

Abstract
Quorum sensing is a bacterial communication process whereby bacteria produce, release, and detect extracellular signaling molecules called autoinducers to coordinate collective behaviors. In the pathogen Vibrio cholerae, the quorum-sensing autoinducer 3,5-dimethyl-pyrazin-2-ol (DPO) binds the receptor and transcription factor VqmA. The DPO-VqmA complex activates transcription of vqmR, encoding the VqmR small RNA, which represses genes required for biofilm formation and virulence factor production. Here, we show that VqmA is soluble and properly folded and activates basal-level transcription of its target vqmR in the absence of DPO. VqmA transcriptional activity is increased in response to increasing concentrations of DPO, allowing VqmA to drive the V. cholerae quorum-sensing transition at high cell densities. We solved the DPO-VqmA crystal structure to 2.0 Å resolution and compared it with existing structures to understand the conformational changes VqmA undergoes upon DNA binding. Analysis of DPO analogs showed that a hydroxyl or carbonyl group at the 2′-position is critical for binding to VqmA. The proposed DPO precursor, a linear molecule, N-alanyl-aminoacetone (Ala-AA), also bound and activated VqmA. Results from site-directed mutagenesis and competitive ligand-binding analyses revealed that DPO and Ala-AA occupy the same binding site. In summary, our structure-function analysis identifies key features required for VqmA activation and DNA binding and establishes that, whereas VqmA binds two different ligands, VqmA does not require a bound ligand for folding or basal transcriptional activity. However, bound ligand is required for maximal activity.
Keywords: quorum sensing; Vibrio cholerae; crystallography; bacteria; gene regulation; 3,5-dimethyl-pyrazin-2-ol (DPO); autoinducer; binding; N-alanyl-aminoacetone (Ala-AA); VqmA
Introduction
Bacteria communicate and orchestrate collective behaviors using a process called quorum sensing (QS).3 QS relies on the production, release, detection, and group-wide response to extracellular signaling molecules called autoinducers (AIs) that accumulate with increasing cell density (reviewed in Ref. 1). In the global pathogen Vibrio cholerae, multiple QS pathways converge to repress biofilm formation and virulence factor production at high cell density (HCD) (1–6). One of the V. cholerae QS pathways is composed of an AI, 3,5-dimethyl-pyrazin-2-ol (DPO), and its partner cytoplasmic receptor, VqmA. VqmA is a transcription factor that, upon binding DPO, activates expression of a gene encoding a small RNA called VqmR (7, 8). VqmR post-transcriptionally controls many target genes (7, 9). Of note, VqmR represses translation of ctx and rtxA, two toxin-encoding genes; vpsT, encoding a component required for biofilm formation; and aphA, encoding the low-cell density (LCD) QS master regulator AphA (7, 9). Thus, analogous to the previously discovered V. cholerae QS systems, the consequence of DPO binding to VqmA at HCD is repression of virulence and biofilm formation.
Homologs of the VqmA receptor are restricted to the Vibrio genus and the vibriophage VP882 (10). By contrast, threonine dehydrogenase (Tdh), an enzyme required for DPO biosynthesis, is highly conserved among bacteria, archaea, and eukarya (11). Consistent with this distribution, with respect to bacteria, DPO is produced by multiple species in different orders (8). Tdh oxidizes l-threonine to 2-amino-3-ketobutyric acid (AKB), which spontaneously decarboxylates to aminoacetone (12). In the proposed mechanism for DPO biosynthesis, aminoacetone is predicted to undergo a condensation reaction with l-alanine to yield N-alanyl-aminoacetone (Ala-AA) (8). This putative linear precursor is subsequently converted to DPO by an unknown mechanism.
In this report, we characterize ligand-controlled activation of VqmA in V. cholerae. In contrast to canonical LuxR-type QS receptor-transcription factors, which typically require binding to their cognate ligands to fold and activate transcription (13–16), here, we find that VqmA does not require DPO for folding or basal transcriptional activity but does require binding DPO to drive maximal activity. These same properties are relevant for the phage VP882-encoded VqmA homolog (VqmAPhage), suggesting that ligand-independent activity is a conserved feature among VqmA proteins. We show that VqmA binds to and is activated by both DPO and the putative DPO intermediate, Ala-AA. We solved the crystal structure of VqmA bound to DPO. Comparison with existing structures (17, 18) allowed us to provide insight into the conformational changes VqmA undergoes upon DNA binding. We were unable to obtain the crystal structure of VqmA bound to Ala-AA because Ala-AA spontaneously cyclizes into DPO under our crystallization conditions. Mutagenesis of the VqmA ligand-binding pocket shows that DPO and Ala-AA likely occupy the same site in VqmA, as substitutions in residues that alter DPO binding and activity have similar effects on Ala-AA binding and activity. Characterization of binding by isothermal titration calorimetry (ITC)-based competition experiments and kinetic studies reveals that VqmA prefers DPO over Ala-AA. Assessment of the activities of a panel of DPO analogs revealed that a 2′-hydroxyl or -carbonyl group is required for binding to VqmA. Taken together, our results show that VqmA is promiscuous with respect to DPO and Ala-AA. Nonetheless, maximal VqmA activity depends on ligand binding. The ability of VqmA to activate gene expression to different levels in the unbound state and when bound to two different ligands could enable greater flexibility in regulating gene expression than exists in canonical LuxR-type QS receptors, which typically have no activity in the apo-state and show strict specificity for a single partner ligand.
Results
VqmA possesses both ligand-independent and ligand-dependent activity
VqmA is a cytoplasmic transcription factor and QS receptor that binds the AI DPO, a hydroxypyrazine that requires the Tdh enzyme for its production (8). The VqmA receptor is restricted to vibrios and one vibriophage. By contrast, canonical LuxR-type cytoplasmic QS receptor-transcription factors exist in thousands of Gram-negative bacterial species, and they bind AIs that are acyl-homoserine lactones (AHLs) (reviewed in Ref. 1). AHL AIs are produced by LuxI-type AI synthases. LuxR-type receptors typically require binding to their cognate AIs to properly fold, become soluble, and activate transcription (13–16). Thus, LuxI-type AI synthases and LuxR-type receptors function as obligate pairs, and mutation of the luxI gene confers the identical defect as does mutation of the luxR gene (19). We wondered whether the Tdh-VqmA pair functions in an analogous manner. To probe this question, we constructed a reporter system to monitor activation of transcription by VqmA. We fused the lux (luciferase) operon to the vqmR promoter (PvqmR-lux) and integrated it into the V. cholerae chromosome at an ectopic locus (lacZ). We measured transcriptional activity in different V. cholerae strains and across different conditions. WT V. cholerae harboring the PvqmR-lux reporter exhibited maximal activity at HCD, which is consistent with our previous report (Fig. 1A) (8). The Δtdh mutant, which is defective for DPO production, elicited ∼10-fold lower activity (Fig. 1A). Restoration of light production to approximately the WT level occurred in the Δtdh mutant following the exogenous addition of 1 μm DPO (Fig. 1A). In the ΔvqmA strain, PvqmR-lux levels were ∼330-fold lower than in the WT and ∼23-fold lower than in the Δtdh strain to which no DPO was added (Fig. 1A). The exogenous addition of DPO did not induce light production in the ΔvqmA strain (Fig. 1A). Thus, unlike what is observed for canonical LuxI-LuxR-type systems, the Δtdh synthase mutant and the ΔvqmA receptor mutant have different phenotypes with respect to QS output.
Figure 1.

VqmA is active in the absence of DPO. A, PvqmR-lux reporter activity for the indicated V. cholerae strains after 6 h of growth in the absence or presence of 1 μm DPO. The cells were at HCD at this time point. Data are represented as mean ± S.D. (error bars) with n = 3 biological replicates. B, SDS-polyacrylamide gel of purified His-VqmA protein produced in WT or tdh::kan E. coli without or with 100 μm DPO added during expression. M, molecular weight marker. C, EMSA showing VqmA binding to PvqmR promoter DNA. 540 pm biotinylated PvqmR DNA and either no protein (lane 1, −), apo-VqmA (lanes 2–8), or holo-VqmA (lanes 9–15). Relative protein concentrations are indicated: 1× = 2 nm and 64× = 130 nm. D, EMSA showing binding of 50 nm apo-VqmA (lanes 2–8) to PvqmR DNA in the absence and presence of the indicated amounts of DPO. Lane 1, as in C. In C and D, quantitation of the percentage of PvqmR DNA bound appears below each lane. RLU, relative light units.
Two possibilities could underpin the differences in the lux phenotypes of the ΔvqmA and Δtdh mutants: either DPO is produced via a Tdh-independent mechanism or VqmA displays basal activity in the absence of its ligand. To investigate the first possibility, we quantified DPO production by WT, Δtdh, and ΔvqmA V. cholerae. Cell-free culture fluids prepared from each strain were supplied to the V. cholerae Δtdh PvqmR-lux reporter strain, and light production was measured (Fig. S1A). Activity present in the preparations was compared with that following the addition of known amounts of synthetic DPO. The EC50 for synthetic DPO is ∼1 μm (8). Using this value, we could estimate that the WT and ΔvqmA culture fluids contained ∼1–2 μm DPO, whereas the amount of DPO in culture fluid from the Δtdh strain was below the 60 nm detection limit of the reporter (Fig. S1A). Thus, if any DPO is produced by a Tdh-independent mechanism(s), it is insufficient to elicit the order-of-magnitude difference in PvqmR activity between the ΔvqmA and Δtdh V. cholerae strains in Fig. 1A. To test the second possibility, that VqmA is expressed and possesses measurable activity in the absence of DPO, we produced VqmA in WT (i.e. tdh+) and tdh::kan Escherichia coli and assessed its solubility. E. coli, like V. cholerae, produces DPO in a Tdh-dependent manner (8). Fig. 1B shows that comparable amounts of soluble VqmA were obtained from the WT and tdh::kan strains, irrespective of the addition of DPO. To eliminate the possibility that a trace amount of DPO is produced by the tdh::kan strain or that some other ligand stabilizes VqmA in tdh::kan E. coli, we produced hexahistidine-tagged VqmA (His-VqmA) via in vitro translation using a cell-free system. In vitro translated His-VqmA protein was readily produced and soluble (Fig. S1B). Thus, we conclude that VqmA does not depend on a ligand to properly fold.
In V. cholerae, VqmA is produced constitutively, whereas vqmR is expressed predominantly at HCD (8). Given our finding that the solubility of VqmA is unaffected by DPO (Fig. 1B), we hypothesized that the VqmA DNA-binding capability could be enhanced by DPO. To examine VqmA DNA binding as a function of ligand occupancy, we purified VqmA from tdh::kan E. coli that had been grown in the presence or absence of exogenously supplied DPO (100 μm). We assessed apo- and holo-VqmA binding to PvqmR DNA using electromobility shift assays (EMSAs). Our results show that, at sufficient protein concentrations, apo-VqmA shifted PvqmR DNA (Fig. 1C), consistent with our finding that VqmA possesses basal activation of PvqmR in the apo-state. We found that 2–8-fold more protein was required to achieve a ∼50% shift of the PvqmR DNA with apo-VqmA compared with holo-VqmA (Fig. 1C). Thus, holo-VqmA has a higher affinity for PvqmR than apo-VqmA. Furthermore, the addition of DPO to apo-VqmA increased DNA binding to PvqmR in a DPO concentration–dependent manner (Fig. 1D). We conclude that the basal DNA-binding activity of apo-VqmA is enhanced when VqmA is bound to DPO. These results explain why the V. cholerae Δtdh mutant exhibits PvqmR-lux expression, whereas the ΔvqmA mutant does not (Fig. 1A).
