

Abstract
The fortuitously discovered antiaging membrane protein αKlotho (Klotho) is highly expressed in the kidney, and deletion of the Klotho gene in mice causes a phenotype strikingly similar to that of chronic kidney disease (CKD). Klotho functions as a co-receptor for fibroblast growth factor 23 (FGF23) signaling, whereas its shed extracellular domain, soluble Klotho (sKlotho), carrying glycosidase activity, is a humoral factor that regulates renal health. Low sKlotho in CKD is associated with disease progression, and sKlotho supplementation has emerged as a potential therapeutic strategy for managing CKD. Here, we explored the structure-function relationship and post-translational modifications of sKlotho variants to guide the future design of sKlotho-based therapeutics. Chinese hamster ovary (CHO)- and human embryonic kidney (HEK)-derived WT sKlotho proteins had varied activities in FGF23 co-receptor and β-glucuronidase assays in vitro and distinct properties in vivo. Sialidase treatment of heavily sialylated CHO-sKlotho increased its co-receptor activity 3-fold, yet it remained less active than hyposialylated HEK-sKlotho. MS and glycopeptide-mapping analyses revealed that HEK-sKlotho is uniquely modified with an unusual N-glycan structure consisting of N,N′-di-N-acetyllactose diamine at multiple N-linked sites, one of which at Asn-126 was adjacent to a putative GalNAc transfer motif. Site-directed mutagenesis and structural modeling analyses directly implicated N-glycans in Klotho's protein folding and function. Moreover, the introduction of two catalytic glutamate residues conserved across glycosidases into sKlotho enhanced its glucuronidase activity but decreased its FGF23 co-receptor activity, suggesting that these two functions might be structurally divergent. These findings open up opportunities for rational engineering of pharmacologically enhanced sKlotho therapeutics for managing kidney disease.
Keywords: fibroblast growth factor receptor (FGFR), signal transduction, glycosidase, glycosylation, sialic acid, pharmacokinetics, acute kidney injury, Klotho, LacdiNAc, mammalian cell expression
Introduction
Chronic kidney disease (CKD),4 a growing public health issue affecting more than 26 million Americans (1–4), is considered a clinical model of “accelerated aging” (5). Progressive deterioration of renal function in CKD patients promotes cellular senescence and premature aging, characterized by hyperphosphatemia, vascular calcification, cardiac hypertrophy, emphysema, and osteopenia. An incidentally discovered anti-aging protein, αKlotho (hereinafter called Klotho) (6), is highly expressed in the kidney, and deletion of the Klotho gene in mice leads to a phenotype with striking similarities to CKD. Abundant evidence from preclinical and clinical studies has revealed that CKD is a state of systemic Klotho deficiency (5, 7–13) and that Klotho deficiency might be an important pathogenic contributor to progression of CKD and development of cardiovascular disease (11, 14–21).
Over the past 2 decades, Klotho biology has been intensely investigated. Molecular functions and physiological roles of Klotho have been found to be complex and multifaceted. The klotho gene encodes a type I single-pass transmembrane protein (6) containing a large extracellular domain composed of two internal repeats (KL1 and KL2) each 450 amino acids long with homology to family 1 glycosidases (22). Two laboratories independently discovered that the Klotho protein forms a complex with fibroblast growth factor (FGF) receptors (23, 24). In the kidney, Klotho serves as a co-receptor for FGF23 to down-regulate the sodium phosphate transporters NaPi2a and NaPi2c and to promote renal phosphate excretion (23–25). Dysfunction in the Klotho/FGF23 axis and associated disordered phosphate metabolism have been suggested as a potential pathological mechanism by which Klotho deficiency may drive CKD progression (11, 15, 26–28).
klotho−/− mice exhibit defects in non-klotho-expressing cells (6), suggesting that Klotho can act at a distance as a humoral factor (29). Indeed, a soluble form of Klotho (sKlotho) can be generated through ectodomain shed by proteases such as ADAM proteases (30) and secretases (31). Studies have shown that sKlotho possesses FGF23 co-receptor function similar to the membrane-bound form (23, 24, 32), suggesting that supplementation of sKlotho could exert therapeutic benefit in CKD by correcting altered phosphate metabolism. Additional activity for Klotho has been ascribed to its weak glycosidase activity (33–36). Based on sequence homology, the catalytic site residues are centered in KL1 and KL2, with each active site lacking one of the two catalytic glutamic acid residues found in family 1 glycosidases (6, 22). Nonetheless, this weak glycosidase activity has been shown to contribute to sKlotho's regulatory function of ion channels and transporters (34–36). sKlotho was shown to directly inhibit NaPi2a via enzymatic modifications of NaPi2a glycans, leading to decreased transporter activity and proteolytic degradation (37). Together, these data suggest that FGF23 co-receptor and FGF23-independent glycosidase activities of sKlotho may be critical in the maintenance of renal health and that enhancement of these functions may represent an attractive strategy to generate a sKlotho-based therapeutic approach to CKD. Moreover, owing to its large size and complex function, Klotho protein is presumably a substrate of various post-translational modifications (PTMs) (38, 39). Better understanding of these PTMs during protein production in mammalian cells should help in comprehending protein quality and protein attributes of sKlotho as a potential supplementation protein therapeutics.
In the present study, we have expressed variants of sKlotho in stable Chinese hamster ovary (CHO) cells and transient human embryonic kidney (HEK) 293 cells. We found that the WT sKlotho derived from these two different mammalian expression systems exhibited differentiated in vitro activities and in vivo pharmacokinetics (PK) properties as well as underwent different types of PTMs, such as N-linked (40–42) and O-linked (43, 44) glycosylation. CHO-sKlotho was heavily modified with sialylation of N-glycans, which showed better in vivo PK properties but surprisingly decreased the coreceptor function. Moreover, HEK-sKlotho was unexpectedly found to be modified with a rare disaccharide N,N′-di-N-acetyllactose diamine (LacdiNAc) N-glycan structure with a moiety of terminal GalNAcβ1–4GlcNAcβ (45–49) at multiple N-linked sites, some being additionally sulfated and fucosylated. A putative sequence motif for GalNAc transfer was identified. In addition, we showed that sKlotho mutant with enhanced glycosidase functions decreased its FGF23 co-receptor function, suggesting that these two well-established functions of sKlotho might be structurally divergent. These results provide new insights into sKlotho's structure-function relationship and PTM biology for guiding future sKlotho therapeutic protein engineering.
Results
Production of human sKlotho in mammalian cells
As shown in Fig. 1A, a secreted form of the full-length ectodomain of Klotho with a V5 tag and a six-histidine tag was expressed by the two most commonly used production methods: a fast-turnaround transient HEK293 system and a high-yield stable CHO system. Expression yield for this sKlotho construct in transient HEK293 was low, with around 1 mg/liter of final purified protein from the conditioned medium. As expected, the final yield of purified sKlotho from the stable CHO expression was higher, around 5 mg/liter. The proteins from both expression approaches were >99% pure as indicated by SDS-PAGE (Fig. 1B). Isoelectro-focused (IEF) gel showed that pI of the stable CHO-sKlotho was much lower than that of the HEK293-derived protein, an indication of much more negatively charged molecules (Fig. 1C). This observation is consistent with the common notion that stable CHO expression produces more N-glycan sialylation in glycoproteins than transient HEK293 expression (50–52). When these proteins were further analyzed by size-exclusion chromatography (Fig. 1D), both CHO- and HEK-sKlotho proteins were eluted slightly later (smaller) than bovine γ-globulin marker (158 kDa), consistent with monomeric species (molecular mass of glycosylated form: ∼130 kDa).
Figure 1.

Production of a secreted full-length extracellular domain of Klotho in mammalian cells. A, amino acid sequence of a secreted full-length ectodomain of human Klotho. The KL1 domain is shown in violet, and the KL2 domain is green. Putative N-linked sites are blue. V5 tag and His6 tag are black. Four residues at highly conserved positions are red. B, SDS-PAGE analysis of purified sKlotho protein expressed in mammalian cells. sKlotho protein in A was expressed in either stable CHO or transient HEK293 and purified as described under “Experimental procedures.” A spliced region in the gel is indicated with a heavy line. C, IEF gel analysis of purified sKlotho protein. D, SEC analysis of purified sKlotho protein. Bio-Rad gel filtration standards indicated by blue arrows are thyroglobulin (bovine, 670 kDa), γ-globulin (bovine, 158 kDa), ovalbumin (chicken, 44 kDa), myoglobin (horse, 17 kDa), and vitamin B12 (1.35 kDa).
Using ultrahigh-resolution MS, we also analyzed the amino acid backbone of intact CHO-sKlotho and HEK-sKlotho after the release of N-glycans with endoglycosidase PNGase F. As shown in Fig. 2, under reducing conditions, both HEK-sKlotho and CHO-sKlotho had nearly identical observed molecular masses (111587.0 and 111587.7 Da, respectively), in agreement with the expected molecular mass of 111587.8 Da, which indicated that the same mature full-length protein was expressed with no protein clipping or side-chain oxidation detected. A small percentage of HEK-sKlotho and CHO-sKlotho contained mucin-type O-linked core-1 glycan (Ser/Thr-GalNAc-Gal), including disialylated (GalNAc(NeuAc)-Gal-NeuAc), or monosialylated (GalNAc-Gal-NeuAc) glycan. Some HEK-sKlotho also had O-linked Ser/Thr-GalNAc glycan.
Figure 2.

Mass spectrometry analysis of CHO-sKlotho (A) and HEK-sKlotho (B). CHO-sKlotho and HEK-sKlotho were digested with PNGase F and subjected to ultrahigh-resolution MS analysis as described under “Experimental procedures.” Structures of mucin-type core-1 O-linked glycans (monosialylated core-1, disialylated core-1) are depicted in addition to Ser/Thr-GalNAc and Ser/Thr-GalNAc-Gal.
In vitro functional characterization of human sKlotho
The biological activities of these purified HEK- and CHO-sKlotho proteins were tested in two in vitro systems. It has been previously reported that the sKlotho can serve as a co-receptor for FGF23 signaling (23, 24). Consistent with this finding, sKlotho induced ERK1/2 phosphorylation in L6 cells expressing FGFR1c. The transient HEK293-derived sKlotho had an EC50 of 0.64 nm, whereas the stable CHO-derived sKlotho had an EC50 of 12.84 nm in this assay (Fig. 3A). Because the primary sequences of these proteins are the same, the potency difference should have resulted from differential PTMs. When tested for enzymatic activity, CHO-sKlotho proteins exhibited weak β-glucuronidase activity (Fig. 3B), consistent with a previous report (33). In contrast to the ERK1/2 phosphorylation assay results, β-glucuronidase activity for the transient HEK-sKlotho was barely detectable.
