

Abstract
Herpesviruses uniquely express two essential nuclear egress-regulating proteins forming a heterodimeric basic structure of the nuclear egress complex (core NEC). These core NECs serve as a hexameric lattice-structured platform for capsid docking and recruit viral and cellular NEC-associated factors that jointly exert nuclear lamina- and membrane-rearranging functions (multicomponent NEC). Here, we report the X-ray structures of β- and γ-herpesvirus core NECs obtained through an innovative recombinant expression strategy based on NEC-hook::NEC-groove protein fusion constructs. This approach yielded the first structure of γ-herpesviral core NEC, namely the 1.56 Å structure of Epstein-Barr virus (EBV) BFRF1–BFLF2, as well as an increased resolution 1.48 Å structure of human cytomegalovirus (HCMV) pUL50-pUL53. Detailed analysis of these structures revealed that the prominent hook segment is absolutely required for core NEC formation and contributes approximately 80% of the interaction surface of the globular domains of NEC proteins. Moreover, using HCMV::EBV hook domain swap constructs, computational prediction of the roles of individual hook residues for binding, and quantitative binding assays with synthetic peptides presenting the HCMV- and EBV-specific NEC hook sequences, we characterized the unique hook-into-groove NEC interaction at various levels. Although the overall physicochemical characteristics of the protein interfaces differ considerably in these β- and γ-herpesvirus NECs, the binding free energy contributions of residues displayed from identical positions are similar. In summary, the results of our study reveal critical details of the molecular mechanism of herpesviral NEC interactions and highlight their potential as an antiviral drug target.
Keywords: herpesvirus, structure-function, energetics, confocal microscopy, crystal structure, alanine scan, Epstein-Barr virus, Human cytomegalovirus, nuclear egress complex, Protein-protein interactions
Introduction
Herpesviruses are major pathogens of humans and animals. They show a worldwide distribution and cause a variety of clinical symptoms and diseases. The range of clinical manifestations can vary substantially as illustrated by the prototypes of the subfamilies α-, β-, and γ-herpesviruses, i.e. herpes simplex virus type 1 (HSV-1),4 human cytomegalovirus (HCMV), and Epstein-Barr virus (EBV) (1). HSV-1 is a neurotropic α-herpesvirus that has been recognized as a ubiquitous pathogen causing a wide range of innocuous diseases including herpes labialis (cold sores), conjunctivitis, genital herpes, epithelial and/or stromal keratitis, and potentially fatal encephalitis in humans. HCMV disease manifestations can range from self-limiting febrile periods to fatal end-organ disease. Specifically, congenital HCMV infection acquired during pregnancy represents a serious medical problem, frequently leading to severe developmental defects and life-threatening HCMV-induced pathology. Both reactivation and reinfection may occasionally occur in HCMV-positive individuals, in most cases either developing in an asymptomatic way or accompanied by mild febrile illness. In immunosuppressed individuals, however, HCMV infection can lead to severe symptoms (2, 3). EBV infection mostly induces acute infectious mononucleosis, which is typically self-limiting, yet EBV is also associated with a number of human cancers such as Burkitt's lymphoma, nasopharyngeal carcinoma, posttransplant B and T cell lymphomas, and gastric cancer (4). To date, no vaccines have been approved for the prevention of these two herpesviral infections. Antiherpesviral drugs are available for application in various clinical settings, but frequently remain unsatisfactory in terms of limited drug compatibility due to adverse side effects or the selection of drug-resistant virus variants.
Herpesvirus-host interaction is closely regulated through the formation of protein-protein complexes, many of which represent rate-limiting determinants of viral replication. The nuclear envelope (NE) represents a physical barrier separating the nucleus from the cytoplasm. As a characteristic feature of herpesviruses, genomic replication starts in the host cell nucleus, where preformed capsids are packaged and exported to the cytoplasm for further virion maturation. The transition of capsids through the NE is a multistep regulatory process, termed nuclear egress. During this process, the NE is reorganized at specific sites and undergoes a decisive phosphorylation-triggered distortion of the nuclear lamina and a docking of viral nuclear capsids to the respective sites of nuclear egress (lamina-depleted areas). The regulation of nuclear egress has first been mechanistically analyzed for cytomegaloviruses (5, 6), followed by the description of a first crystal structure of the core nuclear egress complex (NEC) of HCMV (7, 8) and other herpesviruses (9, 10). Since then, a number of mechanistic details of NEC functionalities have been described (reviewed in Refs. 11–14), but the question, to which extent structural and functional conservation is consistent among members of herpesviral subfamilies, remained poorly answered so far.
Here, we present novel high-resolution X-ray crystal structures of herpesviral core NECs, i.e. the first structure of a γ-herpesviral core NEC, namely of EBV BFRF1 in complex with the hook segment of BFLF2, as well as a considerably increased resolution structure of β-herpesviral HCMV pUL50 in complex with the hook segment of pUL53. The structural investigation is put into context of a multilevel biochemical-functional analysis that focuses in particular on the NEC-specific binding properties. Combined, the data reveal both conserved and individually unique features that are in agreement with the autologous and nonautologous interaction properties.
Results
Conservation of primary sequences of α-, β-, and γ-herpesviral core NEC proteins
Amino acid sequences of core NEC proteins of four herpesviral reference strains selected from the α-, β-, and γ-subfamilies were aligned and analyzed for their degrees of sequence conservation (Table 1). The levels of amino acid identity were generally low, ranging between 11.6 and 34.0% for Orf24/pUL50/pM50/BFRF1 or 12.7 and 32.9% for Orf27/pUL53/pM53/BFLF2 of VZV/HCMV/MCMV/EBV, respectively. Highest levels (around 30%) were found for the human and murine cytomegaloviruses (HCMV and MCMV) homologs within the β-herpesviral subfamily as expected. A closer examination of β-herpesviral sequences revealed a stepwise reduction of conservation levels (Table 2): whereas strains of HCMV showed highly conserved sequences for pUL50 and pUL53 (98.5–99.0 and 98.4–99.5%, respectively), the comparison between HCMVs and primate CMVs (48.0–54.8 and 56.6–63.8%), rodent CMVs (30.5–34.9 and 32.9–37.4%), or the human roseoloviruses HHV-6A, HHV6-B, and HHV-7 (23.3–24.8 and 27.6–31.6%) underlined these low degrees of NEC amino acid identity. Combined, the comparison implies that the functional orthology of herpesviral core NEC proteins may be mostly based on common structural and biochemical properties, but is not mirrored by sequence conservation.
Table 1.
Degree of sequence conservation (% amino acid identities) of four selected herpesviral core NEC proteins
GenbankTM accession numbers used were: VZV Oka, Q4JQV1 and Q4JQU8; HCMV AD169, P16791, and P16794; MCMV Smith, D3XDN8 and D3XDP1; EBV B95-8, P03185 and P0CK47.
| α-Herpesvirus |
β-Herpesviruses |
γ-Herpesvirus |
||
|---|---|---|---|---|
| VZV Orf24 | HCMV pUL50 | MCMV pM50 | EBV BFRF1 | |
| VZV Orf24 | – | 12.6 | 11.6 | 12.6 |
| HCMV pUL50 | 12.6 | – | 34.0 | 15.7 |
| MCMV pM50 | 11.6 | 34.0 | – | 13.9 |
| EBV BFRF1 | 12.6 | 15.7 | 13.9 | – |
| VZV Orf27 | HCMV pUL53 | MCMV pM53 | EBV BFLF2 | |
|---|---|---|---|---|
| VZV Orf27 | – | 12.7 | 16.9 | 18.5 |
| HCMV pUL53 | 12.7 | – | 32.9 | 17.0 |
| MCMV pM53 | 16.9 | 32.9 | – | 15.4 |
| EBV BFLF2 | 18.5 | 17.0 | 15.4 | – |
Table 2.
