression and whether this effect is determined by the Alu sequence, we used a reporter assay designed to compare the effects of three different 3′-UTRs cloned downstream of the luciferase coding sequence (figure 8): (i) a 3′-UTR sequence derived from an allele lacking the insertion (from individual NA11830); (ii) a 3′-UTR sequence derived from an allele containing the AluYa5 insertion (from individual NA12760); (iii) a 3′-UTR sequence derived from the same Alu-containing allele but with a randomly scrambled sequence of the Alu (see electronic supplementary material, data S2 and Material and methods). Reporter plasmids were transfected by electroporation into the LCL GM12831 (NA12831) in order to perform the assays in a cellular environment comparable to that of the eQTL analysis. The results show that attaching any of the three 3′-UTRs to the luciferase coding sequence led to a significant decrease in protein expression compared with a construct without any 3′-UTR, but downregulation was significantly greater for the two Alu-containing constructs, regardless of whether the Alu sequence was scrambled or not (figure 8). These results recapitulate the eQTL data and suggest that the presence of the AluYa5 insertion downregulates gene expression most likely at the post-transcriptional level, but this effect is apparently independent of the Alu sequence.
Figure 8.
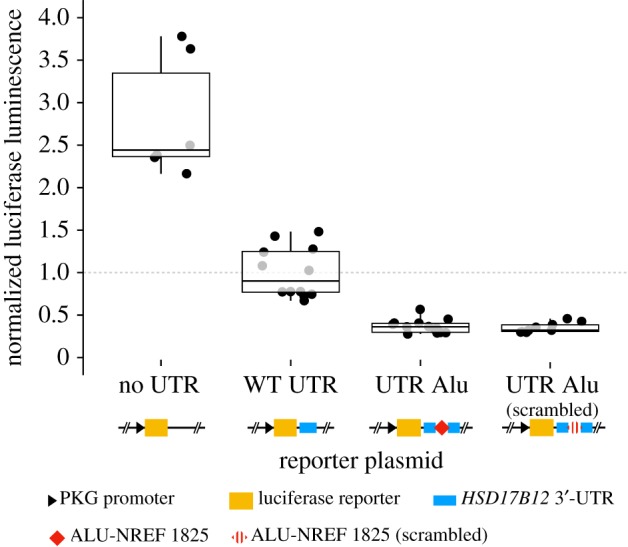
Alu-bearing 3′-UTR haplotype of HSD17B12 shows reduced reporter protein expression levels. Relative luciferase luminescence of transfected LCL (GM11831) with different 3′-UTR construct: ‘no UTR’ = empty plasmid; ‘WT UTR’ = UTR without Alu insertion (from NA11830); ‘UTR Alu’ = UTR with Alu insertion (from NA12760); ‘UTR Alu (scrambled)’ = UTR with a scrambled sequence of the AluYa5 present in NA12760. (Online version in colour.)
3. Discussion
Unfixed TE insertions represent an important class of structural variants between human genomes, but their impact on gene regulation remains poorly characterized [16–19]. To our knowledge, this study is the first to consider the effects of unfixed TEs on gene expression across multiple cell types. We also present the first TE-chromatin accessibility QTL (TE-caQTL) analysis to our knowledge, which sheds light on the impact of recent TE insertions on chromatin state at or near the insertion sites and enables a more direct evaluation of their contribution to cis-regulatory variation.
By leveraging newly predicted genotypes for 2743 TE insertions, we were able to identify 211 cis-TE-eQTL across LCL from 444 individuals and 176 cis-TE-eQTL in iPSC from 188 individuals. A previous analysis of the same LCL dataset using a similar analytical framework reported 53 cis-TE-eQTL (outside the HLA and 17q21.31 loci) [18], including 20 loci that our analyses in LCL also identified. The difference in the results of the two studies, and notably the considerably larger set of TE-eQTL captured by our approach, can be attributed to several important methodological differences. First, we used an improved genotyping pipeline, TypeTE [31], allowing us to consider 860 unfixed TEs present in the reference genome assembly (GRCh37/hg19) [12] that were not included in the dataset used in previous studies [17,18,32] likely due to the unreliability of the original genotypes provided by the 1000 Genome Project [12,31]. Indeed, the genotypes of TEs present in the reference genome were less accurate than that of the non-reference [31], an issue resolved by using TypeTE [31]. We also reprocessed the raw RNA-seq reads originally produced by Lappalainen et al. [24] to match the more recent quantification procedures adopted by the HipSci Consortium (H. Kilpinen 2019, personal communication, www.hipsci.org). Additionally, we employed a recently developed QTL mapping toolkit, QTLtools, which relies on a more robust statistical framework than previous approaches [35]. These improvements likely increased the power of our analysis and enhanced our ability to map TE-eQTL with these datasets.