Considering the differences in the biochemical properties of the VqmA receptor relative to canonical LuxR-type QS receptors, we wanted to examine whether their control of QS outputs also differed. To do this, we measured the effect of DPO binding to VqmA on PvqmR activation, representing the V. cholerae VqmA pathway, and compared the results with the well-characterized LasI-LasR QS system from Pseudomonas aeruginosa. Briefly, in P. aeruginosa, upon binding 3OC12-homoserine lactone (3OC12-HSL), LasR activates transcription of many genes, including, germane to our studies, lasB (20). Here, we introduced the relevant P. aeruginosa and V. cholerae QS components into E. coli (Fig. 2). Specifically, we used Δtdh E. coli as the host for both reporter systems to eliminate native AI production (E. coli does not produce 3OC12-HSL). Into this strain, we introduced a pair of plasmids encoding either arabinose-inducible vqmA and PvqmR-lux or arabinose-inducible lasR and PlasB-lux. Consistent with our finding in Δtdh V. cholerae (Fig. 1A), E. coli producing VqmA in the absence of DPO generated 15-fold more PvqmR reporter activity than a strain lacking the vector harboring arabinose-inducible vqmA (Fig. 2). In response to exogenous DPO, PvqmR-driven light production increased an additional 3-fold. LasR, by contrast, expressed minimal PlasB-lux production in the absence of 3OC12-HSL relative to the no-vector control. In response to 3OC12-HSL, a 143-fold increase in PlasB-lux expression occurred (Fig. 2). Taken together, these results show that both VqmA and LasR are highly activated by their respective AIs. However, VqmA activity, in contrast to LasR activity, is not strictly dependent on an AI. In the opposite vein, the relative activation achieved by 3OC12-HSL-LasR is almost 2 orders of magnitude greater than that for the DPO-VqmA complex.
Figure 2.
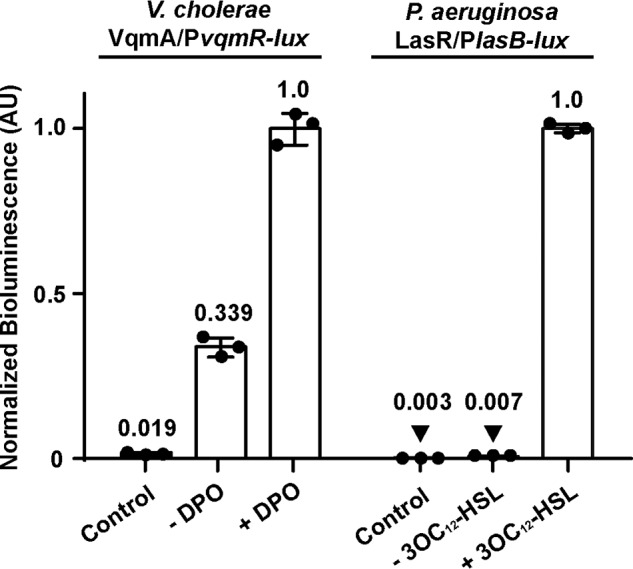
VqmA exhibits activity in the absence of its cognate AI, and the LasR QS receptor does not. Shown is normalized reporter activity from E. coli harboring the VqmA/PvqmR-lux or the LasR/PlasB-lux system. Left, E. coli carrying one plasmid with PvqmR-lux and a second plasmid with pBAD-vqmA. The strain represented by the bar designated Control lacks the pBAD-vqmA plasmid. Right, E. coli carrying one plasmid with PlasB-lux and a second plasmid with pBAD-lasR. The strain represented by the bar designated Control lacks the pBAD-lasR plasmid. All strains were grown in the presence of 0.075% arabinose and in the absence or presence of the designated AI (20 μm DPO or 1 μm 3OC12-HSL). AIs were added to the control strains. Data are represented as mean ± S.D. (error bars) with n = 3 biological replicates. AU, arbitrary units.
We wondered whether the ligand-independent response we observed for V. cholerae VqmA extended to other VqmA proteins, such as that from the phage VP882-encoded VqmA homolog, VqmAPhage. VqmAPhage binds host-produced DPO and, in response, activates expression of the gene, qtip. Qtip promotes host cell lysis (10). Similar to V. cholerae VqmA, VqmAPhage activated Pqtip transcription in the absence of DPO, and activation was enhanced ∼3-fold when DPO was added (Fig. S1C). VqmAPhage shares ∼43% identity with V. cholerae VqmA, and the key residues required for ligand and DNA binding are highly conserved (10). These features likely underlie the similar behaviors in the reporter assays. Thus, the VqmA AI-independent function extends to the phage-encoded system and is likely an inherent property of VqmA proteins more generally.
The proposed linear DPO precursor Ala-AA binds to and activates VqmA
As noted, Tdh is required to convert l-threonine to AKB, which spontaneously decarboxylates to aminoacetone. Aminoacetone is presumed to condense with l-alanine to form the putative DPO precursor Ala-AA (8) (Fig. 3A). Consistent with the placement of threonine upstream and Ala-AA downstream of Tdh in this pathway, we previously showed that the exogenous addition of Ala-AA, but not threonine, restores PvqmR-lux expression in Δtdh V. cholerae to WT levels (8). This finding suggested one of two possibilities. First, in our cell-based assay, the cells convert Ala-AA to DPO, and newly produced DPO drives PvqmR-lux expression; alternatively, Ala-AA could bind to VqmA and induce PvqmR-lux expression without conversion to DPO. To distinguish between these possibilities, we performed EMSAs using apo-VqmA to which we added either threonine, DPO, or Ala-AA (Fig. 3A). As anticipated, provision of l-threonine did not increase VqmA binding relative to when water was added (Fig. 3B). By contrast, inclusion of DPO increased the percentage of PvqmR DNA bound to VqmA ∼2–5 fold (Fig. 3B). Surprisingly, the consequence of adding Ala-AA was indistinguishable from that of DPO (Fig. 3B) (i.e. Ala-AA also drives VqmA to bind PvqmR). In vitro translated His-VqmA behaved identically with respect to the ligands (Fig. S2A). Importantly, we excluded the possibility that Ala-AA is converted to DPO under our EMSA conditions using MS (DPO, 125 m/z; Ala-AA, 145 m/z). In each case, the ligand added, DPO or Ala-AA, was the only ligand detected by MS (Fig. S2B). Taken together, we conclude that both DPO and Ala-AA bind and enhance VqmA DNA binding and transcriptional activity.
Figure 3.

Ala-AA, the proposed linear precursor of DPO, binds to and activates VqmA. A, structures of threonine, Ala-AA, and DPO (both tautomers). B, EMSA of VqmA binding to PvqmR DNA in the presence of threonine, DPO, water, or Ala-AA. Probe and protein concentrations were as in Fig. 1C. C, trypsin digestion of VqmA in the absence (H2O, top) or presence of ligand (DPO, middle; Ala-AA, bottom). M, molecular weight marker; FL, full-length. Time points (min) are indicated above each lane. FL VqmA (∼27 kDa) and trypsin (∼23 kDa) are marked. Asterisks denote VqmA proteolytic cleavage products.
To explore whether binding of DPO and Ala-AA to VqmA drives conformational changes and whether they are the same or different, we performed limited proteolysis. Apo-, DPO-bound, and Ala-AA-bound VqmA were digested with trypsin. The DPO-bound and Ala-AA–bound VqmA proteins were more susceptible to digestion than was apo-VqmA (Fig. 3C). One explanation for these results is that a conformational change occurs upon ligand binding and, moreover, that the change is similar for both ligands. Indeed, in companion experiments, we assessed the binding of DPO and Ala-AA to apo-VqmA using thermal shift analysis. Both ligands caused similar increases (about 6 °C) in thermal stability of VqmA (Fig. S2C). Our data support a model in which both DPO and the linear Ala-AA molecule bind VqmA and effectively activate its transcriptional activity.
The above results show that Ala-AA and DPO have identical activities in both our in vivo cell-based and in vitro protein-based assays. One caution, however, is that Ala-AA is the presumed DPO precursor, but its in vivo role has not been verified. After DPO was discovered and threonine and alanine were shown to be incorporated into DPO, the prediction of Ala-AA as the DPO precursor was put forward based on common chemical reactions in which amino acids and amino-containing compounds participate (8). Here, we aimed to prove or disprove whether Ala-AA is indeed the biological precursor to DPO. To do this, we conducted isotopic labeling experiments to determine whether Ala-AA cyclizes into the final DPO product. We administered deuterium-labeled Ala-AA (d3-Ala-AA) ((S)-2-amino-N-(2-oxopropyl)-3,3,3-d3-propanamide, shown in Fig. S3A) to Δtdh V. cholerae and analyzed incorporation of the deuterium label into DPO by LC-MS. As controls, we also administered d3-Ala-AA to 1) medium alone to eliminate the possibility of spontaneous conversion in the absence of cells; 2) Δtdh ΔvqmA V. cholerae to confirm that binding to VqmA is not required for conversion; and 3) WT V. cholerae to show that the amount of labeled DPO (d3-DPO) extracted is identical between the Tdh+ and Tdh− strains to verify that Tdh is not functioning in any additional downstream step in the pathway. We detected d3-DPO in extracts at ∼5% of the total concentration of added d3-Ala-AA (Fig. S3B). However, analysis of our synthetic preparations of Ala-AA and d3-Ala-AA showed that ∼5% DPO and d3-DPO, respectively, are present (Fig. S3C) as irremovable impurities. The presence of small amounts of DPO demonstrated the facile nature of Ala-AA to undergo cyclization under basic conditions. Thus, in the labeling experiments, the low level of d3-DPO present in the extracts can be attributed to the starting material, and it is not a consequence of enzymatic conversion of d3-Ala-AA to d3-DPO (Fig. S3C). There was no case in which administration of d3-Ala-AA to cells resulted in accumulation of recovered DPO with a 3-Da mass shift above the background level. This result suggests that, at least under our conditions, exogenously supplied Ala-AA is not converted into DPO in vivo (Fig. S3B). We suspect that some other compound could be the precursor to DPO, and we are working to discover its identity. Nonetheless, our findings with Ala-AA are curious, as they show that the VqmA receptor binds to and is activated by an apparently nonpathway substrate. We return to ideas concerning the ramifications of these findings under “Discussion.”
Structural and biochemical characterization of VqmA
To explore the molecular basis underlying VqmA binding to ligands, we solved the crystal structure of VqmA bound to DPO in the absence of DNA to 2.0 Å using multiwavelength anomalous diffraction (Table S1). The structure of the DPO-VqmA complex bound to DNA was reported recently (17). Additionally, during preparation of this manuscript, the structure of DPO-VqmA without DNA was reported with a similar conformation as in our structure (18). Together, the three structures enabled comparisons to discover the conformational changes VqmA undergoes upon DNA binding and, in the context of our work, to gain insight into how VqmA could bind both DPO and Ala-AA.