Figure 3.

In vitro functional characterization of sKlotho produced in mammalian cells. A, sKlotho is active in the FGF23 ERK1/2 activation assay. Purified sKlotho proteins were measured in the ERK1/2 activation assay as described under “Experimental procedures.” The error bars at each data point were calculated from three independent experiments. B, sKlotho possessed weak β-glucuronidase activity. Purified sKlotho proteins were measured in the β-glucuronidase assay as described under “Experimental procedures.” The error bars at each data point were calculated from four independent experiments. Data are shown as mean ± S.E. *, p < 0.05 versus HEK sKlotho, ANOVA.
Pharmacokinetics of HEK- versus CHO- sKlotho
Next, the in vivo PK properties of the HEK- and CHO-sKlotho proteins were evaluated in Sprague–Dawley rats. Following intravenous (IV) administration of HEK-sKlotho at 1 mg/kg, systemic exposure was only measurable at 5 min after dosing (Fig. 4A) and was below the detection limit, ∼1 ng/ml, at all other time points, suggesting extremely high systemic clearance of the protein. In contrast, systemic exposure of CHO-sKlotho was detected for up to 3 days following single-dose IV administration at 0.5 mg/kg (Fig. 4A). The systemic clearance of CHO-sKlotho was estimated to be 15.8 ml/h/kg, the volume of distribution was 120 ml/kg, and terminal half-life was 12.2 h (Table 1). The bioavailability of CHO-sKlotho following a single intraperitoneal (IP) dose was estimated to be ∼47.5%, and the time at which the maximum serum concentration was observed was 2 h post-IP dose.
Figure 4.

Single-dose administration of CHO-sKlotho is protective against IRI in rats. A, pharmacokinetic profiles of HEK- and CHO-sKlotho; B, IRI, serum creatinine; C, IRI, BUN; D, IRI, GFR; E, IRI, albuminuria; F, IRI, renal cortical α-smooth muscle cell actin (scale bar, 200 μm). Data are shown as mean ± S.E. *, p < 0.05 versus vehicle, ANOVA.
Table 1.
Key pharmacokinetic parameters of CHO-sKlotho in rats following a single IV or IP administration at 0.5 mg/kg
Values presented are mean ± S.D. with n = 3. NA, not applicable; CL, systemic clearance; Vc, central volume of distribution; Vss, volume of distribution at steady state; t1/2, terminal half-life; Tmax, time to Cmax; Cmax, observed maximal concentration; AUC(0-∞), area under the concentration-time curve at steady state; F, bioavailability.
| Route | Gender/n | CL | Vc | Vss | t½ | Tmax | Cmax | AUC(0-∞) | F |
|---|---|---|---|---|---|---|---|---|---|
| ml/h/kg | ml/kg | ml/kg | h | h | ng/ml | ng·h/ml | % | ||
| IV | Male/3 | 15.8 ± 1.7 | 47.4 ± 10.7 | 120 ± 15.6 | 12.2 ± 0.9 | 0.083 | 9327 ± 1698 | 31923 ± 3233 | NA |
| IP | Male/3 | NA | NA | NA | 13.0 ± 1.7 | 2 | 1117 ± 473 | 15162 ± 4835 | 47.5 |
In vivo pharmacological effects of CHO-sKlotho
Based on the observation of poor in vivo PK of the HEK-sKlotho protein, exploration of the in vivo pharmacological activity of sKlotho was conducted using the CHO-sKlotho protein in a rat model of renal ischemia-reperfusion injury (IRI). On the first day after surgery, renal IRI resulted in a significant increase in serum creatinine and blood urea nitrogen (BUN) relative to baseline in rats receiving vehicle (Fig. 4, B and C). Decreased kidney function in these animals on day 1 after surgery was also confirmed by a reduction in glomerular filtration rate (GFR) measured transcutaneously (Fig. 4D) and a significant increase in urine albumin/creatinine ratio (UACR) (Fig. 4E). In contrast, single-dose IP administration of the CHO-sKlotho at 0.01 mg/kg 30–60 min after reperfusion prevented or blunted the decline in kidney function on the first day post-surgery, with no significant change in serum BUN, GFR, or UACR relative to baseline and a significant reduction in serum creatinine relative to vehicle (Fig. 4, B–E). Consistent with the transient nature of the reduction in kidney function in this model, functional parameters returned to baseline level by day 7 post-surgery in both groups (Fig. 4, B–E). However, treatment with CHO-sKlotho resulted in decreased renal tissue damage relative to vehicle at 7 days post-surgery, as evidenced by reduced α-smooth muscle actin (α-SMA) staining, an indicator of epithelial to mesenchymal transition (Fig. 4F). These data together indicated that CHO-sKlotho was active in this preclinical model of acute kidney injury (AKI). Further investigation of sKlotho's structure-function relationship and PTM was then undertaken to identify strategies guiding future optimization of sKlotho's pharmacological activity.
Sialidase treatment of CHO-sKlotho
One direction for further investigation was to understand the unexpected result that HEK-sKlotho was found to be 20-fold more active than CHO-sKlotho in the FGF23 co-receptor signaling assay (Fig. 3A). Because these two sKlotho variant proteins share the same primary sequence, this activity difference should be attributed to different PTMs. The most common PTM is N-linked glycosylation. Klotho is a heavily glycosylated protein with eight putative N-linked sites (Fig. 1A), seven of which (except Asn-73) are highly conserved across mammalian species and shown to be fully occupied with N-glycans in the recent published crystal structure (32). The CHO-sKlotho displayed a lower pI than the HEK-sKlotho, as shown in the IEF gel (Fig. 1C). We hypothesized that the pI difference is due to the higher sialylation of the CHO proteins, which could contribute to the FGF23 co-receptor activity difference observed between the two proteins. To test this hypothesis, we treated both HEK- and CHO-sKlotho with sialidase under native condition at room temperature overnight to remove sialic acids and then repurified the digested proteins for testing in the FGF23 signaling assay (Fig. 5A, lanes 5–8, SDS-PAGE). The IEF gel showed that CHO-sKlotho (Fig. 5A, lane 4) has a lower pI than HEK-sKlotho (Fig. 5A, lane 2). After the sialidase treatment, the pI of CHO-sKlotho was significantly increased (Fig. 5A, lane 3 versus lane 4), an indication of the removal of sialic acids (also see data below). The pI of sialidase-treated HEK-sKlotho was similar to that of mock-treated HEK-sKlotho (Fig. 5A, lane 1 versus lane 2). When these proteins were assayed in the FGF23-FGFR1c ERK1/2 activation assay (Fig. 5B), the sialidase-treated or mock-treated HEK-sKlotho proteins were found to have a similar EC50 (0.53 nm versus 0.68 nm), whereas the sialidase-treated CHO-sKlotho became 3-fold more potent than mock-treated CHO-sKlotho (4.0 nm versus 13.6 nm EC50, respectively), demonstrating that there is indeed a sialylation effect on Klotho's FGF23 signaling function.
Figure 5.

Sialidase treatment increased the activity of heavily sialylated CHO-sKlotho by 3-fold but had little effect on the less-sialylated HEK-sKlotho in the FGF23 ERK1/2 activation assay. A, CHO- or HEK-sKlotho proteins (0.5 mg each) were treated or mock-treated with sialidase (500 units; New England Biolabs) overnight at room temperature in G1 buffer, repurified through a nickel-nitrilotriacetic acid column, buffer-exchanged, and concentrated at 0.4 mg/ml in PBS buffer. The protein samples were analyzed by SDS-PAGE or IEF gel. B, sialidase-treated or mock-treated sKlotho proteins were measured in the FGF23 ERK1/2 activation assay as described under “Experimental procedures.” The error bars at each data point were calculated from a triplicated experiment.
Glycan analysis, glycopeptide mapping, and MS analysis on CHO-sKlotho and HEK-sKlotho
It is worth noting that the sialidase-treated CHO-sKlotho protein was still less potent than the HEK-sKlotho (Fig. 5B). To rule out the possibility that the desialylation process is incomplete, we conducted hydrophilic interaction LC (HILIC) released N-glycan profiling analysis on CHO-sKlotho and HEK-sKlotho.
As shown in Fig. 6A, G2 refers to oligosaccharides bearing two galactose residues and no fucose residues. G2F is used when fucose is present. NeuAc refers to carrying N-acetylneuraminic residues. CHO-sKlotho contains heavily sialylated N-linked glycans (∼66.8%) with G2F + NeuAc, G2F + 2NeuAc, G2 + NeuAc, and G2 + 2NeuAc. After sialidase digestion, all sialylated glycan species disappeared, indicating that the treatment was complete and that the remaining potency difference between desialylated CHO-sKlotho and HEK-sKlotho was attributable to factors other than sialylation. HEK-sKlotho comprised few sialylated glycan species, and there were no significant N-glycan profiling changes between before and after sialidase treatment (data not shown). Interestingly, some less common N-glycan species were detected for HEK-sKlotho. As shown in Fig. 6B, some HEK-derived glycan species were sensitive to the digestion of β-N-acetylhexosaminidasef (53), which catalyzes the hydrolysis of terminal GalNAc and GlcNAc residues from oligosaccharides that are β(1–3)-, β(1–4)-, and β(1–6)-linked. The result suggested the presence of the disaccharide LacdiNAc (GalNAcβ1-4GlcNAcβ) (45–49). In contrast, CHO-derived species were not responsive to the β-N-acetylhexosaminidasef digestion (data not shown). This result is consistent with the previous report that LacdiNAc can be synthesized in HEK cells but not CHO cells (54). The presence of LacdiNAc is likely to contribute to the fast clearance of HEK-sKlotho in vivo, similar to the glycoprotein hormone lutropin (55).
Figure 6.