Consequently, we were able to explore in fine detail the regulatory potential of unfixed TEs across two different human cell types. In both LCL and iPSC, we found that most predicted cis-regulatory interactions take place within 250 kb of the gene boundaries, a value consistent with eQTL previously mapped with SNPs [21,33], as well as our own eQTL analysis using SNPs (figure 2a,b). We also noted a trend for p-values to weaken as the distance between eTE and eGene increases (electronic supplementary material, figure S8). These observations are consistent with the current understanding of how distal cis-regulatory elements interact with genes in the human genome [47,48], within topologically associated domains of median length approximately 185 kb [42]. Repeating the analyses with increasing subsamples (electronic supplementary material, figure S5) suggests that many more TE-eQTL remain to be discovered in the human population. This finding is consistent with previous large-scale SNP-eQTL studies [21,25].
Our study provides a first assessment of whether a TE-eQTL discovered in a given cell type may be detected in another. We found that 22.7% of the TE-eQTL identified in LCL were replicated in iPSC, representing nearly 27.2% of the significant TE–gene associations in the latter cell type. The level of statistical significance, as well as the size and direction of the effect on gene expression, were highly correlated across the two cell types, suggesting that their functional impact may be broadly conserved. These findings suggest that a subset of TEs could influence gene expression across multiple cell types and potentially a variety of tissues [21,49]. Indeed, SNP-eQTL analyses across 44 human tissues by the GTEx Consortium suggest that SNPs with cis-regulatory effects fall within one of two broad categories: those with shared effects across tissues and those specific to a single or a few similar tissues [21]. Our results suggest that this dichotomy may also apply to TE insertions, which implies that the phenotypic effect of a TE insertion in a given tissue could be anticipated from data obtained from another tissue.
The diversity of TEs involved in TE-eQTL matches closely that of the starting set of unfixed TEs, which is dominated by Alu insertions (89%). Indeed, 92 and 89% of the eTEs in LCL and iPSC, respectively, were Alu elements, with a variety of effects and positions on their associated genes (figure 3). We observed more biases in the cis-regulatory effects associated with SVA and L1 with regard to the direction of their effects or the cell type (figure 3b), but the relatively small number of SVA and L1 insertions considered does not allow us to draw firm conclusions. Nonetheless, it stands to reason that L1 and SVA might have different regulatory effects in different cell types. Indeed, while SVA is mobilized by the L1 machinery, their expression patterns are only partially overlapping and each family appears to exhibit unique cell type-specific regulatory activities [50–53]. Overall, our findings indicate that the cis-regulatory effects of unfixed TEs may often manifest in multiple cell types, but further efforts are needed to characterize possible cell type-specific interactions.
One of the novelties of our study is to incorporate functional genomics data to investigate further the regulatory potential of unfixed TEs. To our knowledge, we report the first TE-caQTL analysis. We used ATAC-seq data generated previously for 86 of the LCL [40] to map 431 caTEs associated with variation at 656 ATAC peaks. Strikingly, 45.9% of the TEs involved in eQTL were also mapped as caQTL (figure 5a), which brings support to the hypothesis that almost half of TE insertions modulating adjacent gene expression may have an effect through distal chromatin effects, as previously proposed for SNP-eQTL in LCL [54] and iPSC [49]. Furthermore, we found that TEs associated with chromatin accessibility peaks were approximately four times more enriched within regions annotated as enhancers than TE-eQTL, which is consistent with the idea that some of the TE insertions detected in the TE-caQTL analysis could directly modify or act as cis-regulatory elements [55,56]. As a support for this hypothesis, we found that 27 of the caTEs were directly located within their associated ATAC peak and all but two of these insertions correlated with reduced chromatin accessibility. The results of our TE-caQTL analysis indicate that TE insertions within cis-regulatory elements, such as promoters or enhancers, tend to have a disruptive effect on the function of these elements.