Degree of sequence conservation (% amino acid identities) within a selection of 13 β-herpesviral core NEC proteins divided into four homology groups
| Human CMVsa | β-Herpesviruses |
Human roseolod | ||
|---|---|---|---|---|
| Primate CMVsb | Rodent CMVsc | |||
| pUL50 | ||||
| Human CMVs | 98.5–99.0 | 48.0–54.8 | 30.5–34.9 | 23.3–24.8 |
| Primate CMVs | 48.0–54.8 | 47.5–84.1 | 28.7–41.9 | 22.8–34.9 |
| Rodent CMVs | 30.5–34.9 | 28.7–41.9 | 28.7–50.6 | 26.1–33.1 |
| Human roseolo | 23.3–24.8 | 22.8–34.9 | 26.1–33.1 | 52.5–94.2 |
| pUL53 | ||||
| Human CMVs | 98.4–99.5 | 56.6–63.8 | 32.9–37.4 | 27.6–31.6 |
| Primate CMVs | 56.6–63.8 | 55.0–89.9 | 34.2–45.3 | 27.6–42.4 |
| Rodent CMVs | 32.9–37.4 | 34.2–45.3 | 40.1–61.1 | 32.7–35.6 |
| Human roseolo | 27.6–31.6 | 27.6–42.4 | 32.7–35.6 | 60.6–97.0 |
a GenBank accession numbers used were: human CMVs: HCMV strains AD169, P16791, and P16794; Merlin, Q6SW81 and F5HFZ4; TB40, A8T7C7 and A8T7D2; Towne, B9VXL9 and B9VXM2.
b GenBank accession numbers used were: primate CMVs: chimpanzee, Q8QS38 and Q8QS35; rhesus monkey, Q2FAN6 and O71122; simian strain Colburn, G8XTV6 and G8XTV9.
High resolution crystal structures of the NECs of EBV and HCMV
The structure of BFRF1 in complex with the BFLF2 hook segment (termed as BFRF1-BFLF2 complex in the following) represents the first experimentally determined structure of a γ-herpesvirus NEC. The structure was solved at 1.6 Å resolution (Rwork = 21.2%, Rfree = 24.2%) (Table 3). X-ray suitable crystals could be obtained only with a BFRF1::BFLF2 fusion construct, in which the N-terminal hook segment of BFLF2 was linked via a GGSGS linker to the C terminus of BFRF1. Although two loop segments of BFRF1 and some residues from the GGSGS linker were not visible in the electron density maps, the hook segment of BFLF2 could be traced continuously (residues 78 to 110) in the model (Fig. 1A).
Table 3.
Crystallographic data collection and refinement statistics
| Structure | EBV BFRF1::BFLF2 | HCMV pUL50::pUL53 |
|---|---|---|
| Data collection | ||
| Beamline | BESSY II 14.2 | BESSY II 14.2 |
| Wavelength (Å) | 0.9182 | 0.9182 |
| Space group | P6122 | P21 |
| Unit cell parameters (Å, °) | a = b = 59.3, c = 265.4, α = β = 90, γ = 120 | a = 37.3, b = 82.6, c = 63.7, α = γ = 90, β = 95.1 |
| Resolution range (Å) | ||
| Spherical | 47.90–1.56 (1.74–1.56)a | 41.29–1.48 (1.52–1.48)a |
| Ellipsoidal | 2.00 Å (b* x c* direction) | NAb |
| 2.00 Å (b* direction) | ||
| 1.50 Å (c* direction) | ||
| Crystal mosaicity (°) | 0.041 | 0.068 |
| Unique reflections | 24,237 | 63,159 |
| Multiplicity | 36.9 | 4.0 |
| Completeness | ||
| Spherical (%) | 59.4 (10.7) | 98.7 (96.3) |
| Ellipsoidal (%) | 92.3 (68.6) | NA |
| Rmeas (%) | 7.4 (233.9) | 9.0 (92.9) |
| Rpim (%) | 1.2 (43.2) | 4.5 (46.1) |
| 〈I/σI〉 | 26.5 (1.9) | 8.9 (1.59) |
| CC1/2 | 1.000 (0.804) | 0.993 (0.592) |
| Wilson B (Å2) | 43.4 (56.1 in direction of b* x c* and b*; 24.0 in direction of c*)c | 33.7 |
| Refinement | ||
| Rwork/Rfree (%) | 21.2/24.2 | 19.2/22.5 |
| Mean B (Å2) | 45.4 | 42.8 |
| No. of TLS groups | 7 | 14 |
| No. of copies in ASU | 1 | 2 |
| No. of atoms (non-H) | ||
| Protein | 1,743 | 3,235 |
| Ligands | 7 | 0 |
| Solvent | 101 | 247 |
| DPI (Å)d | 0.200 | 0.095 |
| RMSD from ideal geometry | ||
| Bond lengths (Å) | 0.007 | 0.005 |
| Bond angles (°) | 0.800 | 0.785 |
| Ramachandran statistics (%) | ||
| Favored | 97.6 | 98.97 |
| Outliers | 0.00 | 0.00 |
| Clashscoree | 1.7 | 4.6 |
| PDB code | 6T3Z | 6T3X |
Figure 1.
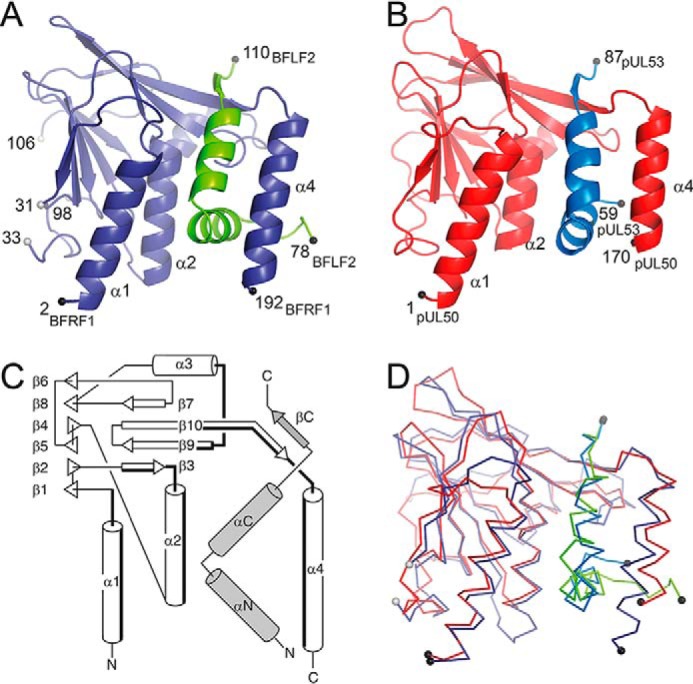
Crystal structures of the EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 complexes. A, ribbon representation of BFRF1 (in prune) in complex with the hook segment of BFLF2 (green). B, ribbon representation of pUL50 (red) in complex with the hook segment of pUL53 (blue). C, common topology plot of the complexes with the secondary structure elements from the hook segment indicated in gray. The secondary structure elements that participate in the binding of the hook segment are lined in bold. D, superposition of the EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 complexes.
An analogous fusion protein construct was also used to obtain a higher resolution crystal structure of HCMV pUL50 in complex with the hook segment of pUL53 (pUL50::pUL53). Here, the hook region of pUL53 was fused via a GGSGSGGS linker to the C terminus of pUL50. The structure of this complex could be solved at 1.5 Å resolution (Rwork = 19.2%, Rfree = 22.5%) thereby considerably extending the resolution of previously available structural data on this complex (PDB entry 5DOB, 2.5 Å resolution, and 5D5N, 2.4 Å resolution) (7, 8) (Fig. 1B). The substantial increase in resolution significantly reduced the coordinate error, and the diffraction precision index (DPI) decreased from 0.27 Å (PDB entry 5D5N) to 0.095 Å in the current structure.
The high resolution structure of the pUL50-pUL53 complex compares well with previously determined structures of the complex obtained by conventional coexpression/copurification strategies (7, 8). The pUL50-pUL53 complex can be superimposed onto PDB entries 5DOB and 5D5N with RMS deviations of 1.5 and 1.4 Å, respectively (180 common Cα positions) (Fig. S1). However, the substantial increase in resolution achieved in the present study not only allowed for a more accurate atom positioning (lower DPI value) but also revealed that the pUL50-pUL53 interface encloses distinct solvent molecules. These participate in hydrogen bond bridges between the two molecules and contribute to the interaction specificity. Many of these solvent molecules went unnoticed in previous analyses.