Released 2-AB–labeled N-glycan analysis of CHO-sKlotho (A) and HEK-sKlotho (B). N-Glycans released from CHO- and HEK-sKlotho were subjected to HILIC-FLD glycan analysis as described under “Experimental procedures” and in Fig. S1. A, CHO-sKlotho is heavily sialylated (black trace) and efficiently desialylated by sialidase A digestion (blue trace). B, HEK-sKlotho was found to contain unique N-glycans with LacdiNAc moieties with both sialyation and sulfation (black trace). Exoglycosidase β-N-acetylhexosaminidasef (New England BioLabs; 16-h incubation at 37 °C prior to chromatography) specifically removed one terminal HexNAc from the LacdiNAc (GalNAcβ1–4GlcNAc)-containing N-glycan structures in various proportions due to interference from terminal fucosylation, sialyation, and sulfation (red trace).
To further confirm the presence of LacdiNAc glycan and ascertain which N-linked site is occupied with LacdiNAc glycan, we did the trypsin glycopeptide-mapping experiment (Table 2). As shown in Fig. 7, LC-MS/MS spectra via positive ion mode higher-energy dissociation (HCD) from glycopeptide mapping (Asn-126) demonstrate that the diagnostic oxonium ion observed at m/z 407.1668 (z = 1) clearly distinguishes N-glycans without (Fig. 7A) and with (Fig. 7B) the LacdiNAc structural determinant, as shown for the isomeric N-glycan structures from CHO-sKlotho and HEK-sKlotho, respectively. Furthermore, the isomeric N-glycan structures in A and B differ in HexNAc composition and attachment (linkage) in the antennal region, and because the individual fragment ions across the mass range display identical m/z values between A and B, only differences in ion relative abundance proportions and the diagnostic m/z 407 oxonium ion can distinguish the two structures, in addition to prior knowledge.
Table 2.
Summary table of predominant N-glycans and O-glycans by amino acid site modified, as analyzed by LC-MS/MS tryptic peptide mapping as described under “Experimental procedures”

Figure 7.

A–C, LC-MS/MS spectra from glycopeptide mapping (Asn-126) demonstrated that the diagnostic oxonium ion observed at m/z 407.1668 (z = 1) clearly distinguishes N-glycans without and with the LacdiNAc structural determinant, as shown for CHO-sKlotho (A) and HEK-sKlotho (B and C), respectively. The glycopeptide fragmentation patterns of the CHO-sKlotho precursor ion, m/z 875.7305 (z = 3), in A and the HEK-sKlotho precursor ion, m/z 875.7305 (z = 3), in B resemble two isomeric N-glycans (HexNAc4Man3Fuc1) following positive ion LC-MS/MS analysis via HCD, given the similar fragment ion mass-intensity information; however, the m/z 407 diagnostic oxonium ion provides clear differentiation of LacdiNAc moiety in B. Furthermore, the fragmentation patterns of the HEK-sKlotho precursor ion, m/z 1059.8028 (z = 3), in C confirmed the peptide (Pep) VLPN126GSAGVPNR from Klotho and the covalent N-linked oligosaccharide composition as HexNAc6Man3Fuc2 with the putative N-linked oligosaccharide structure containing LacdiNAc as shown above C (major and minor fragment ions are assigned in Table S3). D, structural modeling of sKlotho. Motif 1 (REGLRYYRRLLERLR) is part of an α-helix (in red, running left to right). Motif 2 (KRLIKVDGVVTKKRK) is part of an extended loop and not in a helix (in red, running left to right).
In Fig. 7C, the LC-MS/MS spectrum shows the fragmentation pattern of the predominant HEK-sKlotho precursor ion m/z 1059.80 (z = 3) (see Table 2 (N126) and Table S3), which confirmed that the peptide, VLPN126GSAGVPNR, from Klotho is occupied with N-linked glycosylation and that the N-linked oligosaccharide composition is HexNAc6Man3Fuc2 with the putative LacdiNAc structure based on the diagnostic m/z 407 oxonium ion. Likewise, the diagnostic oxonium ions at m/z 350 and 553 further indicate that the putative LacdiNAc structure also contains antennal fucose.
It has been reported that adding a C-terminal 19-amino acid α-helix stretch with several basic amino acids is sufficient to mediate GalNAc transfer to N-linked oligosaccharide (45, 56). As shown in Fig. 7D, there are two sequence stretches with multiple basic residues in Klotho protein. Motif 2 (KRLIKVDGVVTKKRK) is part of an extended loop, not in a helix. Motif 1 ((N126GSAGVPN)REGLRYYRRLLERLR) is part of an α-helix immediately C-terminal to Asn-126. The detection of LacdiNAc at this site supports the consensus sequence prediction for GalNAc transfer (45, 56).
As shown in Table 2, further LC-MS/MS tryptic glycopeptide mapping was performed as described under “Experimental procedures.” Occupancy and structures of N-glycans and O-glycans were determined and summarized (Tables S1–S2, S4–S6, and Figs. S2–S41). Besides Asn-126, the sites of Asn-73, Asn-250, Asn-311, Asn-597, and Asn-661 were also found containing sulfated, sialylated, or fucosylated LacdiNAc glycans in HEK-sKlotho. N-Glycans from CHO-sKlotho do not contain any of these species while being efficiently sialylated (Table 2). These data suggested that the LacdiNAc modification might be attributable to the higher coreceptor activity of HEK-sKlotho. Among the N-linked sites, Asn-73, Asn-311, Asn-597, and Asn-661, were found efficiently occupied with N-glycans in both CHO and HEK-sKlotho, whereas Asn-126 and Asn-250 were 20–30% modified. Two closely localized complex N-glycosylation sites at N574QS and N579HT exist on a single tryptic peptide. This made for inconclusive N-glycan assignments for both CHO- and HEK-sKlotho at Asn-574 and Asn-579 due to challenges in LC-MS/MS fragmentation. Besides occupancy of N-glycans, the glycopeptide mapping has also revealed that Thr-945, Ser-961, and Thr-962 were found modified with O-glycans (Table 2), a stem region of Klotho before the transmembrane domain.
N-Linked glycan mutants of sKlotho and structural modeling analysis
Because N-linked sialylation seems to have a clear impact on sKlotho's FGF23 coreceptor function, we decided to further characterize the putative N-glycosylation sites of sKlotho. We generated eight sKlotho mutants with each single glycosylation site removed individually out of the eight potential sites. The mutants were transiently transfected into HEK cells to see if they affect protein expression and secretion. Conditioned medium and cell pellets were analyzed by immunoblotting. As shown in Fig. 8A, sKlotho expression in cell pellets was similar, but protein secretion into conditioned medium varied among these mutants. Mutation N126Q (lane 2) and N311Q (lane 4) did not affect sKlotho secretion significantly, but N73Q (lane 1), N574Q (lane 5), N597Q (lane 7), or N661Q (lane 8) decreased protein secretion substantially. In particular, N250Q (lane 3) in the KL1 domain and N579Q (lane 6) in the KL2 domain nearly eliminated sKlotho secretion. N-Glycosylation at 73, 250, 574, 579, 597, and 661 locations seemed to be important for proper protein folding of Klotho. When the published structure of sKlotho/FGFR1c/FGF23 complex (32) was analyzed and modeled (Fig. 8B), all four N-linked sites at the KL2 domain appear to be critical. The glycans at Asn-579 sit in the interface between FGFR1c and Klotho, in which negatively charged sialic acids could affect the formation of the co-receptor complex. Sugars at Asn-574 and Asn-597 should be pointing toward the outer surface of the plasma membrane. Sialylated N-glycan at either site could be repelled by the negatively charged cell surface and consequentially affect FGF23 signaling. The fourth site that could affect Klotho co-receptor function is Asn-661, which is near Klotho's binding pocket for FGF23.
Figure 8.

Site mutagenesis and structural modeling analysis further indicated that N-glycans might directly modulate Klotho's protein folding and co-receptor function. A, a single N-linked site mutant of sKlotho was transiently transfected into HEK293 cells as described under “Experimental procedures.” Both conditioned medium (CM) and cell pellets were analyzed by SDS-PAGE and immunoblotted with anti-His4 antibody (Qiagen, Germantown, MD). B, structural modeling of sKlotho/FGFR1c/FGF23 complex. Structure from Protein Data Bank entry 5W21 (32) was modeled with extended sugar chains from Fc glycan in Protein Data Bank entry 5VGP.
“Enzyme-up” EE mutation of sKlotho
Another important question regarding structure-function relationship of Klotho is its hallmark feature of sequence similarities with members of glycosidase family 1. Unlike other glycosidase family 1 members, Klotho does not contain two conserved glutamate residues, one acting as a nucleophile and the other as an acid/base. As shown in Fig. 9A, Klotho KL1 domain has the nucleophilic Glu-381, but the acid/base Glu-206 is replaced by an Asn. Inversely, in the KL2 domain, the acid/base Glu-656 is present, but the nucleophile 839 is replaced by Ser in humans.
Figure 9.

Production of the enzyme-up EE mutant in which two catalytic glutamic acid residues conserved across the glycosidase family members were introduced into sKlotho. A, modeling of sKlotho EE mutant. The Klotho structure (5W21) (32) with active site residues was rendered in a space-filling model (CPK). Mutated residue numbers are indicated in red. The right part of the image is the overlay of the KLrP (2E9L, white peptide backbone and green side chain/ligand carbons) and KL1 (5W21, gray peptide backbone side-chain carbons) active site. B, purification of sKlotho EE mutant from stable CHO production. The sKlotho EE mutant was expressed in stable CHO cells, and conditioned medium was purified as described under “Experimental procedures”. Bio-Rad gel filtration standards (thyroglobulin (bovine, 670 kDa), γ-globulin (bovine, 158 kDa), ovalbumin (chicken, 44 kDa), myoglobin (horse, 17 kDa), and vitamin B12 (1.35 kDa)) are indicated by blue arrows.
To explore the role of the glycosidase activity within the overall activity of Klotho, attempts were made to generate mutant forms of the sKlotho protein that would either enhance or abolish glycosidase activity. To increase the activity, the residues at positions 206 and 839 were mutated to glutamic acids (N203E/S839E) to generate the so-called enzyme-up EE mutant. In this way, both KL domains would have the general acid/base and nucleophilic components on either side of the anomeric center to catalyze the hydrolysis of the substrate glycoside bond. In contrast, the Klotho mutants intended to inactivate the glycosidase activity mutated the existing active site glutamates (residues 381 and 656) to Asp and residues 206 and 839 to Ala to generate the so-called “enzyme-dead” mutant (E381Q/E656Q/N206A/S839A).
