

Abstract
Nuclear accumulation of the small phosphoprotein integrin cytoplasmic domain–associated protein-1 (ICAP1) results in recruitment of its binding partner, Krev/Rap1 interaction trapped-1 (KRIT1), to the nucleus. KRIT1 loss is the most common cause of cerebral cavernous malformation, a neurovascular dysplasia resulting in dilated, thin-walled vessels that tend to rupture, increasing the risk for hemorrhagic stroke. KRIT1's nuclear roles are unknown, but it is known to function as a scaffolding or adaptor protein at cell-cell junctions and in the cytosol, supporting normal blood vessel integrity and development. As ICAP1 controls KRIT1 subcellular localization, presumably influencing KRIT1 function, in this work, we investigated the signals that regulate ICAP1 and, hence, KRIT1 nuclear localization. ICAP1 contains a nuclear localization signal within an unstructured, N-terminal region that is rich in serine and threonine residues, several of which are reportedly phosphorylated. Using quantitative microscopy, we revealed that phosphorylation-mimicking substitutions at Ser-10, or to a lesser extent at Ser-25, within this N-terminal region inhibit ICAP1 nuclear accumulation. Conversely, phosphorylation-blocking substitutions at these sites enhanced ICAP1 nuclear accumulation. We further demonstrate that p21-activated kinase 4 (PAK4) can phosphorylate ICAP1 at Ser-10 both in vitro and in cultured cells and that active PAK4 inhibits ICAP1 nuclear accumulation in a Ser-10–dependent manner. Finally, we show that ICAP1 phosphorylation controls nuclear localization of the ICAP1-KRIT1 complex. We conclude that serine phosphorylation within the ICAP1 N-terminal region can prevent nuclear ICAP1 accumulation, providing a mechanism that regulates KRIT1 localization and signaling, potentially influencing vascular development.
Keywords: cerebral cavernous malformation, cell compartmentalization, KRIT1 (Krev interaction trapped), nuclear translocation, nuclear transport, phosphorylation, ICAP1 (integrin cytoplasmic domain-associated protein-1), nuclear import, nucleocytoplasmic shuttling, phosphoprotein
Introduction
Integrin cytoplasmic domain–associated protein-1 (ICAP1) is a 200-amino acid phosphoprotein composed of an unstructured N-terminal region and a C-terminal phosphotyrosine-binding (PTB)2 domain (Fig. 1). It is ubiquitously expressed and has been implicated in development of bone and the vascular system (1–3). ICAP1 was first identified in screens for proteins that bind the cytoplasmic tail of the integrin adhesion receptor β1 subunit (4, 5). ICAP1 binding inhibits integrin function by competing with integrin-activating proteins (6–8), thereby impacting processes such as focal adhesion assembly, cell adhesion/spreading, and cell migration (2, 8–10). However, the exact cellular roles of ICAP1 remain incompletely understood. For example, ICAP1 is reported to both inhibit (8) and activate (4) cell migration, and integrin-independent ICAP1 functions are suggested by the identification of additional ICAP1-binding partners (9, 11–14). Notably, the cerebral cavernous malformation (CCM) protein Krev/Rap1 interaction trapped-1 (KRIT1) binds tightly to ICAP1 (13, 14).
Figure 1.
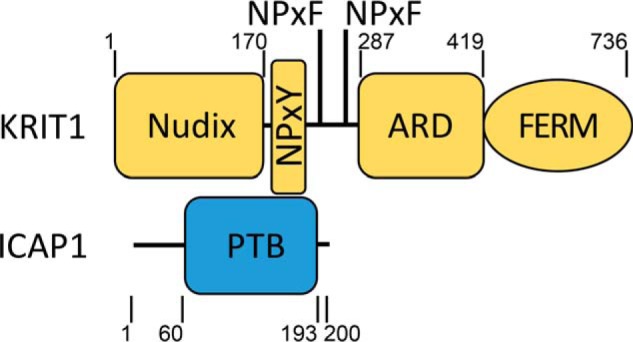
Domain schematic of ICAP1 and KRIT1. ICAP1 contains an unstructured N-terminal region followed by a phosphotyrosine-binding domain (residues 60–193). KRIT1 contains an N-terminal Nudix domain, three NPX(Y/F) motifs, an ankyrin repeat domain (ARD), and a FERM domain. The ICAP1:KRIT1 interaction is mediated by direct ICAP1 PTB domain interactions with the first KRIT1 NPX(Y/F) motif and with an adjacent RR motif (not depicted) (6). Boundaries are indicated by residue numbers.
CCMs are lesions comprised of dilated blood vessels in the neurovasculature. They are characterized by densely packed vascular sinusoids with enlarged capillary channels and thin endothelia (15–17). Endothelial cells within CCMs lack tight junctions and are prone to rupture, increasing the risk of hemorrhage, headaches, epilepsy, seizures, and focal neurological deficits (18). It is estimated that CCMs are present in 0.3–0.5% of the worldwide population (19), affecting >24 million people (15). Loss-of-function mutations in the genes encoding any one of the three proteins making up the CCM protein complex (KRIT1, CCM2, and CCM3) increase the risk of developing CCMs (20), and these three loci account for 70–80% of all cases of familial CCM (15, 21). Importantly, KRIT1 was the first gene linked to CCM, is the most commonly mutated, and is present in >40% of inherited cases (22–25).
KRIT1 is a 736-amino acid multidomain protein consisting of an N-terminal Nudix domain, followed by three protein-binding NPX(Y/F) motifs, four ankyrin repeat domains, and a C-terminal FERM domain (6, 20, 26, 27) (Fig. 1). KRIT1 is essential for vascular development, and KRIT1-null mice die in mid-gestation with vascular defects such as vascular dilation, impaired arterial identity, and narrowing of branchial arch arteries and rostral dorsal aorta (28). However, the molecular mechanisms underlying the roles of KRIT1 in these processes are not fully resolved (28). For example, KRIT1 was first identified as an effector of Rap1 (29), facilitating its function in stabilizing endothelial cell-cell junctions (30, 31). However, KRIT1 also binds and stabilizes CCM2 (27, 32) and ICAP1 (6, 13, 14, 33), further impacting vascular processes like contractility and angiogenesis (1, 34, 35).
Binding of the PTB domain of ICAP1 to the first NPX(Y/F) motif of KRIT1 (Fig. 1) stabilizes both proteins, preventing their proteasomal degradation (1, 32). Although ICAP1 mutations have not been linked to CCM (1), ICAP1 is important for normal vascular development (1, 3), likely in part due to its strong association with KRIT1. ICAP1 overexpression in human umbilical vein endothelial cells inhibits endothelial tube formation and sprouting, whereas ICAP1 knockdown increases these processes (3). Further, ICAP1-deficient endothelial cells grafted onto the flanks of mice display increased sprouting angiogenesis and denser blood vessel network formation (3). Compared with icap1+/+ blood vessels, icap1−/− blood vessels are more dilated, and their surrounding basal lamina structure is disrupted and torn (1). Last, ICAP1 null mice display striking vascular abnormalities, including massive dermal bleeding upon dissection and hemorrhagic kidneys, likely due to increased vessel permeability and dilation of the blood vasculature (1). Thus, both KRIT1 and ICAP1 are important for proper vascular development.
It is well-established that KRIT1 and ICAP1 directly interact, and we previously used X-ray crystallography to reveal the molecular basis for binding of the first KRIT1 NPX(Y/F) motif to the ICAP1 PTB domain (6, 36) (Fig. 1). We also characterized a nuclear localization signal (NLS) in the unstructured region of ICAP1 responsible for its nuclear accumulation (33). We further showed that the ICAP1 NLS is the key determinant of nuclear accumulation of KRIT1 and that this relies on the formation of an ICAP1/KRIT1 complex (33). Thus, signals that regulate ICAP1 nuclear localization will likely impact KRIT1 localization and signaling.
Protein phosphorylation is a well-established regulator of nuclear import (37, 38). ICAP1 contains several phosphorylated serine residues just downstream of its canonical basic NLS (39), suggesting that phosphorylation at these sites might impact ICAP1 localization. Here, we investigate phosphorylation of ICAP1 in regulating its nucleocytoplasmic shuttling. We report that phospho-mimicking mutations at serines 10 and 25, known ICAP1 phosphorylation sites (39), prevent nuclear accumulation of ICAP1 and the ICAP1/KRIT1 complex. ICAP1 Ser-10 lies within a consensus type II p21-activated kinase (PAK) phosphorylation site sequence. Here, we show that activated PAK4 can phosphorylate ICAP1 at Ser-10, preventing the nuclear accumulation of ICAP1 and consequently KRIT1. Therefore, phosphorylation can govern the nucleocytoplasmic distribution of ICAP1 and the ICAP1/KRIT1 complex, presumably impacting their roles in vascular biology and CCM signaling.
Results
ICAP1 phosphorylation site mutants alter its localization
The largely unstructured N-terminal region of ICAP1 preceding the PTB domain contains the short NLS as well as 15 serine residues previously shown by MS to be phosphorylated (39) (Fig. 2A). To investigate whether phosphorylation of these residues influences ICAP1 nuclear localization, we generated phospho-mimicking serine-to-glutamic acid and phospho-blocking serine-to-alanine mutations in N-terminally GFP-tagged ICAP1 (GFP-ICAP1). We expressed these mutants in CHO cells and assessed ICAP1 localization by quantitative microscopy. To facilitate examination of all candidate serine residues, we initially divided residues into five groups (i–v; Fig. 2A) and generated combination mutants of all residues in a given group. CHO cells were transfected with GFP, GFP-ICAP1, or GFP-ICAP1 grouped phosphorylation site mutants, and GFP localization was examined by fluorescence microscopy. As we reported previously (33), WT GFP-ICAP1 predominantly localizes to the nucleus in CHO cells in a manner dependent on the N-terminal 49 ICAP1 residues containing the NLS (Fig. 2B). Strikingly, phospho-mimicking mutations in group i (residues 10–14) or group iii (residues 25 and 28–29) resulted in dramatically decreased ICAP1 nuclear accumulation (Fig. 2B). As expression level can impact localization of recombinant proteins, we attempted to examine cells with comparable GFP signals. Quantitative analysis of GFP nuclear percentage in multiple images from at least three independent experiments using CellProfiler 2.1 (40) revealed that on average, GFP-ICAP1 was ∼65% nuclear, whereas both GFP-ICAP1 SiE and SiiiE phospho-mimicking mutants were significantly less nuclear at ∼38 and ∼50%, respectively (Fig. 2C). These effects are unlikely to be due to global effects on ICAP1 folding, as the mutations all lie in the N-terminal unstructured region and even the complete deletion of this N-terminal region does not prevent the folding of the PTB domain or its interactions with binding partners such as KRIT1 or integrin (6, 33). Immunoblotting confirmed that all GFP-ICAP1 phosphorylation site mutants were expressed at the expected molecular weights (Fig. 2D and Fig. S1A).