The γ-herpesviral EBV core NEC shares a common fold with α- and β-herpesviral NECs
When comparing the structure of the EBV BFRF1-BFLF2 complex to all the structures presently available in the Protein Data Bank, it becomes apparent that its overall conformation resembles that of other NECs (Table S1) (15). Thus, BFRF1 displays the same overall fold as the functionally orthologous proteins of α-herpesviruses (HSV-1 and pseudorabies virus, PRV) as well as β-herpesviruses (HCMV and MCMV) (7–10). At the same time, this fold, which was observed first in MCMV pM50, shares only poor structural resemblance with functionally unrelated proteins (16). Hence, a clear drop in the Z-score is observed between the BFRF1 functional orthologs and unrelated proteins (Table S1). When considering sequence identities and structural similarities, i.e. RMSD values, then it appears that the γ-herpesviral protein BFRF1 is evolutionary equally distant to the α-herpesviral orthologs of HSV-1 and PRV as to the β-herpesviral ortholog of HCMV. At the same time, these orthologous viral proteins share a common fold that is unique among all presently annotated proteins.
BFRF1 and its orthologs contain two β-sheets that form a β-sandwich (Fig. 1C; Table S2). With the exception of the pairing between strands β6 and β8, the two β-sheets are entirely antiparallel. In addition, the structure contains 4 helices (α1 to α4) located at one end of the β-sandwich and oriented at almost a right angle to the planes of the β-sheets. Helices α1, α2, and α4 are oriented parallel to each other and line the binding site for the BFLF2 segment (Fig. 1C). The 33-residue long BFLF2 segment consists of 2 helices (αN and αC) and a short β-strand with an overall hook-like appearance. Hence, the BFRF1-BFLF2 interaction can be described as the hook-into-groove interaction closely resembling the respective pUL50-pUL53 interaction described in our earlier study (8).
The structures of the EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 complex can be superimposed with an RMSD of 2.1 Å (189 common Cα atoms) (Fig. 1D). In this global superposition, the coordinates of BFRF1 and pUL50 (RMSD of 2.1 Å) show similar deviations than the coordinates of BFLF2 and pUL53 (2.1 Å). A different picture arises when superimposing the protomers of the complexes separately. Although BFRF1 and pUL50 still deviate by about 2.0 Å (RMSD), the coordinates of the BFLF2 and pUL53 hook can now be superimposed with an RMSD as low as 1.4 Å. This highlights that BFLF2 and pUL53 share a highly similar hook conformation, which appears to be slightly shifted in the BFRF1-BFLF2 complex compared with pUL50-pUL53.
The EBV core NEC displays a number of structural particularities
The hook-into-groove interactions in EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 share an identical overall topology but also differ in a number of atomic details. The interaction surface area amounts to 1590 Å2 in EBV BFRF1-BFLF2 and is approximately 300 Å2 larger than in HCMV pUL50-pUL53 (1300 Å2). In BFLF2 and pUL53, residues extending over the entire hook segment participate in the interface with BFRF1 or pUL50 (Fig. 2, Fig. S2). In contrast, on the BFRF1 or pUL50 side, only 4 segments contribute to the interaction with the hook segments (Figs. 1C and 2; Fig. S3). These segments can be divided into two groups depending on whether they interact with the inwards-pointing face of the hook (in-pointing interface) or the outwards-pointing face (out-pointing interface) (Fig. 2). Residues contributing to the in-pointing interface are from helix α1 (and an adjacent segment), helix α2, or the loop segment joining helix α3 to strand β9 of BFRF1 and pUL50. In contrast, the out-pointing interface mostly consists of residues located on helix α4 of BFRF1 or pUL50 (Figs. 1C and 2).
Figure 2.

Intermolecular interactions in the EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 complexes. A, schematic representation of the molecular interactions in the BFRF1-BFLF2 complex. The in-pointing interactions of the hook segment are displayed in the upper part and the out-pointing in the lower part of the panel. B, schematic representation of the molecular interactions in the pUL50-pUL53 complex. Intermolecular interactions between two residues were identified using a 3.8-Å interatom distance cut-off.
The levels of sequence identity between the protein segments lining the groove interfaces in BFRF1 and pUL50 (11%, Fig. S3) is considerably lower than that between the BFLF2 and pUL53 hook segments (24%, Fig. S2). Hence, the surface of the hook segment appears to be more conserved then that of the groove in which it is interacting. Moreover, the sequence identity between homologous groove residues appears to be lower than the overall sequence identity between BFRF1 and pUL50 (11 versus 15.7%, Table 1). In contrast, the hook sequence seems to be slightly more conserved than the overall sequence between BFLF2 and pUL53 (24 versus 17.0%, Table 1).
As a consequence of the low sequence identities, the physicochemical properties of the BFRF1-BFLF2 interface differ substantially from those of pUL50-pUL53 (Fig. 3, Fig. S4). Thus, six phenylalanine residues cluster in the in-pointing interface of the BFRF1-BFLF2 complex (Fig. 3, A and B), i.e. Phe-19, Phe-60, and Phe-64 of BFRF1 as well as Phe-87, Phe-88, and Phe-101 of BFLF2. In the pUL50-pUL53 complex, this interface is considerably more polar (Fig. 3, E and F). Ph3–88 and Phe-101 from BFLF2 are exchanged against a histidine (His-65) and tyrosine (Tyr-78), respectively, in pUL53. At the same time, BFRF1 residue Phe-60 is also substituted against a tyrosine (Tyr-57) in pUL50. A similar, albeit less drastic, switch in polarity is also observable in the out-pointing interface of these complexes. Thus, Ile-67 creates a large hydrophobic surface patch in pUL53 (Fig. 3, G and H). In BFLF2, this residue is replaced by a glycine (Gly-90) creating a void at this position. Hence, the surface properties are largely dictated by the polarity of the protein backbone atoms (Fig. 3D). Taken together, the interface sequence conservation is generally very low and considerable differences in surface properties can be observed in both the in-pointing and out-pointing interfaces of the two complexes.
Figure 3.

Details of the interfaces in EBV BFRF1-BFLF2 and HCMV pUL50-pUL53. A and B, inwards-pointing and C and D, outwards-pointing interface in the BFRF1-BFLF2 complex. The interfaces are either viewed from BFLF2 (green sticks) onto the surface of BFRF1 (A and C) or from BFRF1 (prune sticks) onto the surface of BFLF2 (B and D). E and F, inwards-pointing, and G and H, outwards-pointing interface in the pUL50-pUL53 complex. The interfaces are either viewed from pUL53 (blue sticks) onto the surface of pUL50 (E and G) or from pUL50 (red sticks) onto the surface of pUL53 (F and H). The surfaces are colored according to the chemical elements that lie below to the surfaces (white, C; red, O; blue, N and yellow, S). Side chains of residues contributing more than 30 Å2 to the interaction interfaces are shown in stick representations. Selected surface areas are labeled in color according to the underlying residues. See Fig. S4 for a depiction of the entire interaction surfaces.
Functional and energetic characterization of NEC hook sequences
To dissect the contribution of individual residues of the NEC hook proteins to the interaction with the groove proteins, experimental and computational alanine-scanning analyses were performed (Table 4). A set of peptides presenting a complete alanine scan of the HCMV hook peptide (see Table S3 for peptide sequences) was synthesized, and their ability to inhibit the pUL50-pUL53 interaction was compared with the WT peptide. For a range of hook positions, in particular Leu-64, Phe-68, Glu-75, Tyr-78, Leu-79, and Met-82, replacement with alanine resulted in a dramatic loss of inhibitory activity (IC50 > 10 μm), designating these positions as essential contributors to the interaction of the peptide with pUL50.
Table 4.