It is well documented that TEs can affect post-transcriptional gene regulation through many mechanisms, including effects on mRNA splicing, stability or translation [2,4,19,57,58]. While there are many known examples of fixed human TEs with such effects, cases involving unfixed elements have been scarcely described apart from disease-causing insertions [8,9,59]. We confirmed experimentally that the inclusion of an Alu in the 3′-UTR of the gene HSD17B12 (previously reported by Wang et al. [18]) reduces the protein level of a luciferase reporter in LCL. However, this effect appears to be sequence-independent, since a reduction in luciferase expression was observed even when the Alu sequence was scrambled. These results seem to rule out some of the known mechanisms by which (fixed) Alu located in 3′-UTRs affects transcript stability, such as miRNA binding [57] or Staufen-mediated decay [4]. It is possible that the effect merely reflects the elongation of the 3′-UTR caused by the insertion. Indeed, it is known that the RNA helicase Upf1 can sense 3′-UTR size and promote nonsense-mediated decay of abnormally long 3′-UTRs [60]. Also, because we cloned the ‘presence’ and ‘absence’ of 3′-UTR haplotypes from two different LCLs, we cannot rule out that another polymorphism, ‘hitchhiking’ with the Alu insertion, is causing the effect. At the very least, our results indicate that the Alu insertion can act as a reliable marker of HSD17B12 expression. Because the level of HSD17B12 enzyme has been positively correlated to the severity of epithelial ovarian cancer in humans [44], this is a case worth further investigation.
Throughout our analysis, we chose to exclude multiple eQTL mapping within the HLA and the 17q21.31 inversion because there are good reasons to believe these regions of the human genome are prone to yield false positives when it comes to eQTL mapping. While it has been previously shown that relevant QTL can be mapped to the HLA region [3–6], the HLA genes are known for their high level of copy number variation [22], which can be confounding during the quantification of transcript levels with RNA-seq. Indeed, these genes are often discarded from association studies [3,9,10]. It has been shown that the 17q21.31 inversion influences variation in gene expression in a cell type-specific fashion [23]. Moreover, structural variation in this region relative to the reference genome is common in the general population [23], which is likely to hamper TE mapping and/or distort TE genotyping. We found that removing eQTL mapping to these regions, in particular, the HLA locus, removes the enrichment of eGenes for immune-related functions, as previously reported by Wang et al. [18]. When excluding these regions, we found no enrichment for particular gene ontology (GO) terms or pathways among eGenes in either LCL or iPSC.
The example of HSD17B12 and the newly reported case of MAP3K13 (where an Alu insertion could be mapped as both expression and caQTL in LCL), as well as the many other TE-QTL identified in this study, underscore a plausible contribution of TE insertions commonly segregating in the population in human trait variation, including disease susceptibility [9,16,19]. These findings confirm the potential of unfixed TEs to make a non-trivial contribution to human gene expression [5–7]. Our study provides evidence for the persistence of their (putative) cis-regulatory effects across cell types but suggests that some elements have the potential to regulate tissue-specific functions. Moreover, we present the first map of unfixed TEs that correlate with changes in chromatin accessibility in cis, uncovering the importance of this mechanism while its fine details remain to be investigated. A logical extension of this study would be to leverage data produced for a broader range of human tissues, such as those represented in the GTEx initiative [21], to analyse more comprehensively the tissue-specificity of the TE-eQTL identified herein. Complementary genomic assays, such as those measuring DNA methylation levels or nascent RNA transcription, could provide further insight into the mechanisms by which polymorphic TE insertions shape gene expression. Ultimately, the causality of TE insertion variants would need to be tested experimentally through CRISPR-Cas and other manipulative genomic technologies [5]. The data presented here offer a valuable foundation for future studies aimed at illuminating the contribution of TEs to human phenotypic variation.
4. Material and methods
Unless otherwise stated, all statistical analyses were performed using R v. 3.5.1 (R Development Core Team 2018, https://www.r-project.org/).
(a). TE genotypes
The genomic locations of unfixed Alu, LINE1 (L1) and SVA elements were extracted from publicly available datasets. TE insertions in LCL were gathered from the previous analysis of 445 cell lines derived from healthy donors of five populations (CEU, FIN, TSI, GBR and YRI) by the 1000 Genome Project [12]. TE insertions were originally discovered and genotyped in this dataset using MELT v.1 (non-reference insertions) and a collection of structural variant tools [12]. In some cases, these calls were re-genotyped using TypeTE [31] in order to improve their accuracy. TypeTE was used for L1 and SVA insertions present in the reference genome, as well as for both reference and non-reference Alu insertions. In addition, unfixed TE insertions were searched and genotyped de novo in 326 induced human pluripotent stem cells (iPSC), derived from 205 healthy donors as follows. First, whole-genome sequencing data for each cell line was recovered in bam format from the HipSci website (HipSci.org) and unfixed TE insertions were called using MELT v. 2.1.4 [15] using split- and discordant paired-end read information. Since genotype accuracy has substantially improved for non-reference Alu between MELT1 and MELT2 [31], we only re-genotyped L1 and SVA, as well as reference Alu insertions. To analyse the iPSC dataset at the individual level, one genotype file (VCF) per cell line was kept. To match insertions found in both datasets, we intersected the breakpoint positions of the polymorphic insertions discovered in LCL and iPSC using BEDTools (v. 2.28.0 [61]). Two insertions of the same TE type (Alu, L1 or SVA) separated by up to 30 bp were considered identical by descent. After genotyping and individual selection based on RNA-seq (see below), only the loci shared at a minimum insertion frequency of 5% between LCL and iPSC were kept for eQTL mapping.
