

Abstract
Globular protein sequences encode not only functional structures (the native state) but also protein foldability, i.e. a conformational search that is both efficient and robustly minimizes misfolding. Studies of mutations associated with toxic misfolding have yielded insights into molecular determinants of protein foldability. Of particular interest are residues that are conserved yet dispensable in the native state. Here, we exploited the mutant proinsulin syndrome (a major cause of permanent neonatal-onset diabetes mellitus) to investigate whether toxic misfolding poses an evolutionary constraint. Our experiments focused on an invariant aromatic motif (PheB24–PheB25–TyrB26) with complementary roles in native self-assembly and receptor binding. A novel class of mutations provided evidence that insulin can bind to the insulin receptor (IR) in two different modes, distinguished by a “register shift” in this motif, as visualized by molecular dynamics (MD) simulations. Register-shift variants are active but defective in cellular foldability and exquisitely susceptible to fibrillation in vitro. Indeed, expression of the corresponding proinsulin variant induced endoplasmic reticulum stress, a general feature of the mutant proinsulin syndrome. Although not present among vertebrate insulin and insulin-like sequences, a prototypical variant ([GlyB24]insulin) was as potent as WT insulin in a rat model of diabetes. Although in MD simulations the shifted register of receptor engagement is compatible with the structure and allosteric reorganization of the IR-signaling complex, our results suggest that this binding mode is associated with toxic misfolding and so is disallowed in evolution. The implicit threat of proteotoxicity limits sequence variation among vertebrate insulins and insulin-like growth factors.
Keywords: protein folding, protein structure, molecular evolution, hormone, peptide biosynthesis, protein biosynthesis
Introduction
The structure and function of globular proteins are determined by their sequences. Yet the informational content of such sequences must also contain determinants of folding efficiency, providing safeguards against toxic misfolding (1). Such safeguards may be inapparent once the native state is reached (2). In this study, we demonstrate that insulin can in principle exhibit two modes of receptor binding, but only one protects from toxic misfolding. Evidence is provided that the excluded mode would enhance the risk of pancreatic β-cell dysfunction and diabetes mellitus (DM)4 due to endoplasmic reticulum (ER) stress (3).
Physical properties of modern proteins are unrepresentative of polypeptides as a class of heteropolymers (4). Whereas the ground state of a polypeptide may be amyloid (5), evolutionary winnowing has, in almost all cases, established large kinetic barriers protecting the nascent chain from aggregation-coupled misfolding (6, 7). This principle is honored in the breach by human proteotoxic diseases, including monogenic amyloidogenic syndromes (8) (such as the mutant lysozyme syndrome (9)) and common neurodegenerative diseases (10). Such diseases (involving fewer than 20 of the >104 polypeptides in the human proteome (11)) have stimulated interest in features that protect almost all encoded proteins from analogous misfolding (12).
Our study was motivated by the hypothesis that residues may function as critical “safeguards” in protein biosynthesis, but are dispensable in the native state (13). Two classes of safeguards have previously been identified (14). The first, prodomains, functions within the primary translation product but is removed during biosynthesis (15). An example is the connecting (C) domain of proinsulin, which markedly favors native disulfide pairing (16) relative to combination of isolated A and B chains (17). Such internal catalysis enables native folding despite the intrinsic amyloidogenic properties of A and B chain segments (18). The second class of safeguards consists of residues in the mature protein required for foldability but otherwise dispensable once the native state is reached (13). Although such residues may be overlooked in functional screens in vitro, they can be conserved due to critical (yet unseen) roles in erecting kinetic barriers to toxic aggregation (12).
Insulin provides a model for studies of protein evolution (19). Biological selection is strict due to its physiological importance (20). Indeed, large quantities of the hormone must be expressed and stored in β-cells (21). The foldability of proinsulin is nonetheless only precariously maintained; the majority of human cell lines cannot efficiently fold proinsulin (3), highlighting the specialized milieu of the β-cell. Even in β-cells, overexpression of proinsulin (e.g. in response to insulin resistance as often seen in obesity (22)) can induce ER stress (23), ultimately leading to β-cell dysfunction and death (24). This process is central to the natural history of type 2 DM (24). The current pandemic of “diabesity” reflects societal changes rapid on the evolutionary time scale (25).
Given that even wildtype (WT) proinsulin lies near the border of ER stress (21), it is not surprising that mutations might impair foldability at physiological levels of expression (i.e. in the absence of insulin resistance (26)). Such mutations define a monogenic cause of DM, designated the “mutant proinsulin syndrome” (also known as mutant INS diabetes of the young (MIDY) (27)). The first such mutation, CysA7 → Tyr, identified as an autosomal DM locus in the Akita mouse (28), was subsequently observed in human neonatal-onset DM (29). Diverse clinical mutations have subsequently been identified that remove or introduce a Cys, leading in either case to an odd number of thiols (30). The variant proinsulin must therefore contain an unpaired cysteine, in principle mediating aberrant disulfide interchange and inter-molecular disulfide bridges (31). Such a variant proinsulin interferes with the folding, trafficking, and secretion of the WT protein (32), leading in the 1st year of life to unremitting ER stress, progressive β-cell dysfunction, apoptosis, and permanent DM (33).
Of particular interest are DM-associated mutations in proinsulin not involving cysteine. Whereas patients with Cys-related mutations invariably present with neonatal DM, nonCys-related mutations are associated with ages of onset ranging from neonatal to the 3rd decade of life; furthermore, the latter pedigrees typically exhibit incomplete penetrance (34). Such nonCys-related mutations presumably perturb, to varying extents, key structural contacts stabilizing on-pathway folding intermediates (21), such as the B8 φ dihedral angle (35). The present study was broadly stimulated by a mild MIDY mutation, PheB24 → Ser (36). This variant was originally described as an insulinopathy (insulin Los Angeles (37)) due to its isolation in the proband's serum, implying at least partial preservation of prohormone processing and secretion (36). [SerB24]insulin exhibits decreased but non-negligible receptor-binding affinity (38). The decrement is unlikely to explain patient phenotypes. Delayed DM onset correlates with extent of ER stress induced on expression of the variant proinsulins (32).
PheB24, invariant among vertebrate insulins (Fig. S1) and insulin-like growth factors (IGFs), anchors the B20–B23 β-turn in the free hormone (Fig. 1, A and B) (39) and directly contacts the IR (40). In the classical structure of insulin, this turn enables the C-terminal B chain β-strand (residues B24–B28) to pack against the conserved side chains of IleA2, ValA3, LeuB11, ValB12, and LeuB15. These contacts seal the hydrophobic core of the α-helical domain. On insulin self-assembly, the β-strand forms a dimer-related antiparallel β-sheet (Fig. S2) (39, 41). On receptor binding, a conformational change ensues: rotation of the B20–B23 β-turn and adjoining PheB24–PheB25 element leads to detachment of the C-terminal B chain β-strand from the core. The β-strand itself packs in a groove between IR-ectodomain elements L1 and αCT (Fig. S3A), wherein the side chain of PheB24 packs within a nonpolar pocket; the side chains of PheB25 and TyrB26 occupy more peripheral sites (Fig. S3, A and B) (40). Although the B24–B25–B26 “triplet” of aromatic residues is a hallmark of the vertebrate insulin family (20, 42, 43), this motif is not present among invertebrate homologs (44).
Figure 1.

Sequence and binding mode of insulin. A, insulin contains two chains, A and B, connected by two disulfide bridges (A7–B7 and A20–B19); an intra-chain bridge spans residues A6–A11. The “aromatic triplet” comprises residues PheB24, PheB25 (red circles), and TyrB26 (blue circle). The present analogs contain substitution LysB29 → Orn (green circle), which eliminates a tryptic site during semisynthesis. B, ribbon structure of crystallographic T-state insulin protomer (PDB code 4INS). The A chain is shown as a green ribbon and the B chain as an orange ribbon. A type 1 β-turn comprises residues B20–B23 (blue). PheB24 and PheB25 are shown as red sticks, and residue TyrB26 is shown in cyan. C, cartoon representation of insulin-IR binding. Insulin's C-terminal B chain β-strand intercalates between receptor domains αCT and L1; the binding surfaces of B24, B25, and B26 are indicated by orange, purple, and blue-gray semicircles, respectively. D, structure of insulin bound to μIR (PDB code 4OGA). The L1 domain is shown as a blue-gray surface, and the αCT domain is shown as a purple ribbon. The insulin A chain is shown as green ribbon; the B chain is shown as an orange ribbon; residue PheB25 is shown as red sticks; and residues B23–B24 and B26–B29 are shown as orange sticks. E, simulated structure of [GlyB24]insulin bound to μIR, and the color code is as in D. If canonical binding conformations were maintained, the B24-binding pocket in the L1 domain would remain unoccupied as highlighted by the blue box. F, cartoon representation of the register-shift model. Residues PheB24, PheB25, and TyrB26 occupy specialized binding surfaces (color code as in C) on the αCT and L1 domains during IR complexation (left). In the “register shift” model (black arrow), GlyB24 would enable PheB25 to occupy the B24-binding pocket, TyrB26 to pack against the B25-binding surface, and ThrB27 to bind the B26-binding surface.
Substitutions at B24–B26 uncovered distinct and specific side-chain determinants of IR binding. The B24-binding pocket (defined by residues in L1, αCT, and the central B chain α-helix) optimally accepts Phe but can also accommodate cyclohexylalanine (Cha), Met, or branched aliphatic side chain; Tyr, His, and Trp are disfavored despite their aromaticity (39, 45). The B25-binding cleft in the αCT domain by contrast requires aromatic side chains; Phe, Tyr, and Trp each confer high affinity (46, 47). Despite the conservation of TyrB26 (or Phe among IGFs) (48), the solvent-exposed B26-binding surface accommodates diverse charged, polar, or aromatic side chains (42, 48). The striking preservation of the B24–B26 aromatic triplet among vertebrate insulins and IGFs, conserved for more than 500 million years (49), is believed to represent intersecting evolutionary constraints imposed by function, foldability, assembly, and stability.
We address here a long-standing anomaly: the native activity of [GlyB24]insulin (42, 45). Given the structure of the hormone–receptor interface (Fig. 1D) (40), substitution of PheB24 by Gly would seem to leave a destabilizing cavity (blue box in Fig. 1E). How might native affinity and activity be regained? A potential framework is provided by the register-shift model; an alternative receptor-binding mode of insulin in which residue B24 reorients as part of a noncanonical five-residue chain reversal (B20–B24), in turn enabling residues B25–B27 to engage the respective B24-, B25-, and B26-binding pockets of the IR (39, 50, 51). This model posits that the introduction of GlyB24 (or d-amino acid substitutions at B24 (45, 52)) destabilizes the classical four-residue β-turn (B20–B23) (53, 54). Such a shift would (i) preserve insertion of a Phe within the B24-binding pocket (PheB25), (ii) deploy a well-tolerated Tyr within the B25-binding pocket (TyrB26), and (iii) exploit the promiscuity of the solvated B26-binding surface to accommodate a small polar side chain (ThrB27) (illustrated in Fig. 1F).
This study provides mutational evidence in support of the register-shift model and suggests that this alternative mode of binding has been excluded in the evolutionary history of the vertebrate insulin family by toxic protein misfolding. Our results are discussed in relation to molecular dynamics (MD) simulations of [GlyB24]insulin/IR ectodomain complexes based on the crystal structure of insulin bound to its primary IR-binding site (PDB entry 4OGA) (40). Analysis of an excluded evolutionary path highlights the implicit role of PheB24 as a conserved safeguard of native foldability.
Results
Insulin analogs were prepared by trypsin-catalyzed semi-synthesis (55). To eliminate the native LysB29 tryptic site during semi-synthesis, the analogs contained ornithine (Orn) at position B29 (39). Because of the chemical similarities between Lys and Orn, [OrnB29]insulin exhibits native-like biological and biophysical properties. In accordance with the classical studies of Tager and co-workers (42, 45), substitution PheB24 → Gly preserved IR-binding affinity in a competitive displacement assay (Table 1 and Fig. S4A). SerB24 by contrast impaired binding by ∼25-fold. Although similar qualitative trends in biological activity were observed (as assessed in rats rendered diabetic by streptozotocin (STZ) (39)), the activity of the SerB24 analog was less markedly impaired in vivo, presumably due to the compensating effects of delayed clearance from the bloodstream (as documented in human subjects) (56). The activity of [GlyB24,OrnB29]insulin in STZ rats was indistinguishable from that of control [OrnB29]insulin (Fig. S4B).
Table 1.
IR affinities of insulin analogs
| Analog | Kda |
|---|---|
| nm ± S.D. | |
| GlyB24 OrnB29 | 0.06 ± 0.02 |
| SerB24 OrnB29 | 1.91 ± 0.11 |
| OrnB29 | 0.07 ± 0.04 |
| WT human insulin (HI) | 0.08 ± 0.03 |
a Dissociation constants (Kd) for the lectin-purified IR (isoform A) were determined in a competitive binding assay as described previously (111).