Figure 2.

Grouped phospho-mimicking mutants inhibit ICAP1 nuclear localization. A, schematic of ICAP1 noting the boundaries of the NLS sequence, PTB domain, and indicating the five groups (i–v) of serines that were mutated to glutamic acid or alanine. B, representative images of CHO cells expressing WT GFP-tagged ICAP1, or various ICAP1 mutants, fixed 24 h after plating on fibronectin and stained with DAPI (to identify nuclei). Bar, 10 μm. C, the percentage of the total integrated whole-cell GFP intensity found in the nucleus was calculated for each cell using CellProfiler 2.1. The total number of cells (N) in each condition from three independent experiments is indicated. Boxes, 25–50th and 50–75th percentile; whiskers, 10–90th percentile; dots, mean. Statistical significance was determined by a one-way ANOVA with Fisher's LSD test with multiple comparisons. ****, p ≤ 0.0001. D, representative immunoblots indicate expression of individual phosphomutants at the expected size; immunoblotting was against GFP for phosphomutants and vinculin for loading control. Uncropped immunoblots are shown in Fig. S1A.
Next, to assess whether a single serine residue from group i or iii could alter ICAP1 localization, individual GFP-ICAP1 serine phosphorylation site mutants were generated, and their nuclear localizations were assessed as described above. For group i, only the phospho-mimicking S10E mutant reduced ICAP1 nuclear localization, whereas mutations at Ser-11, Ser-12, Ser-13, or Ser-14 had no significant impact on targeting (Fig. 3, A and B). Notably, the single GFP-ICAP1 S10E mutant exhibited a reduction in nuclear localization comparable with that seen in the SiE grouped mutant (∼43% for S10E and ∼41% for SiE compared with ∼65% for WT ICAP1; Fig. 3B). Furthermore, the phospho-blocking S10A mutation, but not mutations at Ser-11, Ser-12, Ser-13, or Ser-14, modestly but significantly enhanced GFP-ICAP1 nuclear localization (Fig. 3, A and B). Together, these data indicate that mutation of Ser-10 modulates ICAP1 nuclear localization.
Figure 3.

Individual phosphomutations at Ser-10 or Ser-25 alter ICAP1 nuclear localization. To assess whether single serine mutations alter ICAP1 localization, individual GFP-ICAP1 serine phospho-mimicking or phospho-blocking mutations from group i (A–C) or group iii (D–F) were generated and expressed in CHO cells. A and D, representative images of CHO cells expressing GFP-tagged ICAP1 constructs stained with DAPI (to identify nuclei) 24 h after plating on fibronectin. Bar, 10 μm. B and E, percentage of GFP intensity in the nucleus. Boxes, 25–50th and 50–75th percentile; whiskers, 10–90th percentile; +, mean. Results are from three independent experiments, and the total number of cells examined (N) is indicated for each condition. ****, statistical significance at p ≤ 0.0001 as determined by a one-way ANOVA with Fisher's LSD test with multiple comparisons. C and F, representative immunoblots (against anti-GFP and anti-vinculin) (IB) indicate expression of individual phosphomutants at the expected size. Uncropped immunoblots are shown in Fig. S1 (B and C).
For individual group iii mutations, only the S25E mutant significantly inhibited GFP-ICAP1 nuclear localization (∼52%), and the inhibition was similar to that seen with the grouped SiiiE mutant (Fig. 3, D and E). The phospho-blocking S25A mutation also enhanced GFP-ICAP1 nuclear targeting (Fig. 3, D and E), consistent with inhibition of nuclear accumulation by phosphorylation of serine 25. Immunoblotting was performed to verify expression and size of all GFP-ICAP1 phosphorylation site mutants (Fig. 3 (C and F) and Fig. S1 (B and C)).
ICAP1 Ser-10 is the major site governing ICAP1 localization
Our phosphorylation site mutation analysis implicates both Ser-10 and Ser-25 in control of ICAP1 nuclear localization. However, the ICAP1 S10E mutation appears to inhibit nuclear accumulation more strongly compared with the S25E mutation (∼43% versus ∼52%; Fig. 3, B versus E). This difference is also evident in the grouped phosphomutant constructs, with ICAP1 SiE exhibiting less nuclear accumulation than ICAP1 SiiiE (Fig. 2, B and C). Furthermore, this trend is also seen with ICAP1 containing a C-terminal mCherry tag (ICAP1-mCherry), indicating that neither the fluorescent tag nor its position with respect to ICAP1 accounts for this result (Fig. 4A and Fig. S2A). Additionally, similar results were also obtained when GFP-ICAP1 constructs were expressed in HeLa cells (Fig. 4B and Fig. S2B).
Figure 4.

Ser-10 is the major site governing ICAP1 nuclear localization. CHO cells (A and C) or HeLa cells (B) were transfected with constructs encoding mCherry, C-terminally mCherry-tagged ICAP1, and phosphomutants (A); GFP, N-terminally GFP-tagged ICAP1, and phosphomutants (B); or GFP, C-terminally tagged ICAP1-GFP, and phosphomutants (C). Cells were plated on fibronectin, fixed 24 h later, and stained with DAPI (to identify nuclei). Nuclear GFP or mCherry signal was calculated, and results from three independent experiments are displayed in box-and-whisker plots. D, CHO cells stably expressing GFP-histone H2B (to identify nuclei) and transiently transfected with constructs expressing mCherry or C-terminal mCherry-tagged ICAP1, ICAP1 phosphomutants, or ICAP1 containing mutations in the NLS (ICAP1 KK6,7AA) were plated on fibronectin-coated glass-bottom dishes (MatTek), stained with HCS CellMask Deep Red Stain (to identify cell boundaries), and imaged live. Nuclear mCherry signal was calculated in three independent experiments and presented in box-and-whisker plots. In all panels, boxes represent 25–50th and 50–75th percentile, whiskers show the 10–90th percentile, and plus signs show the mean. The total number of cells measured in each condition (N) is indicated. Statistical significance was determined by a one-way ANOVA with Fisher's LSD test. **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant. Representative images are shown in Fig. S2.
To further examine whether Ser-10 is the main site governing ICAP1 localization, we generated C-terminal GFP-tagged ICAP1 (ICAP1-GFP) compound (S10A/S25A and S10E/S25E) and mixed (S10A/S25E and S10E/S25A) phosphorylation site mutants and examined their localization. As expected, the double S10A/S25A mutant strongly localized to the nucleus similar to the individual S10A or S25A mutants (Fig. 4C and Fig. S2C). Furthermore, like the individual S10E mutant, the double S10E/S25E mutant exhibited only background GFP levels of nuclear staining (Fig. 4C and Fig. S2C). However, despite the presence of the phospho-mimicking S25E mutation, the mixed S10A/S25E mutant was largely nuclear, comparable with the S10A mutant or S10A/S25A double mutant (Fig. 4C and Fig. S2C). Likewise, despite the phospho-blocking S25A mutation, the mixed S10E/S25A mutant exhibited greatly reduced nuclear localization (Fig. 4C and Fig. S2C). These data suggest that modifications of Ser-10 are the dominant regulators of ICAP1 nuclear localization. However, Ser-25 does influence ICAP1 localization even in the presence of a phospho-mimicking S10E mutation because the S10E/S25A mutant was slightly, but statistically significantly, more nuclear than the S10E/S25E mutant (Fig. 4C and Fig. S2C).
To exclude the possibility that fixation artificially increased nuclear ICAP1 levels, we assessed ICAP1-mCherry localization by live-cell microscopy in cells where the nucleus was marked with GFP-histone H2B. As shown in Fig. 4D and Fig. S2D, data from live cells were comparable with those in fixed cells and supported the importance of Ser-10 and Ser-25. Notably, localization of phosphomimetic ICAP1 S10E/S25E was equivalent to ICAP1 containing mutations inactivating its NLS (ICAP1 K6A/K7A) (33). Thus, both fixed and live fluorescence microscopy implicate Ser-10 and Ser-25 in regulating ICAP1 nuclear localization.
Finally, using lentivirally transduced GFP, ICAP1-GFP, ICAP1-GFP S10A, and ICAP1-GFP S10E, we show that the ICAP1 S10A mutation enhances, whereas the ICAP1 S10E mutation inhibits, ICAP1 nuclear accumulation compared with WT ICAP1 in both CHO (Fig. 5, A and B) and EA.hy926 endothelial-like cells (Fig. 5, D and E). Viral transduction allows visualization of multiple GFP-positive cells in each panel, clearly demonstrating the impact of the Ser-10 phosphomutation on ICAP1 localization (Fig. 5, A and D). Constructs were expressed at the expected size by immunoblotting (Fig. 5, C and F). Therefore, both transiently transfected and lentivirally transduced ICAP1 phosphomutants alter ICAP1 localization.
Figure 5.

Mutations at Ser-10 alter localization of lentivirally transduced ICAP1 in CHO and EA.hy926 cells. CHO cells (A–C) or EA.hy926 cells (D–F) were transduced with lentivirus encoding GFP, WT ICAP1-GFP, ICAP1-GFP S10A, or ICAP1-GFP S10E phosphomutants and selected with hygromycin. A and D, representative images of transduced CHO (A) or EA.hy926 (D) cells stained with DAPI (to identify nuclei) and phalloidin (to identify cell boundaries) 24 h after plating on fibronectin are shown. Bar, 50 μm. The percentage of total GFP signal in the nucleus of transduced CHO (B) or EA.hy926 (E) cells was plotted. Boxes, 25–50 and 50–75 percentile; whiskers, 10–90 percentile; +, mean. Results are from three independent experiments, and the total number of cells examined (N) is indicated for each condition. ****, statistical significance at p ≤ 0.0001 as determined by a one-way ANOVA with Fisher's LSD test with multiple comparisons. C and F, representative anti-GFP immunoblots (IB) indicate expression of individual phosphomutants at the expected size. Vinculin staining was used as a loading control.