The DPO-VqmA complex is a head-to-head dimer with 1:1 protein/ligand stoichiometry. The structure revealed two distinct domains: an N-terminal Per-Arnt-Sim (PAS) ligand-binding domain (LBD) and a C-terminal helix-turn-helix (HTH) DNA-binding domain (DBD) (Fig. 4A). The N-terminal PAS LBD consists of a characteristic five-stranded antiparallel β-sheet core, which forms the DPO-binding pocket along with several α-helices and loops, and this domain contains the bulk of the buried surface area of the dimerization interface (Fig. 4, B and C, respectively). The PAS domain is connected to the C-terminal HTH DBD by a long flexible loop region. The DBD has a stereotypical HTH tetrahelical fold, which is common in transcription factors, including LuxR-type QS receptors (Fig. 4D). Both the LBD PAS and DBD HTH domains are common across biology. However, according to the DALI server (21), which uses structures from the Protein Data Bank to assess structural homologies based on fold rather than amino acid sequence, protein structures containing a domain composition similar to VqmA have not otherwise been reported. The closest VqmA homologs based on fold either contain the PAS domain as part of a signal transduction protein (i.e. TodS and VraR) or the HTH domain as part of a transcription factor (i.e. CviR and VpsT), but not both (Table S2).
Figure 4.

Crystal structure of VqmA bound to DPO. A, 2.0 Å crystal structure of full-length VqmA as a dimer (monomers labeled in red and gold) with two molecules of DPO (green) bound. B, zoomed-in view of one monomer (gold) showing the DPO ligand (green) and its spatial relationship to the residues tested in the mutagenesis studies (Table 1 and Fig. S6). The DPO-VqmA interface is primarily stabilized by residues Phe-67, Phe-99, and Lys-101. C, the N-terminal PAS LBD of VqmA in cartoon format as in A, showing the buried surface area between monomers. The dimer is stabilized by the extensive buried surface region and crossover of two helices (highlighted by the arrows) at the N termini (residues 7–15) of the two monomers. D, the C-terminal HTH DBD of VqmA in cartoon format as in A. Based on solvent accessibility, Lys-185, Lys-193, and Arg-195 are proposed to be involved in DNA binding. E, structural comparison of DPO-VqmA (red) with a previously solved structure of DPO-VqmA bound to DNA (PDB code 6IDE (17); protein in cyan and DNA in orange and blue). F, comparison of the structure of DPO-VqmA solved here (red) with that of a recently reported structure (PDB code 6KJU (18); green).
The VqmA HTH region is enriched in positively charged residues. Within this region, we predicted that the residues Lys-185, Lys-193, and Arg-195 are important for DNA binding due to their solvent accessibility and their conservation among VqmA orthologs (Fig. 4D). Indeed, as judged by EMSA analysis, changing any of these residues to alanine eliminated VqmA DNA binding (Fig. S4). This analysis is consistent with the structure of VqmA bound to DNA, which shows that residue Lys-185 makes direct contact with the DNA backbone (17). We compared our structure of DPO-VqmA with a recently reported structure of DPO-VqmA-DNA (17). Alignment based on the LBD PAS domains reveals a conformational change of ∼30 Å and a rotation of nearly 120° for the DBD (Fig. 4E). Thus, the DBD of the DNA-free VqmA protein adopts an open conformation, apparently conducive to DNA binding, whereas DNA-bound VqmA exhibits a closed conformation. We note that our structure is consistent with a recently reported structure of DPO-VqmA (Fig. 4F) (18), further supporting the above conclusion.
To probe the VqmA ligand binding requirements, we tested a panel of 15 synthesized or commercially available pyrazine, pyrimidine, and pyridine DPO analogs (Fig. 5A). We used the V. cholerae Δtdh PvqmR-lux reporter strain and companion ITC analysis to, respectively, assess activation of and binding to VqmA (Fig. 5B and Table S3, respectively). Surprisingly, given the promiscuity VqmA displays with respect to DPO and Ala-AA binding, VqmA is highly selective in terms of cyclic compounds: VqmA responded most avidly to DPO, and the most potent analogs, capable of inducing maximal lux expression, had EC50 values 6–8-fold higher than that of DPO (Fig. 5C). Our results indicate that a 2′-hydroxyl or 2′-carbonyl group is necessary for binding to VqmA (Table S3) and promoting its transcriptional activity (Fig. 5C). Indeed, compound 10, which differs only from DPO by the lack of the 2′-hydroxyl group, was inactive and showed no binding to VqmA (Fig. 5B and Table S3).
Figure 5.

Structure-activity relationship study of DPO. A, structure of DPO analogs studied here. B, responses of the PvqmR-lux reporter strain to 1 mm DPO and analogs. Shown are compounds exhibiting >25% of the maximal activity (colored bars) and <25% maximal activity (gray bars). C, dose-response analyses of DPO and active analogs from B using the PvqmR-lux reporter strain. Dose responses are depicted as curve fits with the raw data plotted as individual points. Data points in B and C are represented as means ± S.D. (error bars) with n = 3 biological replicates. RLU, relative light units.
Our crystal structure shows that DPO binding is primarily mediated by π-π stacking forces contributed by residues Phe-67 and Phe-99 coupled with hydrogen bonding between the DPO 2′-carbonyl group and residue Lys-101 (Fig. 4B). We note that DPO exists as the enol form in solution (8). Here, we built the keto tautomer of DPO in our crystal structure because it best fits the local density. Probably, the difference between the restricted condition in the crystal and the unrestricted environment in solution drives the preference for a particular tautomer. To verify our structure and to explore how VqmA could bind to both DPO and to Ala-AA, we conducted site-directed mutagenesis and tested binding and activity of DPO and Ala-AA, using an in vitro ITC assay and our in vivo cell-based reporter assay, respectively. We determined that, for WT apo-VqmA, the Kd for DPO is ∼2.3 μm, whereas the Kd for Ala-AA is ∼14 μm (Fig. 6A). As a negative control, we performed ITC on a linear Ala-AA derivative called (2S)-2-amino-N-[(2R)-2-hydroxypropyl]propanamide (AHP) (Fig. S5A) that did not activate VqmA in our PvqmR reporter assay (Fig. S5B), and no binding was detected by ITC (Fig. S5C). Next, we focused on mutations in the DPO-binding site: K23R, Y36F, F67A, D71N, D85N, H87A, H97A, F99A, K101R, F116A, F67A/F99A, and H87A/H97A (Fig. 4B). ITC results reveal that most of these VqmA variants exhibited approximately WT-binding affinity for both DPO and Ala-AA (Table 1). Exceptions were VqmA F67A, VqmA K101R, and VqmA F67A/F99A, which showed no binding to either molecule, indicating that these residues are crucial for binding to both DPO and Ala-AA (Table 1). Notably, VqmA F99A did not bind Ala-AA but retained residual binding to DPO (∼10-fold higher Kd relative to WT), suggesting that the contribution of Phe-99 in the π-π interaction with DPO is less crucial than that of Phe-67. The finding that VqmA K101R was unable to bind DPO coupled with the structure-activity relationship analysis in Fig. 5 suggests that proper hydrogen bonding between Lys-101 and the DPO 2′-hydroxyl/carbonyl group is critical for ligand binding and activity. We suspect that introduction of the bulkier Arg at position 101 prohibits formation of the required hydrogen bond at the 2′-position of DPO. A hydroxypyrimidine (compound 7, Fig. 5A) reduces the basicity of the oxygen on the 2′ moiety of the ligand, which would weaken its interaction with Lys-101 relative to DPO. Indeed, compound 7 had an >800-fold increase in EC50 and exhibited no detectable binding to VqmA (see Fig. 5C and Table S3). Finally, the VqmA Y36F and H97A alterations made the Kd values for DPO and Ala-AA more similar, indicating that these residues are involved in dictating the natural preference VqmA shows for DPO over Ala-AA (Table 1).
Figure 6.

DPO and Ala-AA share the same binding site with DPO being the preferred ligand. A, ITC measurement of the binding affinity of apo-VqmA for DPO and Ala-AA. B, ITC-based competition assay. Left to right, titration of Ala-AA into VqmA prebound with DPO, titration of DPO into VqmA prebound with Ala-AA, titration of Ala-AA into VqmA prebound to DPO with equimolar and 10-fold excess DPO present in solution, and titration of DPO into VqmA prebound with Ala-AA with equimolar and 10-fold excess Ala-AA present in solution. A cartoon schematic of each assay setup is shown above the corresponding ITC results. C, rates of DPO and Ala-AA binding to apo-VqmA derived from stopped-flow binding assays. [Analyte] from low to high is blue, red, green, purple, orange, black, and brown. See “Experimental procedures” for curve fitting in the insets.
Table 1.
Kd values for WT and mutant VqmA proteins for DPO and Ala-AA
| Mutation | DPO | Ala-AA |
|---|---|---|
| μm | μm | |
| WT | 2.3 ± 0.1 | 14 ± 3.0 |
| K23R | 1.2 ± 0.6 | 4.9 ± 0.8 |
| Y36F | 8.8 ± 2.4 | 6.7 ± 2.1 |
| F67A | NDa | ND |
| D71N | 7.6 ± 1.6 | 18 ± 8.0 |
| D85N | 6.1 ± 1.1 | 33 ± 27 |
| H87A | 1.7 ± 0.7 | 22 ± 6.4 |
| H97A | 6.9 ± 1.4 | 5.4 ± 0.7 |
| F99A | 22 ± 7.2 | ND |
| K101R | ND | ND |
| F116A | 7.0 ± 0.7 | 14 ± 6.3 |
| K193A | 2.0 ± 0.8 | 11 ± 2.5 |
| F67A/F99A | ND | ND |
| H87A/H97A | 1.8 ± 1.7 | 16 ± 11 |
a ND, not detectable.
The results from the reporter assay mimic those from the ITC assay (Fig. S6A). Fig. S6B shows that all of the VqmA variant proteins were produced at WT levels. Thus, changes in ligand-driven reporter activity stem from alterations in ligand-binding affinity, not changes in protein stability. Our structure-guided mutagenesis and biochemical analyses suggest that DPO and Ala-AA occupy the same binding site on VqmA. To date, we have been unable to obtain crystal structures of apo-VqmA or VqmA bound to Ala-AA. Apo-VqmA crystals diffract poorly, and Ala-AA, although stable in solution, readily cyclizes under our crystallization conditions (high salt and alkaline pH), independent of VqmA.
Ala-AA and DPO share the same binding site on VqmA, with DPO being the preferred ligand
Based on the surprising result that both Ala-AA and DPO bind to and activate VqmA, we sought to further characterize their interactions with VqmA to substantiate the same-binding-site hypothesis raised from our site-directed mutagenesis experiments. To garner additional evidence that DPO and Ala-AA bind the same site on VqmA, we performed an ITC-based competition assay. We titrated either DPO or Ala-AA into VqmA that had been prebound to the other molecule and assessed binding. We found that binding of the exogenously added molecule occurred no matter which ligand was prebound (Fig. 6B, left two panels), suggesting that the ∼6-fold difference in Kd between the two ligands allows each molecule to displace the other. In a conceptually similar experiment, we performed the same titration in the presence of excess of the prebound ligand. Our rationale is that, if Ala-AA and DPO do share the same binding site in VqmA, the presence of an excess of the ligand that had been prebound should inhibit binding of the subsequently added ligand. In this setup, VqmA showed a clear preference for binding to DPO. Specifically, when either an equal concentration or a 10-fold excess of DPO was provided to DPO-bound VqmA, Ala-AA was prevented from replacing prebound DPO (Fig. 6B, middle two panels). In the opposite case, only when there was at least 10-fold excess Ala-AA could Ala-AA prevent DPO from replacing prebound Ala-AA on VqmA (Fig. 6B, right two panels). These data support the hypothesis that DPO and Ala-AA share the same binding site on VqmA and further corroborate that, between the two ligands, DPO is preferred.