When the “enzyme-dead” mutant was expressed in transient HEK293 or stable CHO, the expression was completely abolished, and no sKlotho protein was produced (data not shown), indicating that these mutations drastically affect Klotho protein folding. For the enzyme-up EE mutant, no protein was produced with transient HEK293 expression either. When this mutant was expressed in the stable CHO system, the clone that exhibited some expression was expanded into a 10-liter culture. Through an extensive multistep purification process, as described under “Experimental procedures,” about 180 μg of the EE mutant was eventually purified with a purity of 65% and behaved as a monomer in analytical size-exclusion chromatography (Fig. 9B). When the EE mutant was assayed for β-glucuronidase activity, the EE mutant was found to be over 25-fold more active than the WT Klotho (Fig. 10A). When the EE mutant was measured in the FGF23-FGFR1c ERK1/2 activation assay along with WT CHO-sKlotho, the EE mutant's activity was nearly 8-fold less than that of the WT (123 nm versus 15.9 nm EC50, respectively; Fig. 10, B and C). These results suggest that Klotho's co-receptor function might be structurally divergent from its role as a glycosidase.
Figure 10.
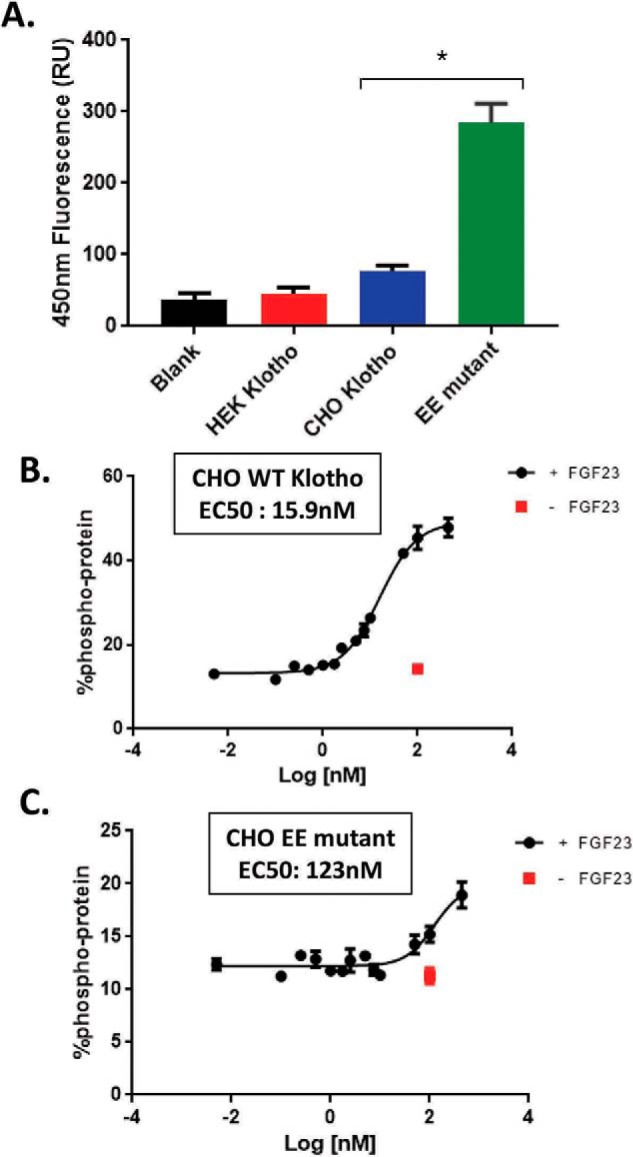
The enzyme-up EE mutant sKlotho had a 25-fold higher β-glucuronidase activity than the WT sKlotho but was 8-fold less active than the WT protein in the FGF23 signaling assay. A, EE mutant sKlotho, along with the stable CHO and transient HEK WT protein, was measured in the β-glucuronidase assay as described under “Experimental procedures.” The error bars at each data point were calculated from a triplicated experiment. Data are shown as mean ± S.E. *, p < 0.05 versus CHO Klotho, ANOVA. Stable CHO-WT sKlotho (B) and EE mutant sKlotho (C) were measured in the FGF23 ERK1/2 activation assay as described under “Experimental procedures.” The error bars at each data point were calculated from a triplicated experiment.
Discussion
The biological function and therapeutic application of the aging suppressor Klotho has been a fascinating research topic since its first discovery more than 20 years ago. This report confirms sKlotho's potential therapeutic effect based on both in vitro (activity assays) and in vivo (PK and AKI models) characterizations. To further explore the therapeutic utility of sKlotho, this study has attempted to uncover novel structure-function relationships and PTM biology of Klotho's extracellular domain. Our data have unexpectedly revealed that sialylation of N-glycans could modulate sKlotho's FGF23-mediated phosphorylation activity. Interestingly, the investigation has also uncovered that sKlotho can be efficiently modified with an unusual N-glycan structure of LacdiNAc and contains a putative GalNAc transfer motif. This is the first report of the detection of this rare PTM on Klotho. In addition, we have found that sKlotho's EE mutant with enhanced glycosidase activity drastically decreased Klotho's FGF23 co-receptor activity, suggesting that these two functions might be structurally divergent. These results in Klotho's PTMs and structure-function relationship not only offer new insights into Klotho biology but also have a potential application in rational engineering for pharmacologically enhanced Klotho therapeutics for kidney disease.
The surprising findings of this study start with the data indicating that sKlotho protein derived from two different production systems had a significant activity difference in vitro. Further investigation revealed that sialidase treatment enhanced the activity of stable CHO proteins, suggesting that sialic acid has a role in regulating Klotho's receptor activity. This result adds another example to a growing list of N-glycan sialylation modifications that could have a direct effect on protein activity (i.e. modulating antibody effector function) (57, 58). To explore the possible mechanism, structural modeling analysis was performed, suggesting that the four N-linked sites at the KL2 domain could possibly contribute to this effect. These sites are located near the interface of either FGF23 or FGFR1c or facing the outer surface of plasma membrane. Asn-579 is particularly interesting because it appears critical for Klotho secretion and KL2 domain folding, yet it sits between FGFR1c and Klotho. N-Glycans at this location could interfere with Klotho activity. A similar situation occurs for immunoglobulin Fc Asn-297 glycans. Although α1,6-fucose of the innermost GlcNAc is part of its typical core N-glycan structure, it interferes with the interaction with glycans at Asn-162 of FcγRIIIa for antibody effector function (59). Systematic mutagenesis combination of Asn-574, Asn-579, Asn-597, and Asn-661 in stable CHO production should allow further study of the importance of the presence of N-glycans on these sites.
Another unanticipated finding in this study is the detection of the uncommon LacdiNAc glycan structure in HEK-sKlotho, which explains its unusually short half-life in rats (Fig. 4A). It has been reported that glycoprotein hormones with the N-glycan terminated with β1,4-linked GalNAc or that with 4-SO4 can be rapidly cleared by asialoglycoprotein receptor and mannose receptor in the liver (60). CHO cells do not produce LacdiNAc glycan structural determinants (54) because they lacking the modifying genes, which is consistent with the lack of detection of this glycan structure in CHO-sKlotho (Figs. 6 and 7 and Table 2). Therefore, the observed high clearance of the HEK-sKlotho is likely due to asialoglycoprotein receptor or mannose receptor–mediated rapid hepatic clearance, whereas the clearance of the CHO-sKlotho might be mainly through slower processes like nonspecific pinocytosis. Glycopeptide-mapping data revealed that N-glycan at Asn-126 contained LacdiNAc structure in the protein sequence region where one potential motif was found to meet the requirements of the reported structural determinant for GalNAc transfer (45, 56), supporting the reported sequence consensus for the modification. Interestingly, LacdiNAc-containing N-glycans were also found in other N-linked sites of sKlotho, suggesting that other motifs might be involved. Proteins without the consensus sequences have been reported to be modified with LacdiNAc glycan (49). Our data demonstrate that Klotho belongs to a handful of LacdiNAc-bearing mammalian glycoproteins, along with the glycoprotein hormone family (61), prolactin-like proteins (62), carbonic anhydrase-6 (61), SorLA/LR11 (63), and others (49). This modification might have physiological consequences for endogenous Klotho, as genes encoding β4GalNAc-T3 (64) and β4GalNAc-T4 (65), accounted for GalNAc transfer activity, have broad issue expression coverage, including in fetal kidney and brain. It is possible that LacdiNAc-modified endogenous Klotho is more active in FGF23 signaling than the nonmodified protein, as our data showing that HEK-sKlotho's coreceptor activity was higher than that of CHO-sKlotho (Fig. 3A). The presence of LacdiNAc glycan in Klotho could increase the interaction with FGFRs or FGF23 through an unknown mechanism. The LacdiNAc modification itself might reflect the protein conformational change of sKlotho, as multiple N-linked sites in HEK-sKlotho were found to be modified. This global change might enhance Klotho's coreceptor function. It would be of great interest to engineer a CHO cell line overexpressing β4GalNAc-T3 or β4GalNAc-T4 (45), which could stably produce sKlotho with LacdiNAc N-glycans fully capped with sialic acids. The resulting new sKlotho variant should not only overcome the short half-life limitation but also possess enhanced FGF23 coreceptor function for therapeutic application.
The other surprising finding from this study is the biochemical evidence suggesting that the two best-established functions of Klotho, glycosidase versus co-receptor for FGF23 signaling, may be structurally divergent. The co-receptor scaffold for FGF23 has been well-demonstrated by biochemical studies and recent structural studies (23–25, 32). As a result of its impaired catalytic machinery and a lack of access to the catalytic center suggested in the structural study (32), Klotho's glycosidase activity has been a matter of debate (6, 32–36). Multiple scenarios have been proposed (33), including substrate-assisted catalysis, the presence of a co-factor, or domain swapping, as half of the catalytic machinery missing in KL1 is present in KL2. The EE mutant generated in this study was poorly expressed; nonetheless, the scale-up efforts in stably expressing CHO cells enabled the generation of enough purified protein for pharmacologic evaluation. Its glucuronidase activity was found to be 25-fold higher than that of the WT protein, demonstrating that the crippled enzyme can indeed be repaired and that the reportedly blocked active site entrance for Klotho is not an impediment relative to other glycosyl hydrolases (32). Unexpectedly, this enzyme-up sKlotho mutant was found to be associated with a drastically decreased in vitro FGF23 co-receptor activity. We have therefore proposed that Klotho's glycosidase function might be structurally divergent from its co-receptor function. An enzymatically crippled WT Klotho might be a structural compromise that allows a delicate balance to accommodate two distinct cellular activities. When the low-expression issue can be resolved, it will be of great interest to generate enough EE mutant protein to further characterize its therapeutic effect in vivo.