ICAP1 Ser-10 and Ser-25 influence localization independently of the ICAP1 PTB domain
The PTB domain accounts for 134 of the 200 amino acids in ICAP1 and contains binding sites for β1 integrins and KRIT1 (6) but is dispensable for nuclear localization (33) (Fig. 6A). However, phosphorylation could theoretically affect either intermolecular or intramolecular interactions with the PTB domain to control nuclear import. To test whether the PTB domain is required for the ability of phosphorylation site mutants to impact ICAP1 localization, we introduced phospho-mimicking and phospho-blocking mutations into the isolated N-terminal region (GFP-ICAP1 1–45). Analysis by quantitative microscopy revealed that the N-terminal region localized similarly to full-length ICAP1 (Fig. 6B), suggesting that the ICAP1 PTB domain does not contain any regions (e.g. nuclear export sequence) that impact ICAP1 localization or mask the NLS. Furthermore, mutations at Ser-10 or Ser-25 had comparable effects on full-length and N-terminal ICAP1 localization (Fig. 6B). These results suggest that phosphorylation at these residues does not alter nuclear localization by changing PTB domain interactions or regulating NLS exposure by releasing the proposed N-terminal region:PTB domain interactions (7). Immunoblots show all constructs expressed at the expected molecular weights (Fig. 6C and Fig. S1D). Overall, our phosphorylation site mutant analysis revealed that phospho-mimicking mutations at Ser-10 and Ser-25 lead to decreased ICAP1 nuclear accumulation, whereas the corresponding phospho-blocking mutations enhance ICAP1 nuclear localization; that Ser-10 is the major site governing ICAP1 localization; and that the effects of phosphomutants on ICAP1 localization do not require the ICAP1 PTB domain.
Figure 6.
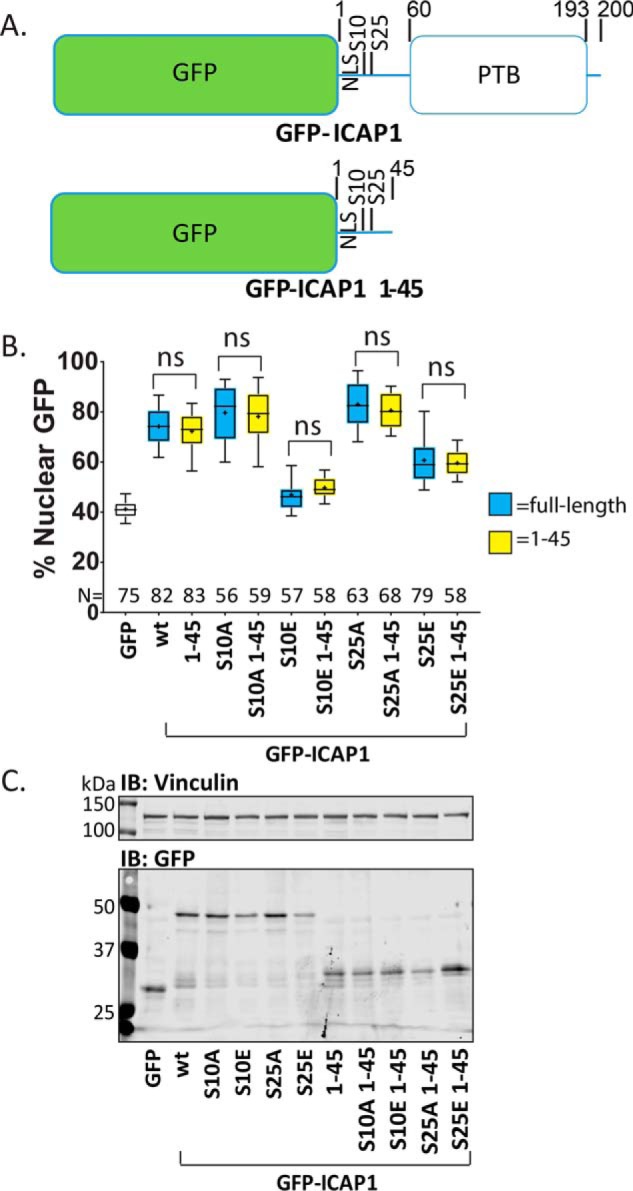
Residues 46–200 do not alter ICAP1 localization. A, schematic depicting GFP-ICAP1 and the truncation mutant GFP ICAP1 1–45. B, percentage of GFP intensity in the nucleus of CHO cells expressing GFP (unfilled box) or phosphomutants of GFP-tagged full-length ICAP1 (blue boxes) or ICAP1 1–45 (yellow boxes) 24 h after plating on fibronectin. Results are from three independent experiments, and the total number of cells examined (N) is indicated for each condition. Boxes, 25–50 and 50–75 percentile; whiskers, 10–90 percentile; +, mean. Statistical analysis was performed using a one-way ANOVA with Fisher's LSD test with multiple comparisons. ns, not significant. C, representative immunoblots (IB) (against anti-GFP and anti-vinculin) indicate construct expression at the expected sizes. Note that GFP (lane 1) runs at a smaller size than GFP-ICAP1 1–45 (lanes 7–11). Uncropped immunoblots are shown in Fig. S1D.
PAK4 phosphorylates ICAP1 at Ser-10
Our analysis suggests that ICAP1 nuclear localization can be inhibited by phosphorylation at Ser-10 and, to a lesser extent, at Ser-25. However, the point mutants we generated could theoretically affect localization independently of phosphorylation (e.g. by disrupting other post-translational modifications). To allow us to test whether authentic phosphorylation was important for localization, we sought kinases that can phosphorylate ICAP1 at these positions. Ser-10 falls within a consensus phosphorylation site motif for type II PAKs (PAK4, -5, and -6) (41, 42) (Fig. 7A). To determine whether type II PAKs could indeed phosphorylate ICAP1, we performed in vitro radiolabel PAK4 kinase assays using recombinant GST-ICAP1 as a substrate. The PAK4 catalytic domain robustly phosphorylated GST-ICAP1. Whereas mutation of Ser-25 alone was without effect, phosphorylation of GST-ICAP1 S10A and S10A/S25A was diminished ∼10-fold (Fig. 7, B and C). These experiments indicate that the PAK4 catalytic domain phosphorylates ICAP1 at Ser-10 but not Ser-25 in vitro.
Figure 7.
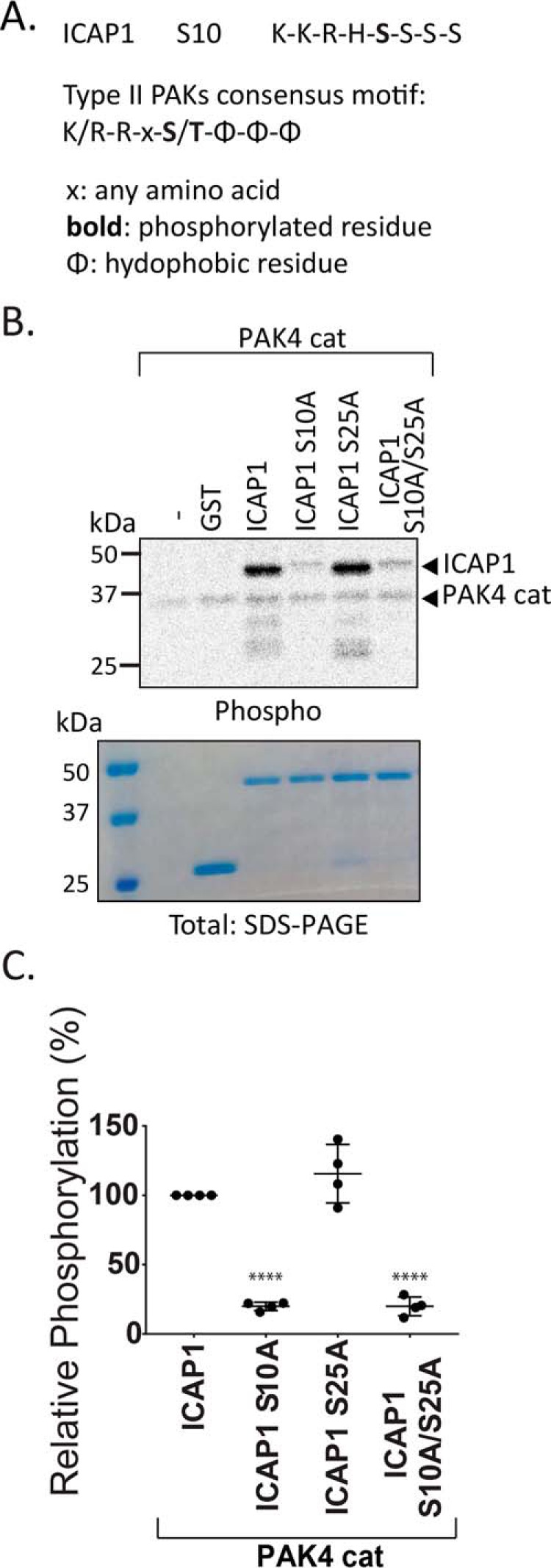
PAK4 catalytic domain phosphorylates ICAP1 Ser-10 but not Ser-25 in vitro. A, alignment of the substrate recognition motif of the type II PAKs and the ICAP1 Ser-10 site. B and C, in vitro kinase assays were performed by incubating GST-ICAP1 or phosphomutants, PAK4 catalytic domain, and [γ-33P]ATP for 30 min. B, representative autoradiograph and corresponding Coomassie-stained gel. C, phosphorylation of GST-ICAP1 mutants was quantified and normalized to WT GST-ICAP1 in four independent experiments. Individual values are represented by filled circles, and bars show mean with S.D. ****, p ≤ 0.0001 with respect to WT GST-ICAP1 as determined by a one-way ANOVA test with Fisher's LSD test with multiple comparisons.