The preference VqmA shows for DPO over Ala-AA in the competitive binding experiments is curious, given that the Kd values of the two ligands for the apoprotein are only ∼6-fold different. To better understand VqmA ligand-binding preferences, we investigated the kinetics of DPO and Ala-AA binding using stopped-flow fluorimetry. When excited by 321-nm light, DPO emission occurs at 392 nm, and the fluorescence is quenched upon binding to VqmA (Fig. S7A). Ala-AA is not fluorescent, but quenching of natural VqmA fluorescence (λex = 280 nm/λem = 340 nm) can be monitored to track Ala-AA binding (Fig. S7B). We found that the kon for DPO and Ala-AA are 7.7 × 104 m−1·s−1 and 1.3 × 104 m−1·s−1, respectively, whereas the calculated koff values are 0.27 s−1 and 0.39 s−1, respectively (Fig. 6C). Thus, the association rate of DPO is ∼6-fold faster than that of Ala-AA, whereas the dissociation rate of DPO is modestly slower, ∼0.7-fold that of Ala-AA. The calculated Kd (koff/kon) values from the stopped-flow experiments are 3.5 μm (DPO) and 30.5 μm (Ala-AA). This result is consistent with our ITC data, because the differences are within an order of magnitude, which is well-accepted for these two methods. Moreover, the relative differences between the affinities of the two ligands for VqmA are consistent between the two methods. Taken together, we conclude that, once DPO is bound to VqmA, the complex does not easily dissociate, which prohibits subsequent binding of Ala-AA to the same binding site. In the reverse case, DPO can readily replace Ala-AA.
Discussion
In this study, we characterize ligand-controlled activation of VqmA, a cytoplasmic QS receptor and transcription factor. We show that VqmA is soluble and properly folded and activates basal-level transcription of its target, vqmR, in the absence of its cognate AI, DPO. We show that the transcriptional activity of VqmA is increased in response to increasing concentrations of DPO, allowing VqmA to drive the V. cholerae QS transition to HCD upon DPO accumulation. A previous study reported that the exogenous addition of DPO to purified apo-VqmA does not increase the binding affinity of VqmA for PvqmR (17). We find that this is not the case, as the DPO-bound VqmA protein has ∼2–8-fold higher affinity for DNA than does apo-VqmA (Fig. 1, C and D). One possible explanation for this discrepancy is that the authors of the previous study purified VqmA from WT E. coli, which is tdh+. As mentioned, WT E. coli makes DPO, and VqmA produced in E. coli binds to it (8). Thus, the authors' putative apo-VqmA could in fact have been holo-VqmA. Consequently, their VqmA was likely saturated with DPO, and the addition of more DPO would not enhance the VqmA DNA-binding properties.
We contrast the behavior of the VqmA receptor to that of a canonical LuxR-type QS AI receptor-transcription factor that is activated only when the ligand is present (Fig. 2). Although we cannot exclude issues arising from using different reporter systems, the dramatic difference in -fold change we observed between the VqmA and LasR receptors in the presence versus absence of their cognate AIs (3-fold for DPO/VqmA and 143-fold for 3OC12-HSL/LasR) leads us to speculate on the biological consequences of the differences between these systems. In particular, the ability of VqmA to function in both a ligand-independent and ligand-dependent manner enables the VqmA pathway to be active across all cell densities. In contrast, because LasR requires its AI for folding and activity, induction of the LasR pathway likely occurs exclusively under conditions when AI is present. We suggest that VqmA proteins are poised to rapidly respond to DPO when it appears. By contrast, canonical LuxR-type receptors require accumulation of cognate AIs for proper folding and activation, therefore delaying QS activation to later in growth. Last, we note that whereas V. cholerae vqmA is expressed constitutively, vqmAPhage is not (10). Although we do not yet know the natural inducer of vqmAPhage, we suspect that its production is regulated. We propose that the DPO-independent activity of VqmAPhage could allow the phage to overcome the requirement for the host to produce the AI and enable lysis provided that sufficient VqmAPhage is present.
The VqmA crystal structures reveal that the DPO-binding pocket is located in the N-terminal PAS domain. Previous reports compared the VqmA fold with that of LasR, TraR, CviR, and other LuxR-type proteins (17, 18), the latter of which, as mentioned, function via simultaneous binding and folding around AHL AIs (13, 15, 16). Whereas this comparison is certainly informative, it is also worth noting that LuxR-type proteins do not contain PAS domains. PAS domains contain characteristic five-stranded antiparallel β-sheet cores, whereas the LBDs of most LuxR-type proteins contain four-stranded antiparallel β-sheet cores. The divergence in folds is exemplified by the relatively low z-scores of LuxR-type proteins compared with the VqmA structure (Table S2). We imagine that the LBDs of canonical LuxR-type proteins constrain their selectivity to AHL molecules. By contrast, PAS domains are one of the most highly represented domains among signaling proteins and are capable of recognizing a wide range of ligands and sensory inputs, such as redox, light, gases, and metabolites (22–24). Within prokaryotic taxa, however, it is unusual for a single PAS domain to bind multiple structurally diverse ligands (23). We are curious to discover what properties conferred by the VqmA PAS domain might enable the receptor to bind both the cyclic DPO and the linear Ala-AA molecules. Finally, despite the prevalence of PAS domains, VqmA represents a unique structure for QS receptors.
Our site-directed mutagenesis and ITC results indicate that DPO and Ala-AA use the same site in VqmA because the identical residues are required for binding and activation by both ligands. Confirmation comes from the competition and kinetic assays. We do note that the enthalpy changes measured in the ITC competition assay (Fig. 6B) are not the same as those that we measured for binding of DPO and Ala-AA to apo-VqmA when there is no competition (Fig. 6A). Although not investigated here, one possibility is that some apo-VqmA remains present when we prebind VqmA to a ligand due to ligand association and disassociation. During the competition ITC, a portion of the ΔH value could stem from ligand binding to the small fraction of apo-VqmA that is present. A second possibility is that ligand replacement in the binding pocket during the competition involves more than simply ligand exchange (e.g. protein breathing).
It is surprising that VqmA binds to the cyclic DPO and the linear Ala-AA molecule using the same pocket. We speculate that Ala-AA adopts a conformation that resembles DPO when it binds to VqmA. Docking studies based on lowest enthalpies also suggest that Ala-AA and DPO bind to the same site on VqmA. Specifically, AutoDock Vina analysis was applied to the PAS domain of our DPO-VqmA structure (25) (Fig. S8). Multiple possible binding sites with different enthalpies were generated by this program. The predicted binding sites of Ala-AA and DPO with the lowest enthalpies are identical, consistent with the experimental DPO-binding site revealed by the DPO-VqmA structure. Moreover, the prediction suggests that Ala-AA binds in a bent configuration. We note that biases might exist in the output of the analysis because the input protein structure is not apo-VqmA.
As mentioned, Ala-AA was a proposed intermediate in DPO production based on the knowledge that threonine and alanine are both substrates in DPO biosynthesis and the proven structure of DPO (8). However, our present work indicates that Ala-AA is likely not the biological DPO precursor. Nonetheless, it is clear that Ala-AA is able to activate VqmA similarly to DPO. It is not simply a matter of the VqmA receptor displaying ligand promiscuity, as we find VqmA is selective for DPO relative to other pyrazine analogs (Fig. 5). This result suggests that the activity conferred by Ala-AA is not coincidental, but likely the actual precursor to DPO is a molecule similar to Ala-AA. Identification of the biological precursor and exploration of its role in VqmA activation are ongoing.
In addition to VqmA, other examples of proteins capable of binding multiple ligands exist, particularly those involved in metabolism. For example, IclR of E. coli binds pyruvate or glyoxylate, leading to activation or repression, respectively, of the operon regulating glyoxylate metabolism (26, 27). This mechanism could enable a single receptor to measure glycolytic flux and efficiently adjust gene expression accordingly. Second, BenM of Acinetobacter binds benzoate and muconate, where muconate is a product of benzoate metabolism (28–30). Simultaneous binding of both molecules leads to transcriptional activation of the benABCDE operon, which further accelerates catabolism of benzoate to muconate (28–30). This arrangement enables Acinetobacter to tightly control the expression of genes required for the degradation of benzoate, only activating them when their functions are required. Distinct from both of these cases, VqmA binds to either DPO or Ala-AA, but not both, to induce maximal transcriptional activity. This result is curious to us, given that QS receptors typically maintain signaling fidelity by distinguishing between different ligands (31–34). This situation appears analogous to that of the V. cholerae AI called CAI-1 ((S)-3-hydroxytridecan-4-one) and its biosynthetic precursor, Ea-CAI-1 (3-aminotridec-2-en-4-one). The study of Wei et al. (35) showed that Ea-CAI-1 is a potent activator of CqsS, but this precursor is likely more sensitive to hydrolysis than CAI-1 and therefore is too unstable to be a satisfactory QS signal. Given that pyrazine molecules are known to be stable compounds (36) and Ala-AA tends to cyclize, we hypothesize that whereas DPO is not an absolute requirement for VqmA-mediated QS, DPO could be the more optimal AI relative to Ala-AA because of its stability and potency.
Finally, we do note that all three V. cholerae QS systems converge to control an overlapping group of targets, including the aphA gene encoding the QS master regulator AphA (6, 9) that is expressed at LCD when AI concentrations are below their detection thresholds. The HCD responses to the CAI-1, AI-2 (4,5-dihydroxy-2,3-pentanedione) (37, 38), and DPO (8) AIs drive repression of aphA. VqmA, because it does not strictly require an AI for activity, activates some vqmR expression at LCD, leading to some repression of aphA. Thus, VqmA-controlled QS could provide a mechanism to attenuate AphA production, and thus alter expression of its downstream targets, prior to when sufficient levels of CAI-1, AI-2, and DPO have accumulated for the transition to the HCD QS state. Subsequently, DPO-VqmA–mediated QS could reinforce repression of aphA once V. cholerae has committed to the HCD QS program. Thus, the VqmA QS circuit could be important, at all cell densities, for fine-tuning the expression of QS target genes and, in turn, collective behaviors.
Experimental procedures
Bacterial strains, plasmids, primers, and reagents
Strains, plasmids, and primers used in this study are listed in Tables S4–S6, respectively. Unless otherwise indicated, V. cholerae and E. coli were grown aerobically in lysogeny broth at 37 °C. M9 medium was supplemented with 0.5% glucose and the indicated amino acids, each at 0.4 mm. Antibiotics were used at the following concentrations: 50 units ml−1 polymyxin B, 200 μg ml−1 ampicillin, and 100 μg ml−1 kanamycin. Primers were obtained from Integrated DNA Technologies. Site-directed mutagenesis of vqmA was performed using PCR with the indicated primers and the cloning strategy described in Table S6. The same primer pairs were used for generating mutants in both the E. coli expression vector (JSS-1057) and the V. cholerae arabinose-inducible vector (KPS-0500). Intramolecular reclosure was performed as described (10) using Q5 High-Fidelity DNA Polymerase (New England Biolabs). QuikChange (Agilent) site-directed mutagenesis was performed as described (31) using PfuUltra Polymerase (Agilent). The parent pET15b-vqmA vector (JSS-1057) was constructed by PCR of pET15b-His-vqmA (KPS-0929) with primers JSO-654 × JSO-655, followed by intramolecular reclosure to remove the His tag. The PvqmR-lux::lacZ suicide vector (JSS-0807) was constructed with primers JSO-383 × JSO-384 on BB-Ec0361 (pCN004) to generate a one-sided PacI-digestable pKAS backbone with flanking lacZ sites. The PvqmR-lux insert was generated using primers JSO-358 × JSO-397 on JSS-743 (PvqmR-lux in a pEVS143-derived vector). The insert and vector were digested with PacI (New England Biolabs), ligated using T4 DNA ligase (New England Biolabs), and transformed into E. coli S17-1 λpir.