This study also found that a number of point mutations impacted sKlotho secretion, including those that cause changes in the catalytic sites and N-linked glycosylation sites. This suggests that all of these residues are critical for the proper folding of the sKlotho protein. The fact that both membrane Klotho and sKlotho are known to be difficult-to-express proteins indicates that even WT Klotho's protein folding is delicate. Six of the eight N-linked glycan sites are important for sKlotho secretion, whereas two of them (each in KL1 and KL2) are nearly essential. Glycan-assisted Klotho protein folding in ER lumen seems to be critical to protein quality control prior to ER exit for surface localization or secretion.
Moreover, additional insights about sKlotho's PTMs were also provided in this study. Both HEK-sKlotho and CHO-sKlotho were found to contain some mucin-type O-linked glycan modifications. Although sKlotho protein has a significant number of Ser or Thr residues, peptide-mapping data identified Thr-945, Ser-961, and Thr-962 near the stem region of Klotho being modified. These positions are right before the transmembrane domain and with a Pro residue nearby, which is thought to increase the rate of O-linked modification (43, 44). It is commonly known that O-linked modification improves protein folding and protects the protein from protease digestion. The significance of these modifications on Klotho biology remains to be determined.
In conclusion, characterization of the sKlotho variants produced in this study revealed that sKlotho underwent multiple PTMs and demonstrated for the first time that sKlotho belongs to a unique group of LacdiNAc-bearing glycoproteins. It would be intriguing to find out whether the endogenous native protein contains this rare type of N-glycan. Various modifications appear to impact sKlotho's in vitro activities and in vivo PK properties. The investigation also indicates that Klotho's FGF23 co-receptor function and β-glucuronidase activity can be modulated through molecular engineering and protein modifications. However, our data also show that it may be challenging to engineer sKlotho proteins with optimal activities in both functions. A further understanding of the specific therapeutic effect of each sKlotho function and its relationship with various PTMs will be important to guide future efforts in the development of sKlotho-based therapeutics for kidney disease.
Experimental procedures
Cell culture and transient HEK293 production
HEK293F cells were grown and maintained in FreeStyleTM 293 medium in a humidified incubator with 5% CO2 at 37 °C. A large-scale transient HEK293F transfection process was used for protein production as described previously (66). Essentially, 1 mg/liter DNA was incubated with 3.5 mg/liter PEI-Max (Sigma) for 20 min before the inoculation into 1.0 × 106/ml of HEK293F cells. Cell culture was continued for 5 days at 37 °C with shaking at 120 rpm. Conditioned medium was harvested and filtered at 0.2 μm prior to the purification process.
Stable CHO cell development and production
CHO-DUKX cells were grown and maintained in a humidified incubator with 7% CO2 at 37 °C. For stable cell line establishment, DNA constructs in pSMED2 vector with a murine cytomegalovirus promoter, encoding human sKlotho-V5-His6 (WT or N206E/S839E), were cloned with XbaI and EcoRI. Native DNA sequences encoding the human mature Klotho extracellular domain (amino acids 1–948) were in-frame fused with N-terminal mouse immunoglobulin heavy-chain signal sequence (MGWSCIILFLVATATGVHS) and C-terminal V5 and His6 tag through PCR. The resulting constructs were linearized with PvuI and transfected into CHO-DUKX cells at 90% confluence in R5CD1 α medium, supplemented with adenosine (10 mg/liter), deoxyadenosine (10 mg/liter), and thymidine (10 mg/liter), and 10% fetal bovine serum (FBS). One day after transfection, cells were placed into R1 α medium with 10% FBS and 50 nm methotrexate as described (67). The stable pools underwent selection for 3 weeks and were then seeded at 2 × 105 cells/ml into serum-free suspension at 37 °C. Stable CHO-DUKX cells were maintained in R5CD1 α medium supplemented with 50 nm methotrexate in suspension with shaking at 120 rpm. During production, cells were seeded in R5CD1 α medium at a cell density of 0.44 × 106/ml at 37 °C for 4 days and then temperature-shifted to 31 °C for an additional 6 days with nutrient feeds. NaHCO3 was added to maintain normal pH when needed during cell culturing. Conditioned medium was harvested after a 10-day production. Conditioned medium was harvested and cleared by centrifugation and 0.2-μm-filtered prior to purification.
Transient HEK293 protein purification
8 liters of conditioned medium from HEK293F expression was buffer-exchanged through tangential flow filters (10 kDa, Pellicon®, Millipore–Sigma, Burlington, MA) into PBS buffer and concentrated 20-fold to around 400 ml. 5 mm imidazole and 250 mm NaCl were added, and the His6-tagged sKlotho protein was captured on a HisTrap column (catalog no. 17-5248-02, GE Healthcare Life Sciences). The column was washed with PBS plus 0.5 m NaCl followed by PBS and eluted using a linear gradient to 0.5 m imidazole. The sKlotho protein was diluted into 20 mm Tris, pH 8.0, and purified on a HiTrap Q HP column (catalog no. 17-0510-01, GE Healthcare Life Sciences) with a linear gradient to 0.5 m NaCl.
Stable CHO protein purification
The sKlotho protein was purified using a three-column purification process of Q-Sepharose FF, HisTrap, and SEC. 10 liters of 0.2-μm-filtered conditioned medium from stable CHO production was adjusted with 2 m Tris base to pH 8.0 and loaded onto an anion-exchange column (Q-Sepharose FF, catalog no. 17-0510-01, GE Healthcare Life Sciences). The anion-exchange column was equilibrated with 5 column volumes (CVs) of 50 mm Tris, pH 8.0. After the conditioned medium was pumped into the protein column, the column was washed with 10 CVs of 50 mm Tris, pH 8.0. The bound protein was then step-eluted with 20% buffer B (50 mm Tris, 1 m NaCl, pH 8.0). The Q-Sepharose FF pool was adjusted to 20 mm imidazole, 300 mm NaCl, pH 8.0, before loading onto a HisTrap HP column (catalog no. 17-5248-02, GE Healthcare Life Sciences). The HisTrap column was gradient-eluted with 100% buffer B containing 500 mm imidazole, 300 mm NaCl, pH 8.0, over 10 CVs. The HisTrap peak pool was concentrated with a 10,000 molecular weight cutoff Amicon® Ultra Filters (UFC5010, Millipore–Sigma), and loaded over a SEC column (Superdex 200 10/300GL, catalog no. 17-5175-01, GE Healthcare Life Sciences). The purity of the SEC pool was confirmed by analytical SEC-HPLC (Superdex 200 10/300GL) and SDS-PAGE. The final pool was 0.2-μm-filtered and stored in a −80 °C freezer. The SDS-PAGE was run with NuPAGETM 4–12% BisTris gel (Thermo Fisher Scientific). Western blotting in Fig. 9B was in NuPAGETM 4–12% BisTris gel and transferred to an iBindTM Card (catalog no. SLF1010, Thermo Fisher Scientific) by an iBindTM device (catalog no. SLF 1000, Thermo Fisher Scientific) and immunoblotted with anti-V5 antibody (1:5000) (catalog no. R960-25, Thermo Fisher Scientific) and anti-mouse horseradish peroxidase (1:400) (catalog no. 62-6520, Thermo Fisher Scientific).
ERK1/2 activation assay
Stably transfected L6 cells expressing human FGFR1c (Biomiga, Inc., San Diego, CA) were maintained in RPMI medium containing 10% FBS and 7 μg/ml puromycin. For the ERK activation assay, cells were grown to 80% confluence (20,000 cells seeded in each well for 48 h) in 96-well plates and serum-starved overnight. Serum-free media include RPMI, 0.1% BSA, and 7 μg/ml puromycin. 100 μg/ml of commercially available FGF23 (catalog no. 2604-FG, R&D Systems) and purified sKlotho variants were co-treated for 1 h. An MSD assay (catalog no. K15107D, Meso Scale Discovery, Rockville, MD) was used to quantify the percentage of phosphoprotein. 75 μl of the lysis buffer, prepared according to the MSD kit, was added to each well to lyse the cells. The MSD assay was then performed, and the results were obtained according to the manufacturer's protocol.
β-Glucuronidase assay
To examine the enzymatic activity of purified sKlotho variants, 4-methylumbelliferyl β-d-glucuronide hydrate (catalog no. M9130, Sigma–Aldrich) was used as substrate. 20 μg of sKlotho protein was added to reaction buffer (0.1 m sodium citrate buffer, pH 5.5, 0.05 m NaCl, 0.01% Tween 20) containing 0.5 mm substrate. The final volume of the assay was 100 μl. The assay was incubated at 37 °C for 2 h. The fluorescence intensities were measured at an excitation wavelength of 360 nm and an emission wavelength of 470 nm. β-Glucuronidase from bovine liver (catalog no. G0251, Sigma–Aldrich) was used as a positive control.
In vivo pharmacokinetic properties of sKlotho
The pharmacokinetic properties of WT sKlotho proteins were evaluated in male Sprague–Dawley rats dosed at 1 mg/kg for the HEK-sKlotho protein and at 0.5 mg/kg for the CHO-sKlotho protein following a single IV or IP administration. Serum levels of either HEK- or CHO-sKlotho were determined using an LC-MSMS method following immunoprecipitation of the protein using a monoclonal anti-human Klotho antibody, MAB5334, obtained from R&D Systems. Following immunoprecipitation of the sKlotho protein from 30–50 μl of serum samples, the intact proteins were further digested as described before (68) and injected into LC-MSMS for quantitative measurement of the Klotho protein using four specific peptides, LQDAYGGWANR, FIMETLK, LWITMNEPYT, and NNFLLPYFTEDEK. All pharmacokinetic parameters were determined from individual animal data using noncompartmental analysis using WinNonlin software (version 5.2, Pharsight).