Computational and experimental alanine scan of the HCMV hook peptide
The numbering and residue type at the individual positions refer to HCMV pUL53.
| Position replaced with Ala | ΔΔGa | IC50b ± S.D. |
|---|---|---|
| kcal/mol | μm | |
| WT | 0 | 0.11 ± 0.04 |
| Leu-59 | 1.42 | 0.89 ± 0.12 |
| Thr-60 | −0.06 | 0.14 ± 0.02 |
| Leu-61 | 1.76 | 0.47 ± 0.01 |
| His-62 | 0.24 | 0.13 ± 0.02 |
| Asp-63 | −0.65 | 0.45 ± 0.01 |
| Leu-64 | 1.22 | >10 |
| His-65 | 1.73 | 0.42 ± 0.01 |
| Asp-66 | −0.34 | 0.12 ± 0.001 |
| Ile-67 | 1.54 | 1.30 ± 0.005 |
| Phe-68 | 1.62 | >10 |
| Arg-69 | 0.78 | 0.15 ± 0.01 |
| Glu-70 | −0.07 | 1.11 ± 0.01 |
| His-71 | 0.64 | 1.16 ± 0.03 |
| Pro-72 | −0.03 | 0.324 ± 0.02 |
| Glu-73 | 0.10 | 0.22 ± 0.002 |
| Leu-74 | 1.09 | 1.87 ± 0.07 |
| Glu-75 | 1.80 | >10 |
| Leu-76 | 0.10 | 0.11 ± 0.003 |
| Lys-77 | 0.65 | 0.37 ± 0.05 |
| Tyr-78 | 2.38 | >10 |
| Leu-79 | 2.18 | >10 |
| Asn-80 | 0.01 | 0.12 ± 0.01 |
| Met-81 | 2.98 | 1.31 ± 0.13 |
| Met-82 | 3.13 | >10 |
| Lys-83 | 0.60 | 0.22 ± 0.001 |
| Met-84 | 0.98 | 0.22 ± 0.06 |
| Ile-86 | 1.69 | 0.53 ± 0.16 |
| Thr-87 | 0.01 | 0.16 ± 0.05 |
a Positive ΔΔG values indicate a destabilization of the complex upon replacement of the respective residue by alanine.
b IC50 (inhibition of pUL50-pUL53 interaction) represent means of at least two experiments.
Similar results were obtained using an in silico alanine-scanning analysis that was performed for the same complex. The reduction of inhibitory activity is here reflected by strong changes in binding-free energy upon mutation of the respective residues to alanine. With respect to the accuracy of the prediction, we noted a good agreement with the experimental alanine-scanning results for the central residues of the hook, whereas some discrepancies were observed for some of the terminal residues (e.g. Leu-59, Leu-61, Met-84, and Ile-86). These discrepancies most likely result from the higher conformational flexibility of terminal residues, which allows the respective alanine mutants to bind with a slightly different backbone conformation. This effect is only detected by the experimental setup, but not by the in silico alanine scan, which was performed using a rigid protein backbone.
The peptide scanning and computational investigations represent the first comprehensive analyses that determine the contribution of all individual amino acids from the pUL53 hook segment to the affinity of the pUL50-pUL53 complex formation. The results are largely in agreement with a previously reported incomplete mutational analysis (17). In the latter, residues Leu-61, Leu-64, Phe-68, His-71, Leu-79, and Met-82 were described as important contributors, with residues Ile-67 and Leu-74 only slightly contributing; however, Glu-75, Tyr-78, and all remaining residues were not addressed. Furthermore, whereas previous alanine replacement studies used variants of the recombinant pUL53 protein, here we show for the first time that an isolated hook peptide retains the hook-into-groove binding specificity without requiring the structural context of the intact pUL53 protein.
The overall good agreement between experimental and computational scanning performed for the pUL50-pUL53 interaction prompted us to generate additional in silico alanine-scanning profiles for the core NECs of HCMV, EBV, PRV, and HSV-1, for which three-dimensional structures have been reported. The results revealed similar energy profiles for this selection of NEC hook peptides (Fig. S5), indicating that the energetics in the hook-into-groove interaction comprise a higher level of conservation than their primary sequences. This observation raised the question whether hook and groove proteins are able to interact in a nonautologous fashion, i.e. through cross-viral interaction between hook and groove proteins derived from different herpesviruses.
Biochemical-functional investigation of herpesviral core NECs at the levels of autologous and nonautologous interactions
In the context of this study, the structural and functional investigation of herpesviral core NEC proteins was focused on β- and γ-herpesviral representatives, the full-length ORFs of which were cloned into a plasmid vector for transient transfection (HCMV UL50 and UL53; EBV BFRF1 and BFLF2). The expression patterns demonstrated reliable and stable products for all proteins. For the comparison of autologous interactions with nonautologous, cross-viral combinations, proteins were pairwise coexpressed in HeLa or 293T cells for analysis in confocal colocalization imaging (Fig. 4) or coimmunoprecipitation (co-IP) assays, respectively. Using immunofluorescence-based confocal imaging, HCMV pUL53 and EBV BFLF2 showed a smooth entirely nuclear distribution when singly expressed (Fig. 4A). Upon coexpression with HCMV pUL50 or EBV BFRF1, which both exert a strict nuclear membrane-anchored rim localization when singly expressed, the autologous NEC partners were effectively recruited to a marked nuclear rim colocalization. This colocalization was not observed in nonautologous combinations between HCMV and EBV (Fig. 4B), indicating that NEC interactions do not occur in a cross-viral manner. This finding was confirmed by co-IP analysis using total cellular lysates derived from the transient expression of the full-length proteins. Strongly positive co-IP signals were exclusively obtained for the interaction between autologous core NEC proteins of HCMV and EBV, but not in nonautologous combinations (data not shown). Thus, using two independent methods, the formation of core NECs is detectable for HCMV pUL50-pUL53 and EBV BFRF1-BFLF2, whereas cross-viral interaction between core NEC proteins of the two viruses was not observed.
Figure 4.

Coexpression of autologous pairs of HCMV and EBV core NEC proteins show perfect nuclear rim colocalization. HeLa cells were used for transient transfections, A, either performed in single transfection or B, cotransfection with constructs coding for HA-tagged pUL50 and BFRF1 or FLAG-tagged pUL53 and BFLF2. Two days post transfection, cells were fixed and used for immunostaining with tag-specific antibodies analyzed by confocal imaging. 4′,6-Diamidino-2-phenylindole (DAPI) counterstaining indicated the morphology of nuclei of the respective cells. Note the smooth entire nuclear distribution of pUL53 or BFLF2 upon single expression (d–f, k–m) or nonautologous coexpression (i–m, n–q), which stood in contrast to the nuclear rim recruitment through the formation of autologous NEC protein pairs (a–d, e–h).
Analysis of a putative formation of chimeric core NECs using HCMV::EBV domain swap constructs
To investigate in greater detail the two core NEC protein pairs of HCMV and EBV, chimeric domain swap constructs were generated. These contained heterologous exchanges within the N-terminal regions of the HCMV pUL53 and EBV BFLF2 proteins (5, 8). Specific coding segments of the extreme N terminus or the more C terminally located segments of the hook structures were fused to the main globular domains of these proteins in the depicted chimeric arrangements (Fig. 5A, constructs termed a–h). In addition to that, a replacement of the main globular domains by the coding sequence of green fluorescent protein (GFP) was performed (constructs termed i–q) to place the NEC-interactive hook element into an inert, nonreactive environment and, in addition, to facilitate the detection of intracellular localization of the expressed constructs. Using these two sets of HCMV::EBV domain swap constructs, co-IP analysis was performed to investigate their potency to interact with their NEC groove counterparts HCMV pUL50 (Fig. 5, C and D) or EBV BFRF1 (Fig. 5, E and F).
Figure 5.