Molecule syntheses
Unless otherwise indicated, all temperatures are expressed in °C. All reactions were conducted at room temperature unless otherwise noted. 1H NMR spectra were recorded on a Varian VXR-400, or a Varian Unity-400 at 400-MHz field strength. Chemical shifts are expressed in ppm (δ units). Coupling constants (J) are in Hz. Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), quin (quintet), or br (broad). The mass spectrum was performed on a Sciex API 100 using electrospray ionization. The LC-MS was performed using a C-18 reverse-phase column (2.1-mm inner diameter, 3.5 μm, 50 mm). The column conditions were 98% water with 0.05% TFA and 2% MeOH to 100% MeOH over 5.5 min. Analytical TLC was used to verify the purity as well as to follow the progress of the reaction(s). Unless otherwise indicated, all final products were at least 95% pure as judged by HPLC/MS. Compounds not mentioned in this section are commercially available (Table S7).
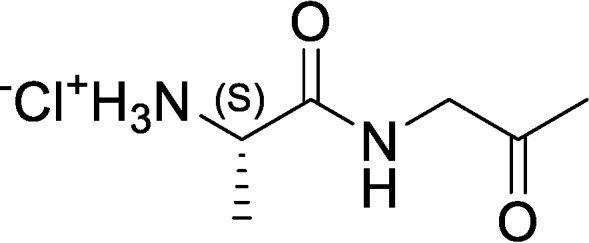
(S)-1-Oxo-1-((2-oxopropyl)amino)propan-2-aminium chloride (Ala-AA)
Structure 1.
Step 1: tert-Butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate
A solution of (tert-butoxycarbonyl)-l-alanine (20.0 g, 105 mmol, 1.0 eq) in DCM (50 ml) was stirred and cooled to 0 °C, and then 4-methylmorpholine (12.8 g, 126 mmol, 13.9 ml, 1.20 eq) was added. The resulting solution was stirred for 5 min, and then isobutyl chloroformate (15.8 g, 116 mmol, 15.2 ml, 1.10 eq) was added dropwise over 15 min. The resulting reaction mixture was stirred at 0 °C for 1 h, and then (R)-1-aminopropan-2-ol (8.34 g, 110 mmol, 8.74 ml, 1.05 eq) was added dropwise over 10 min. The resulting reaction mixture was allowed to warm to room temperature (RT) and stirred for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (DCM/MeOH = 100:0 to 98:2) to afford tert-butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate (13.2 g, 48% yield, 95% purity) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ = 6.83 (s, br, 1H), 5.24 (s, br, 1H), 4.15 (m, 1H), 3.91 (m, 1H), 3.38 (m, 1H), 3.12 (m, 2H), 1.46 (s, 9H), 1.36 (d, J = 7.2, 3H), 1.17 (d, J = 6.4, 3H).
Step 2: tert-Butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl)carbamate
tert-Butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate (8.00 g, 32.4 mmol, 1.00 eq) was dissolved in DCM (65 ml) and cooled to 0 °C. Next, Dess–Martin periodinane (17.9 g, 42.2 mmol, 13.1 ml, 1.30 eq) was added, and the resulting reaction mixture was warmed to RT and stirred for 12 h. The reaction mixture was quenched by the addition of saturated aqueous Na2S2O3 (100 ml) and saturated aqueous NaHCO3 solution (100 ml) and then stirred for 30 min and filtered through a pad of Celite. The filtrate was extracted with DCM (3 × 100 ml). The combined organic layers were washed with brine (100 ml), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (DCM/EtOAc = 100:0 to 20:1) to afford tert-butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl)carbamate (3.70 g, 42% yield, 90% purity) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ = 6.91 (s, br, 1H), 5.12, (s, br, 1H), 4.25 (m, 1H), 4.13 (d, J = 4.0, 2H), 2.19 (s, 3H), 1.44 (s, 9H), 1.36 (d, J = 7.2, 3H).
Step 3: (S)-1-Oxo-1-((2-oxopropyl)amino)propan-2-aminium chloride
tert-Butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl)carbamate (3.70 g, 15.2 mmol, 1.00 eq) was dissolved in HCl/dioxane (4.00 m, 18.9 ml, 5.00 eq), and the resulting reaction mixture was stirred at RT for 2 h, during which time a precipitate appeared. The reaction mixture was filtered to remove liquids, and the filter cake was washed with MTBE (2 × 20 ml) and dried under vacuum. The crude solid was triturated with EtOAc (50 ml) at RT for 2 h, followed by filtration and trituration with MBTE (20 ml) at RT for 2 h to afford (S)-1-oxo-1-((2-oxopropyl)amino)propan-2-aminium chloride (0.50 g, 17% yield, 95% purity) as a yellow solid. 1H NMR (400 MHz, D2O) δ = 4.75 (m, 1H), 4.12 (m, 1H), 2.12 (s, 3H), 1.41 (d, J = 6.4, 3H).
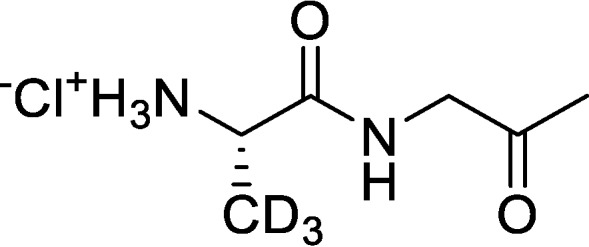
(S)-1-Oxo-1-((2-oxopropyl)amino)propan-3,3,3-d3-2-aminium chloride (d3-Ala-AA)
Structure 2.
Step 1: tert-butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl-3,3,3-d3)carbamate
A solution of (tert-butoxycarbonyl)-l-D3-alanine (2.0 g, 10.6 mmol, 1.0 eq) in DCM (20 ml) was stirred and cooled to 0 °C, and then 4-methylmorpholine (1.28 g, 12.7 mmol, 1.39 ml, 1.20 eq) was added. The resulting solution was stirred for 5 min, and then isobutyl chloroformate (1.58 g, 11.6 mmol, 1.52 ml, 1.10 eq) was added dropwise over 15 min. The resulting reaction mixture was stirred at 0 °C for 1 h, and then (R)-1-aminopropan-2-ol (834 mg, 11.0 mmol, 870 μl, 1.05 eq) was added dropwise over 2 min. The resulting reaction mixture was allowed to warm to RT and stirred for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (DCM/MeOH = 98:2) to afford tert-butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl-3,3,3-d3)carbamate (2.0 g, 68% yield, 90% purity) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 6.66 (s, br, 1H), 5.06 (s, br, 1H), 4.13 (m, 1H), 3.94 (m, 1H), 3.73 (m, 1H), 3.49, (m, 1H), 3.12 (m, 1H), 1.45 (s, 9H), 1.18 (d, J = 6.4, 3H).
Step 2: tert-Butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl-3,3,3-d3)carbamate
tert-Butyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl-3,3,3-d3)carbamate (2.00 g, 7.31 mmol, 1.00 eq) was dissolved in DCM (25 ml) and cooled to 0 °C. Next, Dess–Martin periodinane (4.0 g, 9.5 mmol, 2.94 ml, 1.30 eq) was added, and the resulting reaction mixture was warmed to RT and stirred for 12 h. The reaction mixture was quenched by the addition of saturated aqueous Na2S2O3 (20 ml) and saturated aqueous NaHCO3 solution (20 ml) and then stirred for 30 min and filtered through a pad of Celite. The filtrate was extracted with DCM (3 × 20 ml). The combined organic layers were washed with brine (20 ml), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (DCM/EtOAc = 98:2) to afford tert-butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl-3,3,3-d3)carbamate (1.60 g, 81% yield, 90% purity) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ = 6.82 (s, br, 1H), 4.99, (s, br, 1H), 4.21 (m, 1H), 4.15 (d, J = 4.0, 2H), 2.21 (s, 3H), 1.46 (s, 9H).
Step 3: (S)-1-Oxo-1-((2-oxopropyl)amino)propan-3,3,3-d3-2-aminium chloride
tert-Butyl (S)-(1-oxo-1-((2-oxopropyl)amino)propan-2-yl-3,3,3-d3)carbamate (1.60 g, 5.89 mmol, 1.00 eq) was dissolved in HCl/dioxane (4.00 m, 7.37 ml, 5.00 eq), and the resulting reaction mixture was stirred at RT for 2 h, during which time a yellow precipitate appeared. The reaction mixture was filtered to remove liquids, and the filter cake was washed with MTBE (2 × 20 ml) and dried under vacuum. The crude solid was triturated with EtOAc (50 ml) at RT for 2 h, followed by filtration and trituration with MBTE (20 ml) at RT for 2 h to afford (S)-1-oxo-1-((2-oxopropyl)amino)propan-3,3,3-d3-2-aminium chloride (0.67 g, 60% yield, 95% purity) as a yellow solid. 1H NMR (400 MHz, D2O) δ = 4.75 (s, 2H), 4.15 (s, 1H), 2.16 (s, 3H).
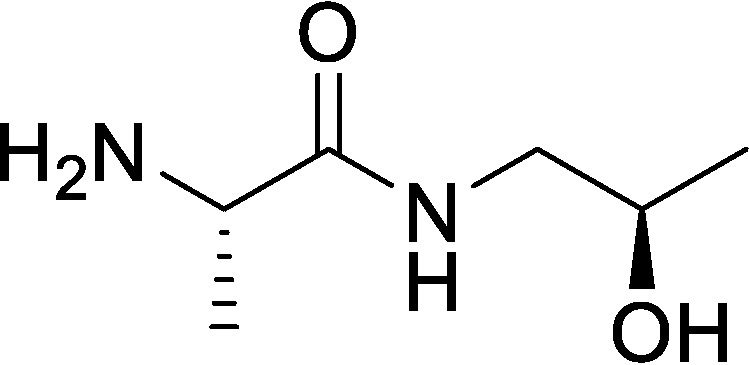
(S)-2-Amino-N-((R)-2-hydroxypropyl)propanamide (AHP)
Structure 3.
Step 1: Benzyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate
(2S)-2-(benzyloxycarbonylamino)propanoic acid (5 g, 22.40 mmol, 1.0 eq) was dissolved in tetrahydrofuran (100 ml), and the resulting solution was cooled to 0 °C under an N2 atmosphere. Next, 4-methylmorpholine (4.53 g, 44.80 mmol, 4.93 ml, 2.0 eq) was added, followed by isobutyl chloroformate (3.06 g, 22.40 mmol, 2.94 ml, 1.0 eq). The resulting reaction mixture was stirred at 0 °C for 1 h. Then (2R)-1-aminopropan-2-ol (2.52 g, 33.60 mmol, 2.65 ml, 1.5 eq) was added to the reaction mixture, and the resulting mixture was allowed to warm to RT and stirred for 11 h. The reaction was concentrated under reduced pressure to remove tetrahydrofuran. The reaction mixture was diluted with H2O (50 ml) and then extracted with EtOAc (2 × 30 ml). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica column chromatography (PE/EtOAc = 1:1 to 0:100) to afford benzyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate (3.0 g, 48% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ = 7.35 (m, 5H), 6.79 (s, br, 1H), 5.62 (d, br, J = 6.0, 1H), 5.10 (m, 2H), 4.24 (m, 1H), 3.88 (s, br, 1H), 3.36 (m, 1H), 3.12 (m, 2H), 1.37 (d, J = 7.2, 3H), 1.15 (d, J = 6.0, 3H); LC-MS calculated for C14H20N2O4: m/z = 280; found: m/z = 281 (M + H).