(b). RNA-seq data processing
Steady-state RNA levels for LCL and iPSC were recovered and normalized as follows. We collected reads from the RNA-seq experiment carried out by Lappalainen et al. [24] in 445 LCL from the Geuvadis repository (https://www.ebi.ac.uk/Tools/geuvadis-das/). Raw reads were quality checked and trimmed using UrQt (v. 1.0.18, [62]); we used a –t quality threshold of 10 and kept the other default parameters. Transcript levels were then quantified with kallisto (v. 0.46.0, [34]) using the reference transcriptome (cDNA) GRCh37.75 from Ensembl. This reference transcriptome and quantification method were used to match the data of 326 iPSC (H. Kilpinen 2019, personal communication, www.hipsci.org). Sample quantifications in transcripts per million (TPM) were then grouped by cell type and normalized. TMM (trimmed mean of M-values) normalization was performed to make transcript level comparable across samples using the script abundance_estimates_to_matrix.pl available with the Trinity distribution v. 2.8.4 [63]. Transcript quantifications were summed for a gene in each cell type and additionally averaged per individual in iPSC. Genes expressed in fewer than 50% of the samples were then discarded (143 436 transcripts discarded in LCL, 141 529 in iPSC). Then, principal component analysis (PCA) using normalized TPM was carried out to identify outlier samples. Individuals with values exceeding three times the standard deviation on each of the first two principal components were removed. After filtering, 444 and 188 samples were kept, respectively, in LCL and iPSC.
(c). TE-eQTL mapping
TE-eQTL were mapped independently in LCL and iPSC datasets using QTLtools v.1.1 [35]. After ensuring that the transcript expression was not primarily structured by population in the LCL dataset, we used QTLtools to perform a new PCA on the final expression matrices. For each cell type, the values of the three first axes were added as covariates to the model, as well as the sex and population of origin for LCL and sex, ethnicity and age for iPSC. cis-eQTL were searched within a 1 Mb window around each transcript using QTLtool cis. Significance was evaluated by running 10 000 permutations of the gene expression matrices and multiple testing was addressed by applying 5% FDR correction, as recommended in the QTLtools manual. The top eTEs (most significant TE insertion significantly associated with a gene expression level) reported by QTLtools were kept for further analysis. Our ability to detect TE-eQTL was evaluated by resampling an increasing number of individuals selected at random (10, 25, 50, 100 and 150) in the TE genotype matrices and re-running the QTLtools cis procedure. Enrichments of TEs in specific gene regions (intergenic, 10 kb upstream, 10 kb downstream, intron, exon, 5′-UTR or 3′-UTR) were calculated for all TEs within the 1 Mb window around a given gene and eTEs reported by QTLtools. Enrichments were calculated by sampling a matching number of 1 bp random genomic intervals (TE breakpoints) in the reference genome using BEDTools 1000 times. The ratio of observed TEs/random breakpoints in a given region was then calculated for each replicate to calculate the fold enrichment.
(d). TE-eQTL sharing between cell types
Sharing of TE-eQTL between cell types was considered significant if two identical gene–TE associations had a p-value below or equal to half the initial FDR threshold (Bonferroni correction). Sharing was also quantified in re-sampled eQTL analyses (increasing sample numbers) using the same criteria.
(e). Conditional analysis of TE- and SNP-eQTL
The regulatory potential of polymorphic TEs was compared with that of SNPs by performing conditional eQTL analysis [35]. The SNP dataset used for this analysis was recovered from the 1000 Genome Project Phase 3 release for LCL [64], while for iPSC, we used individual VCF files available from the HipSci Consortium (these include imputed 1000 Genomes genotypes). To relieve the computational burden of mapping eQTL for all SNPs, we extracted only markers present in the 1 Mb windows where a TE-eQTL had been previously detected. Combining TE and SNP genotypes, eQTL were searched in each cell type using QTLtools cis. A first pass was performed using 10 000 permutations with a FDR threshold of 5%, then, a conditional analysis for each gene was performed in order to rank and assess the independence of the e-variants (variant, either TE or SNP, associated with a gene expression level), using the ‘— —mapping’ option of QTLtools cis.