Single-dose administration of sKlotho after ischemia-reperfusion injury in rats
The study was conducted in accordance with the current guidelines for animal welfare, and all procedures were reviewed and approved by the Pfizer Institutional Animal Care and Use Committee. Male Sprague–Dawley rats were used to evaluate the effect of CHO-sKlotho on kidney structure and function in the ischemia-reperfusion injury model of acute kidney injury. Rats were subjected to renal bilateral clamping for 40 min via a dorsal approach under aseptic conditions and general anesthesia with isoflurane. Vehicle (150 mm NaCl, 10 mm HEPES, pH 7.4) or CHO-sKlotho (0.01 mg/kg IP) was administered at 30–60 min after reperfusion, and rats were followed up for 7 days thereafter. GFR measurement via the transcutaneous method, 24-urine sample collection using metabolic cages, and blood collection were performed before surgery (baseline measures) and at 1, 2, and 7 days post-surgery. At the end of the study, rats were euthanized, and kidneys were collected for immunomorphometry of α-SMA.
Deglycosylation of N-linked glycans and disulfide bond reduction
Protein samples were deglycosylated by PNGase F (New England BioLabs, Ipswich, MA) in the presence of 0.1% SDS (Sigma) and 0.1% Nonidet P-40 (New England Biolabs). The detergents were removed by a Pierce detergent removal spin column (Thermo Fisher Scientific), and protein was further reduced by incubation in 100 mm DTT (Thermo Fisher Scientific) in the presence of 3.5 m guanidine HCl.
Liquid chromatography mass spectrometry
LC-MS analysis was performed using an ultrahigh-resolution ESI-QTOF Bruker maXis II mass spectrometer (Bruker, Billerica, MA) coupled to a Waters H-Class (Waters, Milford, MA) UPLC. The deglycosylated and reduced samples were separated over a Waters BEH C4 (1.7 Å, 2.1 × 150 mm) column maintained at 65 °C with a flow rate of 300 μl/min. Mobile phase A was water with 5% acetonitrile and 0.05% TFA, and mobile phase B was 50:50 acetonitrile/2-propanol and 0.05% TFA. Proteins were eluted from the column using a gradient: 3–20% B in 2.0 min, 20–40% B in 40 min, and 40–90% B in 2 min. The mass spectrometer was run in positive MS only mode scanning from 850 to 4000 m/z, and data were acquired with oTOF control software. The QTOF MS signal was deconvoluted using Maximum Entropy in Bruker Data Analysis version 4.4 (Bruker).
Lys-C/trypsin digestion and LC-MS/MS peptide mapping
The adapted protocol was published previously (69). Protein samples (80 μg) were concentrated into 7 m guanidine HCl in 100 mm Tris-HCl, pH 8.2, via Amicon molecular weight cutoff membranes. Reduction was accomplished with the addition of 5 μl of 0.2 m DTT followed by a 30-min incubation at 37 °C. Alkylation was achieved by adding 20 μl of 200 mm iodoacetic acid followed by incubation in the dark for 30 min. Following denaturation, reduction, and alkylation, the samples were buffer-exchanged into 100 mm Tris, pH 8.2, digestion buffer using Bio-Spin 6 columns (Bio-Rad). Lyophilized Lys-C/trypsin mix was reconstituted to 1 mg/ml with water. Digestions were initiated with the addition of 1 mg/ml Lys-C/trypsin solution to the reduced, alkylated, desalted protein samples, resulting in a 1:5 enzyme/protein (w/w) ratio at 37 °C for 2 h. Aliquots were transferred into autosampler vials and quenched with 10% TFA.
Briefly, chromatographic separation of peptides was carried out on an Agilent 1290 Infinity II UHPLC system equipped with a 2.1 × 150-mm, 2.5-μm Waters XSelect XP CSH C18 column with UV detection at 214 nm. Separation was performed as described (69) with the exception that column temperature was maintained at 60 °C during separation.
LC-MS and LC-MS/MS experiments were performed using a Thermo Fisher Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer operated in the positive ion mode. Peptide accurate masses and sequences were obtained by MS and MS/MS, respectively. Full-scan mass spectra were collected with a resolving power of 60,000. The spray voltage was 3.5 kV, the ion transfer tube temperature was 325 °C, and the sheath and auxiliary gas flow rates were 35 and 10, respectively. Data-dependent fragmentation for glycopeptides with 2–8 charge states was induced by HCD using collision energy of 20% in the Orbitrap or electron transfer dissociation using higher energy in the Orbitrap (EThcD) for glycopeptide sequence analysis. Additional parameters were as follows: 1 microscan/spectrum; 0.7 m/z precursor isolation window; MS injection time of 100 ms; and MS/MS automatic gain control of 1.0 × 105 with maximum MS/MS injection time of 118 ms.
Raw files of Lys-C/trypsin-digested Klotho were searched against both the amino acid sequence and a N-/O-glycan database using ByosTM (Protein Metrics, version 3.5-30) for peptide/glycopeptide identification with a peptide ion m/z tolerance of 5 ppm and a MS/MS fragment ion m/z tolerance of 10 ppm for HCD or EThcD; fixed modifications included carboxymethylated cysteine, and variable modifications included N-linked glycosylation of asparagine and O-linked glycosylation of serine and threonine. ByonicTM allows one N-linked glycan modification on the NX(not P)(S/T) consensus motif per peptide. The glycan database contained ∼330 structures and was manually updated with all expected LacdiNAc moieties. The N-linked glycans were characterized by fragmenting the glycopeptides via HCD, yielding glycan-based fragment ions and oxonium ions. The polypeptide portion of the glycopeptide was sequenced via EThcD. HCD and EThcD spectra were also verified manually using Thermo Xcalibur version 4.1 software.
HILIC for N-glycan profiling
N-Glycan profiling was carried out by a previously published HILIC-fluorescence (FLD) method (70). Briefly, N-glycans were released from the sKlotho protein using PNGase F, labeled with a fluorescent probe, 2-aminobenzamide (2-AB), separated using hydrophilic interaction chromatography with a Waters Xbridge amide HPLC column, and detected using fluorescence. The peaks are identified based on comparison of retention times with peaks in a well-characterized in-house standard. Orthogonal confirmation of sialylated peak identity was achieved by treatment of the released and labeled N-linked glycans with sialidase A (Prozyme) or β-N-acetylhexosaminidasef (New England BioLabs). Sialidase A–treated and β-N-acetylhexosaminidasef-treated samples were incubated for 16 h at 37 °C prior to chromatography. The chromatographic profiles of sialidase A–treated, β-N-acetylhexosaminidasef–treated, and nontreated samples were compared to qualitatively assess the presence or absence of peaks.
LC/MS–N-linked glycan analysis
PNGase F–released and 2-AB–labeled N-glycans were separated by HILIC ultrahigh performance LC with fluorescence optical detection (UHPLC-FLD) using a gradient as described under “HILIC for N-glycan profiling” (70) with the exception that a Waters H-Class Bio Acquity UPLC system was used with a Waters BEH amide (PN 186004802) column at a 0.18 ml/min flow rate. Peak identity was determined by using an on-line ultrahigh-resolution ESI-QTOF Bruker maXis 4G mass spectrometer. The TOF was operated in positive ion mode. Instrument settings for sample analysis were as follows: capillary voltage 4.2 kV, nebulizer gas 1.6 bar, in-source collision energy 0 eV, drying gas 8 liters/min and 180 °C, and collision energy 4 eV. Source parameters were optimized by performing external and internal calibration using Agilent Tuning Mix (part no. G1969-85000). Lock Mass (Agilent G1982-85001) was used to bring mass accuracy to <2 ppm. Mass spectra were acquired from m/z 500–2500. Fluorescence traces from released, 2-AB–labeled N-glycans from the HILIC-FLD and HILIC via UHPLC-FLD/QTOF MS experiments were integrated using Empower (Waters). Corresponding QTOF MS signal was manually annotated using Bruker Data Analysis version 4.4.
Data availability
The mass spectrometry glycomics data have been deposited to the MassIVE repository with the data set identifier MSV000084822.
Author contributions
X. Z., Y. W., M. L., J. C. R., K. M., T. I., J. J. S., R. K., L. L., and V. C. conceptualization; X. Z., S. J., Y. W., and V. C. resources; X. Z., S. J., Y. W., M. L., J. C. R., T. I., Y. Z., E. S., J. C., B. T., K. C., J. J. S., K. J., J. P., J. R. A., S. Y., R. G. V., A. C. O., K. L., Q. Y., W. D., A. S., J. Z., D. F., A. D., R. Z., H. L. Z., R. K., and V. C. data curation; X. Z., S. J., and Y. W. software; X. Z., S. J., Y. W., M. L., J. C. R., K. M., T. I., Y. Z., E. S., J. C., B. T., K. C., K. J., J. P., J. R. A., S. Y., R. G. V., A. C. O., K. L., Q. Y., W. D., A. S., J. Z., D. F., A. D., R. Z., H. L. Z., R. K., L. L., and V. C. formal analysis; X. Z., L. L., and V. C. supervision; X. Z. and V. C. funding acquisition; X. Z., S. J., Y. W., M. L., J. C. R., K. M., and V. C. validation; X. Z., S. J., Y. W., M. L., J. C. R., K. M., T. I., and V. C. investigation; X. Z., S. J., Y. W., J. C. R., and V. C. visualization; X. Z., S. J., Y. W., M. L., Y. Z., E. S., J. C., J. J. S., and V. C. methodology; X. Z., S. J., Y. W., M. L., J. C. R., K. M., Y. Z., J. C., J. P., K. L., A. S., and V. C. writing-original draft; X. Z. and Y. W. project administration; X. Z., S. J., M. L., J. C. R., K. M., S. Y., and V. C. writing-review and editing.
Supplementary Material
Acknowledgments
We thank E. Bennett, W. Stochaj, M. Perreault, I. Korakas, C. Facemire, T. Dickinson, J. Marshall, and W. Somers for helpful discussions. We also thank an anonymous JBC reviewer for pointing out the possibility of LacdiNAc modification on HEK-derived sKlotho.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S6 and Figs. S1–S41.
- CKD
- chronic kidney disease
- α-SMA
- α-smooth muscle actin
- AKI
- acute kidney injury
- BUN
- blood urea nitrogen
- CHO
- Chinese hamster ovary
- EThcD
- electron transfer dissociation
- GFR
- glomerular filtration rate
- HEK
- human embryonic kidney
- HCD
- higher-energy dissociation
- HILIC
- hydrophilic interaction liquid chromatography
- IEF
- isoelectro-focused
- IP
- intraperitoneal
- IRI
- ischemia-reperfusion injury
- IV
- intravenous
- LacdiNAc
- N,N′-di-N-acetyllactose diamine
- PK
- pharmacokinetics
- PTM
- post-translational modification
- SEC
- size-exclusion chromatography
- sKlotho
- soluble Klotho
- UACR
- urine albumin-to-creatinine ratio
- UHPLC
- ultrahigh-performance liquid chromatography
- FLD
- fluorescence optical detection
- PNGase F
- peptide:N-glycosidase F
- NeuAc
- N-acetylneuraminic acid
- FGF
- fibroblast growth factor
- CV
- column volume
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ERK
- extracellular signal–regulated kinase
- ESI
- electrospray ionization
- 2-AB
- 2-aminobenzamide
- ANOVA
- analysis of variance.