Co-IP–based interaction analysis of domain swap constructs between HCMV pUL53 and EBV BFLF2. A, schematic representation of the domain swap constructs generated between pUL53 (blue) and BFLF2 (green); numbers represent the amino acid positions. B, summarized data of the co-IP analysis with domain swap pUL53::BFLF2 proteins in their interaction with HCMV pUL50 or EBV BFRF1, respectively. C-F, 293T cells were transiently transfected with the expression plasmids coding for domain swap proteins as: C and E, FLAG-tagged constructs containing the main globular domains; D and F, GFP fusions replacing the globular domains. At three days post-transfection, cells were lysed and HA-tagged proteins were immunoprecipitated using anti-HA antibodies as indicated. C and D, test for interaction with pUL50-HA. E and F, test for interaction with HA-BFRF1. Co-IP procedures were performed as described previously (32). In each panel, at least three independent experiments have been performed; one representative set of data are shown.
The co-IP experiments conferred detailed insight into the molecular determinants of core NEC interaction. First, the nonchimeric, truncated versions of pUL53 homologs showed a WT-like strength of co-IP interaction with their autologous counterparts (Fig. 5C, lane 3, and E, lane 7; B, constructs a and e). Second, the transfer of the hook structure (combined N-terminal and C-terminal fragments) in fusion to the chimeric main globular domain, i.e. domain swap constructs b and f, determined the interaction with pUL50 or BFRF1, respectively (Fig. 5, C, lane 8; E, lane 4; and B, constructs b and f). Third, constructs b, c, and d retained some property of interaction with pUL50 even in the absence of parts of the autologous hook structural fragment (Fig. 5C, lanes 4–6). Although these faint signals were barely above background, they may suggest that either the main globular domain of pUL53 has a secondary binding interface to its groove counterpart in pUL50, or alternatively, fragmental exchange still allows some low-affinity pUL50 interaction.
This question was further addressed by the use of constructs i–q, in which the main globular domain was replaced by GFP. In this case, no such pUL50-specific co-IP activity of any domain swap construct could be observed (Fig. 5D, lanes 7–13). This finding substantiated our postulate above, in that a secondary globular domain of the pUL50-binding interface apparently contributes, at least to a limited extent, to the pUL53 properties of NEC interaction. Concerning the EBV counterpart BFLF2, interaction with its NEC partner BFRF1 was exclusively seen when the autologous BFLF2 hook element was contained in the constructs (Fig. 5F, lanes 7 and 10). Interestingly, the presence of the amino acid region N-terminal to the hook element, i.e. 1–72 in BFLF2, was not required for interaction, but could be replaced by pUL53(1–49) without impairing the binding activity (Fig. 5, F, lane 10; and B, construct n). Notably, these extreme N-terminal segments of both BFLF2 and pUL53 were found important for the stability of chimeric protein constructs, because another series of N terminally truncated domain swap versions (i.e. lacking amino acids 1–49 of pUL53 or 1–72 of BFLF2, respectively) could not be stably expressed upon transient transfection (data not shown). Another interesting finding was that in these co-IP analyses, the full-length version of BFLF2 showed some faint signals of nonautologous interaction with pUL50 (Fig. 5D, lane 2), whereas the reciprocal combination, full-length pUL53 and BFRF1, did not produce any interaction signal (Fig. 5F, lane 2). This may be explained by some degree of promiscuity in core NEC formation of the EBV hook-type protein BFLF2, which was not exerted by the HCMV counterpart pUL53. An analysis on the single-cell level, by confocal imaging analysis of nuclear rim formation, however, could not confirm a reliable and clear-cut signal of nonautologous BFLF2-pUL50 interaction, thus arguing for a low-affinity association, only reflected by a limited co-IP reactivity. This confocal imaging analysis was also performed with the series of GFP-fused (Fig. S6) and FLAG-tagged (data not shown) domain swap constructs, both yielding results that were entirely compatible with the co-IP findings. Positive signals of nuclear rim recruitment were obtained for construct i in coexpression with pUL50 (Fig. S6B, panels 13–16) and constructs k and n in coexpression with BFRF1 (Fig. S6C, panels 17–20, 29–32). This pattern of colocalization confirmed the abovementioned pattern of co-IP–determined interaction. In essence, these findings provide evidence that the hook-specific sequences of pUL53(50–87) and BFLF2(73–110) represent the major determinants of NEC hook-into-groove interaction, which cannot be replaced by nonautologous domain swap fragments.
Interaction and cross-viral reactivity of HCMV and EBV NEC proteins and derived peptides
The hook-into-groove protein interactions of the HCMV and EBV NECs were further characterized using soluble recombinant groove proteins (pUL50 and BFRF1), in conjunction with synthetic peptides presenting the hook sequences of pUL53 and BFLF2 (Fig. 1, A and B, see Table S3 for peptide sequences). For both autologous peptide-protein interactions (pUL50 with pUL53 hook peptide and BFRF1 with BFLF2 hook peptide, respectively), dose-dependent binding was shown. At the same time, no cross-viral interaction with the nonautologous proteins was detected (Fig. 6, A and B).
Figure 6.

Interaction of soluble herpesviral NEC proteins and peptides. A, direct binding of HCMV pUL50 to the HCMV pUL53 and EBV BFLF2 hook peptides. B, direct binding of EBV BFRF1 to the EBV BFLF2 and HCMV pUL53 hook peptides. C, inhibition of the HCMV pUL50-pUL53 interaction by the HCMV pUL53 and EBV BFLF2 hook peptides. All data points present means of duplicates. All experiments were performed using the same batches of proteins and peptides.
These results could be confirmed using surface plasmon resonance (SPR) spectroscopy, in which KD values in the low and submicromolar range were determined for the autologous hook peptide-groove protein interactions. Interestingly, the pUL50-pUL53 hook peptide interaction (Fig. 7A) appears to be considerably stronger (KD = 228 nm) than the respective EBV-specific interaction (BFRF1-BFLF2 hook peptide) (Fig. 7B, KD = 6.2 μm). It should be noted, however, that this difference does not necessarily indicate a stronger affinity of the HCMV NEC protein interaction, as compared with the respective EBV NEC protein interaction. It may also point to a contribution of additional regions of BFLF2, which are not presented in the BFLF2 hook peptide used here, to the BFRF1-BFLF2 interaction.
Figure 7.

Surface plasmon resonance spectroscopic analysis of HCMV and EBV NEC protein-peptide interactions. A, binding of pUL50 to the pUL53 hook peptide. B, binding of BFRF1 to the BFLF2 hook peptide. C, binding of pUL50 to the BFLF2 hook peptide. D, binding of BFRF1 to the pUL53 hook peptide. For each interaction, at least two independent experiments have been performed; one representative set of sensograms is shown. All experiments were performed using the same batches of proteins and peptides.
As in the ELISA binding assay, no cross-viral interaction was detected in SPR spectroscopy for the nonautologous pairs, i.e. neither binding of pUL50 to the BFLF2 hook peptide (Fig. 7C), nor BFRF1 to the pUL53 hook peptide (Fig. 7D), indicating a high virus selectivity in the NEC protein interactions. This notion was supported by the difference in ability of the pUL53 and BFLF2 hook peptides to interfere with the pUL50-pUL53 interaction (Fig. 6C). The inhibitory activity of the pUL53 hook peptide (IC50 = 110 nm) was approximately 20-fold stronger than that of the BFLF2 hook peptide (IC50 = 2.6 μm). Although emphasizing once again the subfamily specificity of the hook-into-groove interaction, these experiments nevertheless indicate the general feasibility of using a nonautologous hook peptide (BFLF2 hook peptide) to inhibit an autologous NEC interaction (pUL50-pUL53 complex formation), although much higher peptide concentrations are needed.
Discussion
In the present study, significant advances in understanding the herpesviral core NEC complex have been accomplished using NEC groove proteins fused to NEC hook segments. This approach yielded higher-resolution NEC crystal structures and improved the precision of structural information on herpesviral core NECs. Moreover, this strategy enabled for the first time an X-ray structure determination of a γ-herpesviral core NEC, i.e. of the EBV BFRF1-BFLF2 complex. This structure is now available for a comparative analysis with the novel β-herpesviral structures reported here (HCMV), as well as with previously published NEC structures of α- and β-herpesviruses (i.e. HSV-1, PRV, HCMV, and MCMV (7–10, 16)).