Classical SARs underlie specific predictions of the register-shift model
Variant insulins were designed to test the “register-shift” model of [GlyB24]insulin-IR engagement. These analogs exploited the following SARs at respective positions B24–B26.
Position B24
Whereas the B24-binding pocket at the hormone–receptor interface (40) has strict size, polarity, and geometric requirements, analogs with γ-branched aliphatic or small aliphatic residues (Leu, Cha,5 or Phe) at the B24 position retain significant IR affinity. By contrast, the para-OH group of Tyr impairs affinity by more than 10-fold (39).
Position B25
No classical pocket exists at the B25-cognate surface of the receptor. Binding is markedly impaired by a tetrahedral γ-carbon in the B25 side chain (such as in Leu or Cha), highlighting a geometric requirement for trigonal sp2 hybridization at Cγ (46, 47). Shorter side chains lacking a γ-carbon (such as Ala or Ser) likewise lead to low activity.
Position B26
The B26-related IR surface can accommodate diverse (but not all) side chains. Despite broad conservation of TyrB26 among vertebrate insulins (42), small, polar side chains are preferred in vitro (48). Such conservation appears enjoined by the contribution of TyrB26 to native self-assembly (48), a critical feature of insulin storage in the secretory granules of pancreatic β-cells (57).
Given these distinct site-specific SARs, the register-shift mode would lead to successive SAR shifts (Fig. 1, D–F). Whereas a tetrahedral γ-carbon in the B25 side chain (Leu or Cha) would ordinarily impede the binding of insulin, for example, the same modification in the context of a GlyB24 analog might be “rescued” as the variant B25 side chain could then be well-accommodated within the B24-binding pocket. Similarly, whereas SerB26 ordinarily confers high activity, in a shifted complex SerB26 would be directed onto the incompatible B25-binding surface (Fig. S5).
Comparative studies of paired insulin analogs provide critical tests of model
To test such predictions, a series of paired PheB24/GlyB24 analogs were prepared. Probes of respective B24–B26-binding sites were provided by ChaB25, LeuB25, TyrB25, and SerB26 (Fig. S5). The pattern of receptor-binding affinities was found to be in broad agreement with the predictions (Fig. 2 and Table 2, column 3). Particularly striking is the rescue by GlyB24 of otherwise unfavorable B25 substitutions as exemplified by the tetrahedral γ-carbons of LeuB25 and ChaB25. Conversely, introduction of GlyB24 rendered TyrB25 and SerB26 (ordinarily well-tolerated) unfavorable. Whereas this pattern of affinities would seem paradoxical in the context of the native hormone/receptor complex (40), register-shifted SARs provide a unifying and coherent account.
Figure 2.

In vitro competition IR-binding assays. In vitro affinities of insulin analogs containing B24–B26 substitutions for solubilized IR-A were determined by competitive displacement of [125I-TyrA14]insulin. Colored squares represent % tracer bound at increasing concentrations of analog; corresponding colored curves are fitted (see Table 1). Colors are defined within insets.
Table 2.
Bio-activities of register-shift insulin analogs
| Analog | % AUCa ± S.D. | Kdb |
|---|---|---|
| n | nm ± S.D. | |
| LeuB24 OrnB29c | 61 ± 13 (9) | 0.13 ± 0.03 |
| LeuB25 OrnB29c | 78 ± 11 (9) | 4.9 ± 2.50 |
| GlyB24 LeuB25 OrnB29c | 62 ± 13 (9) | 0.48 ± 0.10 |
| ChaB24 OrnB29c | 53 ± 14 (12) | 0.07 ± 0.05 |
| ChaB25 OrnB29 d | 80 ± 10 (10) | 11.00 ± 2.4 |
| GlyB24 ChaB25–OrnB29c | 63 ± 12 (15) | 0.12 ± 0.02 |
| TyrB24 OrnB29c | 68 ± 8 (10) | 1.73 ± 0.57 |
| TyrB25 OrnB29d | 45 ± 10 (10) | 0.03 ± 0.01 |
| GlyB24 TyrB25 OrnB29c | 72 ± 12 (10) | 0.95 ± 0.19 |
| SerB25 OrnB29 | 76 ± 5 (9) | 2.98 ± 0.7 |
| SerB26 OrnB29 | 52 ± 12 (10) | 0.04 ± 0.01 |
| GlyB24 SerB26 OrnB29 | 79 ± 8 (10) | 1.16 ± 0.17 |
| OrnB29 | 61 ± 12 (35) | 0.07 ± 0.04 |
a Values were calculated from experiments in STZ-rendered diabetic rats (Fig. 3).
b Kd values were calculated from in vitro receptor-binding assays shown in Fig. 2.
c Data displayed are statistically significantly different from the B25 analog as determined by two-sided Student's t test with a significance level of 0.05.
d Data displayed are statistically significantly different from B24 analog as determined by two-sided Student's t test with a significance level of 0.05.
To assess the in vivo relevance of these receptor-binding studies, biological activities were tested in STZ rats (Fig. 3 and Table 2, column 2). Pharmacodynamic (PD) results were broadly in accordance with the in vitro data, including GlyB24 rescue of minimally-active [LeuB25]insulin and [ChaB25]insulin analogs. Indeed, the double mutants exhibited activities similar to those of the corresponding B24 analogs. Conversely, addition of GlyB24 to super-active TyrB25 or SerB26 analogs led to impaired function, similar to respective TyrB24 or SerB25 analogs. Relative biological potencies were estimated based on areas under the PD curves (AUCs) as given in Table 2.
Figure 3.

Biological activities in STZ rats. Left-hand panels display averaged glucose-lowering curves as a fraction of initial blood–glucose levels; error bars represent S.E. Initial blood–glucose values averaged 380–410 mg/dl between experiments. Color codes are given in the insets. Right-hand panels display box plots of fractional area under the glucose-lowering curves (AUC). This statistic was calculated over 240 min of time with maintenance of initial blood–glucose level corresponding to 1.0. Lower and upper edges of boxes represent the 1st and 3rd quartiles, respectively; whiskers represent 5th and 95th percentiles. Median values are represented as gray horizontal lines within each box and mean values as white horizontal lines. Individual data points are represented as gray diamonds. AUC values are summarized in Table 2.
Systematic characterization of the complete set of analogs (15 in total) was obtained in cell-based assays. The assays employed human breast cancer cell line MCF-7 (58). Insulin-dependent IR autophosphorylation was assessed via the ratio of phosphorylated IR (p-IR) to total IR in immunoblots (Fig. S6A) (59). Insulin-dependent transcriptional regulation was probed via activation of cyclin D1 and repression of cyclin G2 as measured by q-rtPCR (Fig. S6B) (59). This extensive set of results, consistent with in vitro and rat-based studies, collectively provided evidence for the register-shift model (Fig. 4 and Fig. S7).
Figure 4.
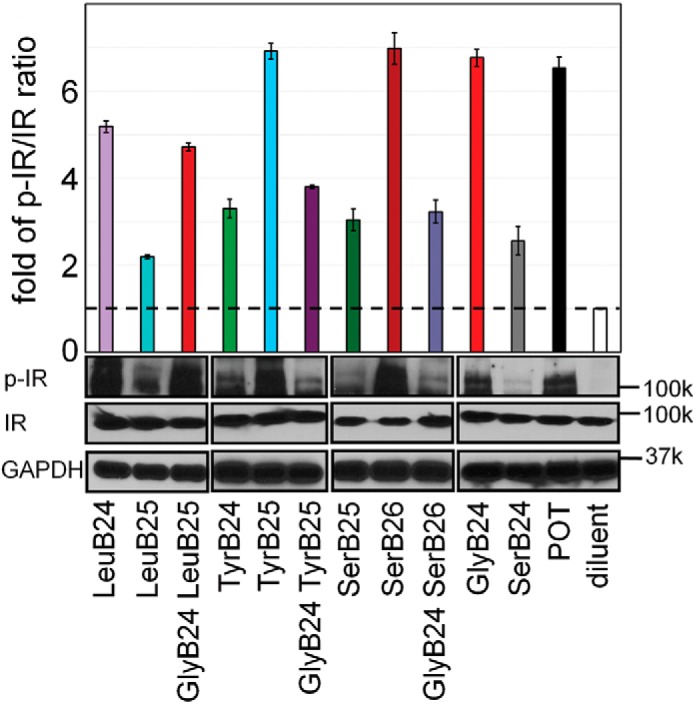
Insulin signaling in a mammalian cell line. Histogram depicts the p-IR/IR ratio (determined by Western blotting) in MCF-7 cells treated with 50 nm insulin analogs or OrnB29 (POT) control. Data are represented as the average fold increase in p-IR/IR in relation to insulin-free diluent. Western blotting bands corresponding to p-IR, IR, and GAPDH-loading control are displayed below the corresponding columns of the histogram. Black boxes indicate separate blots.
Thermodynamic studies highlight the contribution of PheB24 to monomer stability
Thermodynamic unfolding studies, based on circular dichroism (CD)-monitored guanidine titrations (60, 61), demonstrated that GlyB24 attenuates stability (ΔΔGu 1.3(±0.2) kcal/mol relative to parent [OrnB29]insulin (Fig. 5, A and B, and Table 3)). SerB24 and LeuB24 were likewise destabilizing (ΔΔGu 1.3(±0.2) and 1.3(±0.2) kcal/mol, respectively; positive values represent loss of stability in the difference ΔGu[mutant] − ΔGu[parent], where the parent analog is [OrnB29]insulin). These thermodynamic decrements are in accordance with the structural role of PheB24 at the edge of the hydrophobic core (39); in this context truncation of the B24 side chain would lead to a destabilizing crevice (39). That ChaB24 is also destabilizing (1.1(±0.2) kcal/mol) suggests that the aromaticity of PheB24 may also make a specific contribution, presumably via weakly polar interactions (39).
Figure 5.

Biophysical characterization of [GlyB24]insulin. A, CD spectra of [GlyB24,OrnB29]insulin, [SerB24,OrnB29]insulin, and control analogs. Color code is given in the inset. B, guanidine denaturation assays as monitored by ellipticity at 222 nm; color code is the same as in A. Stabilities and calculated thermodynamic parameters are given in Table 3. C, gel-filtration elution profiles of insulin analogs in the absence of zinc or phenolic ligand. The color code is given in the inset. D and E, visible absorption spectra and hexamer dissociation kinetics. D, Co2+ d–d bands of WT insulin (black), [OrnB29]insulin (blue), insulin lispro (green), [GlyB24,OrnB29]insulin (red), and [SerB24,OrnB29]insulin (orange); this band provides a signature of the R6 (or Rf) hexameric state. The amplitude of [GlyB24,OrnB29]insulin was attenuated in relation to WT insulin, [OrnB29]insulin, and lispro. E, sequestration of Co2+ by EDTA; WT insulin, [OrnB29]insulin, insulin lispro, [GlyB24,OrnB29]insulin, and [SerB24,OrnB29]insulin; color code is as in D. Hexamer-dissociation half-lives are given in Table 4. F, transmission electron micrograph of mature insulin fibrils. The black bar indicating 100 nm is given for scale. G, assessment of fibrillation lag times of insulin analogs. Box plot of lag times prior to onset of fibrillations: of [GlyB24, OrnB29]insulin, and [SerB24,OrnB29]insulin are shown in relation to controls. Data were collected under accelerated conditions at 37 °C (see “Experimental procedures”). The upper and lower boundaries of boxes represent 1st and 3rd quartiles, respectively. Upper and lower whiskers demarcate 1.5 times the interquartile range. Mean lag times are represented by gray horizontal lines, and median lag times by black horizontal lines. Lag times of individual replicates are represented by black diamonds.
Table 3.
Thermodynamic stabilities of insulin analogs
| Analog | ΔGua | Cmidb | mc |
|---|---|---|---|
| kcal mol−1 | m | kcal mol−1 m−1 | |
| WT insulin | 3.4 ± 0.1 | 5.0 ± 0.20 | 0.7 ± 0.03 |
| OrnB29 | 3.6 ± 0.2 | 4.9 ± 0.21 | 0.7 ± 0.03 |
| GlyB24 OrnB29 | 2.3 ± 0.1 | 4.6 ± 0.28 | 0.5 ± 0.03 |
| SerB24 OrnB29 | 2.3 ± 0.1 | 4.5 ± 0.10 | 0.5 ± 0.01 |
a Data are from two-state modeling of CD-guanidine titrations performed at 25 °C and pH 7.4.
b Cmid is the guanidine denaturant concentration at which 50% of the protein is in the unfolded state.
c The m value is the slope of unfolding free energy ΔGu versus the molar concentration of denaturant.