Step 2: (S)-2-Amino-N-((R)-2-hydroxypropyl)propanamide
To a solution of benzyl ((S)-1-(((R)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate (1 g, 3.57 mmol, 1.0 eq) in MeOH (10 ml) was added Pd(OH)2/C (1 g, 20% by weight) under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (15 p.s.i.) at RT for 2 h. The reaction mixture was flushed three times with N2 to degas and then filtered through a pad of Celite to remove the palladium, and the filter cake was washed with MeOH (3 × 10 ml). The filtrate was concentrated under reduced pressure to give a residue. The residue was purified by alumina column chromatography (EtOAc/MeOH = 100:0 to 10:1) to afford (S)-2-amino-N-((R)-2-hydroxypropyl)propanamide (500 mg, 95% yield) as a white solid. 1H NMR (400 MHz, MeOH-d4) δ = 3.83 (m, 1H), 3.42 (q, J = 6.8, 1H), 3.24 (dd, J = 4.8, 8.8, 1H), 3.12 (dd, J = 6.4, 8.8, 1H), 1.27 (d, J = 6.8, 3H), 1.15 (d, J = 6.4, 3H); LC-MS calculated for C6H14N2O2: m/z = 146; found: m/z = 147 (M + H).
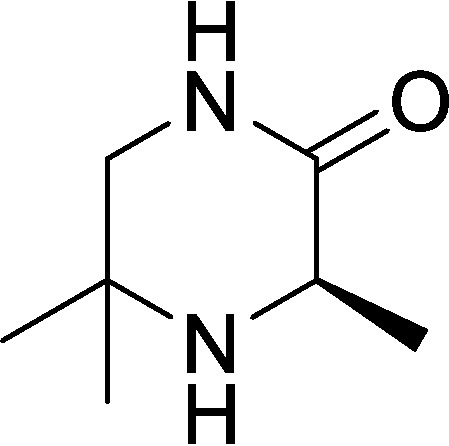
(R)-3,5,5-Trimethylpiperazin-2-one (compound 1)
Structure 4.
Step 1: Methyl (2-methyl-1-(tritylamino)propan-2-yl)-l-alaninate
2-Methyl-N1-trityl-propane-1,2-diamine (2.2 g, 6.66 mmol, 1.0 eq) and Et3N (1.01 g, 9.99 mmol, 1.39 ml, 1.5 eq) were dissolved in DCM (50 ml) and cooled to 0 °C. Next, methyl (2S)-2-(trifluoromethyl-sulfonyloxy)propanoate (1.57 g, 6.66 mmol, 1.0 eq) was added, and the resulting reaction mixture was allowed to stir for 1 h at 0 °C and then warmed to RT and stirred for 12 h. The reaction mixture was diluted with NaHCO3 (20 ml) and extracted with EtOAc (3 × 20 ml). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE/EtOAc = 100:1 to 50:1) to afford methyl (2-methyl-1-(tritylamino)propan-2-yl)-l-alaninate (1.4 g, 50% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 7.46 (d, J = 7.5, 6H), 7.23 (m, 6H), 7.13 (m, 3H), 3.55 (s, 3H), 2.90 (q, J = 7.1, 1H), 1.95 (m, 2H), 1.09 (s, 3H), 1.04 (d, J = 7.1, 3H), 0.96 (m, 3H).
Step 2: (R)-3,5,5-Trimethylpiperazin-2-one
A solution of methyl (2-methyl-1-(tritylamino)propan-2-yl)-l-alaninate (1.2 g, 2.88 mmol, 1.0 eq) in DCM (7.5 ml) was cooled to 0 °C, and then TFA (5 ml) was added dropwise. The resulting reaction mixture was warmed to RT and stirred for 2 h. The mixture was concentrated under reduced pressure to afford a residue. The residue was diluted with NaOH (10%, 20 ml) and then extracted with DCM (3 × 30 ml). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE/EtOAc = 1:1 to 0:100) to afford a yellow solid. The solid was triturated for 10 min with n-pentane (10 ml), filtered, and dried under vacuum to afford (R)-3,5,5-trimethylpiperazin-2-one (180 mg, 44% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ = 5.92 (s, br, 1H), 3.62 (q, J = 6.8, 1H), 3.29 (d, J = 11.6, 1H), 3.08 (dd, J = 4.5, 11.6, 1H), 1.39 (d, J = 6.8, 3H), 1.31 (s, 3H), 1.21 (s, 3H); LC-MS calculated for C7H14N2O: m/z = 142; found: m/z = 143 (M + H).
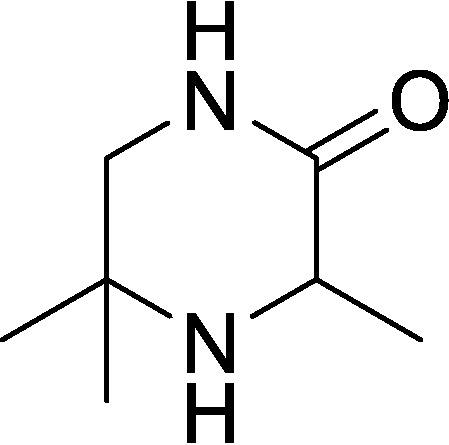
3,5,5-Trimethylpiperazin-2-one (compound 2)
Structure 5.
Step 1: Methyl 2-(trifluoromethylsulfonyloxy)propanoate
A solution of methyl 2-hydroxypropanoate (5.0 g, 48.03 mmol, 4.59 ml, 1.0 eq) and 2,6-dimethylpyridine (5.15 g, 48.03 mmol, 5.59 ml, 1.0 eq) in DCM (50 ml) was cooled to −10 °C, degassed, and purged with N2 three times, and then trifluoromethylsulfonyl trifluoromethanesulfonate (16.26 g, 57.63 mmol, 9.51 ml, 1.2 eq) was added dropwise at −10 °C under N2. The resulting reaction mixture was stirred at −10 °C for 30 min under N2 atmosphere. The reaction mixture was quenched by the addition of aqueous hydrochloric acid (0.5 m, 50 ml) and stirred at −10 °C for another 30 min, and then the reaction mixture was transferred to a separatory funnel, and the layers were separated. The organic layer was washed with brine (30 ml), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE/EtOAc = 100:0 to 10:1) to afford methyl 2-(trifluoromethylsulfonyloxy)propanoate (6.03 g, 53% yield) as a brown oil. 1H NMR (400 MHz, CDCl3) δ = 5.25 (q, J = 7.0, 1H), 3.87 (s, 3H), 1.73 (d, J = 7.0, 3H).
Step 2: Methyl (2-methyl-1-(tritylamino)propan-2-yl)-alaninate
2-Methyl-N1-trityl-propane-1,2-diamine (5.0 g, 15.13 mmol, 1.0 eq) and Et3N (1.53 g, 15.13 mmol, 2.11 ml, 1.0 eq) were dissolved in DCM (50 ml) and cooled to 0 °C. Next, methyl 2-(trifluoromethyl-sulfonyloxy)propanoate (3.57 g, 15.13 mmol, 1.0 eq) was added, and the resulting reaction mixture was allowed to stir for 1 h at 0 °C and then warmed to RT and stirred for 6 h. The reaction mixture was diluted with NaHCO3 (50 ml) and extracted with EtOAc (3 × 30 ml). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE/EtOAc = 100:1 to 10:1) to afford methyl (2-methyl-1-(tritylamino)propan-2-yl)-alaninate (4.3 g, 61% yield) as a white solid. 1H NMR (400 MHz, MeOH-d4) δ = 7.48 (m, 6H), 7.26 (m, 6H), 7.17 (m, 3H), 3.59 (s, 3H), 3.05 (q, J = 7.2, 1H), 2.04 (m, 1H), 1.96 (m, 1H), 1.11 (d, J = 7.2, 3H), 1.08 (s, 3H), 1.03 (s, 3H).
Step 3: 3,5,5-Trimethylpiperazin-2-one
A solution of methyl (2-methyl-1-(tritylamino)propan-2-yl)-alaninate (3.0 g, 7.2 mmol, 1.0 eq) in DCM (15 ml) was cooled to 0 °C, and then TFA (10 ml) was added dropwise. The resulting reaction mixture was warmed to RT and stirred for 2 h. The mixture was concentrated under reduced pressure to afford a residue. The residue was diluted with NaOH (10%, 20 ml) and then extracted with DCM (3 × 30 ml). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by silica gel column chromatography (PE/EtOAc = 1:1 to 0:100) to afford a yellow solid. The solid was triturated for 10 min with n-pentane (10 ml), filtered, and dried under vacuum to afford 3,5,5-trimethylpiperazin-2-one (168 mg, 16% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ = 6.11 (s, br, 1H), 3.61 (q, J = 6.8, 1H), 3.27 (d, J = 11.6, 1H), 3.08 (dd, J = 4.5, 11.6, 1H), 1.39 (d, J = 6.8, 3H), 1.31 (s, 3H), 1.19 (s, 3H); LC-MS calculated for C7H14N2O: m/z = 142; found: m/z = 143 (M + H).
Bioluminescence assays
Assays were performed as described (8) with the following modifications. V. cholerae reporter strains were grown overnight and back-diluted to OD600 = 0.005 in M9 medium supplemented with threonine and leucine. 150 μl of culture was transferred to the wells of a 96-well plate, followed by the addition of 50 μl of cell-free culture fluids. Synthetic molecule standards were prepared in M9 medium, and 50 μl of these preparations were added to the V. cholerae reporter strains in place of cell-free fluids. AI dose-response assays were performed as described (10) with the following modifications. 198-μl aliquots of cell cultures were dispensed into a 96-well plate with 2-μl aliquots of serially diluted synthetic compounds. Control wells received 2 μl of solvent (water or 10% NaOH).
EMSAs
EMSAs were performed as described (10). If not specified otherwise, the highest concentration of VqmA assessed was 130 nm. 2-Fold serial dilutions of protein were used to generate lower concentrations. Biotinylated PvqmR DNA (540 pm) was used in all EMSAs. The percentage of PvqmR DNA bound was calculated using the gel analyzer tool in ImageJ.
Production of cell-free translated protein
Cell-free translated His-VqmA was produced using the S30 T7 High-Yield Protein Expression System (Promega) according to the manufacturer's protocol with the following amendments. 1.5 μg of purified plasmid DNA, encoding His-VqmA under the T7 promoter, was used as the template; 40 units of murine RNase inhibitor (New England Biolabs) was added; five reactions were pooled together; and the reactions were incubated (translated) for up to 1.5 h at 37 °C. After translation, the His-VqmA was purified using MagneHis Ni-Particles (Promega).
VqmA/PvqmR, LasR/PlasB, and VqmAPhage/Pqtip reporter assays
For each system being tested, Δtdh E. coli harboring one plasmid encoding the arabinose-inducible receptor-transcription factor (VqmA, LasR, or VqmAPhage) and a second plasmid containing the cognate target promoter (PvqmR, PlasB, or Pqtip, respectively) fused to lux (10, 39) was used. In parallel, a control strain for each system was used that carried the reporter plasmid alone (PvqmR-lux, PlasB-lux, or Pqtip-lux). Each strain was grown overnight and back-diluted 1:500 into fresh lysogeny broth containing 0.075% arabinose and either synthetic AI (20 μm DPO or 1 μm 3OC12-HSL) or an equivalent volume of solvent (water for DPO and DMSO for 3OC12-HSL). 200-μl aliquots of each culture were dispensed into 96-well plates, and the plates were incubated at 30 °C with shaking. OD600 and bioluminescence were measured at regular intervals for 20 h. The bars in Fig. 2 and Fig. S1C represent the time points with the largest differences in relative light units (RLU) between the AI-receiving and -nonreceiving samples for each system. The same time points were used for the reporter-only strains, which received both arabinose and AI.