References
- 1. Coresh J., Selvin E., Stevens L. A., Manzi J., Kusek J. W., Eggers P., Van Lente F., and Levey A. S. (2007) Prevalence of chronic kidney disease in the United States. JAMA 298, 2038–2047 10.1001/jama.298.17.2038 [DOI] [PubMed] [Google Scholar]
- 2. Levey A. S., Atkins R., Coresh J., Cohen E. P., Collins A. J., Eckardt K. U., Nahas M. E., Jaber B. L., Jadoul M., Levin A., Powe N. R., Rossert J., Wheeler D. C., Lameire N., and Eknoyan G. (2007) Chronic kidney disease as a global public health problem: approaches and initiatives—a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 72, 247–259 10.1038/sj.ki.5002343 [DOI] [PubMed] [Google Scholar]
- 3. Radhakrishnan J., Remuzzi G., Saran R., Williams D. E., Rios-Burrows N., Powe N., CDC-CKD Surveillance Team, Brück K., Wanner C., Stel V. S., European CKD Burden Consortium, Venuthurupalli S. K., Hoy W. E., Healy H. G., Salisbury A., et al. (2014) Taming the chronic kidney disease epidemic: a global view of surveillance efforts. Kidney Int. 86, 246–250 10.1038/ki.2014.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grams M. E., Plantinga L. C., Hedgeman E., Saran R., Myers G. L., Williams D. E., Powe N. R., and CDC CKD Surveillance Team (2011) Validation of CKD and related conditions in existing data sets: a systematic review. Am. J. Kidney Dis. 57, 44–54 10.1053/j.ajkd.2010.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stenvinkel P., and Larsson T. E. (2013) Chronic kidney disease: a clinical model of premature aging. Am. J. Kidney Dis. 62, 339–351 10.1053/j.ajkd.2012.11.051 [DOI] [PubMed] [Google Scholar]
- 6. Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T., Ohyama Y., Kurabayashi M., Kaname T., Kume E., Iwasaki H., Iida A., Shiraki-Iida T., Nishikawa S., Nagai R., and Nabeshima Y. I. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 10.1038/36285 [DOI] [PubMed] [Google Scholar]
- 7. Hu M. C., Shi M., Zhang J., Quiñones H., Griffith C., Kuro-o M., and Moe O. W. (2011) Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 22, 124–136 10.1681/ASN.2009121311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim H. R., Nam B. Y., Kim D. W., Kang M. W., Han J. H., Lee M. J., Shin D. H., Doh F. M., Koo H. M., Ko K. I., Kim C. H., Oh H. J., Yoo T. H., Kang S. W., Han D. S., and Han S. H. (2013) Circulating α-klotho levels in CKD and relationship to progression. Am. J. Kidney Dis. 61, 899–909 10.1053/j.ajkd.2013.01.024 [DOI] [PubMed] [Google Scholar]
- 9. Pavik I., Jaeger P., Ebner L., Wagner C. A., Petzold K., Spichtig D., Poster D., Wüthrich R. P., Russmann S., and Serra A. L. (2013) Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol. Dial. Transplant. 28, 352–359 10.1093/ndt/gfs460 [DOI] [PubMed] [Google Scholar]
- 10. Mencke R., Olauson H., and Hillebrands J. L. (2017) Effects of Klotho on fibrosis and cancer: a renal focus on mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 121, 85–100 10.1016/j.addr.2017.07.009 [DOI] [PubMed] [Google Scholar]
- 11. Lu X., and Hu M. C. (2017) Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis. 3, 15–23 10.1159/000452880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erben R. G., and Andrukhova O. (2017) FGF23-Klotho signaling axis in the kidney. Bone 100, 62–68 10.1016/j.bone.2016.09.010 [DOI] [PubMed] [Google Scholar]
- 13. Sakan H., Nakatani K., Asai O., Imura A., Tanaka T., Yoshimoto S., Iwamoto N., Kurumatani N., Iwano M., Nabeshima Y., Konishi N., and Saito Y. (2014) Reduced renal α-Klotho expression in CKD patients and its effect on renal phosphate handling and vitamin D metabolism. PLoS ONE 9, e86301 10.1371/journal.pone.0086301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu M. C., Shi M., Cho H. J., Adams-Huet B., Paek J., Hill K., Shelton J., Amaral A. P., Faul C., Taniguchi M., Wolf M., Brand M., Takahashi M., Kuro-O M., Hill J. A., and Moe O. W. (2015) Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol 26, 1290–1302 10.1681/ASN.2014050465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuro-O M. (2017) The FGF23 and Klotho system beyond mineral metabolism. Clin. Exp. Nephrol. 21, 64–69 10.1007/s10157-016-1357-6 [DOI] [PubMed] [Google Scholar]
- 16. Hu M. C., Kuro-o M., Moe O. W. (2013) Renal and extrarenal actions of Klotho. Semin. Nephrol. 33, 118–129 10.1016/j.semnephrol.2012.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitagawa M., Sugiyama H., Morinaga H., Inoue T., Takiue K., Ogawa A., Yamanari T., Kikumoto Y., Uchida H. A., Kitamura S., Maeshima Y., Nakamura K., Ito H., and Makino H. (2013) A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PLoS ONE 8, e56695 10.1371/journal.pone.0056695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grabner A., and Faul C. (2016) The role of fibroblast growth factor 23 and Klotho in uremic cardiomyopathy. Curr. Opin. Nephrol. Hypertens. 25, 314–324 10.1097/MNH.0000000000000231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu M. C., Shi M., Gillings N., Flores B., Takahashi M., Kuro-O M., and Moe O. W. (2017) Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 91, 1104–1114 10.1016/j.kint.2016.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie J., Yoon J., An S. W., Kuro-o M., Huang C. L. (2015) Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J. Am. Soc. Nephrol. 26, 1150–1160 10.1681/ASN.2014040325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang K., Wang C., Nie L., Zhao X., Gu J., Guan X., Wang S., Xiao T., Xu X., He T., Xia X., Wang J., and Zhao J. (2015) Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. J. Am. Soc. Nephrol. 26, 2434–2446 10.1681/ASN.2014060543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henrissat B., and Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 10.1016/S0959-440X(97)80072-3 [DOI] [PubMed] [Google Scholar]
- 23. Kurosu H., Ogawa Y., Miyoshi M., Yamamoto M., Nandi A., Rosenblatt K. P., Baum M. G., Schiavi S., Hu M. C., Moe O. W., and Kuro-o M. (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281, 6120–6123 10.1074/jbc.C500457200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K., Fujita T., Fukumoto S., and Yamashita T. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 10.1038/nature05315 [DOI] [PubMed] [Google Scholar]
- 25. Goetz R., Nakada Y., Hu M. C., Kurosu H., Wang L., Nakatani T., Shi M., Eliseenkova A. V., Razzaque M. S., Moe O. W., Kuro-o M., Mohammadi M. (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. U.S.A. 107, 407–412 10.1073/pnas.0902006107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu M. C., Kuro-o M., Moe O. W. (2013) Klotho and chronic kidney disease. Contrib. Nephrol. 180, 47–63 10.1159/000346778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu M. C., Kuro-o M., and Moe O. W. (2014) αKlotho and vascular calcification: an evolving paradigm. Curr. Opin. Nephrol. Hypertens. 23, 331–339 10.1097/01.mnh.0000447024.97464.a3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Erben R. G. (2016) Update on FGF23 and Klotho signaling. Mol. Cell. Endocrinol. 432, 56–65 10.1016/j.mce.2016.05.008 [DOI] [PubMed] [Google Scholar]
- 29. Dalton G. D., Xie J., An S. W., and Huang C. L. (2017) New insights into the mechanism of action of soluble klotho. Front. Endocrinol. (Lausanne) 8, 323 10.3389/fendo.2017.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen C. D., Podvin S., Gillespie E., Leeman S. E., and Abraham C. R. (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U.S.A. 104, 19796–19801 10.1073/pnas.0709805104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bloch L., Sineshchekova O., Reichenbach D., Reiss K., Saftig P., Kuro-o M., Kaether C. (2009) Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 583, 3221–3224 10.1016/j.febslet.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen G., Liu Y., Goetz R., Fu L., Jayaraman S., Hu M. C., Moe O. W., Liang G., Li X., and Mohammadi M. (2018) α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553, 461–466 10.1038/nature25451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tohyama O., Imura A., Iwano A., Freund J. N., Henrissat B., Fujimori T., and Nabeshima Y. (2004) Klotho is a novel β-glucuronidase capable of hydrolyzing steroid β-glucuronides. J. Biol. Chem. 279, 9777–9784 10.1074/jbc.M312392200 [DOI] [PubMed] [Google Scholar]
- 34. Chang Q., Hoefs S., van der Kemp A. W., Topala C. N., Bindels R. J., and Hoenderop J. G. (2005) The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493 10.1126/science.1114245 [DOI] [PubMed] [Google Scholar]
- 35. Cha S. K., Ortega B., Kurosu H., Rosenblatt K. P., Kuro-O M., and Huang C. L. (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl. Acad. Sci. U.S.A. 105, 9805–9810 10.1073/pnas.0803223105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cha S. K., Hu M. C., Kurosu H., Kuro-o M., Moe O., and Huang C. L. (2009) Regulation of renal outer medullary potassium channel and renal K+ excretion by Klotho. Mol. Pharmacol. 76, 38–46 10.1124/mol.109.055780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu M. C., Shi M., Zhang J., Pastor J., Nakatani T., Lanske B., Razzaque M. S., Rosenblatt K. P., Baum M. G., Kuro-o M., Moe O. W. (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 24, 3438–3450 10.1096/fj.10-154765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong X., and Wright J. F. (2013) Biological insights into therapeutic protein modifications throughout trafficking and their biopharmaceutical applications. Int. J. Cell Biol. 2013, 273086 10.1155/2013/273086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walsh G., and Jefferis R. (2006) Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 24, 1241–1252 10.1038/nbt1252 [DOI] [PubMed] [Google Scholar]