Although the side chain of PheB25 is flexible in NMR-derived structures of insulin (as indicated by motional narrowing) (62), its substitution by LeuB25 or ChaB25 was also observed to impair thermodynamic stability (although to a lesser extent than the B24 substitutions): ΔΔGu 0.7(±0.2) and 0.9(±0.2) kcal/mol, respectively. Such destabilization may in part reflect a solvation free-energy penalty (63). In contrast to the above functional rescue, co-introduction of GlyB24 accentuated the destabilizing effects of LeuB25 and ChaB25. The combination of GlyB24 and LeuB25 impaired stability (ΔΔGu 1.9(±0.2) kcal/mol), whereas the combination of GlyB24 and ChaB25 kcal/mol impaired stability by 1.5(±0.2) kcal/mol (Fig. S8 and Table S1). These additive perturbations are uncorrelated with effects on activity.
PheB24 stabilizes native self-assembly and metal-ion coordination
Protein self-association (in the absence of zinc ions or phenolic ligands) was probed by size-exclusion chromatography (SEC) (48). A range of elution times and peak shapes was observed in relation to WT insulin (earliest eluting) and engineered monomer (KP-insulin; latest eluting) (Fig. 5C); molecular masses were inferred in reference to these standards (Fig. S9 and Table 4). Whereas WT insulin and [OrnB29]insulin were essentially dimeric under these conditions (inferred molecular masses 6.8 and 6.6 kDa, respectively), the SerB24 and GlyB24 analogs exhibited elution profiles intermediate between monomer and dimer (molecular masses in each case 4.9 kDa). Such variant dimers are presumably destabilized by a cavity at the dimer interface, comprising the contiguous volumes occupied by PheB24 and its dimer-related mate.
Table 4.
Self-association properties of insulin analogs
| Analog | Hexamer lifetimes (t½) | Calculated SEC massa |
|---|---|---|
| min ± S.D. | kDa | |
| WT insulin | 8.2 ± 1.3 | 6.8 |
| Insulin lispro | 4.6 ± 0.3 | 4.2 |
| OrnB29 | 8.2 ± 0.8 | 6.6 |
| SerB24 OrnB29 | 2.3 ± 0.2 | 4.9 |
| GlyB24 OrnB29 | 1.2 ± 0.2 | 4.9 |
a Proteins were made 0.6 mm in a zinc-free buffer and applied to the SEC column as described under “Experimental procedures”; masses were calculated from Fig. S9.
The kinetic stability of the metal ion-stabilized insulin hexamer (Fig. S10) was evaluated by a Co2+ sequestration assay (64). This approach, pertaining to the R6 hexamer (65), provides disassembly rates (or hexamer lifetimes; Table 4) (48). The hexamer lifetime of [GlyB24,OrnB29]insulin (t½ = 1.2(±0.2) min) was profoundly reduced relative to [OrnB29]insulin (8.2(±0.8) min), WT insulin (8.2(±1.3) min), or even the rapidly-dissociating analog insulin lispro6 (KP) (4.6(±0.3) min). The hexamer lifetime of [SerB24,OrnB29]insulin was also reduced, but ∼2-fold greater than that of [GlyB24,OrnB29]insulin. PheB24 therefore contributes to a kinetic barrier that locks the R6 insulin hexamer (Fig. 5, D and E.)
A qualitative probe for the dynamic stability of the tetrahedral Co2+-coordination site within the R6 hexamer is provided by the magnitude of the 574 nm d–d absorbance band; this spectral feature would be absent in an octahedral site. The Co2+ d–d band was attenuated in the GlyB24 analog relative to the other proteins, including [SerB24,OrnB29]insulin and insulin lispro (Fig. 5, D and E, and Table 4). These data suggest that the GlyB24-analog hexamer contains a distorted coordination site and/or that this site exhibits local dynamic interconversion between tetrahedral and octahedral structures. Either mechanism would represent a transmitted perturbation as the mutation site (B24) is distant from the HisB10-related site of metal-ion coordination (66).
PheB24 protects insulin from fibrillation, a toxic misfolding process
Susceptibilities of the GlyB24 and SerB24 analogs to the formation of insoluble amyloid fibrils (Fig. 5F) were evaluated (in the absence of metal ions) relative to WT insulin, [OrnB29]insulin, and insulin lispro. Aliquots were agitated at a protein concentration of 60 μm at 37 °C. Lag times were measured using thioflavin T (ThT) as a fluorescent probe of cross-β assembly (19). The B24 analogs exhibited shorter lag times than the control samples (Fig. 5G and Table 5). Comparison of corresponding single and double mutants demonstrated that GlyB24 also accelerated the fibrillation of analogs containing substitutions at positions B25 or B26 (Fig. S11 and Table S2; data obtained at room temperature using “gentle sloshing” in glass vials). We envision that, under native conditions, the B24 substitutions enhance subpopulations of amyloidogenic partial folds as components of a conformational equilibrium (67).
Table 5.
Fibrillation lag times of insulin analogs
| Analog | Fibrillation lag timea |
|---|---|
| h ± S.D. (n) | |
| WT insulin | 8.3 ± 3.4 (8) |
| OrnB29 | 14.0 ± 1.4 (7) |
| GlyB24 OrnB29 | 3.3 ± 0.3 (7) |
| SerB24 OrnB29 | 2.7 ± 0.3 (6) |
| Insulin lispro | 6.2 ± 2.8 (7) |
a Fibrillation lag times pertain to zinc-free WT insulin and analogs (in a monomer–dimer equilibrium); each protein was made 60 μm in phosphate-buffered saline (pH 7.4). Samples were agitated via continuous shaking at 37 °C. Time of initial fluorescence, defined as a 2-fold increase over baseline in ThT fluorescence, provided a criterion for onset of fibrillation.
B24 mutations in proinsulin impair cellular folding efficiency and induce ER stress
How GlyB24 affects the biosynthesis and secretability of proinsulin (the single chain precursor of insulin (68)) was evaluated in transiently-transfected HEK293T cells (Fig. S12) (69). Nascent proteins were pulse-labeled with [35S]cysteine/methionine for 30 min followed by a 2-h chase. Oxidative folding and secretion of [GlyB24]proinsulin were evaluated using nonreducing Tris–Tricine–urea–SDS-PAGE (Fig. 6A, the c and m refer to the cells and chase media, respectively) (3). [GlyB24]Proinsulin exhibited impaired folding efficiency (relative to WT) as demonstrated by a weaker band corresponding to native proinsulin and greater relative prominence of bands corresponding to non-native disulfide isomers. In accordance with previous studies (32), SerB24 led to similar qualitative perturbations, but with less marked attenuation of the native band in the cellular fraction (Fig. S13).
Figure 6.

Cell-based studies of proinsulin biosynthesis. A, nonreducing SDS-PAGE of cell lysates (lanes labeled c) and chase medium (lanes labeled m) of HEK293 cells transiently transfected with [GlyB24]proinsulin, WT-proinsulin, or empty vector (control). The figure displays nonconsecutive lanes from the same gel. B, proinsulin mutants secreted into chase medium quantified and normalized against WT-proinsulin. * indicates reduced statistically significant (p < 0.05) secretion compared with WT-proinsulin; ** indicates a statistically significant decrease in secretion compared with WT and [SerB24]proinsulin. C, intracellular distribution of B24 proinsulin variants. INS1 cells were transiently transfected to express an ER-targeted RFP-KDEL (ER-RFP, red) and a proinsulin construct containing GFP within the C domain (ProIns-GFP, green). The scale bar indicates a length of 10 μm. GlyB24 and SerB24 variants displayed an increased degree of co-localization with ER-RFP in relation to that of WT-proinsulin, which was largely localized in peripheral puncta consistent with secretory granules. D, activation of the unfolded protein response in HEK293 cells by proinsulin variants as monitored by expression of a BiP-luciferase reporter. # and †, respectively, indicate increased BiP-luciferase activation compared with WT control and with WT, [GlyB24]proinsulin, and [SerB24]proinsulin at a statistically significant level.
Additional experiments were performed to assess protein trafficking and secretion. The variant proinsulins exhibited preferential secretion of non-native disulfide isomers (upper bands in lanes “m” in Fig. 6A and Fig. S13). Radiolabeled WT or variant proinsulin in cell lysates and post-chase media was quantified; relative secretion efficiencies of [GlyB24]proinsulin and [SerB24]proinsulin were 25 and 40%, respectively (Fig. 6B). Further insight was obtained through fluorescence studies of variant proinsulins containing green fluorescent protein (GFP) within the C domain, as described previously (33). This chimeric construct is shown in schematic form in Fig. S14.
In rat INS1 β-cells, WT-proinsulin–GFP chimera exited from the ER were stored in the secretory granules and exhibited a punctate secretory–granule pattern that did not co-localize with an ER marker (Fig. 6C, red). Although previously, the [CysB24]proinsulin–GFP (a mutation associated with neonatal-onset DM) was severely misfolded and completely retained in the ER (Fig. S15), SerB24 and GlyB24 were partially exported from the ER (Fig. 6C). It is possible that partial ER exit of the B24 variants in these assays was due to co-expression of WT rat proinsulins, which might act in trans to rescue trafficking of the human chimeric variants (70).
Our cell-based studies could be calibrated in relation to MIDY variants. Whereas SerB24 represents a mild mutation (with onset in the 3rd decade of life and with variable penetrance (26, 71)), a negative control was provided by CysB24: its secretion efficiency was ∼2% of WT (Fig. 6B). Because induction of ER stress is central to the pathogenesis of the mutant proinsulin syndrome (72), we next exploited a resident ER quality-control sensor (binding immunoglobulin protein; BiP), constructed as a luciferase fusion protein, to probe the extent of proinsulin misfolding (Fig. S16). Luciferase activity was stimulated in the order WT < GlyB24, SerB24 < CysB24 (Fig. 6D). In addition to its the structural roles highlighted above (i.e. receptor binding, native-state stability, self-assembly, and protection from fibrillation), PheB24 thus contributes to the folding efficiency of proinsulin in the ER and its secretability.
Together, these cell-based studies suggest that potential inheritance of GlyB24 in a vertebrate would enhance the risk of β-cell dysfunction, rationalizing its evolutionary exclusion despite the native biological function of the mature hormone. Comparison with MIDY mutations CysB24 (neonatal) and SerB24 (onset in early adulthood) suggests that in humans a GlyB24 variant INS gene would lead to DM onset in adolescence.
Discussion
The Anfinsen paradigm posits that the sequence of a protein determines its structure and stability (73). The “Levinthal paradox” (74), highlighting the complementary requirement for folding efficiency, has long stimulated investigation into biophysical mechanisms by which a folding chain avoids the need for an exhaustive conformational search (75). This study was motivated by the notion that the informational context of protein sequences can either be overt (apparent in the native structure), implicit (critical to folding efficiency but dispensable in the native state, once obtained), or cryptic (safeguards against off-pathway events) (76). Studies of diseases of protein misfolding (77) have provided insight into implicit determinants of foldability and the evolution of cryptic safeguards against toxic misfolding (5). These concepts have gained prominence as cross-β assembly, the core structure of amyloid (67), was recognized as a general thermodynamic ground state for polypeptides as a class of heteropolymers (1).
Insulin provides an attractive model for interdisciplinary investigation of protein foldability due to its small size, structural richness, deep evolutionary history, and therapeutic importance (78). To this end, this study has focused on an invariant aromatic residue (PheB24) that makes critical contributions to structure, assembly, and function. A site of clinical mutations associated with DM (30, 36), position B24, illustrates the remarkable compression of information that is possible within the sequence of a globular protein. The rigorous conservation of PheB24 in vertebrates presumably reflects the intersection of multiple distinct constraints, from biosynthesis to function, that have governed the evolution of insulin-related sequences for >500 million years (Fig. S1).
Anomalous structure–activity relationships
A starting point for this study was provided by a long-standing enigma: the native activity of [GlyB24]insulin. Originally regarded as anomalous by Tager and co-workers (42, 45), the analog's high activity stands in contrast to the impaired function of mutant insulins containing diverse l-amino acid substitutions at B24 (39). This seeming paradox was deepened by the enhanced IR-binding affinities of nonstandard analogs containing d-amino acid substitutions at this position (45, 52). Such SARs motivated the hypothesis that PheB24 functions as a site of conformational change on receptor binding (39, 51). Detachment of the B24–B28 segment from the α-helical core of insulin was proposed both to liberate the “aromatic triplet” (PheB24–PheB25–TyrB26) as a conserved receptor-binding motif and to expose part of the underlying core to enable its further receptor engagement (80, 81). Such extended SARs were supported by residue-specific photocross-linking studies (“photomapping mutagenesis” (82)).
The B chain detachment model envisaged the following: (a) the classical structure of insulin (wherein the B24–B28 β-strand packs against the hormone's α-helical core (41)) represents an inactive conformation, and (b) GlyB24 or the corresponding d-amino acid substitutions destabilize this auto-inhibited state to enable native IR binding (45, 52). Supported by the low activities of cross-linked (83) or single-chain (80, 84) insulin analogs, this hypothesis received direct experimental support from crystallographic studies of insulin bound to fragments of the IR ectodomain (40, 85). Indeed, the aromatic triplet was observed to pack within a groove between ectodomain elements L1 and αCT; their displacement indeed enables receptor engagement by conserved nonpolar side chains in the central B chain α-helix and N-terminal A chain α-helix (86). In addition, recent structures of insulin bound to the intact IR ectodomain (obtained by cryo-electron microscopic (cryo-EM) single-particle image reconstruction) have revealed that binding of the detached B24–B28 β-strand is coupled to large-scale reorganization of domains within the dimeric assembly, a plausible first step in transmembrane signal propagation (87).