To address whether PAK4 could phosphorylate ICAP1 at Ser-10 in cells, we performed phosphate affinity gel analysis, which relies on the reduced migration of phosphorylated proteins when fractionated by SDS-PAGE in the presence of polyacrylamide-bound Phos-tagTM reagent (43). To maximize the potential mobility shift, we initially used GFP-ICAP1 residues 1–45 because this construct targets to the nucleus like full-length ICAP1 (Fig. 6B) (44, 45). To investigate PAK4-mediated phosphorylation, CHO cells were co-transfected with mCherry or hyperactivate mCherry-PAK4 S445N mutant (46) and GFP-tagged ICAP1 constructs. GFP-tagged proteins were isolated from cell lysates, mock-treated or λ-protein phosphatase–treated, and fractionated by Phos-tagTM or standard SDS-PAGE and immunoblotted with an anti-GFP antibody (Fig. 8A and Fig. S3A). In Phos-tagTM gels, line scans depicting signal intensity exhibited at least three GFP-ICAP1 1–45 species, which we interpret as an unphosphorylated and two slower-migrating phosphorylated forms (Fig. 8B). λ-Protein phosphatase treatment resulted in loss of most of the upper two bands, supporting this interpretation (Fig. 8 (A and B) and Fig. S3A). Notably, when mCherry-PAK4 was co-expressed with GFP-ICAP1 1–45, the intensity of the intermediately migrating band (phosphospecies 1) was substantially increased, indicating enrichment of this phosphospecies (Fig. 8 (A and B) and Fig. S3A). This enrichment required Ser-10, as phosphospecies 1 was largely absent in the GFP-ICAP1 1–45 S10A mutant (Fig. 8 (A and B) and Fig. S3A). Multiple independent replicates confirmed the dependence of phosphospecies 1 on the ICAP1 Ser-10 site (Fig. 8C). These data strongly suggest that when co-expressed, PAK4 phosphorylates Ser-10 of ICAP1.
Figure 8.

Activated PAK4 phosphorylates ICAP1 Ser-10 in cells. Phos-tagTM gel mobility shift analyses were performed in CHO cells co-expressing mCherry, mCherry-PAK4 S445N, and either GFP, GFP-ICAP1 1–45, or GFP-ICAP1 1–45 S10A (A–C) or ICAP1-Spot or ICAP1-Spot S10A (D–F) in the presence and absence of λ-protein phosphatase (λPP). A–C, GFP-nanotrap pulldowns were resolved by Phos-tagTM PAGE and by standard SDS-PAGE (as indicated) and analyzed by immunoblotting (IB) against GFP (A). Input samples were also assessed by standard SDS-PAGE to evaluate expression levels of all constructs. B, line scans (outlined by blue boxes) depicting signal intensities of mobility shift bands of GFP-ICAP1 1–45 or GFP-ICAP1 S10A 1–45 from A in conditions with mCherry, mCherry-PAK4 S445N alone, or mCherry-PAK4 S445N with λ-protein phosphatase. C, percentages of the signal in each of the mobility shift bands of GFP-ICAP1 1–45 or GFP-ICAP1 S10A 1–45 were calculated by dividing the signal intensity of each band by the total amount of ICAP1. Data are represented as mean ± S.D. from independent preparations. D–F Spot-Trap® agarose pulldowns were resolved by Phos-tagTM PAGE and by standard SDS-PAGE and analyzed by immunoblotting against ICAP1 (D). Input samples were also assessed by standard SDS-PAGE to evaluate expression levels of all constructs. E, line scans (outlined by blue boxes) depicting signal intensities of mobility shift bands of ICAP1-Spot or ICAP1-Spot S10A from D in conditions with mCherry, mCherry-PAK4 S445N alone, or mCherry-PAK4 S445N with λ-protein phosphatase. F, percentages of the signal in each of the mobility shift bands of ICAP1-Spot or ICAP1-Spot S10A were calculated by dividing the signal intensity of each band by the total amount of ICAP1. Data are represented as mean ± S.D. from independent preparations. Uncropped immunoblots are shown in Fig. S3.
To test whether PAK4 phosphorylates full-length ICAP1 in cells, we used ICAP1 with a short C-terminal Spot-tag® (ChromoTek). Phos-tagTM gel analysis was performed on ICAP1 isolated from CHO cells co-expressing mCherry or mCherry-PAK4 S445N mutant (46) and C-terminal Spot-tagged® ICAP1 (ICAP1-Spot) or ICAP1-Spot S10A. Line scans revealed two ICAP1-Spot species, likely a lower unphosphorylated species and a slower-migrating phosphospecies that was largely lost upon phosphatase treatment (Fig. 8 (D–F) and Fig. S3B). When mCherry-PAK4 S445N was co-expressed with ICAP1-Spot, the signal of the phosphorylated band was significantly enriched, whereas that of the unphosphorylated band decreased (Fig. 8 (D–F) and Fig. S3B). However, when ICAP1-Spot S10A was co-expressed with mCherry-PAK4 S445N compared with mCherry, no changes in the ratio of phosphorylated to unphosphorylated species was detected, indicating that the enrichment of the phosphorylated species seen with PAK4 S445N and WT ICAP1 is specific to the Ser-10 site (Fig. 8 (D–F) and Fig. S3B). Therefore, Phos-tagTM analysis using both GFP-ICAP1 1–45 and full-length ICAP1-Spot constructs specifically revealed a phosphorylated species in the presence of activated PAK4 that was largely absent with the corresponding S10A mutant.
PAK4 inhibits ICAP1 nuclear accumulation in a Ser-10–dependent manner
Having shown that PAK4 phosphorylates ICAP1 on Ser-10, we tested whether activated PAK4 could alter ICAP1 localization in cells. CHO cells were co-transfected with GFP or GFP-PAK4 S445N and WT or mutant ICAP1-mCherry, and nuclear localization was evaluated in double-positive cells. Notably, GFP-PAK4 S445N expression significantly reduced ICAP1-mCherry nuclear localization but had no effect on mCherry localization (Fig. 9, A and B). Importantly, GFP-PAK4 S445N did not alter the enhanced nuclear localization of ICAP1-mCherry S10A/S25A or ICAP1-mCherry S10A (Fig. 9, A and B). These data are consistent with PAK4-mediated phosphorylation of ICAP1 at Ser-10 inhibiting ICAP1 nuclear accumulation and blockade of phosphorylation by S10A mutations. Interestingly, GFP-PAK4 S445N did modestly, but significantly, inhibit nuclear localization of ICAP1-mCherry S25A, consistent with the results for the ICAP1 S10E/S25A mixed phosphorylation site mutant (compare Fig. 9B with Fig. 4C). This further supports a model where Ser-10 is the major phosphorylation site governing ICAP1 localization but in which modification of Ser-25 can modulate this effect.
Figure 9.

Activated PAK4 or -6 prevents ICAP1 nuclear accumulation. A–D, CHO cells expressing mCherry, ICAP1-mCherry, or ICAP1-mCherry phospho-blockers and GFP or GFP-PAK4 (A–C) or PAK6 (D) constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. A, representative images; bar, 10 μm. B–D, percentage of mCherry intensity in the nucleus of mCherry and GFP double-positive cells. Results are from 4–6 independent experiments, and the total number of cells examined (N) is indicated for each condition. Boxes, 25–50th and 50–75th percentile; whiskers, 10–90th percentile; +, mean. Statistical analysis was performed using a one-way ANOVA with Fisher's LSD test with multiple comparisons. ****, p ≤ 0.0001; **, p ≤ 0.01; ns, not significant. Representative full immunoblots indicating construct expression at the expected sizes are shown in Fig. S5 (A–C).
The preceding data suggest that GFP-PAK4 S445N reduces ICAP1-mCherry nuclear accumulation by phosphorylating ICAP1 at Ser-10. Imaging indicates that PAK4 is predominantly cytoplasmic (Fig. 9A), suggesting that phosphorylation occurs in the cytosol. To exclude potential kinase-independent effects of PAK4, we used hyperactive, WT, and kinase-dead PAK4 to investigate whether effects on ICAP1 localization depend on PAK4 kinase activity. In separate experiments, we confirmed the relative kinase activities of these constructs by co-transfecting with the type II PAK substrate, Pacsin-1, and blotting for phospho-Pacsin-1, as described previously (47). We found that GFP-PAK4 S445N was more active than WT PAK4, but that kinase-inactive GFP-PAK4 K350M mutant (48) failed to phosphorylate Pacsin-1 (Fig. S4, A and B). Analysis of ICAP1 localization showed that kinase-inactive PAK4 had no significant effect on ICAP1-mCherry nuclear localization, whereas WT PAK4 produced a slight, but significant, reduction in ICAP1-mCherry nuclear localization that was enhanced with the activating S445N mutation (Fig. 9C). None of the PAK4 constructs impacted localization of ICAP1-mCherry S10A/S25A or of mCherry alone (Fig. 9C). Thus, the use of PAK4 mutants with altered kinase activity and ICAP1 S10A mutants supports a model where PAK4-mediated phosphorylation of Ser-10 inhibits ICAP1 nuclear localization.
PAK6 also inhibits ICAP1 nuclear accumulation
The similarity in substrate specificity between type II PAKs suggests that other type II PAKs should also modulate ICAP1 localization. Consistent with this, analysis of PAK6 showed that hyperactive PAK6 P52L (47) but not kinase-dead PAK6 K436M (51) led to a statistically significant decrease in ICAP1-mCherry localization but had no effect on ICAP1-mCherry S10A/S25A localization (Fig. 9D). The effect of PAK6 P52L on ICAP1 localization was less pronounced than that of PAK4 S445N, but this is likely due to differences in kinase activity, as PAK4 S445N led to a higher level of Pacsin-1 phosphorylation than PAK6 P52L did upon co-expression (Fig. S4, A and B). Taken together, our data further support the hypothesis that phosphorylation of ICAP1 on Ser-10 inhibits ICAP1 nuclear localization.