- 40. Kornfeld R., and Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 10.1146/annurev.bi.54.070185.003215 [DOI] [PubMed] [Google Scholar]
- 41. Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H. (eds) (2017) Essentials of Glycobiology, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor Laboratory, NY: [PubMed] [Google Scholar]
- 42. Moremen K. W., Tiemeyer M., and Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 10.1038/nrm3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brockhausen I., Schachter H., and Stanley P. (2009) O-GalNAc glycans. in Essentials of Glycobiology, 2nd Ed. (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 44. Van den Steen P., Rudd P. M., Dwek R. A., and Opdenakker G. (1998) Concepts and principles of O-linked glycosylation. Crit. Rev. Biochem. Mol. Biol. 33, 151–208 10.1080/10409239891204198 [DOI] [PubMed] [Google Scholar]
- 45. Miller E., Fiete D., Blake N. M., Beranek M., Oates E. L., Mi Y., Roseman D. S., and Baenziger J. U. (2008) A necessary and sufficient determinant for protein-selective glycosylation in vivo. J. Biol. Chem. 283, 1985–1991 10.1074/jbc.M708160200 [DOI] [PubMed] [Google Scholar]
- 46. Manzella S. M., Hooper L. V., and Baenziger J. U. (1996) Oligosaccharides containing β1,4-linked N-acetylgalactosamine, a paradigm for protein-specific glycosylation. J. Biol. Chem. 271, 12117–12120 10.1074/jbc.271.21.12117 [DOI] [PubMed] [Google Scholar]
- 47. Kawar Z. S., Haslam S. M., Morris H. R., Dell A., and Cummings R. D. (2005) Novel poly-GalNAcβ1–4GlcNAc (LacdiNAc) and fucosylated poly-LacdiNAc N-glycans from mammalian cells expressing β1,4-N-acetylgalactosaminyltransferase and α1,3-fucosyltransferase. J. Biol. Chem. 280, 12810–12819 10.1074/jbc.M414273200 [DOI] [PubMed] [Google Scholar]
- 48. Nimtz M., Conradt H. S., and Mann K. (2004) LacdiNAc (GalNAcβ1–4GlcNAc) is a major motif in N-glycan structures of the chicken eggshell protein ovocleidin-116. Biochim. Biophys. Acta 1675, 71–80 10.1016/j.bbagen.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 49. Bonar D., and Hanisch F. G. (2014) Trefoil factor family domains represent highly efficient conformational determinants for N-linked N,N′-di-N-acetyllactosediamine (LacdiNAc) synthesis. J. Biol. Chem. 289, 29677–29690 10.1074/jbc.M114.596049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Croset A., Delafosse L., Gaudry J. P., Arod C., Glez L., Losberger C., Begue D., Krstanovic A., Robert F., Vilbois F., Chevalet L., and Antonsson B. (2012) Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J. Biotechnol. 161, 336–348 10.1016/j.jbiotec.2012.06.038 [DOI] [PubMed] [Google Scholar]
- 51. Wang M., Ishino T., Joyce A., Tam A., Duan W., Lin L., Somers W. S., Kriz R., and O'Hara D. M. (2015) Faster in vivo clearance of human embryonic kidney than Chinese hamster ovary cell derived protein: role of glycan mediated clearance. J. Biosci. Bioeng. 119, 657–660 10.1016/j.jbiosc.2014.11.008 [DOI] [PubMed] [Google Scholar]
- 52. Zhong X., Ma W., Meade C. L., Tam A. S., Llewellyn E., Cornell R., Cote K., Scarcelli J. J., Marshall J. K., Tzvetkova B., Figueroa B., DiNino D., Sievers A., Lee C., Guo J., et al. (2019) Transient CHO expression platform for robust antibody production and its enhanced N-glycan sialylation on therapeutic glycoproteins. Biotechnol. Prog. 35, e2724 10.1002/btpr.2724 [DOI] [PubMed] [Google Scholar]
- 53. Wong-Madden S. T., and Landry D. (1995) Purification and characterization of novel glycosidases from the bacterial genus Xanthomonas. Glycobiology 5, 19–28 10.1093/glycob/5.1.19 [DOI] [PubMed] [Google Scholar]
- 54. Do K. Y., Do S. I., and Cummings R. D. (1997) Differential expression of LacdiNAc sequences (GalNAcβ1–4GlcNAc-R) in glycoproteins synthesized by Chinese hamster ovary and human 293 cells. Glycobiology 7, 183–194 10.1093/glycob/7.2.183 [DOI] [PubMed] [Google Scholar]
- 55. Fiete D., Srivastava V., Hindsgaul O., and Baenziger J. U. (1991) A hepatic reticuloendothelial cell receptor specific for SO4-4GalNAcβ1,4GlcNAcβ1,2Manα that mediates rapid clearance of lutropin. Cell 67, 1103–1110 10.1016/0092-8674(91)90287-9 [DOI] [PubMed] [Google Scholar]
- 56. Mengeling B. J., Manzella S. M., and Baenziger J. U. (1995) A cluster of basic amino acids within an α-helix is essential for α-subunit recognition by the glycoprotein hormone N-acetylgalactosaminyltransferase. Proc. Natl. Acad. Sci. U.S.A. 92, 502–506 10.1073/pnas.92.2.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaneko Y., Nimmerjahn F., and Ravetch J. V. (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313, 670–673 10.1126/science.1129594 [DOI] [PubMed] [Google Scholar]
- 58. Scallon B. J., Tam S. H., McCarthy S. G., Cai A. N., and Raju T. S. (2007) Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol. Immunol. 44, 1524–1534 10.1016/j.molimm.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 59. Mizushima T., Yagi H., Takemoto E., Shibata-Koyama M., Isoda Y., Iida S., Masuda K., Satoh M., and Kato K. (2011) Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells 16, 1071–1080 10.1111/j.1365-2443.2011.01552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mi Y., Lin A., Fiete D., Steirer L., and Baenziger J. U. (2014) Modulation of mannose and asialoglycoprotein receptor expression determines glycoprotein hormone half-life at critical points in the reproductive cycle. J. Biol. Chem. 289, 12157–12167 10.1074/jbc.M113.544973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith P. L., and Baenziger J. U. (1988) A pituitary N-acetylgalactosamine transferase that specifically recognizes glycoprotein hormones. Science 242, 930–933 10.1126/science.2460923 [DOI] [PubMed] [Google Scholar]
- 62. Manzella S. M., Dharmesh S. M., Cohick C. B., Soares M. J., and Baenziger J. U. (1997) Developmental regulation of a pregnancy-specific oligosaccharide structure, NeuAcα2,6GalNAcβ1,4GlcNAc, on select members of the rat placental prolactin family. J. Biol. Chem. 272, 4775–4782 10.1074/jbc.272.8.4775 [DOI] [PubMed] [Google Scholar]
- 63. Fiete D., Mi Y., Oats E. L., Beranek M. C., and Baenziger J. U. (2007) N-Linked oligosaccharides on the low density lipoprotein receptor homolog SorLA/LR11 are modified with terminal GalNAc-4-SO4 in kidney and brain. J. Biol. Chem. 282, 1873–1881 10.1074/jbc.M606455200 [DOI] [PubMed] [Google Scholar]
- 64. Sato T., Gotoh M., Kiyohara K., Kameyama A., Kubota T., Kikuchi N., Ishizuka Y., Iwasaki H., Togayachi A., Kudo T., Ohkura T., Nakanishi H., and Narimatsu H. (2003) Molecular cloning and characterization of a novel human β 1,4-N-acetylgalactosaminyltransferase, β4GalNAc-T3, responsible for the synthesis of N,N′-diacetyllactosediamine, GalNAcβ1–4GlcNAc. J. Biol. Chem. 278, 47534–47544 10.1074/jbc.M308857200 [DOI] [PubMed] [Google Scholar]
- 65. Gotoh M., Sato T., Kiyohara K., Kameyama A., Kikuchi N., Kwon Y. D., Ishizuka Y., Iwai T., Nakanishi H., and Narimatsu H. (2004) Molecular cloning and characterization of β1,4-N-acetylgalactosaminyltransferases IV synthesizing N,N′-diacetyllactosediamine. FEBS Lett. 562, 134–140 10.1016/S0014-5793(04)00219-4 [DOI] [PubMed] [Google Scholar]
- 66. Zhong X., Kieras E., Sousa E., D'Antona A., Baber J. C., He T., Desharnais J., Wood L., Luxenberg D., Stahl M., Kriz R., Lin L., Somers W., Fitz L. J., and Wright J. F. (2013) Pyroglutamate and O-linked glycan determine functional production of anti-IL17A and anti-IL22 peptide-antibody bispecific genetic fusions. J. Biol. Chem. 288, 1409–1419 10.1074/jbc.M112.417717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhong X., Cooley C., Seth N., Juo Z. S., Presman E., Resendes N., Kumar R., Allen M., Mosyak L., Stahl M., Somers W., and Kriz R. (2012) Engineering novel Lec1 glycosylation mutants in CHO-DUKX cells: molecular insights and effector modulation of N-acetylglucosaminyltransferase I. Biotechnol. Bioeng. 109, 1723–1734 10.1002/bit.24448 [DOI] [PubMed] [Google Scholar]
- 68. Palandra J., Finelli A., Zhu M., Masferrer J., and Neubert H. (2013) Highly specific and sensitive measurements of human and monkey interleukin 21 using sequential protein and tryptic peptide immunoaffinity LC-MS/MS. Anal. Chem. 85, 5522–5529 10.1021/ac4006765 [DOI] [PubMed] [Google Scholar]
- 69. Marzilli L. A., Stephens E., Philip S., English A. M., and Rouse J. C. (2017) Minimizing method-induced deamidation and isomerization during antibody characterization to ensure optimal understanding of product quality attributes. LCGC Current Trends in Mass Spectrometry Special Issue 15, 6–14 [Google Scholar]
- 70. Shang T. Q., Saati A., Toler K. N., Mo J., Li H., Matlosz T., Lin X., Schenk J., Ng C. K., Duffy T., Porter T. J., and Rouse J. C. (2014) Development and application of a robust N-glycan profiling method for heightened characterization of monoclonal antibodies and related glycoproteins. J. Pharm. Sci. 103, 1967–1978 10.1002/jps.24004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry glycomics data have been deposited to the MassIVE repository with the data set identifier MSV000084822.