Register-shift model
In the hormone/ectodomain complex, the side chain of PheB24 inserts within a classical nonpolar pocket, with boundaries formed by aromatic and aliphatic side chains in L1, αCT, and the central B chain α-helix (40). With the exception of GlyB24 and d-amino acid substitutions (above), the dimensions and nonpolar character of this pocket rationalizes SARs at B24 (39). The crystallographic and cryo-EM structures further defined the binding sites of PheB25 and TyrB26 (40, 88). Respective SARs at these sites differ markedly from those at B24 in accordance with their distinctive structural features.
Our experimental strategy exploited these differences to test the register-shift model. The essential idea, first envisaged by Kaarsholm and co-workers (50) and made explicit by Brzozowski and co-workers (51), posits that the C-terminal segment of the insulin B chain may undergo a shift in alignment relative to the B24-, B25-, and B26-binding sites in the receptor. In this model, GlyB24 or d-amino acid substituents would not themselves engage the receptor; rather, their destabilizing effects would enable “slippage” of the B chain such that residues B25–B27 would align with the B24–B26 receptor sites. The side chain of PheB25 would thus replace PheB24 in the B24-binding pocket; TyrB26 would replace PheB25 at an adjoining surface of αCT, and ThrB27 would replace TyrB26 at a peripheral surface near the L1–αCT interface. The latter, although a seemingly nonconservative substitution, would be in accordance with the intrinsic compatibility of the B26-binding surface of the ectodomain for small polar side chains (48).
The register-shift model made specific yet counter-intuitive predictions that GlyB24 could rescue the function of otherwise unfavorable substitutions at B25 or B26 and, conversely, render unfavorable substitutions at these positions otherwise compatible with high activity. Thus, whereas a tetrahedral γ-carbon (such as in the aliphatic side chains of Leu or Cha) is ordinarily excluded at residue B25, such side chains could readily dock within the B24-related pocket via a register shift and hence be rescued as a mechanism-based example of second-site compensation. Conversely, Phe and Tyr are each well-accommodated at the B25-binding site (and indeed IGFs contain Tyr at this position (89)). GlyB24 would be predicted to impair the activity of a TyrB25 analog by forcing its insertion into the B24-binding pocket (in which the para-OH group is unfavorable (39)). Together, these results are in accordance with such reasoning and so provide strong evidence in support of the register-shift hypothesis.
Systematic screening of analog activities in human breast cancer cell line MCF-7 (assessinginsulin-dependent IR autophosphorylation and downstream transcriptional regulation) provided additional evidence in support of the register-shift model. This complete set of activities (15 analogs in total) corroborated selected measurements of receptor-binding affinities and in vivo potencies. Together, these results suggest that the native and register-shift modes of insulin binding lead to similar structural reorganization of the IR ectodomain and mechanisms of transmembrane signal propagation (90).
To explore potential structural mechanisms underlying the proposed register shift, two models of the interface between [GlyB24]insulin and the IR ectodomain were constructed. Modeling was based on the crystal structure of WT insulin in complex with the primary IR binding site (PDB code 4OGA) (40). Relative to the WT complex (Fig. 7A), a naïve model of the [GlyB24]insulin complex was first constructed without a shift, i.e. PheB24 was substituted by Gly in the context of the native structure (Fig. 7B). In this model, such a binding mode would leave a cavity in the space ordinarily occupied by the B24 aromatic ring with an average volume of 27.3(±0.5) Å3 across the simulation. Such a large cavity would presumably incur a free-energy penalty (91); an estimate of this penalty (based on the use of graph-based signatures assessing the change in residue environments proposed by Blundell and co-workers (92)) would be 2.12 kcal/mol. Although such a model illustrates how the WT complex could accommodate a large–to–small substitution without transmitted conformational changes, it appears inconsistent with the present functional studies of insulin analogs.
Figure 7.

Molecular models of insulin and variant [GlyB24]insulin in complex to the IR. Structural representations before (A–C) and in a representative model after (D–F) 250 ns of MD simulation of native insulin (A and D), [GlyB24]insulin without a register-shift (B and E), and with register-shift (C and F) leading to PheB25 occupying the B24-binding pocket. In all models, the L1 domain is shown as a blue-gray surface, and the αCT domain is shown as a purple ribbon; the insulin A chain is shown as green ribbon; the insulin B chain is shown as orange ribbon, and B chain residues GlyB20–ThrB30 are depicted as orange sticks, with pink spheres depicting the cavity within the B24 pocket formed due to the absence of PheB24.
To allow for structural rearrangements near the site of substitution, the modeling procedure next exploited the unoccupied space in the WT structure near the native B20–B23 β-turn. This space would readily accommodate a five-residue noncanonical turn (Gly–Glu–Arg–Gly–Gly) without steric clash such that the side chain of PheB25 can orient toward and partially fill the B24-binding pocket (Fig. 7C). The root mean square deviation between this model and the WT complex (after MD simulation) is 1.9 Å. In this model TyrB26 inserts into the B25-binding cleft (between αCT residues Val715 and Pro718), thereby mimicking the role of PheB25 in WT complexes. MD simulations of all three binding modes (WT insulin in the native complex (Fig. 7D), [GlyB24]insulin in the naïve model (Fig. 7E), and [GlyB24]insulin in a shifted model (Fig. 7F)) indicated that such modes were stable throughout simulations. Whereas in the unshifted mode only the main-chain atoms of GlyB24 can mitigate the otherwise empty B24-binding pocket, in the register-shifted model this potential space is more efficiently filled by the side chain of PheB25(pink spheres in Fig. 7). Estimates of residual cavity volumes in the respective models predicted the register-shift mode reduces the potential B24-related cavity by 12.9(±0.4) Å3 (Fig. S17).
We note in passing that the first mode, wherein the native PheB24-binding pocket would largely be empty, native affinity might be in part rescued from the full cavity penalty (91) by a presumed reduced free-energy cost of conformational change in the more flexible variant insulin. Although this scheme would be in accordance with general biophysical principles, it would not rationalize the present pattern of second-site effects. In particular, an empty B24-binding pocket could not rationalize why GlyB24 rendered TyrB25 unfavorable or rescued the activity of ChaB25 through second-site compensation. In the future, such entropic effects could be addressed via free-energy molecular dynamics simulations. It would also be of interest to obtain a definitive structure of a [GlyB24]insulin analog bound to the IR ectodomain, such that the status of the B24-binding pocket (empty or filled by PheB25) could be directly visualized.
We emphasize that both models assume “molecular parsimony” as potential large-scale reorganization of the hormone–receptor interface (including changes in domain–domain contacts within the dimeric ectodomain) was not considered. These more complex possibilities, if realized, would underscore the essential insight that two modes of hormone binding are possible, but one is disallowed in evolution (see below).
Evolutionary implications
Our rodent studies demonstrated that the register-shift mode of receptor engagement not only permits IR binding but also can mediate metabolic regulation in a vertebrate. Indeed, [GlyB24,OrnB29]insulin was as effective as its parent [OrnB29]insulin for the short-term treatment of DM. This observation was not self-evident based on receptor-binding studies, as it was a priori possible either (a) that a shifted mode of receptor engagement could have been less active in signal propagation or (b) that the impaired stability of the GlyB24 analog could have led to its premature degradation in vivo. Given the observed maintenance of biological activity in vivo, however, we were motivated to consider why GlyB24 has apparently been disallowed in the evolution of vertebrate insulins and IGFs. This question relates to possible evolutionary constraints beyond prominence of PheB24 in the structure and function of insulin's native state. A precedent for cryptic functions of insulin residues was previously provided by PheB1, shown to be critical to the folding efficiency of proinsulin in mammalian cells, but dispensable for structure and function of the mature hormone once its folding is achieved (93).
Implicit or cryptic functions of PheB24 were probed through studies of the kinetics of insulin fibrillation in vitro (39, 94) and through cell-based studies of the nascent foldability of variant proinsulins (69). GlyB24 analogs exhibited shorter lag times prior to onset of fibrillation, suggesting that the heightened presence of conformational substrates (which may constitute a small fraction of protein molecules at any one time) are amenable to non-native aggregation en route to formation of an amyloidogenic nucleus (95). The side chain of PheB24 may thus serve to reduce the stability or accessibility of such conformational excursions relative to the predominant (native) conformation. Because in general fibrillation lag times have been found not to correlate with thermodynamic stabilities (ΔGu relative to the unfolded state), this role of PheB24 presumably relates to amyloidogenic partial folds (39). Analogous concepts were introduced by Dobson and co-workers (96) in the analysis of human lysozyme variants associated with pathological amyloid deposits as a prototypical disease of toxic extracellular protein misfolding.
WT insulin is itself exquisitely susceptible to fibrillation. Such misfolding is ordinarily avoided in vivo through storage of the hormone as zinc-coordinated hexamers (97). The hexamers in turn crystallize with calcium to form protective microcrystals within the β-cell's secretory granules (57). An elaborate machinery of zinc import has co-evolved within the glucose-regulated β-cell granules (57). Such native self-assembly sequesters the flexible insulin monomer and presumably dampens conformational fluctuations that could otherwise nucleate cross-β polymer assembly (39). These studies have shown that substitution of PheB24 by Gly impairs native self-assembly in the absence of metal ions and reduces the kinetic stability of R6 hexamers (containing Co2+ as an isomorphic replacement for Zn2+) once formed. The intrinsic structural role of PheB24 to protect the insulin monomer from non-native aggregation is thus accompanied by a second protective function in promoting protective native self-assembly.
An insulin species variant (in the South American rodent Octadon degus) forms senile amyloid plaques in pancreatic islets; these are proposed to impair β-cell viability leading to late-onset DM (98, 99). The variant B chain sequence contains ProB27–HisB28, which impairs dimerization (98) (in a manner similar to insulin lispro; KP (100)), and substitution HisB10 → Asn, which blocks binding of zinc ions. This species variant is thus less protected from fibrillation than other vertebrate insulins. The continued survival of this species (and thus existence of the variant insulin as an extant vertebrate sequence) presumably reflects the post-reproductive onset of DM.
PheB24 as a cryptic determinant of foldability
Proinsulin, the single-chain biosynthetic precursor of insulin (16, 101), ordinarily enables efficient disulfide pairing in the ER within β-cells. Diverse clinical mutations have been identified in the insulin gene that impair this process (30), leading to chronic ER stress, β-cell dysfunction, and eventual β-cell death (78). This syndrome (MIDY) is a major cause of permanent neonatal-onset diabetes. Whereas the majority of such mutations add or remove a cysteine (thus giving rise to an odd number of reactive thiol groups in the nascent polypeptide chain), mutations have also been identified unrelated to Cys. Such mutations presumably identify sites critical to the mechanism of disulfide pairing (21).
MIDY mutations span a broad range of folding defects, from severe to subtle (78). The mutation PheB24 → Ser, originally identified by Tager and co-workers (insulin Los Angeles (37)), moderately impairs the foldability of proinsulin and is associated with variable onset of diabetes in the 3rd decade of life (32). In our cell-based studies, GlyB24 was at least as perturbing as SerB24 with respect to fidelity of disulfide pairing, efficiency of folding, induction of ER stress, and extent of secretion into the medium. Prior to the discovery of insulin for the treatment of insulin-deficient DM, onset during the reproductive years (even with variable genetic penetrance) is likely to have been sufficient to reduce reproductive fitness and so purge such a sequence variation from extant insulin genes.
Previous fluorescence-imaging studies of β-cell lines expressing proinsulin variants have defined a range of potential defects in protein biosynthesis and/or subcellular trafficking. At one extreme, MIDY mutations resulting in an odd number of cysteines (including classical Akita allele CysA7 → Tyr) typically induce severe defects in nascent folding, leading to ER sequestration and ER-associated degradation (70). By contrast, a “zip code” variant compatible with native folding (HisB10 → Asp; proinsulin Providence) readily exits the ER, but altered trafficking in the Golgi apparatus leads to storage largely within a constitutive pool of secretory granules lacking prohormone convertases and the machinery of glucose-dependent exocytosis (102). Although [AspB10]insulin exhibits enhanced affinity for the IR and augmented thermodynamic stability, the patients were found to exhibit elevated levels of circulating [AspB10]proinsulin due to its aberrant constitutive secretion7 (102).
Clinical mutations in proinsulin may exhibit a range of intermediate cellular phenotypes depending on the specific amino acid substitution. Fluorescence images have thus revealed partial or near-complete sequestration of the variant proinsulin within the ER with varying degrees of impaired Golgi trafficking (103). Within this spectrum of mutations, the most severe (exemplified by GlyB23 → Val, perturbing a β-turn adjoining cystine B19–A20) are associated with neonatal-onset DM (70); less destabilizing substitutions (such as LeuB6 → Met adjoining cystine B7–A7) are associated with disease onset in early adulthood (akin to maturity-onset diabetes of the young) (104). The pattern of fluorescence conferred by GlyB24 (as probed by the C domain–inserted GFP in a proinsulin chimera) would predict onset in the 2nd or 3rd decade of life should this mutation be observed in future patients.