ICAP1 phospho-mimicking mutants and PAK4 S445N inhibit KRIT1 nuclear accumulation
We have previously shown that ICAP1 directs KRIT1 to the nucleus (33). This process requires the ICAP1:KRIT1 interaction and depends on the ICAP1 NLS (33). Therefore, we asked whether mutation of ICAP1 phosphorylation sites would also alter KRIT1 localization. CHO cells co-expressing GFP, GFP-KRIT1, or GFP-KRIT1 R179A/R185A/N192A/Y195A (KRIT1; KRIT1 mutant defective in binding ICAP1) (33) and mCherry, ICAP1-mCherry, or ICAP1-mCherry phosphomutants were examined by fluorescence microscopy, and nuclear GFP and mCherry signals were calculated. As expected, in the presence of co-expressed GFP, GFP-KRIT1, or GFP-KRIT1, ICAP1-mCherry was largely nuclear, whereas S10A/S25A mutations further enhanced ICAP1 nuclear localization, and S10E/S25E mutations inhibited ICAP1 nuclear accumulation (Fig. 10, A and B). However, when GFP-KRIT1 localization was examined, ICAP1 substantially increased KRIT1 nuclear localization (Fig. 10, A and C). This was further increased by ICAP1-mCherry S10A/S25A, whereas ICAP1-mCherry S10E/S25E retained KRIT1 in the cytoplasm (Fig. 10, A and C). Thus, KRIT1 localization is driven by ICAP1. This effect requires ICAP1:KRIT1 interaction, as GFP-KRIT1 (33) that fails to bind ICAP1 does not follow ICAP1 localization and instead remains largely cytoplasmic (Fig. 10C). Altogether, these data suggest that phosphorylation of ICAP1 could alter KRIT1 localization in a manner dependent on their direct association.
Figure 10.

Serine phosphorylation of ICAP1 impairs KRIT1 nuclear accumulation. A–D, CHO cells expressing mCherry, ICAP1-mCherry, or ICAP1-mCherry phosphomutants and GFP, GFP-tagged KRIT1, or a KRIT1 mutant defective in binding ICAP1 (KRIT1; GFP-KRIT1 R179A/R185A/N192A/Y195A) were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. A, representative images; bar, 10 μm. B and C, the percentage of mCherry (B) or GFP (C) signal in the nucleus of GFP and mCherry double-positive cells. Results are from three independent experiments, and the total number of cells examined (N) is indicated for each condition. Boxes, 25–50th and 50–75th percentile; whiskers, 10–90th percentile; +, mean. Statistical analysis was performed using a one-way ANOVA with Fisher's LSD test with multiple comparisons. ****, p ≤ 0.0001. D, CHO cells co-expressing FLAG, C-terminally triple FLAG-tagged ICAP1 (ICAP1-FLAG), or ICAP1-FLAG S10A, with mCherry or mCherry-PAK4 S445N, and GFP or GFP-KRIT1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI (to identify nuclei). D, nuclear GFP was calculated in double GFP- and mCherry-positive cells in four independent experiments and presented as in C. Representative full immunoblots illustrating construct expression are shown in Fig. S5 (D and E).
To study whether phosphorylation inhibits nuclear accumulation of the ICAP1/KRIT1 complex, we examined nuclear localization of CHO cells co-expressing C-terminally triple FLAG-tagged ICAP1 (ICAP1-FLAG) or ICAP1-FLAG S10A, and mCherry or mCherry-PAK4 S445N, and GFP or GFP-KRIT1. As expected, cells expressing mCherry, ICAP1-FLAG, and GFP-KRIT1 shifted GFP-KRIT1 more strongly nuclear (∼63%, compared with ∼45% nuclear GFP-KRIT1 with mCherry and FLAG) (Fig. 10D). Importantly, with mCherry-PAK4 S445N and ICAP1-FLAG, GFP-KRIT1 became more diffusely localized (∼55% nuclear) (Fig. 10D). Further, this shift in KRIT1 localization depends on the ICAP1 Ser-10 site, as the effect of PAK4 S445N was prevented when KRIT1 was co-expressed with ICAP1 S10A (Fig. 10D). This experiment suggests that phosphorylation at ICAP1 Ser-10 can prevent KRIT1 nuclear accumulation.
Discussion
ICAP1 plays critical roles in vascular biology (1, 3). Further, ICAP1 binds, stabilizes, and regulates the localization of KRIT1 (1, 4, 5, 33), a protein essential for vascular development and whose loss results in CCM (24, 25, 28). We have shown that the NLS in ICAP1 drives both ICAP1 and KRIT1 nuclear localization (33). Now, using ICAP1 phosphorylation site mutants and PAK4, a kinase that we show can phosphorylate ICAP1, we find that phosphorylation at Ser-10 impairs ICAP1 nuclear accumulation and that this in turn impacts nuclear localization of KRIT1.
Regulation of ICAP1 by phosphorylation
ICAP1 is composed of a PTB domain, which mediates interactions with both integrin β1 and KRIT1 (Fig. 1), and a preceding unstructured region (residues 1–60) (6, 33), which contains the NLS and is rich in serine and threonine residues. Mass spectroscopic analysis of ICAP1 co-purified with KRIT1 from U2OS cells revealed numerous phosphorylation sites in the N-terminal region (39), but the role of this phosphorylation was unknown. As phosphorylation at sites close to, or within, an NLS is a well-established mechanism to inhibit nuclear accumulation (37, 38), we used ICAP1 phospho-mimicking mutations to assess the potential impact of ICAP1 phosphorylation on localization. Of the 15 potential serine phosphorylation sites examined (39), only phospho-mimicking mutations at Ser-10, and to a lesser extent at Ser-25, significantly inhibited nuclear localization. Furthermore, co-expressing ICAP1 with activated PAK4 (a kinase that we show to be capable of phosphorylating ICAP1 Ser-10) also inhibited nuclear accumulation of ICAP1. Our results were consistent across multiple cell types, were unaffected by location or identity of fluorescent protein tags, were evident in fixed and live cells, and could be obtained in transiently transfected or stably transduced cells, strongly supporting a model where phosphorylation generally inhibits ICAP1 nuclear import. However, one caveat of our study is that it relies on exogenously expressed, tagged ICAP1. We have been unable to identify an anti-ICAP1 antibody that is effective in immunofluorescence staining protocols or one that recognizes a single band in immunoblots, complicating attempts to follow endogenous ICAP1. Our efforts to study localization of endogenous ICAP1 by subcellular fractionation (not shown) were limited by the low expression level of endogenous ICAP1 and the lack of good anti-ICAP1 antibodies. We note the importance of investigating endogenous proteins in future studies with improved reagents and approaches.
Kinases regulating ICAP1 nuclear localization
Our mutagenic data suggest that phosphorylation at Ser-10 or Ser-25 inhibits ICAP1 nuclear accumulation. Phosphorylation at these sites had been identified in cells by MS (39), but the kinases responsible had not been identified. Although the kinases phosphorylating Ser-25 remain unknown, our data establish that PAK4, a type II PAK, can phosphorylate ICAP1 at Ser-10 in vitro and when co-overexpressed in cells. Furthermore, active PAK4 and the closely related PAK6, but not their kinase-defective mutants, inhibit ICAP1 nuclear accumulation in a manner dependent on Ser-10. We initially tested PAK4 because the residues flanking Ser-10 in ICAP1 conform to a preferred type II PAK substrate motif (41, 42). Indeed, despite the presence of 14 other Ser and Thr residues in the ICAP1 N-terminal region, we found that PAK4 almost exclusively phosphorylates Ser-10. Our data establish that PAK4 activity can inhibit nuclear localization of ICAP1 via modification of Ser-10 but does not allow us to conclude whether PAK4 (or other type II PAKs) is the physiological ICAP1 kinase. Indeed, this may be cell type–specific. However, like ICAP1 and KRIT1, PAK4 is important for proper vascular development. PAK4 null mice die at embryonic day 11.5, and embryos examined at embryonic day 10.5 display vascular abnormalities in the developing heart, including thinning of the myocardium and dilation of the atrium and sinus venosus (52). Deletion of PAK4 in the precursors of the secondary heart field also produced heart defects in adult mice (53). PAK4 substrates important for its effects on the vasculature are not clearly defined, raising the possibility that PAK-mediated inhibition of nuclear accumulation of ICAP1 and KRIT1 might have an important role. As both Ser-10 and Ser-25 lie in serine-rich regions, ICAP1 phosphorylation at nearby sites by other kinases may alter Ser-10 or Ser-25 phosphorylation and hence indirectly modulate nuclear translocation.
Mechanisms by which phosphorylation may regulate ICAP1 localization
We found that the effect of phosphorylation on nuclear localization of ICAP1 was independent of its PTB domain. This effectively rules out the possibility that Ser-10 or Ser-25 phosphorylation acts by triggering N-terminal region:PTB domain interactions that mask the NLS. Whereas NLS masking by phosphorylation-induced conformational changes within the N-terminal sequence are theoretically possible, this region is predicted to be largely unstructured, suggesting more direct effects of phosphorylation on nuclear import. ICAP1 contains a classic basic NLS at residues 6–9 (KKHR) that is likely to be recognized by the acidic, hydrophobic NLS-binding groove of importin-α (IMP-α), a nuclear import receptor that binds classic NLSs to facilitate nuclear transport (37, 38, 54). Given that Ser-10 is the residue immediately following the NLS, its phosphorylation may disrupt the interaction between the NLS and IMP-α, preventing ICAP1 nuclear import. However, we note that despite falling within a stretch of five serine residues, only modification of Ser-10 alters nuclear accumulation, showing that the effect is specific to one residue. Furthermore, mutations at Ser-25, which is even more distant from the NLS, also affect nuclear localization. Rather than inhibiting binding to nuclear import machinery, phosphorylation may alternatively inhibit nuclear import by facilitating binding to cytoplasmic anchors. Further studies will be required to distinguish between these possibilities.
Potential nuclear roles of ICAP1
Although ICAP1 has now been observed in the nucleus by several groups (32, 33, 55, 56), its role there is not well-established. It has been reported that ICAP1 increases cellular proliferation by activating c-Myc transcription and that this effect requires the NLS (55), but it is unclear whether this is a direct consequence of ICAP1 nuclear compartmentalization. ICAP1 nuclear translocation also recruits KRIT1 to the nucleus (33), and we have shown here that phosphorylation of ICAP1 prevents KRIT1 accumulation in the nucleus. KRIT1 has been observed at perichromatin fibrils and the nucleolar dense fibrillar component, sites of active transcription (57), but its function at these locations is unknown. However, loss of KRIT1 expression results in altered gene expression (58–65), notably including a decrease in expression of angiogenesis-related (58–62, 65) and antioxidant (63, 64) genes. Whether nuclear KRIT1 is involved in modulating the expression of these genes is yet to be determined, as CCM proteins can also influence cytoplasmic signaling cascades that activate ERK5, hence altering gene transcription (60, 66). Further investigation of the nuclear functions of ICAP1 and KRIT1 is a priority, and the ability to direct ICAP1 and the ICAP1/KRIT1 complex to distinct cellular compartments by phosphorylation should aid these studies.