Our structural investigations focused on the interaction of the NEC groove protein with the hook segment of its autologous NEC counterpart. As the hook segment contributes ∼80% of the interaction surface in the pUL50-pUL53 complex (7, 8), as well as of NEC complex pUL50-pUL53 homologs (9, 10), we propose that the structures reported here, although representing truncated complexes, are appropriate tools to study the structural basis of core NEC protein interactions.
With regard to the biochemical-functional properties of the HCMV- and EBV-specific core NECs, the study provides a characterization on three levels, involving independent approaches, i.e. a co-IP and colocalization-based protein biochemical analysis of virological aspects, including HCMV::EBV hook domain swap constructs, a bioinformatic investigation revealing the energetic landscape of core NEC interactions, as well as a quantitative interaction analysis using synthetic peptides. The data re-emphasize our earlier statements in the following points: (i) NEC protein primary sequences are poorly conserved, (ii) X-ray crystal structures of core NEC proteins generally show a high degree of conservation, but in structural details also individually unique features, (iii) biochemical NEC binding properties, intranuclear rim recruitment, and inhibition by synthetic hook peptides points to virus-specificity (in particular a lack of cross-viral core NEC interaction between herpesviruses of different subfamilies), and (iv) inhibition of NEC protein interactions could evolve into a potential novel antiherpesviral therapeutic strategy.
The co-IP data presented here support our earlier notion that the hook and groove segments of all herpesviral core NECs so far analyzed strictly determine their heterodimeric interaction. Our experimental strategy of domain swap analysis clearly underlined that the freedom to exchange individual elements of the hook segment in a nonautologous manner is narrow, at least between herpesviruses belonging to different subfamilies. This may be unexpected considering the high degree of structural conservation and orthology, which includes distantly related members of α-, β-, and γ-herpesviruses. However, although the hook-into-groove pattern of core NEC interaction is generally conserved, the present study emphasized the existing determinants of subfamily specificity.
Our in vitro characterization of the hook-into-groove interaction further underlines that the hook segment represents the major binding determinant in the core NEC complex. Using synthetic peptides that present the hook regions of the HCMV and EBV hook proteins, we could show that the interaction with the respective groove proteins is independent of the groove protein globular domains. This is illustrated by the affinity of the pUL53 hook peptide for pUL50 (KD = 228 nm), which is essentially identical to the previously reported affinity of the entire pUL53 protein (KD = 290 nm (17)). On the other hand, the co-IP experiments indicate that an additional region in the pUL53 globular domain can contribute to complex formation with pUL50 even in the absence of the autologous hook segment. This is in line with the previously published structure of the pUL50-pUL53 complex, which showed that regions outside the pUL53 hook segment are also involved in the interaction with pUL50 (7, 8, 18). Considering the relatively low affinity of the BFLF2 hook peptide for BFRF1 (KD = 6.2 μm), it may be possible that regions other than the hook segment of BFLF2 also contribute to the interaction of the protein with BFRF1. This, however, remains to be demonstrated in a structure of BFRF1 in complex with the entire globular domain of BFLF2, which is not available yet.
The significance ascribed to the hook segment in the complexes studied here highlights the hook-into-groove interaction as a target for potential NEC inhibitors. The development of such inhibitors, however, will remain challenging. In addition to the fairly large hook-into-groove interface (1590 Å2 in EBV, 1300 Å2 in HCMV), the energetics of the interactions appear to be rather complex. Hot spot residues are distributed over the entire hook segment (19), as evidenced by our alanine-scanning analysis, revealing that residues that contribute most to the pUL50-pUL53 interaction are located in both the N-terminal (αN) and C-terminal (αC) hook helices. Furthermore, these hot spot residues are involved in both in-pointing and out-pointing interfaces. The same also holds true for the BFRF1-BFLF2 interaction, as predicted by the computational alanine-scanning analysis.
In addition to virus-specific properties, however, the study also highlights a number of shared features in the analyzed core NECs of herpesviruses from different subfamilies, which may serve as a starting point for structure-based inhibitor development. Notably, the structural features of the hook-into-groove interaction appear to be well-conserved across herpesviruses, despite the low degree of sequence identity. Furthermore, hook residues that are crucial for the interaction with the groove proteins are predicted to be located in similar regions of the hook proteins, indicating a potential shared target of herpesviruses for antiviral drugs. Finally, synthetic hook peptides covering the HCMV pUL53 and EBV BFLF2 hook sequences may serve as a starting point in the search for small molecule NEC inhibitors. In conclusion, the results presented here provide further important insights into the molecular mechanism of herpesviral core NEC interactions. These will stimulate the exploration of NEC proteins as antiherpesviral targets.
Experimental procedures
Protein production and purification
Coding sequences for pUL50, pUL53, BFRF1, and the fusion proteins pUL50::pUL53 and BFRF1::BFLF2 were cloned into the pET28b vector (Merck Millipore). All constructs contain a N-terminal hexa-histidine extension and a thrombin-cleavage site (MGSSHHHHHHSSGLVPRGSH). In the case of pUL50, residues 1 to 175 of the open reading frame (ORF) of UL50 (Uniprot entry P16791 (20)) were incorporated into the vector; in the case of pUL53, residues 50 to 292 of ORF-UL53 (Uniprot entry P16794) were incorporated. In the case of the pUL50::pUL53 fusion protein, residues 1 to 171 of UL50 were fused via a GGSGSGGS linker to residues 59 to 87 of ORF-UL53. The pUL50::pUL53 fusion protein, pUL50 and pUL53 contain in addition a 3-residue-long extension (MAS) at their N terminus. In the case of the BFRF1::BFLF2 fusion protein, residues 1 to 192 of ORF-BFRF1 (Uniprot entry V5KTU9) were fused via a GGSGS linker to residues 78 to 110 of ORF-BFLF2 (Uniprot entry K9UT32). First, a fragment covering residues 78 to 328 of BFLF2 was fused to BFRF1. Subsequently, a STOP codon was inserted after BFLF2 residue 110, using site-directed mutagenesis, to yield the final construct. In the case of BFRF1 (1 to 192) by itself, a STOP codon was inserted after BFRF1 residue 192 in the above fusion construct. All primers are listed in Table S4.
Plasmids were transformed into Escherichia coli BL21(DE3), using TB medium supplemented with 50 μg/ml of kanamycin for selection and growth at 20 °C. Protein production was induced with 0.25–1.0 mm isopropyl 1-thio-β-d-galactopyranoside. After harvesting and disruption of cells, using a sonicator or high pressure homogenizer, recombinant proteins were purified using a HisTrap affinity chromatography step, followed by thrombin cleavage. Thrombin cleavage was omitted for pUL50 and BFRF1. As a result of thrombin cleavage, the protein products started with a prepended N-terminal extension with the sequence GSHMAS (pUL53, pUL50::pUL53) or GSH (BFRF1::BFLF2). The proteins were further purified using gel filtration chromatography and, depending on the presence of contaminants, with an additional ion exchange chromatography step.
Unless stated otherwise, proteins from multiple batches were used in the different experiments. No variations in protein properties were observed between batches. At the same time, this has not been studied in detail here.
Crystallization and crystal structure determinations
The truncated fusion proteins HCMV pUL50::pUL53 and EBV BFRF1::BFLF2 were screened for successful crystallization with the sitting drop technique. The proteins were dissolved in a buffer consisting of 50 mm Tris-HCl, 150 mm NaCl, pH 7.5, and concentrated to values between 10 and 15 mg/ml. Diffraction quality crystals of pUL50::pUL53 were obtained at 4 °C with 20% PEG 4000, 10% propanol, 100 mm HEPES, pH 7.5, as a reservoir solution. Diffraction quality crystals of BFRF1::BFLF2 were obtained at 4 °C with 0.2 m sodium malonate, pH 4.5, 20% PEG 3350 as a reservoir solution. Crystals of HCMV pUL50::pUL53 were briefly transferred into Paratone-N oil (Hampton Research) before flash-freezing and prior to data collection at 100 K. In the case of EBV BFRF1::BFLF2, the cryoprotectant solution consisted of reservoir solution supplemented with 20% glycerol.