PheB24 thus plays a role in the ER of β-cells (to enhance the foldability of nascent proinsulin) that is distinct from its subsequent roles in the post-Golgi network (to enable productive trafficking) and in glucose-regulated secretory granules (to promote native self-assembly). These aspects of biosynthesis are unrelated to the function of PheB24 in the mature hormone (as a key contact at a conserved hormone–receptor interface). The multiple roles of this invariant residue thus illustrates the remarkable compression of coding information that is possible within a protein sequence. GlyB24 honors in the breach the importance of cell-biological mechanisms and pathological processes not discernable in the three-dimensional structures of proteins, however beautiful and compelling as molecular architectures.
GlyB24 is unique among potential amino acid mutations at B24 in that this variant insulin would retain native activity should its biosynthesis be feasible. Our results nonetheless predict significant defects in the folding, trafficking, and secretion of the variant proinsulin. It would be of future interest to test this prediction in an engineered mouse. To this end, a CRISPR/Cas9-directed GlyB24 variant could define the extent of associated β-cell dysfunction and so inform which evolutionary constraints govern its exclusion among vertebrate insulin sequences. Because the mouse contains two insulin genes (four per diploid genome), such a mouse model may also require a deletion of one WT gene (per haploid genome) to mimic the gene dosage in a human heterozygous patient with the mutant proinsulin syndrome.
Concluding remarks
The complex conformational “life cycle” of the insulin molecule—from its nascent folding within pancreatic β-cells to receptor engagement at target tissues—imposes evolutionary constraints that may be overt, implicit, or cryptic. Safeguards against proteotoxic misfolding, as exemplified by the PheB24-anchored B20–B23 β-turn, can make independent contributions to the biological function of the native state (40). Multidisciplinary dissection of such a safeguard, made possible here by a collaborative team, may in the future enable a deeper understanding of the informational context of insulin sequences. Such studies may define a subset of MIDY mutations amenable to compensation by chemical chaperones as an opportunity for precision medicine beyond insulin replacement therapy (105).
The PheB24-anchored protective hinge (40) is a conserved functional element of vertebrate insulins and insulin-like growth factors. A broader view of the space of insulin-like sequences would encompass invertebrate as well as vertebrate homologs. The genome of Caenorhabditis elegans, for example, encodes 38 putative insulin-like proteins with divergent features relative to vertebrate insulins (106). Remarkably, despite containing multiple core and surface substitutions, each individually disallowed in vertebrate insulin, the structure of one such nematode protein closely resembles human insulin (107). We envision that multisite compensation in this broader sequence space has markedly enlarged the fitness landscape (34). Second-site compensation among the present set of GlyB24 human insulin analogs provides proof of principle that this landscape can include novel modes of hormone–receptor recognition leading to transmembrane signaling. Although excluded in vertebrate evolution evolution, these alternative mechanisms of signaling may in principle be exploited in the design of therapeutic analogs (108).
Experimental procedures
Preparation of insulin analogs
Analogs were prepared by trypsin-catalyzed semi-synthesis using an insulin fragment, des-octapeptide (B23–B30)-insulin and modified octapeptides as described (55). The fragment was made by tryptic cleavage of human insulin and purified by reverse-phase HPLC (rp-HPLC). Octapeptides were prepared by solid-phase synthesis (109). The resulting 51-residue insulin analogs were purified by preparative C4 rp-HPLC (Higgins Analytical Inc., Proto 300 C4 with 10-μm particle size and dimensions 250 × 20 mm), and their purity was assessed by analytical C4 rp-HPLC (Higgins Analytical Inc., Proto 300 C4 with 5-μm particle size and dimensions 250 × 4.6 mm). Predicted molecular masses were in each case verified using an Applied Biosystems 4700 proteomics analyzer MALDI-TOF.
Circular dichroism spectroscopy
Far-UV spectra were obtained from 200 to 250 nm on an AVIV spectropolarimeter equipped with an automated syringe-driven titration unit (39). Helix-sensitive wavelength of 222 nm was used as a probe of protein denaturation by guanidine-HCl. Thermodynamic parameters were obtained by application of a two-state model (61). In brief, data were fit by nonlinear least squares to a two-state-model as shown in Equation 1,
| (Eq. 1) |
where x is the concentration of guanidine hydrochloride, and A and B represent respective estimates of the baseline ellipticities of the protein in its unfolded and native states as extrapolated to zero guanidine concentration (61). Simultaneous fitting of pre- and post-transition baselines avoided artifacts of linear plots of G versus concentration of denaturant (110).
Receptor binding assays
Affinities for IR-A were measured by a competitive-displacement scintillation-proximity assay (48). The assay employed detergent-solubilized holoreceptor with C-terminal streptavidin-binding protein tag. The receptor was purified by sequential wheat-germ agglutinin and streptactin-affinity chromatography from detergent lysates of polyclonal stably-transfected 293PEAK cell lines expressing each receptor. To obtain analog dissociation constants, competitive binding data were analyzed by nonlinear regression by the method of Wang (111).
Rodent assays
Male Lewis rats (mean body mass ∼300 g) were rendered diabetic by STZ. To test potencies, the analogs were made 10 μg per 100 μl in a formulation buffer (16 mg/ml glycerin, 1.6 mg/ml meta-cresol, 0.65 mg/ml phenol, and 3.8 mg/ml sodium phosphate (pH 7.4)) and injected intravenously into tail veins (39). WT insulin or analogs were each re-purified by rp-HPLC, lyophilized, dissolved in Lilly diluent at the same maximum protein concentration, and re-quantitated by analytical C4 rp-HPLC to ensure uniformity. Dilutions were made using the above buffer.
Rats were injected at time t0. Blood was obtained from the clipped tip of the tail at time t0 and every 10 min for the 1st h, every 20 min for the 2nd h, every 30 min for the 3rd h, and every hour thereafter to a final time of 5 h. Efficacy of WT insulin or analog to reduce the blood glucose concentration was calculated using the following: (a) the rate of change in blood glucose concentration over 240 min following initial injection, and (b) the integrated area between the x axis and the curve representing fractional blood–glucose level with relation to initial blood–glucose concentration (AUC). Statistical significance was assessed using a Student's t test.
Animals used in this study were housed at the Association for Assessment and Accreditation of the Laboratory Animal Care (AAALAC)-accredited facilities of Case Western Reserve University (CWRU) School of Medicine. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) Office at CWRU, which provided Standard Operating Procedures and reference materials for animal use (in accordance with the NIH Guide for the Care and Use of Laboratory Animals). The animal health program for all laboratory animals was directed by the CWRU Animal Resource Center. Animal care and use were further monitored for Training and Compliance issues by Veterinary Services.
Signaling assays in mammalian cell culture
Signaling activities of the insulin analogs were tested in MCF-7 human breast adenocarcinoma cells (which express IR-A, IR-B, and high levels of IGF-1 receptor (58, 90)). MCF-7 cells (American Type Culture Collection, Manassas, VA) were cultured in Eagle's minimum essential medium supplemented with 10% FBS, 1% penicillin/streptomycin, and sodium pyruvate (1 mm). 24-h serum-starving protocol using appropriate culture medium except FBS was applied at 70–75% cell confluence. After serum-starving, serum-free medium containing selected concentrations of insulin analogs was added to each well (control wells received medium with no added insulin). Media were removed after 15 min, followed by cell lysis using RIPA buffer with protease and phosphatase inhibitors (Cell Signaling Technology, Danvers, MA). Protein concentrations in cell lysates were determined with a BCA assay kit (Thermo Fisher Scientific) for use in immunoblotting. Cells cultured for q-rtPCR assays (see below) were, after 24 h of serum starvation, treated with either a protein-free medium or medium supplemented with an insulin analog medium for 8 h.
Quantitative real-time PCR transcriptional assay
Mitogenic gene transcription was measured in triplicate by q-rtPCR with samples prepared according to the one-step q-rtPCR kit (Bio-Rad). The following sets of primers (5′–3′) were used for MCF-7 cell-based assays and the following primers (5′–3′) were used: cyclin D1, AATGACCCCGCACGATTTC and TCAGGTTCAGGCCTTGCAC; cyclin G2, ATCGTTTCAAGGCGCACAG and CAACCCCCCTCAGGTATCG; GAPDH, AGCCGAGCCACATCGCT and TGGCAACAATATCCACTTTACCAGAGT; and TFIID, GCACAGGAGCCAAGAGTGAA and TCACAGCTCCCCACCATGTT.
Insulin-stimulated protein phosphorylation
Cell lysates were dissolved in Laemmli buffer (Bio-Rad) with 10% β-mercaptoethanol, heated at 100 °C for 8 min (for MCF-7 p-IR/IR), and then centrifuged at 9300 × g for 1 min (Biofuge A; Baxter Scientific, Soddy Daisy, TN). Samples were loaded into 10% MiniPROTEAN TGX gels (Bio-Rad). Proteins were transferred to polyvinylidene difluoride membrane and then blocked in 5% BSA for 1 h. All antibodies were purchased from Cell Signaling Technology unless otherwise stated. Membranes were incubated overnight at 4 °C with insulin receptor β (4B8) rabbit monoclonal antibody (mAb) or an equal mixture of phospho-insulin receptor β (Tyr1150/1151) rabbit mAb; phospho-insulin receptor (Tyr1158) polyclonal antibody (Thermo Fisher Scientific); phospho-insulin receptor (Tyr1334) polyclonal antibody (Thermo Fisher Scientific); phospho-insulin receptor β (Tyr1345) rabbit mAb; and anti-insulin receptor (phospho-Tyr972) antibody (Abcam, Cambridge, UK). Dilutions for these antibodies were 1:5000 in 5% BSA. After the primary antibody incubation, membranes were washed with 1×TBS with 0.1% Tween 20 and incubated in goat anti-rabbit HRP-conjugated secondary antibody diluted 1:10,000 in 5% BSA for 1–2 h at room temperature. All membranes were washed three times with 1×TBS, 0.1% Tween 20 and incubated in HRP substrate (Merck Millipore, Billerica, MA) for 45 s and developed.
Assessment of fibril formation
Physical stability was assessed by propensity to form fibrils. Insulin or analogs were made 60 μm in phosphate-buffered saline (PBS; pH 7.4) with 0.02% sodium azide and 16 μm ThT. Samples were plated in a Costar® plate (250 μl/well) and incubated at 37 °C with continuous shaking at 1096 cycles/min in a Biotek (Winooski, VT) Synergy H1® plate reader. ThT fluorescence at 480 nm was assessed (following excitation at 450 nm) at 15-min intervals. The time of initial THT fluorescence defined the lag time.
Fibrillation lag times of active [GlyB24]insulin analogs containing Cha or Leu at the B25 position were assessed in relation to control analogs at room temperature in a milder assay designed to simulate a patient-carried insulin pump. Analogs were made 60 μm in PBS containing 0.01% sodium azide as an antimicrobial agent. The insulin solutions were gently rocked at 25 °C in glass vials containing a liquid–air interface. Aliquots, taken at regular intervals, were frozen to enable later analysis of ThT fluorescence (19). For a given sample tube, the assay was terminated on the 2nd day following the appearance of cloudiness in the solution.
Assessment of hexamer stability
Visual absorption spectroscopy was employed to probe formation and disassembly of phenol-stabilized R6 Co2+-substituted insulin hexamers (112). WT insulin or analogs were made 0.6 mm in a buffer containing 50 mm Tris-HCl (pH 7.4), 50 mm phenol, 0.2 mm CoCl2, and 1 mm NaSCN. Samples were incubated overnight at room temperature. Spectra (450–700 nm) were obtained to monitor tetrahedral Co2+ coordination with its signature peak absorption band at 574 nm (64). To determine the rate of Co2+ release from the hexamers, metal-ion sequestration was initiated at 25 °C by addition of an aliquot of EDTA (stored at 50 mm at pH 7.4) to a final concentration of 2 mm. Attenuation of the 574-nm absorption band was monitored on a time scale of seconds to hours. Kinetic data were consistent with monoexponential decay and were fit using Kaleidagraph® software allowing for calculation of kinetic half-life (t½).
Assessment of insulin self-assembly
Oligomeric states of the insulin analogs were monitored by HPLC SEC (48). Analogs were made 0.6 mm in PBS. Samples (10 μl) were applied through a Waters 717 autosampler onto a Zenix-C SEC-150 column (Sepax Technologies) with a nominal fractionation range of 0.5–150 kDa. Proteins were fractionated at a flow rate of 1 ml/min using a Waters Binary HPLC system. Elution was monitored at 215 and 280 nm using a dual-λ Waters 2487 absorbance detector. The mobile phase consisted of 10 mm Tris-HCl (pH 7.4) and 140 mm NaCl (48). Data acquisition and processing utilized Waters HPLC Empower software. The column was calibrated for apparent molecular-mass determination by fractionating standard proteins individually on the column (see Fig. S9 legend).
