

Version Changes
Revised. Amendments from Version 1
The manuscript was revised according to the reviewers’ comments, as follows: 1. Methods: Preparation of structural model. The details of the starting dimeric model were included. 2. Table 1 now includes a caption to make it clearer. Single-letter amino-acid codes were added. The original residue ID in the SARS-CoV enzyme was included for comparison. The list of dimerisation residues was revised. 3. Methods:Virtual screening. More details were given to the Drugs-lib and its content. The options of defining the grid centre with active-site residues (of each chain from the dimeric model) were included. 4. Methods:Virtual screening. Now includes a description of how the top list was assembled from individual screening results, with multiple stereoisomers of a compound merged. 5. Results:Virtual screening. The full range of binding energies of all screening results, and the mean scores are given for comparison. 6. Table 3 and Table S2. A column was added to indicate the compounds’ molecular weights. The ‘Hits’ column was revised to show the number of occurrences of a compound (different stereoisomers, each has a unique ZINC15 ID) found in the top-scoring positions, out of the total number of stereoisomers of that compound. At the bottom of the tables, a ‘Reference’ section was added indicating the mean binding energies of each screen; as well as the binding energies of lopinavir and ritonavir. 7. Results: Assessment. More discussion and additional reference were made on hesperidin. 8. Discussion. The discussion of lopinavir/ritonavir now included their scores and the comparison with the top scorers. The results of the latest clinical trial were included, with reference. 9. Data: the DOI to the extended data was updated (Table S2 was updated). 10. Minor changes: updated with the latest statistics and additional references.
Abstract
We prepared the three-dimensional model of the SARS-CoV-2 (aka 2019-nCoV) 3C-like protease (3CL pro) using the crystal structure of the highly similar (96% identity) ortholog from the SARS-CoV. All residues involved in the catalysis, substrate binding and dimerisation are 100% conserved. Comparison of the polyprotein PP1AB sequences showed 86% identity. The 3C-like cleavage sites on the coronaviral polyproteins are highly conserved. Based on the near-identical substrate specificities and high sequence identities, we are of the opinion that some of the previous progress of specific inhibitors development for the SARS-CoV enzyme can be conferred on its SARS-CoV-2 counterpart. With the 3CL pro molecular model, we performed virtual screening for purchasable drugs and proposed 16 candidates for consideration. Among these, the antivirals ledipasvir or velpatasvir are particularly attractive as therapeutics to combat the new coronavirus with minimal side effects, commonly fatigue and headache. The drugs Epclusa (velpatasvir/sofosbuvir) and Harvoni (ledipasvir/sofosbuvir) could be very effective owing to their dual inhibitory actions on two viral enzymes.
Keywords: COVID-19, SARS, 2019-nCoV, 3C-like protease, drug repurpose, antiviral, coronavirus, virtual screening, molecular modelling, ledipasvir, velpatasvir, Hepatitis C virus, HCV
Introduction
On 7 January 2020, a new coronavirus, 2019-nCoV (now officially named SARS-CoV-2) was implicated in an alarming outbreak of a pneumonia-like illness COVID-19, originating from Wuhan City, Hubei, China. Human-to-human transmission was first confirmed in Guangdong, China 1. The World Health Organisation has declared this a global public health emergency — on 15 February 2020, there are more than 65,000 confirmed cases reported, and the death toll is over 1500. In the height of the crisis, this virus is spreading at a rate and scale far worse than previous coronaviral epidemics. By the time we finished revising this article (1 April 2020), it is a pandemic with more than 850,000 infected and total deaths of more than 42,000 affecting more than 180 countries/regions.
It was immediately evident from its genome that the coronavirus is evolutionarily related (80% identity) to the beta-coronavirus implicated in the severe acute respiratory syndrome (SARS), which originated in bats and was causative of a global outbreak in 2003. The momentum of research on developing antiviral agents against the SARS-CoV carried on after the epidemic subsided. Despite this, no SARS treatment has yet come to fruition; however, knowledge acquired from the extensive research and development efforts may be of use to inform the current therapeutic options.