DPO extraction from cell-free culture fluids
V. cholerae strains were grown overnight and back-diluted to OD600 = 0.005 in M9 medium supplemented with leucine. The cultures were grown to OD600 ∼1.0, followed by incubation with 100 μm synthetic DPO or 100 μm synthetic d3-Ala-AA for 15 min. DPO, d3-DPO, and d3-Ala-AA were extracted from cell-free culture fluids by SPE (HyperSep Hypercarb SPE cartridges, Thermo Scientific). Chromatographic separation was achieved with increasing concentrations of acetonitrile (0, 10, 20, 30, 50, and 100%). The relative quantities of DPO, d3-DPO, and d3-Ala-AA were determined by LC-MS (described below). Samples were back-diluted 1:10 in water prior to column injection. Standards containing synthetic DPO, Ala-AA, or d3-Ala-AA were prepared in 2% acetonitrile.
Ala-AA conversion to DPO
10 μm synthetic Ala-AA or 10 μm synthetic DPO was incubated with 50 μm apo-VqmA in EMSA buffer. Each single component was also diluted into EMSA buffer as a control. Following incubation for 15 min, excess and nonprotein-bound small molecules were removed by size-exclusion chromatography (Zeba), and the resulting protein was heated to 70 °C for 10 min. Denatured protein was removed by centrifugation (13,000 × g, 10 min), and supernatants were assessed for Ala-AA or DPO by LC-MS (described below).
LC-MS to detect DPO, Ala-AA, d3-DPO, and d3-Ala-AA
LC-MS was performed with modifications (10). Samples and standards were loaded onto a C18 column (1 mm × 100 mm; ACE 3 C18 AQ, Mac-Mod) using a Shimadzu HPLC system and PAL autosampler (20 μl/injection) at a flow rate of 70 μl min−1. Full scan MS data were acquired with an LTQ-Orbitrap XL mass spectrometer (Thermo) at a resolution of 60,000 in profile mode from the m/z range of 110–200. DPO, Ala-AA, d3-DPO, and d3-Ala-AA were detected by performing XIC at m/z values of 125.0714 (C6H9N2O), 145.0977 (C6H13N2O2), 128.0903 (C6H6d3N2O), and 148.1165 (C6H10d3N2O2), respectively, each with a mass accuracy of ±10 ppm. Caffeine (2 μm in 50% acetonitrile with 0.1% formic acid) was delivered as a lock mass using an HPLC pump (LC Packing) with a flow splitter and at an effective flow rate of 20 μl min−1 through a tee at the column outlet. Raw files were imported into Skyline version 4.2.0 (40), and peak areas for each molecule were extracted using the small-molecule workflow.
Protein expression and purification
V. cholerae vqmA was cloned into pET15b and overexpressed in Δtdh BL21 (DE3) E. coli cells using 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h. Cells were pelleted at 3,300 × g for 15 min and resuspended in lysis buffer (20 mm Tris-HCl, pH 8, 150 mm NaCl, 1 mm DTT). Cells were lysed using sonication and subjected to centrifugation at 32,000 × g for 30 min. The supernatant was loaded onto a heparin column (GE Healthcare) and eluted by a linear gradient from buffer A (20 mm Tris-HCl, pH 8, 1 mm DTT) to buffer B (20 mm Tris-HCl, pH 8, 1 m NaCl, 1 mm DTT). Peak fractions were pooled, concentrated, and loaded onto a Superdex-200 size-exclusion column (GE Healthcare) in gel filtration buffer (20 mm Tris-HCl, pH 8, 150 mm NaCl, and 1 mm DTT). Proteins were concentrated to 30 mg/ml, flash-frozen, and stored at −80 °C. All VqmA mutants exhibited production and solubility levels similar to WT VqmA, suggesting that they are correctly folded, although this was not confirmed explicitly.
Crystallization and structure determination
The DPO-VqmA complex was formed by mixing 1 mm purified apo-VqmA with 10-fold excess synthetic DPO prior to crystallization. Crystals were obtained by the hanging drop vapor diffusion method from drops containing a 1:1 mixture of protein/precipitant (0.1 m Tris-HCl, pH 7.3, 16% PEG3350, and 0.2 m sodium malonate) at 22 °C. The trays were seeded using cat whiskers (Spark Bassler). Crystals were cryoprotected in a solution containing the precipitant plus 10% (v/v) glycerol. Crystals belonged to the P 212121 space group with unit cell dimensions a = 50.4 Å, b = 84.6 Å, c = 111.6 Å. A data set with heavy atom derivatives was obtained by soaking the crystals in cryoprotectant supplemented with 10 mm KAu(CN)2 (Hampton Research) for 10 min at RT as reported (41). Crystals were subsequently flash-frozen and stored in liquid N2. Diffraction data were collected at the NSLS II (National Synchrotron Light Source) beamlines FMX and AMX. Data were processed using HKL-3000, and experimental phases were determined using single-wavelength anomalous dispersion (42). Experimental phases were calculated using SHELX (43). The data sets were subjected to density modification using PHENIX (44–46), and the VqmA structure was built manually using Coot (47) over multiple iterations with further refinement in PHENIX. Images were generated using PyMOL.
Proteolytic cleavage time course
Apo-VqmA at 5 μm was incubated in the presence of 10 μm synthetic DPO, 10 μm synthetic Ala-AA, or H2O, in a final volume of 90 μl for 15 min at RT. A 10-μl aliquot was mixed with stop solution (2× SDS loading buffer with 5 mm phenylmethylsulfonyl fluoride and 5 mm EDTA) to represent the zero time point. Trypsin was added to the preparations at a final concentration of 171 nm and incubated at RT for 1, 2, 3, 5, 10, 15, and 20 min. At each time point, 10 μl of the reactions were added to 10 μl of stop solution. Samples were heated to 95 °C and subjected to SDS-PAGE.
Thermal shift assay
Thermal shift analyses of VqmA were performed as described (31). In brief, VqmA was diluted to 5 μm in reaction buffer (20 mm Tris-HCl, pH 8, 150 mm NaCl) containing either 10 μm synthetic DPO, 10 μm synthetic Ala-AA, or an equivalent volume of water in an 18-μl reaction (final volume). The mixtures were incubated at RT for 15 min. 2 μl of 200× SYPRO Orange in reaction buffer were added to the samples immediately before analysis. Samples were subjected to a linear heat gradient of 0.05 °C s−1, from 25 to 99 °C in a Quant Studio 6 Flex System (Applied Biosystems) using the melting curve setting. Fluorescence was measured using the ROX reporter setting.
ITC
A MicroCal PEAQ-ITC (Malvern) instrument was used to measure the binding affinities between VqmA proteins and ligands at 25 °C. All synthetic ligands for ITC were prepared in gel filtration buffer (described above). For measurement of Kd, 500 μm ligand was titrated into 50 μm apo-VqmA or mutant VqmA proteins using continuous stirring at 750 rpm. For the ITC-based competition assay, 500 μm synthetic Ala-AA was titrated into 50 μm DPO-VqmA in the presence of 0, 50, or 500 μm synthetic DPO, and in the opposite case, 500 μm DPO was titrated into 50 μm Ala-AA-VqmA in the presence of 0, 50, or 500 μm Ala-AA. The initial and subsequent 18 injections were 0.4 and 2 μl, respectively. Heat changes during the titration were measured by the PEAQ-ITC Control software (MicroCal). Data were fitted and evaluated by the PEAQ-ITC Analysis software (MicroCal).
Stopped-flow fluorimetry
A SX20 stopped-flow spectrometer (Applied Photophysics) was used for the kinetic study of ligand interactions with VqmA at 25 °C. DPO and VqmA have fluorescence spectra of λex = 321 nm/λem = 392 nm and λex = 280 nm/λem = 340 nm, respectively. For DPO binding to VqmA, the kobs was measured by recording the decrease in DPO fluorescence using a 390-nm emission interference filter. The concentration of VqmA was fixed at 2.5 μm, whereas DPO was supplied at concentrations from 1 to 20 μm. For Ala-AA binding to VqmA, the kobs was measured by recording the decrease in VqmA fluorescence using a 302-nm emission interference filter. The concentration of Ala-AA was fixed at 7.5 μm, and VqmA, from 3 to 37.5 μm, was supplied. Typically, 5–10 traces were recorded and averaged for each data point. Data were fitted with an exponential decay equation using GraphPad Prism version 6. Formation of complexes follows the equation, kobs = kon × [analyte] + koff. By plotting kobs versus [analyte], kon and koff are obtained from the slope and y-axis intercept, respectively.
Western blot analysis
Western blot analyses probing for FLAG fusions were performed as reported previously (8). V. cholerae cells were collected and diluted 2-fold in Laemmli sample buffer, giving a final concentration of 0.01 OD/μl. Following denaturation for 15 min at 95 °C, 0.1 OD600 equivalents were subjected to SDS-gel electrophoresis. RNAPα was used as loading control. Signals were visualized using an ImageQuant LAS 4000 imager (GE Healthcare).
Docking analysis
Docking analyses were performed as described (25). In brief, AutoDockTools version 1.5.6 was used to generate the pdbqt files, and AutoDock Vina version 1.1.2 was used to assess possible ligand-binding sites in the VqmA PAS domain (25, 48). The lowest-enthalpy results are shown in Fig. S8.
Author contributions
X. H., O. P. D., J. E. S., J. E. P., and B. L. B. conceptualization; X. H., O. P. D., J. E. S., J. E. P., J. C., and B. R. H. resources; X. H., O. P. D., J. E. S., J. E. P., and B. R. H. data curation; X. H., O. P. D., J. E. S., J. E. P., J. C., and B. L. B. formal analysis; X. H., O. P. D., J. E. S., J. E. P., and B. L. B. validation; X. H., O. P. D., J. E. S., J. E. P., J. C., and B. L. B. investigation; X. H., O. P. D., J. E. S., and J. E. P. visualization; X. H., O. P. D., J. E. S., J. E. P., J. C., and B. R. H. methodology; X. H., O. P. D., J. E. S., J. E. P., and B. L. B. writing-original draft; X. H., O. P. D., J. E. S., J. E. P., B. R. H., and B. L. B. writing-review and editing; B. L. B. supervision; B. L. B. funding acquisition; B. L. B. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Phillip Jeffrey of the Macromolecular Crystallography Core Facility, Dr. Saw Kyin of the Proteomics and Mass Spectrometry Core Facility, and Dr. Venu Vandasvasi of the Biophysics Core Facility, all at Princeton University, for expert assistance. We thank Dr. Frederick Hughson for thoughtful discussions. We thank the Bassler group for insight into the research. This research used resources FMX and AMX of the National Synchrotron Light Source II, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by NIGMS, National Institutes of Health, through Biomedical Technology Research Resource P41 Grant P41GM111244 and by DOE Office of Biological and Environmental Research Grant KP1605010. The authors received expert technical support from Dr. Martin Fuchs and Dr. Jean Jakoncic of the NSLS II FMX and AMX beamlines.