(f). TE-caQTL mapping
To investigate potential cis-effects of TE insertions on chromatin accessibility, we collected ATAC-seq data for 85 GBR individuals included in our LCL dataset [40]. Normalized ATAC peak levels were used as a response variable to search for TE-caQTL with QTLtools, using the genotypes of unfixed TEs for these LCL. As for eQTL mapping, we used population and sex of each cell line as covariates and report the best significant TE per normalized ATAC peak as provided by QTLtools in a cis-window of 1 Mb. The resulting caTEs (significant TE in caQTL) were then searched for enrichment in regulatory regions by intersecting their genomic coordinates with the Segway/ChromHMM combined regulatory track from ENCODE generated for the LCL GM12878.
(g). Luciferase reporter assays
(i). UTR amplification
We evaluated the regulatory potential of a candidate AluYa5 insertion within the 3′-UTR of the gene HSD17B12 using dual-luciferase reporter assay. First, three LCL corresponding to one homozygote for the insertion (GM12760), one homozygote for the absence (GM11830) and one heterozygote (GM12831) were cultured in RMPI (Gibco) supplemented with 15% FBS (Gibco) at 37°C and 5% CO2. Cells were passaged and refreshed with new medium every 3 to 4 days, before reaching approximately 1 million cells per millilitre of culture. 3′-UTR sequences with and without Alu insertion were amplified from homozygote individuals by PCR as follows. DNA was extracted using the Qiagen© DNeasy Blood and Tissue kit following the manufacturer's instructions. For PCR amplification, no more than 1 µg of DNA template was used per sample in a total volume of 25 µl. Five microlitres of Q5 High Fidelity Master Mix (2×) was added with 0.3 µl of each primer (F: 5′-AAACGAGCTCGC-TAGTCAAACCTGCCTTCTTGGA-3′ and R: 5′-CGACTCTAG-ACTCGACTGTCCAGGTCATTGTGGTG-3′). The primers have 15 bp homology with the reporter plasmid (in bold type), as needed for inFusion-HD cloning (Takara Bio). The mixture was amplified after 30 s initial denaturation for 25 cycles (10 s denaturation at 98°C, 30 s annealing at 60°C and 20 s elongation at 72°C) followed by 2 min of final elongation. Additionally, a construct similar in nucleotide composition to the UTR amplified from GM12760 was generated with a scrambled Alu sequence instead of the original TE (electronic supplementary material, data S2). These three sequences were, respectively, named ‘WT UTR’ (GM12830, no Alu in UTR), ‘Alu UTR’ (GM12760, Alu in UTR) and ‘Alu UTR scrambled’ (UTR identical to GM12760 but Alu sequence scrambled). In order to assess successful amplification of the UTRs, the band corresponding to the expected PCR product (1551 bp for ‘WT UTR’ and 1825 bp for ‘Alu’) was subject to Sanger sequencing using the F- and R-m13 primers flanking the insert, as well as two internal primers (H3Pint1-F: 5′-CAGACACACTGCAATTTACAAAGA-3′ and H3Pint1-R: 5′-ACGGCCTTAATTTCAATCACCA-5′) to fill the gap. PCR products with the expected sequences were then kept for In-Fusion cloning into the reporter plasmid.
(ii). In-Fusion cloning into reporter plasmid
The artificially generated ‘scrambled’ sequence was also amplified using the same PCR conditions as the natural UTRs to add the 15 bp flanking sequences matching the cloning site of the receiving vector. PCR products were then cloned into a pmirGlo dual-luciferase miRNA target expression vector (Promega). This plasmid contains a multiple cloning site downstream of the luciferase gene, terminated by a polyadenylation signal. As a transfection control, the plasmid also contained a Renilla reporter gene whose expression should not be affected by the sequence cloned downstream from the luciferase. Cloning was done using the In-Fusion HD Cloning kit (Takara Bio) following the provider's documentation. Plasmids were transformed into competent alpha-5 Escherichia coli (New England Biolabs) and cultured on LB-agar plates with 1% ampicillin overnight at 37°C. Next, 10 clones per condition were extracted using a Miniprep kit (Qiagen) and successful insertion of the constructs was assessed by performing double digestion of the plasmid by BamHI and EcoRI. Clones with the expected product size (‘WT UTR’: 5681 and 3188 bp; ‘UTR Alu’ and ‘UTR Alu scrambled’: 5955 and 3188 bp) were then selected for the luciferase reporter assay and amplified by MaxiPrep (Qiagen) upon transfection.