Cell-based proinsulin folding and secretion
HEK293T cells were plated into 6- or 12-well plates 1 day before transfection. A total of 1–2 μg of plasmid DNA was transfected using Lipofectamine (Invitrogen®). At 48 h post-transfection, cells were pulse-labeled with [35S]Cys/Met and chased for 120 min. A proteinase inhibitor mixture was added to cell lysates and chase media. Samples were pre-cleared with Zysorbin and immunoprecipitated with anti-insulin antibodies (113) and protein-A–agarose overnight at 4 °C. Anti-insulin immunoprecipitates were boiled for 5 min in gel sample buffer (1% SDS, 12% glycerol, and 0.0025% Serva Blue in 50 mm Tris (pH 6.8)) and analyzed using Tris–Tricine–urea–SDS-PAGE under nonreducing conditions (69).
BiP-driven luciferase assay
Mouse Min6 cells were plated into 24-well plates 1 day before transfection. Using Lipofectamine 2000 (Invitrogen), cells were co-transfected with BiP–firefly–luciferase reporter plasmid (72), CMV–Renilla–luciferase plasmid (Promega), and human WT or mutant proinsulin at a DNA ratio of 1:2:5, respectively. At 48 h post-transfection, cell extracts were prepared for the dual-luciferase reporter assay (Promega) with BiP-luciferase normalized to Renilla luciferase activity.
Confocal microscopy of transiently transfected β-cells
INS1 cells were co-transfected with plasmids encoding (a) GFP-tagged proinsulin–WT or mutants and (b) RFP fused with KEDL sequence. At 48 h post-transfection, the cells were fixed with 3.7% formalin in PBS for 20 min, mounted with Prolong Gold with DAPI (Invitrogen), and imaged by epifluorescence in an Olympus FV500 confocal microscope. For immunofluorescence, transfected cells were fixed with 3.7% formalin in PBS for 20 min, permeabilized with TBS containing 0.4% Triton X-100, blocked with TBS containing 3% bovine serum albumin (BSA) and 0.2% Triton X-100, and then stained with primary rabbit anti-calnexin at 4 °C overnight. Thereafter, sections were rinsed and incubated with secondary antibodies conjugated to Alexa Fluor 488 or 568 (Invitrogen). Slides were mounted with Prolong Gold with DAPI (Invitrogen) and imaged by epifluorescence in an Olympus FV500 confocal microscope (114).
Molecular modeling
Models of variant interfaces between [GlyB24]insulin and the IR ectodomain were constructed, as described previously (40), using the MODELLER (version 9.X) program (115). Both the monomeric NMR structure of insulin (PDB code 2KJJ) and the crystal structure of insulin in complex with the micro-receptor and the IR-A isoform αCT (PDB code 4OGA) were used as templates, modeling only the L1 and αCT IR domains in complex with [GlyB24]insulin. Models were generated both with the alignment against the templates leading to a void generated in the B24 pocket (GlyB24 aligned at the B24 position) and with the B chain register-shift leading to PheB25 occupying the B24 pocket. Glycosylation is accounted for via the use of a single N-linked N-acetyl-d-glucosamine carbohydrate at each of the IR asparagine residues 16, 25, and 111. From the 50 models created, the model with the lowest MODELLER objective function was used in subsequent MD simulations.
MD simulations were performed, as described previously (40), using the GROMACS (version 5.1.2) (116) suite of programs and the CHARMM36 (117, 118) force field. The simulation consisted of an initial steepest descent minimization, a short 50-ps positionally restrained MD holding the protein fixed, and finally unrestrained MD for 250 ns. Each complex was placed within a cubic box extending 10 Å beyond all atoms, with the remaining volume solvated using the TIP3P water molecule with sodium and chloride ions to an ionic strength of 0.1 m. The temperature and pressure of the systems were maintained using the velocity rescaling (119) thermostat at 300 K, and the Berendsen barostat (120) at 1.0 bar, updated every 0.1 and 0.5 ps, respectively. A cutoff of 12 Å was used to account for nonbonded interactions and the particle-mesh Ewald method (102) to account for long-range electrostatics applying a grid width of 1.2 Å and a sixth-order spline interpolation. All bond lengths were constrained with the P-LINCS algorithm, which allowed a time step of 2 fs.
Modeling analysis
The DUET (121) and mCSM (92) webservers were used to estimate in silico the effect of the GlyB24 mutation on the interaction between insulin and the IR primary binding site. Cavity analysis was subsequently performed using the trj_cavity tool (122) across all trajectories, defining cavities with a minimum volume of 50 Å3, and then were completely buried (i.e. in all six dimensions surrounding each voxel).
Author contributions
N. K. R., M. L., N. F. P., B. J. S., P. A., and M. A. W. conceptualization; N. K. R. and L. H. data curation; N. K. R., M. L., and L. H. validation; N. K. R., M. L., B. D., Y.-S. C., N. A. S., L. R., J. S., H. G., Y. Y., N. F. P., B. J. S., and F. I.-B. investigation; N. K. R. and M. A. W. writing-original draft; M. L., N. F. P., P. A., F. I.-B., and M. A. W. formal analysis; M. L., N. F. P., J. W., P. A., and F. I.-B. methodology; M. L., Y.-S. C., L. H., N. F. P., B. J. S., P. A., F. I.-B., and M. A. W. writing-review and editing; N. F. P., P. A., F. I.-B., and M. A. W. supervision; N. F. P., P. A., F. I.-B., and M. A. W. funding acquisition.
Supplementary Material
Acknowledgments
We thank M. C. Lawrence, J. G. Menting, and V. Pandyarajan for helpful discussion; K. Carr, R. Grabowski, P. Macklis, and M. Swain for assistance with rat studies; L. Whittaker for advice regarding IR-binding assays; and P. DeMeyts (Novo Nordisk) for the gift of 125I-radiolabeled insulin.
This work was supported in part by National Institutes of Health Grants R01 DK040949 (to M. A. W.) and R01 DK48280 (to P. A.). M. A. W. has equity in Thermalin, Inc. (Cleveland, OH), where he serves as Chief Innovation Officer; he has also been a consultant to Merck Research Laboratories and DEKA Research & Development Corp. N. F. B. P. is a consultant to Thermalin, Inc. F. I.-B. has equity in Thermalin, Inc., and is a consultant to COVANCE, Sanofi, and Novo Nordisk. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We dedicate this article to the memory of Prof. P. G. Katsoyannis (Mt. Sinai Medical Center, New York).
This article contains Figs. S1–S17, Tables S1 and S2, and supporting Refs. 1–3.
Cha designates cyclohexylalanine, a nonplanar aliphatic analog of Phe.
Insulin lispro, containing substitutions ProB28 → Lys and LysB29 → Pro, is the active component of Humalog®, a prandial analog formulation (Eli Lilly and Co.) (100). The Lys–Pro switch (“KP”) destabilizes the dimer interface (100).
HisB10 → Asp also augments mitogenicity and affinity for the type 1 IGF-1 receptor; long-term administration in rats induces mammary tumors (79).
- DM
- diabetes mellitus
- αCT
- C-terminal domain of IR α-subunit
- μIR
- domain-minimized insulin micro-receptor
- BiP
- binding immunoglobulin protein
- Cha
- cyclohexylalanine
- cryo-EM
- cryogenic electron microscopy
- ER
- endoplasmic reticulum
- IGF
- insulin-like growth factor
- IR
- insulin receptor
- L1
- leucine-rich domain of IR
- MIDY
- mutant INS diabetes of the young
- Orn
- ornithine
- PD
- pharmacodynamics
- PDB
- Protein Data Bank
- p-IR
- phospho-IR
- rp-HPLC
- reverse-phase high-performance liquid chromatography
- q-rtPCR
- quantitative real-time–polymerase chain reaction
- SAR
- structure–activity relationships
- SEC
- size-exclusion chromatography
- TBS
- Tris-buffered saline
- ThT
- thioflavin T
- WT
- wildtype (WT insulin pertains to the human sequence unless otherwise stated)
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- AUC
- area under the curve
- STZ
- streptozotocin
- CWRU
- Case Western Reserve University
- DAPI
- 4′,6-diamidino-2-phenylindole
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 10.1038/nature02261 [DOI] [PubMed] [Google Scholar]
- 2. Simkovsky R., and King J. (2006) An elongated spine of buried core residues necessary for in vivo folding of the parallel β-helix of P22 tailspike adhesin. Proc. Natl. Acad. Sci. U.S.A. 103, 3575–3580 10.1073/pnas.0509087103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu M., Hodish I., Haataja L., Lara-Lemus R., Rajpal G., Wright J., and Arvan P. (2010) Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 21, 652–659 10.1016/j.tem.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pedersen J. S., and Otzen D. E. (2008) Amyloid a state in many guises: survival of the fittest fibril fold. Protein Sci. 17, 2–10 10.1110/ps.073127808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dobson C. M. (1999) Protein misfolding, evolution and disease. Trends Biochem. Sci. 24, 329–332 10.1016/S0968-0004(99)01445-0 [DOI] [PubMed] [Google Scholar]
- 6. Betts S., and King J. (1999) There's a right way and a wrong way: in vivo and in vitro folding, misfolding and subunit assembly of the P22 tailspike. Structure 7, R131–R139 10.1016/S0969-2126(99)80078-1 [DOI] [PubMed] [Google Scholar]
- 7. Westermark P., Bellotti V., Obici L., Kisilevsky R., Merlini G., Sipe J. D., Baskakov I. V., Nilsson M. R., Macraild C. A., and Howlett G. J. (2005) Amyloid Proteins: The Beta Sheet Conformation and Disease. (John D. S., ed) pp. 385–491, Wiley-VCH Verlag GmbH & Co, Weinheim, Germany [Google Scholar]
- 8. Stefani M., and Dobson C. M. (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699 10.1007/s00109-003-0464-5 [DOI] [PubMed] [Google Scholar]
- 9. Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D. C., and Ballabio A. (2008) A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129 10.1093/hmg/ddm289 [DOI] [PubMed] [Google Scholar]
- 10. Murphy R. M. (2002) Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 4, 155–174 10.1146/annurev.bioeng.4.092801.094202 [DOI] [PubMed] [Google Scholar]
- 11. Monsellier E., and Chiti F. (2007) Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 8, 737–742 10.1038/sj.embor.7401034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fink A. L. (2005) Natively unfolded proteins. Curr. Opin. Struct. Biol. 15, 35–41 10.1016/j.sbi.2005.01.002 [DOI] [PubMed] [Google Scholar]
- 13. Thomas G. J. Jr., Becka R., Sargent D., Yu M. H., and King J. (1990) Conformational stability of P22 tailspike proteins carrying temperature-sensitive folding mutations. Biochemistry 29, 4181–4187 10.1021/bi00469a022 [DOI] [PubMed] [Google Scholar]
- 14. Kim P. S., and Baldwin R. L. (1982) Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 51, 459–489 10.1146/annurev.bi.51.070182.002331 [DOI] [PubMed] [Google Scholar]
- 15. Subbian E., Williamson D. M., and Shinde U. (2015) Protein folding mediated by an intramolecular chaperone: energy landscape for unimolecular pro-subtilisin E maturation. Adv. Biosci. Biotechnol. 6, 73 10.4236/abb.2015.62008 [DOI] [Google Scholar]
- 16. Steiner D. F., and Oyer P. E. (1967) The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc. Natl. Acad. Sci. U.S.A. 57, 473–480 10.1073/pnas.57.2.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C. C., and Tsou C. L. (1991) The insulin A and B chains contain sufficient structural information to form the native molecule. Trends Biochem. Sci. 16, 279–281 10.1016/0968-0004(91)90114-B [DOI] [PubMed] [Google Scholar]
- 18. Eisenberg D., and McLachlan A. D. (1986) Solvation energy in protein folding and binding. Nature 319, 199–203 10.1038/319199a0 [DOI] [PubMed] [Google Scholar]
- 19. Yang Y., Petkova A., Huang K., Xu B., Hua Q. X., Ye I. J., Chu Y. C., Hu S. Q., Phillips N. B., Whittaker J., Ismail-Beigi F., Mackin R. B., Katsoyannis P. G., Tycko R., and Weiss M. A. (2010) An Achilles' heel in an amyloidogenic protein and its repair. Insulin dynamics, misfolding, and therapeutic design. J. Biol. Chem. 285, 10806–10821 10.1074/jbc.M109.067850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conlon J. M. (2001) Evolution of the insulin molecule: insights into structure–activity and phylogenetic relationships. Peptides 22, 1183–1193 10.1016/S0196-9781(01)00423-5 [DOI] [PubMed] [Google Scholar]
- 21. Weiss M. A. (2013) Diabetes mellitus due to the toxic misfolding of proinsulin variants. FEBS Lett. 587, 1942–1950 10.1016/j.febslet.2013.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenfield J. R., and Campbell L. V. (2004) Insulin resistance and obesity. Clin. Dermatol. 22, 289–295 10.1016/j.clindermatol.2004.01.011 [DOI] [PubMed] [Google Scholar]