Implications for understanding ICAP1- and KRIT1-dependent vascular defects and CCM disease
Both ICAP1 and KRIT1 play roles in the vasculature. ICAP1 null mice display vascular abnormalities such as increased vessel permeability and dilation (1), whereas KRIT1 knockout mice are embryonic lethal with primarily vascular defects (28). Further, loss of KRIT1 results in CCM, and ICAP1 function is likely impaired in CCM, given ICAP1's tight association with KRIT1 (1, 13, 33). Whereas much of the focus on KRIT1 in regulating endothelial cell function has been on cytoplasmic or cell-cell junctional roles of KRIT1 (30, 61, 67), our observation of kinase-regulated ICAP1-mediated translocation of KRIT1 to the nucleus suggests that nuclear functions of KRIT1 should also be considered.
Experimental procedures
Antibodies/cDNA
Anti-GFP (Rockland, catalogue no. 600-101-215), anti-ICAP-1 (R&D systems, catalogue no. AF6805), anti-DsRed (Clontech, catalogue no. 632496), anti-FLAG® (Sigma–Aldrich, catalogue no. F1804), Myc tag (Cell Signaling, catalogue no. 2276), anti-phospho-PACSIN1 (Millipore, catalogue no. ABS39), and anti-vinculin (VCL, Sigma, V-9131) antibodies were purchased. WT and mutant ICAP1 (6, 33), KRIT1 (6, 33), and PAK (68) cDNAs previously used for protein expression/purification were subcloned into pmCherry-C1 (Clontech), pEGFP-C1 (Clontech), and pV1900 3× FLAG (gift from Benjamin Turk, Yale University School of Medicine). Additional mutations were introduced by QuikChange site-directed mutagenesis (Stratagene). Lentiviral ICAP1 and histone H2B expression constructs were generated by subcloning into CMV-pLENTI-Hygro (Addgene, plasmid 17454, gift from Eric Campeau). All constructs used were authenticated by DNA sequencing.
Cell culture
CHO cells were cultured in Dulbecco modified Eagle's medium containing 9% fetal bovine serum, 1% sodium pyruvate, and 1% nonessential amino acids (Gibco). HEK 293T and HeLa cells were cultured in Dulbecco's modified Eagle's medium containing 9% fetal bovine serum. EA.hy926 cells were cultured in Dulbecco's modified Eagle's medium containing 9% fetal bovine serum, 1% sodium pyruvate, 1% nonessential amino acids, and 1× HAT supplement (Gibco). All lines were confirmed to be mycoplasma-free by testing with the MycoAlertTM mycoplasma detection kit (Lonza).
Lentiviral knockdown and expression
Lentiviruses were produced by co-transfecting packaging vectors psPAX2 (viral proteins Gag and Rev under the SV40 promoter; Addgene plasmid 12260, a gift from D. Trono, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) and pMD2.G (viral protein VSV-G expressed under the CMV promoter; Addgene plasmid 12259, a gift from D. Trono) into HEK 293T cells with the pLENTI-Hygro construct. Viral supernatant was collected 48 and 72 h after transfection and filtered with a 0.45-μm filter.
To generate polyclonal knockdown or expression lines, cells were incubated with viral supernatant and 8 μg/ml Polybrene for 24 h. Cells were either analyzed 48–72 h post-infection or selected with 50 μg/ml hygromycin (CMV-pLENTI-Hygro) as appropriate.
Sample preparation and immunoblotting
Unless otherwise stated, transfected lines were lysed in radioimmune precipitation assay buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing cOmplete protease inhibitor mixture tablets (Roche Applied Science) for 30 min, boiled in 4× Laemmli sample buffer for 5 min at 95 °C, and loaded onto a 12% SDS-polyacrylamide gel. Samples were transferred to 0.45-μm nitrocellulose membranes, and membranes were blocked for >1 h in 5% BSA in TBS-T. Immunoblotting was performed with primary antibodies diluted in 5% BSA in TBS-T (1:1000 of α-GFP, 1:1000 of α-DsRed, 1:1000 of α-FLAG, 1:10,000 of α-vinculin) fluorescent secondary antibodies (IR Dye800 or IR Dye680; LI-COR Biosciences, diluted 1:20,000 in 5% BSA in TBS-T) and scanned on the Odyssey CLx IR imaging system.
Microscopy
CHO, HeLa, or EA.hy926 cells were transfected with GFP, mCherry, and/or V1900 3× FLAG expression vectors using linear polyethyleneimine, molecular weight 25,000 (Polysciences, Inc.). 24 h later, cells were plated on coverslips coated with 5 μg/ml fibronectin (Sigma-Aldrich). 24 h after replating, cells were fixed in 4% paraformaldehyde in PBS, pH 7.4, for 15 min, washed with PBS, stained with 1:2000 Alexa Fluor 647 phalloidin (Thermo Fisher Scientific) in PMZ-T Buffer for 30 min or with 0.2 μg/ml HCS CellMaskTM Deep Red Stain in PBS following Triton® X-100 permeabilization, washed with PBS, and mounted using ProLong Diamond with 25 ng/ml DAPI added (Invitrogen). Images were acquired using Nikon Eclipse Ti-S with a 20×, 40×, or 100× objective using the μManager acquisition software (69). A minimum of 40 cells (from more than three replicates) were analyzed for each single-expression experiment, and a minimum of 75 cells (from more than three replicates) were analyzed for each double-expression experiment. For live-imaging experiments, cells stably infected with GFP-histone H2B were plated on glass-bottom dishes (MatTek) and stained with HCS Cell Mask Deep Red Stain (Thermo Fisher Scientific) prior to imaging using a Nikon Ti-2 Eclipse microscope (Nikon, Tokyo, Japan) with a Prime95B RoHS cMOS camera (pixel size = 110 nm) (Photometrics, Tucson, AZ). We verified by immunoblotting that exogenous proteins were of the expected size. Quantification of ICAP1/KRIT1 nuclear/cytoplasmic localization was performed using CellProfiler 2.1 (40). Nuclear GFP and/or mCherry was quantified as the integrated intensity of the fluorophore within the nucleus, as defined by DAPI staining. The outer boundary of the cell was determined by the Watershed-Image method within CellProfiler 2.1 using the Phalloidin or HCS CellMaskTM fluorescence signal (40). Data were charted, and statistical analyses were performed using GraphPad Prism.
In vitro kinase assays
Previously used pGEX-6P-1 WT ICAP1 (6, 33) and pGEX-6P-1 ICAP1 phosphomutants (generated via QuikChange) were transformed in BL21 (DE3) RosettaTM (Novagen) cells and were cultivated at 37 °C to an A600 of 0.6. ICAP1 expression was induced by 0.1 mm isopropyl β-d-1-thiogalactopyranoside at 16 °C for 20 h. Cell pellets were then lysed in 50 mm Tris-HCl, pH 7.9, 100 mm NaCl, 5% glycerol, 0.1 mm tris(2-carboxyethyl) phosphine, and 1× cOmpleteTM EDTA-free protease inhibitor mixture (Roche Applied Science). GST-ICAP1 was purified from the lysate on Sepharose-4B GSH beads. GST-ICAP1 was further purified by size-exclusion chromatography on a SuperdexTM S200 16/600 pg column (GE Healthcare) in 50 mm Tris-HCl, pH 8.2, 100 mm NaCl, 5% glycerol, 0.1 mm tris(2-carboxyethyl) phosphine and concentrated using Amicon® Ultra-4 (Millipore) centrifugal filters. Purified N-terminally hexahistidine-tagged PAK4 catalytic domain prepared as described (70) was kindly provided by Chad Miller (Yale University).
ICAP1 kinase reactions contained 100 nm kinase and 1 μm purified ICAP1 in kinase assay buffer (50 mm Tris, pH 7.5, 10 mm DTT, 2.5 mm MgCl2). Reactions were started by the addition of ATP (10 μm, with 0.20 μCi/μl [γ-33P]ATP), quenched with 4× Laemmli sample buffer after a 30-min incubation at 30 °C, and fractionated by 12% acrylamide SDS-PAGE. Gels were dried, and radiolabel incorporation was detected by phosphorimaging (Bio-Rad Molecular Imager FX Pro Plus).
Phos-tagTM
CHO cells were co-transfected with GFP or Spot® and mCherry expression constructs using Lipofectamine® 2000 (Thermo Fisher Scientific), following the manufacturer's instructions. 24 h later, cells were washed with PBS, scraped, and lysed directly in a plate with radioimmune precipitation assay buffer containing 1× cOmpleteTM EDTA-free protease inhibitor mixture (Roche Applied Science) and 1× PhosSTOPTM phosphatase inhibitor mixture (Roche Applied Science). Cells were sheared five times with a 23-gauge needle, sonicated briefly, and centrifuged at maximum speed. The supernatant was incubated with GFP-nanotrap (44, 45) or Spot-Trap® agarose (ChromoTek) for 2 h at 4 °C with rotation. Beads were washed two times and then subjected to λ-protein phosphatase or mock treatment (LambdaPP, New England Biolabs) following the manufacturer's instructions. 2× Laemmli sample buffer was added to beads, boiled for 5 min, and loaded onto 12.5% SuperSep Phos-tagTM precast gels (Wako Pure Chemical Industries, Ltd.). Electrophoresis was performed at 90 V until the bromphenol blue dye reached the bottom of the gel. Gels were soaked in transfer buffer (25 mm Tris, 192 mm glycine, 20% (v/v) MeOH) with 10 mm EDTA for 10 min, repeating twice with buffer exchanges. Gels were then soaked in transfer buffer without EDTA for 10 min and then transferred to polyvinylidene difluoride membranes. After transfer, the membranes were soaked in TBS-T for 1 h, blocked with 3% BSA in TBS-T for 1 h, and probed with α-GFP or α-ICAP-1 antibody in 3% BSA in TBS-T.