This work was supported by the Howard Hughes Medical Institute, National Institutes of Health Grant 5R37GM065859, and National Science Foundation Grant MCB-1713731 (to B. L. B.); a Jane Coffin Childs Memorial Fund for Biomedical Research Postdoctoral Fellowship (to J. E. P.); NIGMS, National Institutes of Health, Grant T32GM007388 (to O. P. D.); and a Charlotte Elizabeth Procter Fellowship provided by Princeton University and a National Defense Science and Engineering Graduate Fellowship supported by the Department of Defense (to J. E. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S7 and Figs. S1–S8.
The atomic coordinates and structure factors (code 6UGL) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- QS
- quorum sensing
- DPO
- 3,5-dimethyl-pyrazin-2-ol
- Ala-AA
- N-alanyl-aminoacetone
- AI
- autoinducer
- HCD
- high cell density
- LCD
- low cell density
- Tdh
- threonine dehydrogenase
- AKB
- 2-amino-3-ketobutyric acid
- ITC
- isothermal titration calorimetry
- AHL
- acyl-homoserine lactone
- 3OC12-HSL
- 3OC12-homoserine lactone
- EMSA
- electromobility shift assay
- d3-Ala-AA
- (S)-2-amino-N-(2-oxopropyl)-3,3,3-d3-propanamide
- PAS
- Per-Arnt-Sim
- LBD
- ligand-binding domain
- HTH
- helix-turn-helix motif
- DBD
- DNA-binding domain
- AHP
- (2S)-2-amino-N-[(2R)-2-hydroxypropyl]propanamide
- CAI-1
- (S)-3-hydroxytridecan-4-one
- Ea-CAI-1
- 3-aminotridec-2-en-4-one
- AI-2
- 4,5-dihydroxy-2,3-pentanedione
- CDCl3
- deuterated chloroform
- DMSO-d6
- deuterated dimethylsulfoxide
- PE
- petroleum ether
- EtOAc
- ethyl acetate
- DCM
- dichloromethane
- MeOH
- methanol
- MTBE
- methyl tert-butyl ether
- Et3N
- triethylamine
- RT
- room temperature
- OD
- optical density
- PDB
- Protein Data Bank.
References
- 1. Papenfort K., and Bassler B. L. (2016) Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 10.1038/nrmicro.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Waters C. M., Lu W., Rabinowitz J. D., and Bassler B. L. (2008) Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 190, 2527–2536 10.1128/JB.01756-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammer B. K., and Bassler B. L. (2003) Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol. 50, 101–104 10.1046/j.1365-2958.2003.03688.x [DOI] [PubMed] [Google Scholar]
- 4. Jung S. A., Chapman C. A., and Ng W. L. (2015) Quadruple quorum-sensing inputs control Vibrio cholerae virulence and maintain system robustness. PLoS Pathog. 11, e1004837 10.1371/journal.ppat.1004837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu J., Miller M. B., Vance R. E., Dziejman M., Bassler B. L., and Mekalanos J. J. (2002) Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 99, 3129–3134 10.1073/pnas.052694299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller M. B., Skorupski K., Lenz D. H., Taylor R. K., and Bassler B. L. (2002) Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110, 303–314 10.1016/S0092-8674(02)00829-2 [DOI] [PubMed] [Google Scholar]
- 7. Papenfort K., Förstner K. U., Cong J. P., Sharma C. M., and Bassler B. L. (2015) Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 112, E766–E775 10.1073/pnas.1500203112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Papenfort K., Silpe J. E., Schramma K. R., Cong J. P., Seyedsayamdost M. R., and Bassler B. L. (2017) A Vibrio cholerae autoinducer-receptor pair that controls biofilm formation. Nat. Chem. Biol. 13, 551–557 10.1038/nchembio.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herzog R., Peschek N., Fröhlich K. S., Schumacher K., and Papenfort K. (2019) Three autoinducer molecules act in concert to control virulence gene expression in Vibrio cholerae. Nucleic Acids Res. 47, 3171–3183 10.1093/nar/gky1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silpe J. E., and Bassler B. L. (2019) A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Cell 176, 268–280.e13 10.1016/j.cell.2018.10.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mukherjee J. J., and Dekker E. E. (1987) Purification, properties, and N-terminal amino acid sequence of homogeneous Escherichia coli 2-amino-3-ketobutyrate CoA ligase, a pyridoxal phosphate-dependent enzyme. J. Biol. Chem. 262, 14441–14447 [PubMed] [Google Scholar]
- 13. Zhu J., and Winans S. C. (2001) The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc. Natl. Acad. Sci. U.S.A. 98, 1507–1512 10.1073/pnas.98.4.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuster M., Urbanowski M. L., and Greenberg E. P. (2004) Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc. Natl. Acad. Sci. U.S.A. 101, 15833–15839 10.1073/pnas.0407229101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu J., and Winans S. C. (1999) Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc. Natl. Acad. Sci. U.S.A. 96, 4832–4837 10.1073/pnas.96.9.4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swem L. R., Swem D. L., O'Loughlin C. T., Gatmaitan R., Zhao B., Ulrich S. M., and Bassler B. L. (2009) A quorum-sensing antagonist targets both membrane-bound and cytoplasmic receptors and controls bacterial pathogenicity. Mol. Cell 35, 143–153 10.1016/j.molcel.2009.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu H., Li M., Guo H., Zhou H., Li B., Xu Q., Xu C., Yu F., and He J. (2019) Crystal structure of the Vibrio cholerae VqmA-ligand-DNA complex provides insight into ligand-binding mechanisms relevant for drug design. J. Biol. Chem. 294, 2580–2592 10.1074/jbc.RA118.006082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu H., Li M., Peng C., Yin Y., Guo H., Wang W., Xu Q., Zhou H., Xu C., Yu F., and He J. (2019) Large conformation shifts of Vibrio cholerae VqmA dimer in the absence of target DNA provide insight into DNA-binding mechanisms of LuxR-type receptors. Biochem. Biophys. Res. Commun. 520, 399–405 10.1016/j.bbrc.2019.10.063 [DOI] [PubMed] [Google Scholar]
- 19. Seed P. C., Passador L., and Iglewski B. H. (1995) Activation of the Pseudomonas aeruginosa lasI gene by LasR and the Pseudomonas autoinducer PAI: an autoinduction regulatory hierarchy. J. Bacteriol. 177, 654–659 10.1128/JB.177.3.654-659.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Storey D. G., Ujack E. E., Rabin H. R., and Mitchell I. (1998) Pseudomonas aeruginosa lasR transcription correlates with the transcription of lasA, lasB, and toxA in chronic lung infections associated with cystic fibrosis. Infect. Immun. 66, 2521–2528 10.1128/IAI.66.6.2521-2528.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holm L., and Laakso L. M. (2016) Dali server update. Nucleic Acids Res. 44, W351–W355 10.1093/nar/gkw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henry J. T., and Crosson S. (2011) Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 65, 261–286 10.1146/annurev-micro-121809-151631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Möglich A., Ayers R. A., and Moffat K. (2009) Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17, 1282–1294 10.1016/j.str.2009.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor B. L., and Zhulin I. B. (1999) PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 63, 479–506 10.1128/MMBR.63.2.479-506.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lorca G. L., Ezersky A., Lunin V. V., Walker J. R., Altamentova S., Evdokimova E., Vedadi M., Bochkarev A., and Savchenko A. (2007) Glyoxylate and pyruvate are antagonistic effectors of the Escherichia coli IclR transcriptional regulator. J. Biol. Chem. 282, 16476–16491 10.1074/jbc.M610838200 [DOI] [PubMed] [Google Scholar]
- 27. Maloy S. R., and Nunn W. D. (1982) Genetic regulation of the glyoxylate shunt in Escherichia coli K-12. J. Bacteriol. 149, 173–180 10.1128/JB.149.1.173-180.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collier L. S., Gaines G. L. 3rd, Neidle E. L. (1998) Regulation of benzoate degradation in Acinetobacter sp. strain ADP1 by BenM, a LysR-type transcriptional activator. J. Bacteriol. 180, 2493–2501 10.1128/JB.180.9.2493-2501.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bundy B. M., Collier L. S., Hoover T. R., and Neidle E. L. (2002) Synergistic transcriptional activation by one regulatory protein in response to two metabolites. Proc. Natl. Acad. Sci. U.S.A. 99, 7693–7698 10.1073/pnas.102605799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harwood C. S., and Parales R. E. (1996) The β-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50, 553–590 10.1146/annurev.micro.50.1.553 [DOI] [PubMed] [Google Scholar]
- 31. McCready A. R., Paczkowski J. E., Henke B. R., and Bassler B. L. (2019) Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc. Natl. Acad. Sci. U.S.A. 116, 245–254 10.1073/pnas.1817239116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ke X., Miller L. C., and Bassler B. L. (2015) Determinants governing ligand specificity of the Vibrio harveyi LuxN quorum-sensing receptor. Mol. Microbiol. 95, 127–142 10.1111/mmi.12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gerdt J. P., Wittenwyler D. M., Combs J. B., Boursier M. E., Brummond J. W., Xu H., and Blackwell H. E. (2017) Chemical interrogation of LuxR-type quorum sensing receptors reveals new insights into receptor selectivity and the potential for interspecies bacterial signaling. ACS Chem. Biol. 12, 2457–2464 10.1021/acschembio.7b00458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eberhard A., Widrig C. A., McBath P., and Schineller J. B. (1986) Analogs of the autoinducer of bioluminescence in Vibrio fischeri. Arch. Microbiol. 146, 35–40 10.1007/BF00690155 [DOI] [PubMed] [Google Scholar]
- 35. Wei Y., Perez L. J., Ng W. L., Semmelhack M. F., and Bassler B. L. (2011) Mechanism of Vibrio cholerae autoinducer-1 biosynthesis. ACS Chem. Biol. 6, 356–365 10.1021/cb1003652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurihara T., Watanabe Y., Takayama K., and Ohta A. (1991) Stability of naturally occurring pyrazines and construction route of pyrazine rings. J. Heterocyclic Chem. 28, 1557–1560 10.1002/jhet.5570280616 [DOI] [Google Scholar]
- 37. Schauder S., Shokat K., Surette M. G., and Bassler B. L. (2001) The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 41, 463–476 10.1046/j.1365-2958.2001.02532.x [DOI] [PubMed] [Google Scholar]
- 38. Chen X., Schauder S., Potier N., Van Dorsselaer A., Pelczer I., Bassler B. L., and Hughson F. M. (2002) Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415, 545–549 10.1038/415545a [DOI] [PubMed] [Google Scholar]
- 39. Paczkowski J. E., Mukherjee S., McCready A. R., Cong J. P., Aquino C. J., Kim H., Henke B. R., Smith C. D., and Bassler B. L. (2017) Flavonoids suppress Pseudomonas aeruginosa virulence through allosteric inhibition of quorum-sensing receptors. J. Biol. Chem. 292, 4064–4076 10.1074/jbc.M116.770552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 10.1093/bioinformatics/btq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joyce M. G., Radaev S., and Sun P. D. (2010) A rational approach to heavy-atom derivative screening. Acta Crystallogr. D Biol. Crystallogr. 66, 358–365 10.1107/S0907444909053074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- 43. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 10.1107/S0108767307043930 [DOI] [PubMed] [Google Scholar]
- 44. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., et al. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 10.1016/j.ymeth.2011.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Echols N., Grosse-Kunstleve R. W., Afonine P. V., Bunkóczi G., Chen V. B., Headd J. J., McCoy A. J., Moriarty N. W., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Adams P. D. (2012) Graphical tools for macromolecular crystallography in PHENIX. J. Appl. Crystallogr. 45, 581–586 10.1107/S0021889812017293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.