- 23. Sun J., Cui J., He Q., Chen Z., Arvan P., and Liu M. (2015) Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Aspects Med. 42, 105–118 10.1016/j.mam.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kahn S. E., Cooper M. E., and Del Prato S. (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383, 1068–1083 10.1016/S0140-6736(13)62154-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zimmet P., Alberti K. G., and Shaw J. (2001) Global and societal implications of the diabetes epidemic. Nature 414, 782–787 10.1038/414782a [DOI] [PubMed] [Google Scholar]
- 26. Raile K., O'Connell M., Galler A., Werther G., Kühnen P., Krude H., and Blankenstein O. (2011) Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur. J. Endocrinol. 165, 255–260 10.1530/EJE-11-0208 [DOI] [PubMed] [Google Scholar]
- 27. Vinik A., and Bell G. (1988) Mutant insulin syndromes. Horm. Metab. Res. 20, 1–10 10.1055/s-2007-1010736 [DOI] [PubMed] [Google Scholar]
- 28. Izumi T., Yokota-Hashimoto H., Zhao S., Wang J., Halban P. A., and Takeuchi T. (2003) Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 52, 409–416 10.2337/diabetes.52.2.409 [DOI] [PubMed] [Google Scholar]
- 29. Herbach N., Rathkolb B., Kemter E., Pichl L., Klaften M., de Angelis M. H., Halban P. A., Wolf E., Aigner B., and Wanke R. (2007) Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2C95S mutant mice. Diabetes 56, 1268–1276 10.2337/db06-0658 [DOI] [PubMed] [Google Scholar]
- 30. Edghill E. L., Flanagan S. E., Patch A. M., Boustred C., Parrish A., Shields B., Shepherd M. H., Hussain K., Kapoor R. R., Malecki M., MacDonald M. J., Støy J., Steiner D. F., Philipson L. H., Bell G. I., et al. (2008) Insulin mutation screening in 1044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 57, 1034–1042 10.2337/db07-1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu M., Li Y., Cavener D., and Arvan P. (2005) Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J. Biol. Chem. 280, 13209–13212 10.1074/jbc.C400475200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu M., Haataja L., Wright J., Wickramasinghe N. P., Hua Q. X., Phillips N. F., Barbetti F., Weiss M. A., and Arvan P. (2010) Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on nonmutant proinsulin transport. PLoS One 5, e13333 10.1371/journal.pone.0013333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu M., Hodish I., Rhodes C. J., and Arvan P. (2007) Proinsulin maturation, misfolding, and proteotoxicity. Proc. Natl. Acad. Sci. U.S.A. 104, 15841–15846 10.1073/pnas.0702697104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu M., Sun J., Cui J., Chen W., Guo H., Barbetti F., and Arvan P. (2015) INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol. Aspects Med. 42, 3–18 10.1016/j.mam.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hua Q. X., Nakagawa S., Hu S. Q., Jia W., Wang S., and Weiss M. A. (2006) Toward the active conformation of insulin. Stereospecific modulation of a structural switch in the B chain. J. Biol. Chem. 281, 24900–24909 10.1074/jbc.M602691200 [DOI] [PubMed] [Google Scholar]
- 36. Shoelson S. E., Polonsky K. S., Zeidler A., Rubenstein A. H., and Tager H. S. (1984) Human insulin B24 (Phe → Ser), secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J. Clin. Invest. 73, 1351–1358 10.1172/JCI111338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shoelson S., Fickova M., Haneda M., Nahum A., Musso G., Kaiser E. T., Rubenstein A. H., and Tager H. (1983) Identification of a mutant human insulin predicted to contain a serine-for-phenylalanine substitution. Proc. Natl. Acad. Sci. U.S.A. 80, 7390–7394 10.1073/pnas.80.24.7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shoelson S., Haneda M., Blix P., Nanjo A., Sanke T., Inouye K., Steiner D., Rubenstein A., and Tager H. (1983) Three mutant insulins in man. Nature 302, 540–543 10.1038/302540a0 [DOI] [PubMed] [Google Scholar]
- 39. Pandyarajan V., Smith B. J., Phillips N. B., Whittaker L., Cox G. P., Wickramasinghe N., Menting J. G., Wan Z. L., Whittaker J., Ismail-Beigi F., Lawrence M. C., and Weiss M. A. (2014) Aromatic anchor at an invariant hormone–receptor interface function of insulin residue B24 with application to protein design. J. Biol. Chem. 289, 34709–34727 10.1074/jbc.M114.608562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Menting J. G., Yang Y., Chan S. J., Phillips N. B., Smith B. J., Whittaker L. J., Wickramasinghe N. P., Whittaker L., Pandyarajan V., Wan Z. L., Yadav S. P., Carroll J. M., Strokes N., Roberts C. T. Jr., Ismail-Beigi F., et al. (2014) A structural hinge in insulin enables its receptor engagement. Proc. Natl. Acad. Sci. U.S.A. 111, E3395–E3404 10.1073/pnas.1412897111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. M., Hubbard R. E., Isaacs N. W., and Reynolds C. D. (1988) The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319, 369–456 10.1098/rstb.1988.0058 [DOI] [PubMed] [Google Scholar]
- 42. Mirmira R. G., Nakagawa S. H., and Tager H. S. (1991) Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J. Biol. Chem. 266, 1428–1436 [PubMed] [Google Scholar]
- 43. Conlon J. M. (2000) Molecular evolution of insulin in non-mammalian vertebrates. Am. Zool. 40, 200–212 10.1093/icb/40.2.200 [DOI] [Google Scholar]
- 44. Grönke S., Clarke D. F., Broughton S., Andrews T. D., and Partridge L. (2010) Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6, e1000857 10.1371/journal.pgen.1000857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mirmira R. G., and Tager H. S. (1989) Role of the phenylalanine B24 side chain in directing insulin interaction with its receptor: Importance of main chain conformation. J. Biol. Chem. 264, 6349–6354 [PubMed] [Google Scholar]
- 46. Nakagawa S. H., and Tager H. S. (1986) Role of the phenylalanine B25 side chain in directing insulin interaction with its receptor. Steric and conformational effects. J. Biol. Chem. 261, 7332–7341 [PubMed] [Google Scholar]
- 47. Mirmira R. G., and Tager H. S. (1991) Disposition of the phenylalanine B25 side chain during insulin-receptor and insulin-insulin interactions. Biochemistry 30, 8222–8229 10.1021/bi00247a019 [DOI] [PubMed] [Google Scholar]
- 48. Pandyarajan V., Phillips N. B., Rege N., Lawrence M. C., Whittaker J., and Weiss M. A. (2016) Contribution of TyrB26 to the function and stability of insulin. Structure–activity relationships at a conserved hormone–receptor interface. J. Biol. Chem. 291, 12978–12990 10.1074/jbc.M115.708347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chan S. J., and Steiner D. F. (2000) Insulin through the ages: phylogeny of a growth promoting and metabolic regulatory hormone. Am. Zool. 40, 213–222 [Google Scholar]
- 50. Ludvigsen S., Olsen H. B., and Kaarsholm N. C. (1998) A structural switch in a mutant insulin exposes key residues for receptor binding. J. Mol. Biol. 279, 1–7 10.1006/jmbi.1998.1801 [DOI] [PubMed] [Google Scholar]
- 51. Žáková L, Kletvíková E., Veverka V., Lepsík M., Watson C. J., Turkenburg J. P., Jirácek J, and Brzozowski A. M. (2013) Structural integrity of the B24 site in human insulin is important for hormone functionality. J. Biol. Chem. 288, 10230–10240 10.1074/jbc.M112.448050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobayashi M., Ohgaku S., Iwasaki M., Maegawa H., Shigeta Y., and Inouye K. (1982) Supernormal insulin: [d-PheB24]insulin with increased affinity for insulin receptors. Biochem. Biophys. Res. Commun. 107, 329–336 10.1016/0006-291X(82)91708-9 [DOI] [PubMed] [Google Scholar]
- 53. Hua Q. X., Shoelson S. E., and Weiss M. A. (1992) Nonlocal structural perturbations in a mutant human insulin: sequential resonance assignment and 13C-isotope-aided 2D-NMR studies of [PheB24 → Gly]insulin with implications for receptor recognition. Biochemistry 31, 11940–11951 10.1021/bi00162a037 [DOI] [PubMed] [Google Scholar]
- 54. Olsen H. B., Ludvigsen S., and Kaarsholm N. C. (1996) Solution structure of an engineered insulin monomer at neutral pH. Biochemistry 35, 8836–8845 10.1021/bi960292+ [DOI] [PubMed] [Google Scholar]
- 55. Inouye K., Watanabe K., Morihara K., Tochino Y., Kanaya T., Emura J., and Sakakibara S. (1979) Enzyme-assisted semisynthesis of human insulin. J. Am. Chem. Soc. 101, 751–752 10.1021/ja00497a051 [DOI] [Google Scholar]
- 56. Ribel U., Hougaard P., Drejer K., and Sørensen A. R. (1990) Equivalent in vivo biological activity of insulin analogues and human insulin despite different in vitro potencies. Diabetes 39, 1033–1039 10.2337/diab.39.9.1033 [DOI] [PubMed] [Google Scholar]
- 57. Dodson G., and Steiner D. (1998) The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8, 189–194 10.1016/S0959-440X(98)80037-7 [DOI] [PubMed] [Google Scholar]
- 58. Listov-Saabye N., Jensen M. B., Kiehr B., Hansen E. W., Svendsen J. E., Lundby A., Holm G. M., and Oleksiewicz M. B. (2009) MCF-7 human mammary adenocarcinoma cells exhibit augmented responses to human insulin on a collagen IV surface. J. Appl. Toxicol. 29, 470–477 10.1002/jat.1428 [DOI] [PubMed] [Google Scholar]
- 59. Glidden M. D., Aldabbagh K., Phillips N. B., Carr K., Chen Y. S., Whittaker J., Phillips M., Wickramasinghe N. P., Rege N., Swain M., Peng Y., Yang Y., Lawrence M. C., Yee V. C., Ismail-Beigi F., and Weiss M. A. (2018) Crystal structure and function of an ultra-stable single chain insulin analog. J. Biol. Chem. 293, 47–68 10.1074/jbc.M117.808626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pandyarajan V., Phillips N. B., Cox G. P., Yang Y., Whittaker J., Ismail-Beigi F., and Weiss M. A. (2014) Biophysical optimization of a therapeutic protein by non-standard mutagenesis. Studies of an iodo-insulin derivative. J. Biol. Chem. 289, 23367–23381 10.1074/jbc.M114.588277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sosnick T. R., Fang X., and Shelton V. M. (2000) Application of circular dichroism to study RNA folding transitions. Methods Enzymol. 317, 393–409 10.1016/S0076-6879(00)17026-0 [DOI] [PubMed] [Google Scholar]
- 62. Hua Q. X., Jia W., and Weiss M. A. (2011) Conformational dynamics of insulin. Front. Endocrinol. 2, 48 10.3389/fendo.2011.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jørgensen A. M., Kristensen S. M., Led J. J., and Balschmidt P. (1992) Three-dimensional solution structure of an insulin dimer. A study of the B9(Asp) mutant of human insulin using nuclear magnetic resonance, distance geometry and restrained molecular dynamics. J. Mol. Biol. 227, 1146–1163 10.1016/0022-2836(92)90527-Q [DOI] [PubMed] [Google Scholar]
- 64. Birnbaum D. T., Kilcomons M. A., DeFelippis M. R., and Beals J. M. (1997) Assembly and dissociation of human insulin and LysB28ProB29-insulin hexamers: a comparison study. Pharm. Res. 14, 25–36 10.1023/A:1012095115151 [DOI] [PubMed] [Google Scholar]
- 65. Chodera J. D., and Mobley D. L. (2013) Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 42, 121–142 10.1146/annurev-biophys-083012-130318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. C., Mercola D. A., and Vijayan M. (1971) Atomic positions in rhombohedral 2-zinc insulin crystals. Nature 231, 506–511 10.1038/231506a0 [DOI] [PubMed] [Google Scholar]
- 67. Jiménez J. L., Nettleton E. J., Bouchard M., Robinson C. V., Dobson C. M., and Saibil H. R. (2002) The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201 10.1073/pnas.142459399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Steiner D. F., Clark J. L., Nolan C., Rubenstein A. H., Margoliash E., Aten B., and Oyer P. E. (1969) Proinsulin and the biosynthesis of insulin. Recent Prog. Horm. Res. 25, 207–282 10.1016/b978-0-12-571125-8.50008-9 [DOI] [PubMed] [Google Scholar]
- 69. Liu M., Ramos-Castañeda J., and Arvan P. (2003) Role of the connecting peptide in insulin biosynthesis. J. Biol. Chem. 278, 14798–14805 10.1074/jbc.M212070200 [DOI] [PubMed] [Google Scholar]