(iii). Luciferase assay
‘WT UTR’, ‘UTR Alu’ and ‘UTR Alu scrambled’ were transfected into the LCL GM11831 for reporter assay, as well as a pGFP reporter plasmid to evaluate transfection efficiency. For each condition and each replicate, 50 million cells were transfected with 125 µg µl−1 of plasmid by electroporation using the Neon Transfection System (Life Technologies). Reaction took place in 100 µl tips, applying three pulses of 1200 V for 20 ms each. Transfection efficiency was approximately 30%, and luciferase signal was above background (pGFP cells) by two orders of magnitude for the conditions tested. Reporter assay was performed using the Dual-Glo Luciferase assay system (Promega) following the manufacturer's instructions. For each experiment, the average blank (pGFP) value was subtracted from each luminescence signal. Additionally, the luciferase signal was normalized for each replicate by the Renilla luminescence, and the ratio of luciferase/Renilla was finally normalized by the average ratio of the ‘WT UTR’ construct.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We would like to warmly thank Mitchell Lokey and Kathleen Gordon for their sustained help during this study. We are grateful to Kathleen Burns and Lindsay Payer for sharing preliminary data and insights to this project. We also thank Andrew Grimson, Ciáran Daly and Jessica West for their help and providing reagent for the luciferase assay. The authors are grateful to two anonymous referees whose comments helped enhance the quality of the manuscript.
Data accessibility
This article has no additional data.
Authors' contributions
C.F. and C.G. designed the experiments and wrote the manuscript. C.G. conducted the statistical and experimental analyses. C.G. and C.F. interpreted the data. N.A.Z. performed and analysed the luciferase reporter assay.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by grants nos R35 GM122550, R01 GM059290 and U01 HG009391 from the National Institutes of Health to C.F.
References
- 1.de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7, e1002384 ( 10.1371/journal.pgen.1002384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feschotte C. 2008. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 9, 397–405. ( 10.1038/nrg2337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck CR, Garcia-Perez JL, Badge RM, Moran JV. 2011. LINE-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet. 12, 187–215. ( 10.1146/annurev-genom-082509-141802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elbarbary RA, Lucas BA, Maquat LE. 2016. Retrotransposons as regulators of gene expression. Science 351, aac7247 ( 10.1126/science.aac7247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuong EB, Elde NC, Feschotte C. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86. ( 10.1038/nrg.2016.139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuentes DR, Swigut T, Wysocka J. 2018. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife 7, e35989 ( 10.7554/eLife.35989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deniz Ö, Frost JM, Branco MR. 2019. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 20, 417–431. ( 10.1038/s41576-019-0106-6) [DOI] [PubMed] [Google Scholar]
- 8.Kazazian HH, Moran JV. 2017. Mobile DNA in health and disease. N. Engl. J. Med. 377, 361–370. ( 10.1056/NEJMra1510092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Payer LM, Burns KH. 2019. Transposable elements in human genetic disease. Nat. Rev. Genet. 20, 760–772. ( 10.1038/s41576-019-0165-8) [DOI] [PubMed] [Google Scholar]
- 10.Mills RE, Bennett EA, Iskow RC, Devine SE. 2007. Which transposable elements are active in the human genome? Trends Genet. 23, 183–191. ( 10.1016/j.tig.2007.02.006) [DOI] [PubMed] [Google Scholar]
- 11.Stewart C. et al. 2011. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 7, e1002236 ( 10.1371/journal.pgen.1002236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sudmant PH. et al. 2015. An integrated map of structural variation in 2504 human genomes. Nature 526, 75–81. ( 10.1038/nature15394) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witherspoon DJ, Zhang Y, Xing J, Watkins WS, Ha H, Batzer MA, Jorde LB. 2013. Mobile element scanning (ME-scan) identifies thousands of novel Alu insertions in diverse human populations. Genome Res. 23, 1170–1181. ( 10.1101/gr.148973.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feusier J, Witherspoon DJ, Scott Watkins W, Goubert C, Sasani TA, Jorde LB. 2017. Discovery of rare, diagnostic AluYb8/9 elements in diverse human populations. Mob. DNA 8, 9 ( 10.1186/s13100-017-0093-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, Mills RE, 1000 Genomes Project Consortium, Devine SE. 2017. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929. ( 10.1101/gr.218032.116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payer LM. et al. 2017. Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl Acad. Sci. USA 114, E3984–E3992. ( 10.1073/pnas.1704117114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Norris ET, Jordan IK. 2017. Human retrotransposon insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front. Microbiol. 8, 1418 ( 10.3389/fmicb.2017.01418) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Rishishwar L, Mariño-Ramírez L, Jordan IK. 2016. Human population-specific gene expression and transcriptional network modification with polymorphic transposable elements. Nucleic Acids Res. 45, 2318–2328. ( 10.1093/nar/gkw1286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payer LM, Steranka JP, Ardeljan D, Walker J, Fitzgerald KC, Calabresi PA, Cooper TA, Burns KH. 2019. Alu insertion variants alter mRNA splicing. Nucleic Acids Res. 47, 421–431. ( 10.1093/nar/gky1086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Signor SA, Nuzhdin SV. 2018. The evolution of gene expression in cis and trans. Trends Genet. 34, 532–544. ( 10.1016/j.tig.2018.03.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguet F. et al. 2017. Genetic effects on gene expression across human tissues. Nature 550, 204–213. ( 10.1038/nature24277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majewski J, Pastinen T. 2011. The study of eQTL variations by RNA-seq: from SNPs to phenotypes. Trends Genet. 27, 72–79. ( 10.1016/j.tig.2010.10.006) [DOI] [PubMed] [Google Scholar]
- 23.Gilad Y, Pritchard JK, Thornton K. 2009. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 25, 463–471. ( 10.1016/j.tig.2009.09.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lappalainen T. et al. 2013. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. ( 10.1038/nature12531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryois J. et al. 2014. Cis and trans effects of human genomic variants on gene expression. PLoS Genet. 10, e1004461 ( 10.1371/journal.pgen.1004461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stranger BE. et al. 2012. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8, e1002639 ( 10.1371/journal.pgen.1002639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang C. et al. 2017. The impact of structural variation on human gene expression. Nat. Genet. 49, 692–699. ( 10.1038/ng.3834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alkan C, Coe BP, Eichler EE. 2011. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376. ( 10.1038/nrg2958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alm J, Ohnmeiss TE, Lanza J, Vriesenga L. 1990. Preference of cabbage white butterflies and honey bees for nectar that contains amino acids. Oecologia 84, 53–57. ( 10.1007/BF00665594) [DOI] [PubMed] [Google Scholar]
- 30.Chaisson MJP. et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 10, 1784 ( 10.1038/s41467-018-08148-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goubert C, Thomas J, Payer LM, Kidd JM, Feusier J, Watkins WS, Burns KH, Jorde LB, Feschotte C. 2019. TypeTE: a tool to genotype mobile element insertions from whole genome resequencing data. bioRχiv, 791665 ( 10.1101/791665) [DOI] [PMC free article] [PubMed]
- 32.Rishishwar L, Wang L, Wang J, Yi SV, Lachance J, Jordan IK. 2018. Evidence for positive selection on recent human transposable element insertions. Gene 675, 69–79. ( 10.1016/j.gene.2018.06.077) [DOI] [PubMed] [Google Scholar]
- 33.Kilpinen H. et al. 2017. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375. ( 10.1038/nature22403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. ( 10.1038/nbt.3519) [DOI] [PubMed] [Google Scholar]
- 35.Delaneau O, Ongen H, Brown AA, Fort A, Panousis NI, Dermitzakis ET. 2017. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 8, 15452 ( 10.1038/ncomms15452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castelli EC, Paz MA, Souza AS, Ramalho J, Mendes-Junior CT. 2018. Hla-mapper: an application to optimize the mapping of HLA sequences produced by massively parallel sequencing procedures. Hum. Immunol. 79, 678–684. ( 10.1016/j.humimm.2018.06.010) [DOI] [PubMed] [Google Scholar]
- 37.Orenbuch R, Filip I, Comito D, Shaman J, Pe'er I, Rabadan R. 2019. arcasHLA: high resolution HLA typing from RNAseq. Bioinformatics 2019, btz474 ( 10.1093/bioinformatics/btz474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian L, Quitadamo A, Lin F, Shi X. 2015. Methods for population-based eQTL analysis in human genetics. Tsinghua Sci. Technol. 19, 624–634. ( 10.1109/TST.2014.6961031) [DOI] [Google Scholar]
- 39.Reich DE. et al. 2001. Linkage disequilibrium in the human genome. Nature 411, 199–204. ( 10.1038/35075590) [DOI] [PubMed] [Google Scholar]