High resolution diffraction data sets from crystals of the HCMV and EBV fusion proteins were collected at the MX beamlines of the BESSY synchrotron Berlin (21). Data were processed with program XDS (22). Initial phases were obtained with the molecular replacement technique with program PHENIX_MRAGE using the previously determined structure of the pUL50-pUL53 complex as a search model (PDB entry code 5D5N) (8, 23, 24). The structures were completed using either the PHENIX program AUTOBUILD or manually corrected using the program COOT (23, 25). The structures were refined to convergence by the use of PHENIX.REFINE software (23). The crystals of BFRF1::BFLF2 diffracted highly anisotropically, and the web server STARANISO (36) was used to generate an ellipsoidally truncated dataset for refinement (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi).5 Although the dataset extended to 1.5 Å resolution in direction of the c*-axis, the diffraction is limited to 2.0 Å in the a*-b* plane. Paired refinement was used to evaluate the information content of high resolution shells (26) (Table S5). Crystallographic data collection and refinement statistics for pUL50::pUL53 and BFRF1::BFLF2 are summarized in Table 3.
The structures were superimposed and compared either using program LSQKAB from the CCP4 program suite or the DALI webserver (15, 27). All illustrations of 3D structures were produced with PyMOL (28) and residues resulting from cloning artifacts (N-terminal extension, artificial linker residues) were omitted. The DPI was calculated using a web-based server (29).
Antibodies
Antibodies used were: mAb-HA (Clone 7, H9658, Sigma), pAb-HA (Signalway Eurogentec), mAb-HA-HRP (12013819001, Roche), mAb-FLAG (F1804, Sigma), pAb-FLAG (F7425, Sigma-Aldrich), mAb-FLAG-HRP (A8592, Sigma-Aldrich), mAb-GFP (11814460001, Roche); anti-mouse Alexa 555 (A-21422, ThermoFisher Scientific), and anti-rabbit Alexa 488 (A-11008, ThermoFisher Scientific).
Plasmids and transfection
Transient transfection in 293T cells was performed using polyethylenimine-DNA complexes (Sigma-Aldrich) as described previously (30). HeLa cells were transfected by the use of Lipofectamine 2000 (ThermoFisher Scientific) according to the manufacturer's instructions. The following plasmids were used for transfection: pDsRed1-N1 (BD Biosciences, Clontech), pcDNA-UL50-HA, pcDNA-UL53-Flag (31), and pcDNA-HA-BFRF1, pcDNA-Flag-BFLF2 (kindly provided by Chung-Pei Lee; National Taipei University of Nursing and Health Sciences, Taiwan). Expression plasmids pcDNA-BFRF1-HA and pcDNA-BFLF2-Flag were generated by standard PCR amplification of the respective template DNA isolated from EBV-infected lymphocytes (strain B95-8). Domain swap constructs were generated using pcDNA-UL53-Flag and pcDNA-BFLF2-Flag as a template. Oligonucleotide primers used for PCR were purchased from Biomers; their sequences are given in Table S4. After cleavage with the corresponding restriction enzymes, PCR products were inserted into the eukaryotic expression vector pcDNA3.1(+) (Life Technologies).
Cell culture
Human embryonic kidney epithelial cells HEK 293T and cervix carcinoma epithelial cells HeLa (ATCC) were cultivated at 37 °C, 5% CO2 and 80% humidity using Dulbecco's modified Eagle's medium (11960044, ThermoFisher Scientific). Cell culture medium was supplemented with 1× GlutaMAXTM (35050038, ThermoFisher Scientific), 10 μg/ml of gentamicin, and 10% fetal bovine serum (FBS, F7524, Sigma-Aldrich).
Indirect immunofluorescence assay and confocal laser-scanning microscopy
HeLa were grown on coverslips, 2 days after transfection cells were fixed with 4% paraformaldehyde solution (10 min, room temperature) and permeabilized by incubation with 0.2% Triton X-100 solution (15 min, 4 °C). Indirect immunofluorescence staining was performed by incubation with primary antibodies as indicated for 60 min at 37 °C, followed by incubation with dye-conjugated secondary antibodies (anti-rabbit Alexa 488 and anti-mouse Alexa 555) for 30 min at 37 °C. Cells were mounted with Vectashield Mounting Medium containing 4′,6-diamidino-2-phenylindole and analyzed using a TCS SP5 confocal laser-scanning microscope (Leica Microsystems). Images were processed using the LAS AF software (Leica Microsystems) and Photoshop CS5.
Co-IP
For co-IP analysis, 293T cells were seeded into 10-cm dishes with a density of 5 × 106 cells and used for transient transfection with expression plasmids. Two to three days post-transfection, co-IP was performed as described previously (32). Antibody-coupled Dynabeads (25 μg/ml, 10002D, ThermoFisher Scientific) were used to obtain specific immunoprecipitates and co-IP samples were further analyzed by Western blotting.
Bioinformatic protein analysis
Energetic analyses were done using the PSSM algorithm of the program Fold-X (version 5) (33). The analysis was performed for the following complexes from HCMV (PDB entry 6T3X, present study), EBV (6T3Z, present study), HSV-1 (4ZXS (9)), and PRV (4Z3U (9)).
Peptide synthesis
HCMV pUL53 and EBV BFLF2 hook peptides (see Table S3), as well as peptides presenting the alanine scan of the HCMV pUL53 hook peptide, were synthesized as C-terminal amides by Fmoc/tBu-based solid-phase synthesis, as previously described (34). Biotin was introduced by coupling of Fmoc-Lys(biotin). Crude peptides were purified by preparative HPLC, and purified peptides were then characterized by analytical HPLC with online electrospray ionization MS detection (LC-MS). Stock solutions of purified peptides were prepared at 2.5 mm in 50% acetonitrile/water.
Direct-binding assay
High binding Immulon microtiter plates were coated overnight at 4 °C with NeutrAvidin (ThermoFisher; 4 μg/ml in 0.1 m sodium carbonate buffer, pH 9.5). Unspecific binding was blocked with 1% BSA in 0.1 m phosphate buffer, pH 7.2. Plates were then incubated with biotinylated peptide (2.5 μm) for 3 h, followed by incubation with His-tagged groove protein solution (pUL50 and BFRF1, respectively) at serial dilutions, starting from 2.5 and 10 μg/ml, respectively, for 1 h. Bound protein was detected using anti-His-HRP conjugate (Sigma; 1:10,000). All proteins and antibodies were in 0.1 m phosphate buffer, pH 7.2, containing 0.1% BSA and 0.01% Tween 20. Plates were washed four times with 0.01% Tween 20 in 0.1 m phosphate buffer, pH 7.2, after each incubation step. Plates were developed with OPD (1 mg/ml) in the presence of 0.03% H2O2 for approximately 10 min in the dark. After the reaction was stopped with 2 m H2SO4, absorbance was read at 492 nm.
Competitive binding assay
High binding Immulon microtiter plates were coated with pUL53 (2 μg/ml) as described above. After blocking with BSA, plates were incubated with peptide solutions in serial dilutions, starting at 10 μm, followed by His-tagged pUL50 (0.16 μg/ml). His-tagged pUL50 was detected and plates developed as described above for the direct binding assay. IC50 values were determined using the program GraphPad. Inhibition was calculated according to the following formula,
| (Eq. 1) |
in which “100%” means a sample without peptide, “blank1” means a sample without pUL53, and “blank2” means a sample without pUL53 and without peptide.
SPR spectroscopy
All SPR measurements were performed using a Biacore X100 instrument in conjunction with the Biotin CAPture Kit (both GE Life Science). Biotinylated peptides were immobilized on the chip at 25 nm according to the manufacturer's instructions. The analytes (pUL50 and BFRF1) were added at 2-fold serial dilutions, starting at 1 (pUL50) and 8 μm (BFRF1), respectively.