- 70. Wright J., Birk J., Haataja L., Liu M., Ramming T., Weiss M. A., Appenzeller-Herzog C., and Arvan P. (2013) Endoplasmic reticulum oxidoreductin-1a (Ero1a) improves folding and secretion of mutant proinsulin and limits mutant proinsulin-induced ER stress. J. Biol. Chem. 288, 31010–31018 10.1074/jbc.M113.510065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Støy J., Edghill E. L., Flanagan S. E., Ye H., Paz V. P., Pluzhnikov A., Below J. E., Hayes M. G., Cox N. J., Lipkind G. M., Lipton R. B., Greeley S. A., Patch A. M., Ellard S., Steiner D. F., et al. (2007) Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. U.S.A. 104, 15040–15044 10.1073/pnas.0707291104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tirasophon W., Welihinda A. A., and Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Anfinsen C. B. (1973) Principles that govern the folding of protein chains. Science 181, 223–230 10.1126/science.181.4096.223 [DOI] [PubMed] [Google Scholar]
- 74. Karplus M. (1997) The Levinthal paradox: yesterday and today. Fold. Des. 2, S69–75 10.1016/S1359-0278(97)00067-9 [DOI] [PubMed] [Google Scholar]
- 75. Sali A., Shakhnovich E., and Karplus M. (1994) How does a protein fold. Nature 369, 248–251 10.1038/369248a0 [DOI] [PubMed] [Google Scholar]
- 76. Yu M. H., and King J. (1984) Single amino acid substitutions influencing the folding pathway of the phage P22 tail spike endorhamnosidase. Proc. Natl. Acad. Sci. U.S.A. 81, 6584–6588 10.1073/pnas.81.21.6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dobson C. M. (2001) Protein folding and its links with human disease. Biochem. Soc. Symp. 2001, 1–26 [PubMed] [Google Scholar]
- 78. Liu M., Weiss M. A., Arunagiri A., Yong J., Rege N., Sun J., Haataja L., Kaufman R. J., and Arvan P. (2018) Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 20, 28–50 10.1111/dom.13378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hansen B. F., Kurtzhals P., Jensen A. B., Dejgaard A., and Russell-Jones D. (2011) Insulin X10 revisited: a super-mitogenic insulin analogue. Diabetologia 54, 2226–2231 10.1007/s00125-011-2203-8 [DOI] [PubMed] [Google Scholar]
- 80. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Bing X., and Markussen J. (1991) X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule. A completely inactive analogue. J. Mol. Biol. 220, 425–433 10.1016/0022-2836(91)90022-X [DOI] [PubMed] [Google Scholar]
- 81. Hua Q. X., Shoelson S. E., Kochoyan M., and Weiss M. A. (1991) Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354, 238–241 10.1038/354238a0 [DOI] [PubMed] [Google Scholar]
- 82. Xu B., Huang K., Chu Y. C., Hu S. Q., Nakagawa S., Wang S., Wang R. Y., Whittaker J., Katsoyannis P. G., and Weiss M. A. (2009) Decoding the cryptic active conformation of a protein by synthetic photoscanning: insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 284, 14597–14608 10.1074/jbc.M900087200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nakagawa S. H., and Tager H. S. (1989) Perturbation of insulin-receptor interactions by intramolecular hormone cross-linking. Analysis of relative movement among residues A1, B1, and B29. J. Biol. Chem. 264, 272–279 [PubMed] [Google Scholar]
- 84. Hua Q. X., Hu S. Q., Jia W., Chu Y. C., Burke G. T., Wang S. H., Wang R. Y., Katsoyannis P. G., and Weiss M. A. (1998) Mini-proinsulin and mini-IGF-I: homologous protein sequences encoding non-homologous structures. J. Mol. Biol. 277, 103–118 10.1006/jmbi.1997.1574 [DOI] [PubMed] [Google Scholar]
- 85. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., et al. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 10.1038/nature11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Weiss M. A., and Lawrence M. C. (2018) A thing of beauty: structure and function of insulin's “Aromatic Triplet”. Diabetes Obes. Metab. 20, 51–63 10.1111/dom.13402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. De Meyts P. (2015) Insulin/receptor binding: the last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. Bioessays 37, 389–397 10.1002/bies.201400190 [DOI] [PubMed] [Google Scholar]
- 88. Scapin G., Dandey V. P., Zhang Z., Prosise W., Hruza A., Kelly T., Mayhood T., Strickland C., Potter C. S., and Carragher B. (2018) Structure of the insulin receptor–insulin complex by single-particle cryo-EM analysis. Nature 556, 122–125 10.1038/nature26153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Upton Z., Yandell C. A., Degger B. G., Chan S. J., Moriyama S., Francis G. L., and Ballard F. J. (1998) Evolution of insulin-like growth factor-I (IGF-I) action: in vitro characterization of vertebrate IGF-I proteins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 121, 35–41 10.1016/S0305-0491(98)10111-6 [DOI] [PubMed] [Google Scholar]
- 90. Knudsen L., Hansen B. F., Jensen P., Pedersen T. Å., Vestergaard K., Schäffer L., Blagoev B., Oleksiewicz M. B., Kiselyov V. V., and De Meyts P. (2012) Agonism and antagonism at the insulin receptor. PLoS ONE 7, e51972 10.1371/journal.pone.0051972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Eriksson A. E., Baase W. A., Zhang X. J., Heinz D. W., Blaber M., Baldwin E. P., and Matthews B. W. (1992) Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255, 178–183 10.1126/science.1553543 [DOI] [PubMed] [Google Scholar]
- 92. Pires D. E., Ascher D. B., and Blundell T. L. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 10.1093/bioinformatics/btt691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu M., Hua Q.-X., Hu S.-Q., Jia W., Yang Y., Saith S. E., Whittaker J., Arvan P., and Weiss M. A. (2010) Deciphering the hidden informational content of protein sequences: foldability of proinsulin hinges on a flexible arm that is dispensable in the mature hormone. J. Biol. Chem. 285, 30989–31001 10.1074/jbc.M110.152645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vinther T. N., Norrman M., Ribel U., Huus K., Schlein M., Steensgaard D. B., Pedersen T. Å., Pettersson I., Ludvigsen S., Kjeldsen T., Jensen K. J., and Hubálek F. (2013) Insulin analog with additional disulfide bond has increased stability and preserved activity. Protein Sci. 22, 296–305 10.1002/pro.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brange J., Andersen L., Laursen E. D., Meyn G., and Rasmussen E. (1997) Toward understanding insulin fibrillation. J. Pharm. Sci. 86, 517–525 10.1021/js960297s [DOI] [PubMed] [Google Scholar]
- 96. Dumoulin M., Canet D., Last A. M., Pardon E., Archer D. B., Muyldermans S., Wyns L., Matagne A., Robinson C. V., Redfield C., and Dobson C. M. (2005) Reduced global cooperativity is a common feature underlying the amyloidogenicity of pathogenic lysozyme mutations. J. Mol. Biol. 346, 773–788 10.1016/j.jmb.2004.11.020 [DOI] [PubMed] [Google Scholar]
- 97. Attri A. K., Fernández C., and Minton A. P. (2010) Self-association of Zn-insulin at neutral pH: investigation by concentration gradient–static and dynamic light scattering. Biophys. Chem. 148, 23–27 10.1016/j.bpc.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hellman U., Wernstedt C., Westermark P., O'Brien T. D., Rathbun W. B., and Johnson K. H. (1990) Amino acid sequence from degu islet amyloid–derived insulin shows unique sequence characteristics. Biochem. Biophys. Res. Commun. 169, 571–577 10.1016/0006-291X(90)90369-X [DOI] [PubMed] [Google Scholar]
- 99. Nishi M., and Steiner D. F. (1990) Cloning of complementary DNAs encoding islet amyloid polypeptide, insulin, and glucagon precursors from a New World rodent, the degu, Octodon degus. Mol. Endocrinol. 4, 1192–1198 10.1210/mend-4-8-1192 [DOI] [PubMed] [Google Scholar]
- 100. Ciszak E., Beals J. M., Frank B. H., Baker J. C., Carter N. D., and Smith G. D. (1995) Role of C-terminal B chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure 3, 615–622 10.1016/S0969-2126(01)00195-2 [DOI] [PubMed] [Google Scholar]
- 101. Steiner D. F., Cho S., Bayliss C., and Hallund O. (1968) On the isolation and some properties of bovine proinsulin. Diabetes 17, 309 [PubMed] [Google Scholar]
- 102. Chan S. J., Seino S., Gruppuso P. A., Schwartz R., and Steiner D. F. (1987) A mutation in the B chain coding region is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc. Natl. Acad. Sci. U.S.A. 84, 2194–2197 10.1073/pnas.84.8.2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Park S. Y., Ye H., Steiner D. F., and Bell G. I. (2010) Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem. Biophys. Res. Commun. 391, 1449–1454 10.1016/j.bbrc.2009.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Meur G., Simon A., Harun N., Virally M., Dechaume A., Bonnefond A., Fetita S., Tarasov A. I., Guillausseau P. J., Boesgaard T. W., Pedersen O., Hansen T., Polak M., Gautier J. F., Froguel P., Rutter G. A., and Vaxillaire M. (2010) Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 59, 653–661 10.2337/db09-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vetere A., Choudhary A., Burns S. M., and Wagner B. K. (2014) Targeting the pancreatic β-cell to treat diabetes. Nat. Rev. Drug Discov. 13, 278–289 10.1038/nrd4231 [DOI] [PubMed] [Google Scholar]
- 106. Pierce S. B., Costa M., Wisotzkey R., Devadhar S., Homburger S. A., Buchman A. R., Ferguson K. C., Heller J., Platt D. M., Pasquinelli A. A., Liu L. X., Doberstein S. K., and Ruvkun G. (2001) Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 15, 672–686 10.1101/gad.867301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hua Q. X., Nakagawa S. H., Wilken J., Ramos R. R., Jia W., Bass J., and Weiss M. A. (2003) A divergent INS protein in Caenorhabditis elegans structurally resembles human insulin and activates the human insulin receptor. Genes Dev. 17, 826–831 10.1101/gad.1058003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Menting J. G., Gajewiak J., MacRaild C. A., Chou D. H., Disotuar M. M., Smith N. A., Miller C., Erchegyi J., Rivier J. E., Olivera B. M., Forbes B. E., Smith B. J., Norton R. S., Safavi-Hemami H., and Lawrence M. C. (2016) A minimized human insulin-receptor-binding motif revealed in a Conus geographus venom insulin. Nat. Struct. Mol. Biol. 23, 916–920 10.1038/nsmb.3292 [DOI] [PubMed] [Google Scholar]
- 109. Barany G., and Merrifield R. B. (1980) in The Peptides (Gross E., and Meienhofer J., eds) pp. 273–284, Academic Press, New York [Google Scholar]
- 110. Pace C. N., and Shaw K. L. (2000) Linear extrapolation method of analyzing solvent denaturation curves. Proteins 2000, Suppl. 4, 1–7 [DOI] [PubMed] [Google Scholar]
- 111. Wang Z. X. (1995) An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule FEBS Lett. 360, 111–114 10.1016/0014-5793(95)00062-E [DOI] [PubMed] [Google Scholar]
- 112. Roy M., Brader M. L., Lee R. W., Kaarsholm N. C., Hansen J. F., and Dunn M. F. (1989) Spectroscopic signatures of the T to R conformational transition in the insulin hexamer. J. Biol. Chem. 264, 19081–19085 [PubMed] [Google Scholar]
- 113. Arunagiri A., Haataja L., Pottekat A., Pamenan F., Kim S., Zeltser L. M., Paton A. W., Paton J. C., Tsai B., Itkin-Ansari P., Kaufman R. J., Liu M., and Arvan P. (2019) Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife 8, e44532 10.7554/eLife.44532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Haataja L., Snapp E., Wright J., Liu M., Hardy A. B., Wheeler M. B., Markwardt M. L., Rizzo M., and Arvan P. (2013) Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J. Biol. Chem. 288, 1896–1906 10.1074/jbc.M112.420018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6. 1–5.6. 32 10.1002/0471250953.bi0506s47 [DOI] [PubMed] [Google Scholar]
- 116. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 117. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Guvench O., Mallajosyula S. S., Raman E. P., Hatcher E., Vanommeslaeghe K., Foster T. J., Jamison F. W. 2nd., and Mackerell A. D. Jr. (2011) CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 7, 3162–3180 10.1021/ct200328p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101–14107 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 120. Berendsen H. J., Postma J. P. M., van Gunsteren W. F., DiNola A., and Haak J. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 10.1063/1.448118 [DOI] [Google Scholar]
- 121. Pires D. E., Ascher D. B., and Blundell T. L. (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319 10.1093/nar/gku411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Paramo T., East A., Garzón D., Ulmschneider M. B., and Bond P. J. (2014) Efficient characterization of protein cavities within molecular simulation trajectories: trj_cavity. J. Chem. Theory Comput. 10, 2151–2164 10.1021/ct401098b [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.