- 40.Kumasaka N, Knights AJ, Gaffney DJ. 2018. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 51, 128–137. ( 10.1038/s41588-018-0278-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun X, Wang X, Tang Z, Grivainis M, Kahler D, Yun C, Mita P, Fenyö D, Boeke JD. 2018. Transcription factor profiling reveals molecular choreography and key regulators of human retrotransposon expression. Proc. Natl Acad. Sci. USA 115, E5526–E5535. ( 10.1073/pnas.1722565115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao SSP. et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. ( 10.1016/j.cell.2014.11.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q, Li X, Cui K, Liu C, Wu M, Prochownik EV, Li Y. 2019. The MAP3K13-TRIM25-FBXW7α axis affects c-Myc protein stability and tumor development. Cell Death Differ. ( 10.1038/s41418-019-0363-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kemiläinen H, Huhtinen K, Auranen A, Carpén O, Strauss L, Poutanen M. 2018. The expression of HSD17B12 is associated with COX-2 expression and is increased in high-grade epithelial ovarian cancer. Oncology 94, 233–242. ( 10.1159/000485624) [DOI] [PubMed] [Google Scholar]
- 45.Kemiläinen H. et al. 2016. The hydroxysteroid (17β) dehydrogenase family gene HSD17B12 is involved in the prostaglandin synthesis pathway, the ovarian function, and regulation of fertility. Endocrinology 157, 3719–3730. ( 10.1210/en.2016-1252) [DOI] [PubMed] [Google Scholar]
- 46.Rantakari P, Lagerbohm H, Kaimainen M, Suomela J-P, Strauss L, Sainio K, Pakarinen P, Poutanen M. 2010. Hydroxysteroid (17β) dehydrogenase 12 is essential for mouse organogenesis and embryonic survival. Endocrinology 151, 1893–1901. ( 10.1210/en.2009-0929) [DOI] [PubMed] [Google Scholar]
- 47.He B, Chen C, Teng L, Tan K. 2014. Global view of enhancer–promoter interactome in human cells. Proc. Natl Acad. Sci. USA 111, E2191–E2199. ( 10.1073/pnas.1320308111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mifsud B. et al. 2015. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606. ( 10.1038/ng.3286) [DOI] [PubMed] [Google Scholar]
- 49.Banovich NE. et al. 2018. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res. 28, 122–131. ( 10.1101/gr.224436.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Philippe C, Vargas-Landin DB, Doucet AJ, van Essen D, Vera-Otarola J, Kuciak M, Corbin A, Nigumann P, Cristofari G. 2016. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife 5, e13926 ( 10.7554/eLife.13926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gianfrancesco O, Bubb VJ, Quinn JP. 2016. SVA retrotransposons as potential modulators of neuropeptide gene expression. Neuropeptides 64, 3–7. ( 10.1016/j.npep.2016.09.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwon Y-J, Choi Y, Eo J, Noh Y-N, Gim J-A, Jung Y-D, Lee J-R, Kim H-S. 2013. Structure and expression analyses of SVA elements in relation to functional genes. Genomics Inform. 11, 142–148. ( 10.5808/GI.2013.11.3.142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bodega B, Orlando V. 2014. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 31, 67–73. ( 10.1016/j.ceb.2014.09.002) [DOI] [PubMed] [Google Scholar]
- 54.Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. 2016. RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. ( 10.1126/science.aad9417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Souza FSJ, Franchini LF, Rubinstein M. 2013. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol. Biol. Evol. 30, 1239–1251. ( 10.1093/molbev/mst045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sundaram V, Wang T. 2017. Transposable element mediated innovation in gene regulatory landscapes of cells: re-visiting the ‘gene-battery’ model. Bioessays 40, 1700155 ( 10.1002/bies.201700155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen LL, Yang L. 2017. ALUternative regulation for gene expression. Trends Cell Biol. 27, 480–490. ( 10.1016/j.tcb.2017.01.002) [DOI] [PubMed] [Google Scholar]
- 58.Chen LL, DeCerbo JN, Carmichael GG. 2008. Alu element-mediated gene silencing. EMBO J. 27, 1694–1705. ( 10.1038/emboj.2008.94) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hancks DC, Kazazian HH. 2012. Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev. 22, 191–203. ( 10.1016/j.gde.2012.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hogg JR, Goff SP. 2010. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 143, 379–389. ( 10.1016/j.cell.2010.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. ( 10.1093/bioinformatics/btq033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Modolo L, Lerat E. 2015. UrQt: an efficient software for the unsupervised quality trimming of NGS data. BMC Bioinf. 16, 137 ( 10.1186/s12859-015-0546-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grabherr MG. et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. ( 10.1038/nbt.1883) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Auton A. et al. 2015. A global reference for human genetic variation. Nature 526, 68–74. ( 10.1038/nature15393) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article has no additional data.