The viral genome encodes more than 20 proteins, among which are two proteases (PL pro and 3CL pro) that are vital to virus replication; they cleave the two translated polyproteins (PP1A and PP1AB) into individual functional components. The 3-chymotrypsin-like protease (3CL pro, aka main protease, M pro) is considered to be a promising drug target. Tremendous effort has been spent on studying this protein in order to identify therapeutics against the SARS-CoV in particular and other pathogenic coronaviruses (e.g. MERS-CoV, the Middle East respiratory syndrome coronavirus) in general because they share similar active sites and enzymatic mechanisms. The purpose of this study is to build a molecular model of the 3CL pro of the SARS-CoV-2 and to carry out virtual screening to identify readily usable therapeutics. It was not our intention, however, to comment on other structure-based drug design research as these will not be timely for the current epidemic.
Methods
Analysis of protein sequences
The translated polyprotein (PP1AB) sequence was obtained from the annotation of the GenBank entry of the SARS-CoV-2 genome (accession number MN908947). By comparing this sequence with the SARS-CoV PP1AB sequence (accession number ABI96956), the protease cleavage sites and all mature protein sequences were obtained. Sequence comparison and alignment were performed with BLASTp.
Preparation of structural model
The high-resolution apo-enzyme structure of SARS-CoV 3CL pro (PDBID: 2DUC) 2 was employed as the template. The functional SARS-CoV 3CL pro is a dimer, therefore the SARS-CoV-2 enzyme was also constructed as a dimeric model, preserving all intermolecular interactions. The variant residues ( Table 1) were “mutated” in silico by SCWRL4 3, followed by manual adjustment to ensure that the best side-chain rotamer was employed ( Table 2). The rebuilt model was subjected to steepest descent energy minimisation by Gromacs 2018.4 using the Gromos 54A7 forcefield, with a restraint force constant of 1000 kJ mol -1 nm -2 applied on all backbone atoms and all atoms of the vital residues ( Table 1). Accessible surface area of residues were calculated with areaimol of the CCP4 suite v7.0.
Table 1. Important residues of 3CL pro from SARS-CoV (conserved) and the SARS-CoV-2 variant residues.
The residues that play functional roles in SARS-CoV 3CL pro are listed on the top three rows. These are absolutely conserved in the SARS-CoV-2 protein. The variant residues found in the SARS-CoV-2 protein are listed in the bottom row, with the SARS-CoV residues in brackets.
| Residue Number | Reference | |
|---|---|---|
| SARS-CoV | ||
| Catalytic | H41, C145 | 6 |
| Substrate binding | H41, M49, G143,
S144, 163–167, 187–192 |
2, 7 |
| Dimerisation | R4, M6, S10, G11,
E14, N28, S139, F140, S147, E166, E290, R298 |
8– 12 |
| SARS-CoV-2 | ||
| Variant positions | V35(T), S46(A),
N65(S), V86(L), K88(R), A94(S), F134(H), N180(K), V202(L), S267(A), A285(T), L286(I) |
This work |
Table 2. In silico mutagenesis to make the SARS-CoV-2 3CL pro.
The 12 variant residues with reference to the SARS-CoV enzyme are shown with the respective treatment of rotamer. “A” and “B” refers to the individual chains of the dimeric model. Both chains are in the crystal asymmetric unit and are not identical. The rotamer symbol (bracketed) is defined according to the conventions of Richardson 13, followed by its respective rank of popularity. ‘ASA’: accessible surface area (average of A and B chains) of the residue in the SARS-CoV 3CL pro structure, in Å 2 and in % relative to the ASA of a residue X in the Gly-X-Gly conformation.