Author contributions
Y. A. M., H. S., J. E., and M. M. conceptualization; Y. A. M., S. Häge, S. A., T. H., S. W., S. D., S. A. W., M. C., S. Holst, J. L., E. S., H. S., J. E., and M. M. formal analysis; Y. A. M., S. Häge, S. A., T. H., S. W., S. D., K. H., S. A. W., C. E.-S., M. C., S. Holst, J. L., E. S., H. S., J. E., and M. M. investigation; Y. A. M., S. Häge, H. S., J. E., and M. M. writing-original draft; Y. A. M., S. Häge, H. S., J. E., and M. M. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to Jens Milbradt for long-term cooperation in the NEC project (FAU, Erlangen), Chung-Pei Lee (National Taipei University of Nursing and Health Sciences, Taipei, Taiwan) for providing helpful reagents, Christina Wangen and Regina Müller for excellent technical assistance (FAU, Erlangen), Andrea Decker and Johannes Schweininger (FAU, Erlangen) for assistance with recombinant protein production, and Manfred Weiss and the MX team from HZB Berlin for help with diffraction data collection at BESSY synchrotron. The coordinates of the EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 complexes have been deposited with the Protein Data Bank with accession codes 6T3Z and 6T3X, respectively.
This work was supported by Deutsche Forschungsgemeinschaft Grants MA 1289/8-1, EI 423/4-1, and MU 1477/10-1 (to Y. A. M., J. E., and M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6 and Tables S1–S5.
The atomic coordinates and structure factors (codes 6T3X and 6T3Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HSV-1
- herpes simplex virus type 1
- HCMV
- human cytomegalovirus
- EBV
- Epstein-Barr virus
- NE
- nuclear envelope
- NEC
- nuclear egress complex
- DPI
- diffraction precision index
- PRV
- pseudorabies virus
- RMSD
- root mean square deviation
- co-IP
- co-immunoprecipitation
- SPR
- surface plasmon resonance
- HA
- hemagglutinin
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- HRP
- horseradish peroxidase.
References
- 1. Mocarski E. S., Shenk T., Griffiths P. D., and Pass R. F. (2013) Cytomegaloviruses. in Fields Virology (Knipe D. M., and Howley P. M., eds) 6th Ed., pp. 1960–2014, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Griffiths P., and Reeves M. (2017) Cytomegalovirus. in Clinical Virology (Richman D. D., Whitley R. J., and Hayden F. G., eds) 4th Ed., pp. 481–510, ASM Press, Washington, D. C. [Google Scholar]
- 3. Tsutsui Y. (2009) Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. (Kyoto) 49, 47–55 [DOI] [PubMed] [Google Scholar]
- 4. Jha H. C., Banerjee S., and Robertson E. S. (2016) The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5, e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milbradt J., Auerochs S., Sevvana M., Muller Y. A., Sticht H., and Marschall M. (2012) Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J. Biol. Chem. 287, 24004–24016 10.1074/jbc.M111.331207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milbradt J., Webel R., Auerochs S., Sticht H., and Marschall M. (2010) Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 285, 13979–13989 10.1074/jbc.M109.063628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lye M. F., Sharma M., El Omari K., Filman D. J., Schuermann J. P., Hogle J. M., and Coen D. M. (2015) Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 34, 2937–2952 10.15252/embj.201592651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walzer S. A., Egerer-Sieber C., Sticht H., Sevvana M., Hohl K., Milbradt J., Muller Y. A., and Marschall M. (2015) Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J. Biol. Chem. 290, 27452–27458 10.1074/jbc.C115.686527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bigalke J. M., and Heldwein E. E. (2015) Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 34, 2921–2936 10.15252/embj.201592359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeev-Ben-Mordehai T., Weberruß M., Lorenz M., Cheleski J., Hellberg T., Whittle C., El Omari K., Vasishtan D., Dent K. C., Harlos K., Franzke K., Hagen C., Klupp B. G., Antonin W., Mettenleiter T. C., and Grünewald K. (2015) Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep. 13, 2645–2652 10.1016/j.celrep.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailer S. M. (2017) Venture from the interior-herpesvirus pUL31 escorts capsids from nucleoplasmic replication compartments to sites of primary envelopment at the inner nuclear membrane. Cells 6, e46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bigalke J. M., and Heldwein E. E. (2015) The great (nuclear) escape: new insights into the role of the nuclear egress complex of herpesviruses. J. Virol. 89, 9150–9153 10.1128/JVI.02530-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hellberg T., Paßvogel L., Schulz K. S., Klupp B. G., and Mettenleiter T. C. (2016) Nuclear egress of herpesviruses: the prototypic vesicular nucleocytoplasmic transport. Adv. Virus Res. 94, 81–140 10.1016/bs.aivir.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 14. Marschall M., Muller Y. A., Diewald B., Sticht H., and Milbradt J. (2017) The human cytomegalovirus nuclear egress complex unites multiple functions: recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 27, 10.1002/rmv.1934 [DOI] [PubMed] [Google Scholar]
- 15. Holm L., and Laakso L. M. (2016) Dali server update. Nucleic Acids Res. 44, W351–W355 10.1093/nar/gkw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leigh K. E., Sharma M., Mansueto M. S., Boeszoermenyi A., Filman D. J., Hogle J. M., Wagner G., Coen D. M., and Arthanari H. (2015) Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. U.S.A. 112, 9010–9015 10.1073/pnas.1511140112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sam M. D., Evans B. T., Coen D. M., and Hogle J. M. (2009) Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 83, 2996–3006 10.1128/JVI.02441-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diewald B., Socher E., Soldner C. A., and Sticht H. (2018) Conformational dynamics of herpesviral NEC proteins in different oligomerization states. Int. J. Mol. Sci. 19, 2908 10.3390/ijms19102908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cunningham B. C., and Wells J. A. (1993) Comparison of a structural and a functional epitope. J. Mol. Biol. 234, 554–563 10.1006/jmbi.1993.1611 [DOI] [PubMed] [Google Scholar]
- 20. The UniProt Consortium. (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 10.1093/nar/gkw1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., and Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 10.1107/S0909049512006395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 10.1107/S0021889893005588 [DOI] [Google Scholar]
- 23. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rose P. W., Prlić A., Altunkaya A., Bi C., Bradley A. R., Christie C. H., Costanzo L. D., Duarte J. M., Dutta S., Feng Z., Green R. K., Goodsell D. S., Hudson B., Kalro T., Lowe R., et al. (2017) The RCSB Protein Data Bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 45, D271–D281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 10.1126/science.1218231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeLano W. (2003) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 29. Kumar K. S. D., Gurusaran M., Satheesh S. N., Radha P., Pavithra S., Thulaa Tharshan K. P. S., Helliwell J. R., and Sekar K. (2015) Online_DPI: a web server to calculate the diffraction precision index for a protein structure. J. Appl. Crystallogr. 48, 939–942 10.1107/S1600576715006287 [DOI] [Google Scholar]
- 30. Schregel V., Auerochs S., Jochmann R., Maurer K., Stamminger T., and Marschall M. (2007) Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Gen. Virol. 88, 395–404 10.1099/vir.0.82393-0 [DOI] [PubMed] [Google Scholar]
- 31. Milbradt J., Auerochs S., and Marschall M. (2007) Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 88, 2642–2650 10.1099/vir.0.82924-0 [DOI] [PubMed] [Google Scholar]
- 32. Sonntag E., Hamilton S. T., Bahsi H., Wagner S., Jonjic S., Rawlinson W. D., Marschall M., and Milbradt J. (2016) Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 97, 1676–1685 10.1099/jgv.0.000495 [DOI] [PubMed] [Google Scholar]
- 33. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., and Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groß A., Röodel K., Kneidl B., Donhauser N., Mössl M., Lump E., Münch J., Schmidt B., and Eichler J. (2015) Enhancement and induction of HIV-1 infection through an assembled peptide derived from the CD4 binding site of gp120. ChemBioChem 16, 446–454 10.1002/cbic.201402545 [DOI] [PubMed] [Google Scholar]
- 35. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., and Richardson D. C. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tickle I. J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2018) STARANISO Global Phasing Ltd., Cambridge, United Kingdom [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.