Author contributions
V. L. S., K. M. D., and D. A. C. conceptualization; V. L. S., B. S., and D. A. C. funding acquisition; V. L. S. investigation; V. L. S., and B. S. methodology; V. L. S. writing-original draft; V. L. S., B. S., K. M. D., and D. A. C. writing-review and editing; K. M. D. and D. A. C. supervision; D. A. C. project administration.
Supplementary Material
Acknowledgments
We thank Benjamin Turk and Titus Boggon (Department of Pharmacology, Yale University School of Medicine), as well as past and present members of the Calderwood laboratory, for helpful insights and discussion. We further thank Chad Miller and Nicholas Zeringo (Turk laboratory) for assistance with in vitro kinase assays.
This work was supported by National Institutes of Health Grants R01NS085078 (to D. A. C.) and GM007324 and F31HL143831 (to V. L. S.) and American Heart Association Grant 18POST33960340 (to B. S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- PTB
- phosphotyrosine-binding
- CCM
- cerebral cavernous malformation
- NLS
- nuclear localization signal
- PAK
- p21-activated kinase
- CHO
- Chinese hamster ovary
- DAPI
- 4′,6-diamidino-2-phenylindole
- ANOVA
- analysis of variance
- LSD
- least significant difference.
References
- 1. Faurobert E., Rome C., Lisowska J., Manet-Dupé S., Boulday G., Malbouyres M., Balland M., Bouin A.-P., Kéramidas M., Bouvard D., Coll J.-L., Ruggiero F., Tournier-Lasserve E., and Albiges-Rizo C. (2013) CCM1–ICAP-1 complex controls β1 integrin–dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 202, 545–561 10.1083/jcb.201303044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brunner M., Millon-Frémillon A., Chevalier G., Nakchbandi I. A., Mosher D., Block M. R., Albigès-Rizo C., and Bouvard D. (2011) Osteoblast mineralization requires β1 integrin/ICAP-1–dependent fibronectin deposition. J. Cell Biol. 194, 307–322 10.1083/jcb.201007108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brütsch R., Liebler S. S., Wüstehube J., Bartol A., Herberich S. E., Adam M. G., Telzerow A., Augustin H. G., and Fischer A. (2010) Integrin cytoplasmic domain–associated protein-1 attenuates sprouting angiogenesis. Circ. Res. 107, 592–601 10.1161/CIRCRESAHA.110.217257 [DOI] [PubMed] [Google Scholar]
- 4. Zhang X. A., and Hemler M. E. (1999) Interaction of the integrin β1 cytoplasmic domain with ICAP-1 protein. J. Biol. Chem. 274, 11–19 10.1074/jbc.274.1.11 [DOI] [PubMed] [Google Scholar]
- 5. Chang D. D., Wong C., Smith H., and Liu J. (1997) ICAP-1, a novel β1 integrin cytoplasmic domain–associated protein, binds to a conserved and functionally important NPXY sequence motif of β1 integrin. J. Cell Biol. 138, 1149–1157 10.1083/jcb.138.5.1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu W., Draheim K. M., Zhang R., Calderwood D. A., and Boggon T. J. (2013) Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 10.1016/j.molcel.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Millon-Frémillon A., Brunner M., Abed N., Collomb E., Ribba A.-S., Block M. R., Albigès-Rizo C., and Bouvard D. (2013) Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1) negatively regulates 1 integrins. J. Biol. Chem. 288, 20248–20260 10.1074/jbc.M113.455956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bouvard D., Vignoud L., Dupé-Manet S., Abed N., Fournier H.-N., Vincent-Monegat C., Retta S. F., Fassler R., and Block M. R. (2003) Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1. J. Biol. Chem. 278, 6567–6574 10.1074/jbc.M211258200 [DOI] [PubMed] [Google Scholar]
- 9. Degani S., Balzac F., Brancaccio M., Guazzone S., Retta S. F., Silengo L., Eva A., and Tarone G. (2002) The integrin cytoplasmic domain-associated protein ICAP-1 binds and regulates Rho family GTPases during cell spreading. J. Cell Biol. 156, 377–387 10.1083/jcb.200108030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millon-Frémillon A., Bouvard D., Grichine A., Manet-Dupé S., Block M. R., and Albiges-Rizo C. (2008) Cell adaptive response to extracellular matrix density is controlled by ICAP-1–dependent β1-integrin affinity. J. Cell Biol. 180, 427–441 10.1083/jcb.200707142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fournier H.-N., Dupé-Manet S., Bouvard D., Lacombe M.-L., Marie C., Block M. R., and Albiges-Rizo C. (2002) Integrin cytoplasmic domain-associated protein 1 (ICAP-1) interacts directly with the metastasis suppressor nm23-H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J. Biol. Chem. 277, 20895–20902 10.1074/jbc.M200200200 [DOI] [PubMed] [Google Scholar]
- 12. Stroeken P. J. M., Alvarez B., Van Rheenen J., Wijnands Y. M., Geerts D., Jalink K., and Roos E. (2006) Integrin cytoplasmic domain-associated protein-1 (ICAP-1) interacts with the ROCK-I kinase at the plasma membrane. J. Cell Physiol. 208, 620–628 10.1002/jcp.20699 [DOI] [PubMed] [Google Scholar]
- 13. Zawistowski J. S., Serebriiskii I. G., Lee M. F., Golemis E. A., and Marchuk D. A. (2002) KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 11, 389–396 10.1093/hmg/11.4.389 [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., Clatterbuck R. E., Rigamonti D., Chang D. D., and Dietz H. C. (2001) Interaction between krit1 and icap1α infers perturbation of integrin β1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953–2960 10.1093/hmg/10.25.2953 [DOI] [PubMed] [Google Scholar]
- 15. Cavalcanti D. D., Kalani M. Y. S., Martirosyan N. L., Eales J., Spetzler R. F., and Preul M. C. (2012) Cerebral cavernous malformations: from genes to proteins to disease: clinical article. J. Neurosurg. 116, 122–132 10.3171/2011.8.JNS101241 [DOI] [PubMed] [Google Scholar]
- 16. McCormick W. F. (1966) The pathology of vascular (“arteriovenous”) malformations. J. Neurosurg. 24, 807–816 10.3171/jns.1966.24.4.0807 [DOI] [PubMed] [Google Scholar]
- 17. Wong J. H., Awad I. A., and Kim J. H. (2000) Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery 46, 1454–1459 10.1097/00006123-200006000-00027 [DOI] [PubMed] [Google Scholar]
- 18. Clatterbuck R. E., Eberhart C. G., Crain B. J., and Rigamonti D. (2001) Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 71, 188–192 10.1136/jnnp.71.2.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otten P., Pizzolato G. P., Rilliet B., and Berney J. (1989) [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. Neurochirurgie 35, 82–83, 128–131 [PubMed] [Google Scholar]
- 20. Draheim K. M., Fisher O. S., Boggon T. J., and Calderwood D. A. (2014) Cerebral cavernous malformation proteins at a glance. J. Cell Sci. 127, 701–707 10.1242/jcs.138388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Denier C., Labauge P., Bergametti F., Marchelli F., Riant F., Arnoult M., Maciazek J., Vicaut E., Brunereau L., Tournier-Lasserve E., and Société Française de Neurochirurgie (2006) Genotype–phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 60, 550–556 10.1002/ana.20947 [DOI] [PubMed] [Google Scholar]
- 22. Cavé-Riant F., Denier C., Labauge P., Cécillon M., Maciazek J., Joutel A., Laberge-Le Couteulx S., and Tournier-Lasserve E. (2002) Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with cerebral cavernous malformations. Eur. J. Hum. Genet. 10, 733–740 10.1038/sj.ejhg.5200870 [DOI] [PubMed] [Google Scholar]
- 23. Riant F., Cecillon M., Saugier-Veber P., and Tournier-Lasserve E. (2013) CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 14, 133–141 10.1007/s10048-013-0362-0 [DOI] [PubMed] [Google Scholar]
- 24. Marchuk D. A., Gallione C. J., Morrison L. A., Clericuzio C. L., Hart B. L., Kosofsky B. E., Louis D. N., Gusella J. F., Davis L. E., and Prenger V. L. (1995) A locus for cerebral cavernous malformations maps to chromosome 7q in two families. Genomics 28, 311–314 10.1006/geno.1995.1147 [DOI] [PubMed] [Google Scholar]
- 25. Kurth J. H., Zabramski J. M., and Dubovsky J. (1994) Genetic linkage of the familial cavernous malformation (CM) gene to chromosome 7q. Am. J. Hum. Genet. 55, Suppl. 3 [Google Scholar]
- 26. Zhang R., Li X., and Boggon T. J. (2015) Structural analysis of the KRIT1 ankyrin repeat and FERM domains reveals a conformationally stable ARD–FERM interface. J. Struct. Biol. 192, 449–456 10.1016/j.jsb.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher O. S., Liu W., Zhang R., Stiegler A. L., Ghedia S., Weber J. L., and Boggon T. J. (2015) Structural basis for the disruption of the cerebral cavernous malformations 2 (CCM2) interaction with Krev interaction trapped 1 (KRIT1) by disease-associated mutations. J. Biol. Chem. 290, 2842–2853 10.1074/jbc.M114.616433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whitehead K. J., Plummer N. W., Adams J. A., Marchuk D. A., and Li D. Y. (2004) Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131, 1437–1448 10.1242/dev.01036 [DOI] [PubMed] [Google Scholar]
- 29. Serebriiskii I., Estojak J., Sonoda G., Testa J. R., and Golemis E. A. (1997) Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 15, 1043–1049 10.1038/sj.onc.1201268 [DOI] [PubMed] [Google Scholar]
- 30. Glading A., Han J., Stockton R. A., and Ginsberg M. H. (2007) KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell–cell junctions. J. Cell Biol. 179, 247–254 10.1083/jcb.200705175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J. J., Stockton R. A., Gingras A. R., Ablooglu A. J., Han J., Bobkov A. A., and Ginsberg M. H. (2011) A mechanism of Rap1-induced stabilization of endothelial cell–cell junctions. Mol. Biol. Cell 22, 2509–2519 10.1091/mbc.e11-02-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zawistowski J. S., Stalheim L., Uhlik M. T., Abell A. N., Ancrile B. B., Johnson G. L., and Marchuk D. A. (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 10.1093/hmg/ddi256 [DOI] [PubMed] [Google Scholar]
- 33. Draheim K. M., Huet-Calderwood C., Simon B., and Calderwood D. A. (2017) Nuclear localization of integrin cytoplasmic associated protein-1 influences β1 integrin activation and recruits Krev/interaction trapped-1 to the Nucleus. J. Biol. Chem. 292, 1884–1898 10.1074/jbc.M116.762393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stockton R. A., Shenkar R., Awad I. A., and Ginsberg M. H. (2010) Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 207, 881–896 10.1084/jem.20091258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lisowska J., Rödel C. J., Manet S., Miroshnikova Y. A., Boyault C., Planus E., De Mets R., Lee H.-H., Destaing O., Mertani H., Boulday G., Tournier-Lasserve E., Balland M., Abdelilah-Seyfried S., Albiges-Rizo C., and Faurobert E. (2018) The CCM1-CCM2 complex controls complementary functions of ROCK1 and ROCK2 that are required for endothelial integrity. J. Cell Sci. 131, jcs216093 10.1242/jcs.216093 [DOI] [PubMed] [Google Scholar]
- 36. Liu W., and Boggon T. J. (2013) Cocrystal structure of the ICAP1 PTB domain in complex with a KRIT1 peptide. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 494–498 10.1107/S1744309113010762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harreman M. T., Kline T. M., Milford H. G., Harben M. B., Hodel A. E., and Corbett A. H. (2004) Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J. Biol. Chem. 279, 20613–20621 10.1074/jbc.M401720200 [DOI] [PubMed] [Google Scholar]
- 38. Nardozzi J. D., Lott K., and Cingolani G. (2010) Phosphorylation meets nuclear import: a review. Cell Commun. Signal. 8, 32 10.1186/1478-811X-8-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J., Sherman N. E., Fox J. W., and Ginsberg M. H. (2011) Phosphorylation sites in the cerebral cavernous malformations complex. J. Cell Sci. 124, 3929–3932 10.1242/jcs.095471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., and Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rennefahrt U. E. E., Deacon S. W., Parker S. A., Devarajan K., Beeser A., Chernoff J., Knapp S., Turk B. E., and Peterson J. R. (2007) Specificity profiling of Pak kinases allows identification of novel phosphorylation sites. J. Biol. Chem. 282, 15667–15678 10.1074/jbc.M700253200 [DOI] [PubMed] [Google Scholar]
- 42. Ha B. H., Morse E. M., Turk B. E., and Boggon T. J. (2015) Signaling, regulation, and specificity of the type II p21-activated kinases. J. Biol. Chem. 290, 12975–12983 10.1074/jbc.R115.650416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteomics 5, 749–757 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 44. Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M. C., and Leonhardt H. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell Proteomics 7, 282–289 10.1074/mcp.M700342-MCP200 [DOI] [PubMed] [Google Scholar]
- 45. Kadry Y. A., Huet-Calderwood C., Simon B., and Calderwood D. A. (2018) Kindlin-2 interacts with a highly-conserved surface of ILK to regulate focal adhesion localization and cell spreading. J. Cell Sci. 131, jcs221184 10.1242/jcs.221184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qu J., Cammarano M. S., Shi Q., Ha K. C., de Lanerolle P., and Minden A. (2001) Activated PAK4 regulates cell adhesion and anchorage-independent growth. Mol. Cell Biol. 21, 3523–3533 10.1128/MCB.21.10.3523-3533.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gao J., Ha B. H., Lou H. J., Morse E. M., Zhang R., Calderwood D. A., Turk B. E., and Boggon T. J. (2013) Substrate and inhibitor specificity of the type II p21-activated kinase, PAK6. PLoS ONE 8, e77818 10.1371/journal.pone.0077818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abo A., Qu J., Cammarano M. S., Dan C., Fritsch A., Baud V., Belisle B., and Minden A. (1998) PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 17, 6527–6540 10.1093/emboj/17.22.6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deleted in proof.
- 50. Deleted in proof.
- 51. Schrantz N., da Silva Correia J., Fowler B., Ge Q., Sun Z., and Bokoch G. M. (2004) Mechanism of p21-activated kinase 6-mediated inhibition of androgen receptor signaling. J. Biol. Chem. 279, 1922–1931 10.1074/jbc.M311145200 [DOI] [PubMed] [Google Scholar]
- 52. Qu J., Li X., Novitch B. G., Zheng Y., Kohn M., Xie J.-M., Kozinn S., Bronson R., Beg A. A., and Minden A. (2003) PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol. Cell Biol. 23, 7122–7133 10.1128/MCB.23.20.7122-7133.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nekrasova T., and Minden A. (2012) Role for p21-activated kinase PAK4 in development of the mammalian heart. Transgenic Res. 21, 797–811 10.1007/s11248-011-9578-7 [DOI] [PubMed] [Google Scholar]
- 54. Wagstaff K. M., and Jans D. A. (2006) Intramolecular masking of nuclear localization signals: analysis of importin binding using a novel AlphaScreen-based method. Anal. Biochem. 348, 49–56 10.1016/j.ab.2005.10.029 [DOI] [PubMed] [Google Scholar]
- 55. Fournier H.-N., Dupé-Manet S., Bouvard D., Luton F., Degani S., Block M. R., Retta S. F., and Albiges-Rizo C. (2005) Nuclear translocation of integrin cytoplasmic domain-associated protein 1 stimulates cellular proliferation. Mol. Biol. Cell 16, 1859–1871 10.1091/mbc.e04-08-0744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Francalanci F., Avolio M., De Luca E., Longo D., Menchise V., Guazzi P., Sgrò F., Marino M., Goitre L., Balzac F., Trabalzini L., and Retta S. F. (2009) Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp. Cell Res. 315, 285–303 10.1016/j.yexcr.2008.10.006 [DOI] [PubMed] [Google Scholar]
- 57. Marzo S., Galimberti V., and Biggiogera M. (2014) Unexpected distribution of KRIT1 inside the nucleus: new insight in a complex molecular pathway. Eur. J. Histochem. 58, 2358 10.4081/ejh.2014.2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou Z., Tang A. T., Wong W.-Y., Bamezai S., Goddard L. M., Shenkar R., Zhou S., Yang J., Wright A. C., Foley M., Arthur J. S. C., Whitehead K. J., Awad I. A., Li D. Y., Zheng X., and Kahn M. L. (2016) Cerebral cavernous malformations arise from endothelial gain of MEKK3–KLF2/4 signalling. Nature 532, 122–126 10.1038/nature17178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cuttano R., Rudini N., Bravi L., Corada M., Giampietro C., Papa E., Morini M. F., Maddaluno L., Baeyens N., Adams R. H., Jain M. K., Owens G. K., Schwartz M., Lampugnani M. G., and Dejana E. (2016) KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 8, 6–24 10.15252/emmm.201505433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou Z., Rawnsley D. R., Goddard L. M., Pan W., Cao X.-J., Jakus Z., Zheng H., Yang J., Arthur J. S. C., Whitehead K. J., Li D., Zhou B., Garcia B. A., Zheng X., and Kahn M. L. (2015) The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 32, 168–180 10.1016/j.devcel.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lopez-Ramirez M. A., Fonseca G., Zeineddine H. A., Girard R., Moore T., Pham A., Cao Y., Shenkar R., de Kreuk B.-J., Lagarrigue F., Lawler J., Glass C. K., Awad I. A., and Ginsberg M. H. (2017) Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J. Exp. Med. 214, 3331–3346 10.1084/jem.20171178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lopez-Ramirez M. A., Pham A., Girard R., Wyseure T., Hale P., Yamashita A., Koskimäki J., Polster S., Saadat L., Romero I. A., Esmon C. T., Lagarrigue F., Awad I. A., Mosnier L. O., and Ginsberg M. H. (2019) Cerebral cavernous malformations form an anticoagulant vascular domain in humans and mice. Blood 133, 193–204 10.1182/blood-2018-06-856062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goitre L., DiStefano P. V., Moglia A., Nobiletti N., Baldini E., Trabalzini L., Keubel J., Trapani E., Shuvaev V. V., Muzykantov V. R., Sarelius I. H., Retta S. F., and Glading A. J. (2017) Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 7, 8296 10.1038/s41598-017-08373-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Goitre L., Balzac F., Degani S., Degan P., Marchi S., Pinton P., and Retta S. F. (2010) KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 5, e11786 10.1371/journal.pone.0011786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DiStefano P. V., Kuebel J. M., Sarelius I. H., and Glading A. J. (2014) KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 289, 33054–33065 10.1074/jbc.M114.582304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fisher O. S., Deng H., Liu D., Zhang Y., Wei R., Deng Y., Zhang F., Louvi A., Turk B. E., Boggon T. J., and Su B. (2015) Structure and vascular function of MEKK3–cerebral cavernous malformations 2 complex. Nat. Commun. 6, 7937 10.1038/ncomms8937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Glading A. J., and Ginsberg M. H. (2010) Rap1 and its effector KRIT1/CCM1 regulate β-catenin signaling. Dis. Model. Mech. 3, 73–83 10.1242/dmm.003293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morse E. M., Sun X., Olberding J. R., Ha B. H., Boggon T. J., and Calderwood D. A. (2016) PAK6 targets to cell-cell adhesions through its N-terminus in a Cdc42-dependent manner to drive epithelial colony escape. J. Cell Sci. 129, 380–393 10.1242/jcs.177493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stuurman N., Edelstein A. D., Amodaj N., Hoover K. H., and Vale R. D. (2010) Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. Chapter 14, Unit 14.20 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller C. J., Lou H. J., Simpson C., van de Kooij B., Ha B. H., Fisher O. S., Pirman N. L., Boggon T. J., Rinehart J., Yaffe M. B., Linding R., and Turk B. E. (2019) Comprehensive profiling of the STE20 kinase family defines features essential for selective substrate targeting and signaling output. PLoS Biol. 17, e2006540 10.1371/journal.pbio.2006540 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.