| Residue | Rotamer | ASA, Å
2
(%) |
Remarks on
replacement |
|---|---|---|---|
| T35V | AB: (t-), top | 19 (14%) | conservative |
| A46S | A: (t-), 3rd; B:
(p-), top |
73 (63%) | A chain disordered,
rotamer chosen to minimise steric clash |
| S65N | AB: (m-20), top | 38 (28%) | |
| L86V | A: (m), 2nd; B:
(t), top |
0 (0%) | A chain rotamer to
avoid clash |
| R88K | A: (mtpt), 9th;
B: (mtpp), 19th |
81 (33%) | AB: real-space
refined with good fit to arginine densities |
| S94A | not applicable | 64 (51%) | |
| H134F | AB: (m-85), top | 57 (29%) | occupy similar but
larger space |
| K180N | AB: (m-20), top | 102 (50%) | |
| L202V | AB: (p), 3rd | 22 (12%) | avoid steric clash |
| A267S | AB: (m), 2nd | 0 (0%) | avoid steric clash |
| T285A | not applicable | 68 (44%) | at dimeric interface |
| I286L | (mt), top | 75 (46%) | at dimeric interface |
Virtual screening
MTiOpenScreen web service 4 was used for screening against its library of 7173 purchasable drugs (Drugs-lib), with 4574 unique compounds and their stereoisomers. Each library entry is identified with the name of the compound as well as an ZINC15 ID. The target binding site grid centre was specified by the active-site residues. At the MTiOpenScreen interface, the ‘Mode’ was set to ‘List of residues’ and these residues were specified: H41, M49, G143, S144, C145, H163, H164, M165, E166, L167, D187, R188, Q189, T190, A191 and Q192. The active sites on chain A and chain B, each derived from the catalytically-active dimeric model, were screened independently with AutoDock Vina 5.
When the crystal structure was released, it was stripped of its inhibitor and subjected to a screening.
The results returned from MTiOpenScreen is a list of 4500 target:ligand docking combinations (1500 ligands, each with 3 binding modes) ranked by binding energies. We listed the top 10 scorers of each chain as results. Stereoisomers of a compound (with the same drug name but unique ZINC15 IDs) that appear in the top list are collected together and presented as hits. The top ranking candidates for chains A and B were examined visually in PyMOL (version 1.7.X) 14.
An earlier version of this article can be found on ChemRxiv (DOI: 10.26434/chemrxiv.11831103.v2).
Results
High sequence homology with SARS-CoV
The first available genome was GenBank MN908947, now NCBI Reference Sequence NC_045512. From it, the PP1AB sequence of SARS-CoV-2 was extracted and aligned with that of SARS-CoV. The overall amino-acid sequence identity is very high (86%). The conservation is noticeable at the polyprotein cleavage sites. All 11 3CL pro sites 2 are highly conserved or identical ( Extended data 15, Table S1), inferring that their respective proteases have very similar specificities. The 3CL pro sequence of SARS-CoV-2 has only 12 out of 306 residues different from that of SARS-CoV (identity = 96%).
Conserved sequence identity among SARS-CoV-2
We compared the polyprotein PP1AB and the 3CL pro sequences among all 11 SARS-CoV-2 genomes (GenBank MN908947, MN938384, MN975262, MN985325, MN988668, MN988669, MN988713, MN994467, MN994468, MN996527 and MN996528) that were available on 1 February 2020. With reference to MN908947 (NC_045512), among the 7096 residues, there is only one variable residue in each of MN975262 (in NSP-4), MN994467 (in NSP-2), MN994468 (in NSP-13), MN996527 (in NSP-16); and two in MN988713 (in NSP-1 and NSP-3). The remaining five have no difference. To summarise, all SARS-CoV-2 3CL pro sequences and all their cleavage junctions on their polyproteins are 100% conserved.
3D model of the SARS-CoV-2 3CL pro
The amino acids that are known to be important for the enzyme’s functions are listed in Table 1. Not unexpectedly, none of the 12 variant positions are involved in major roles. Therefore, we are confident to prepare a structural model of the SARS-CoV-2 3CL pro by molecular modelling ( Extended data 15, Figure S1), which will be immediately useful for in silico development of targeted treatment. After we submitted the first draft of this study, the crystal structure of SARS-CoV-2 3CL pro was solved and released (PDB ID 6LU7) 16, which confirms that the predicted model is good within experimental errors ( Extended data 15, Figure S2).
Virtual screening for readily available drugs
The list of 1500 results has Autodock Vina binding energies ranging from -10.1 to -7.6 (mean = -8.2) kcal mol -1 from chain A active site; and -8.7 to -6.5 (mean = -7.1) kcal mol -1 for that of chain B. When examined in molecular graphics 14, all solutions were found to fit into their respective active sites convincingly. The binding energies of chain A complexes were generally higher than those of chain B by approximately 1.4 kcal mol -1 among the top scorers ( Table 3). This presumably demonstrates the intrinsic conformational variability between the A- and B-chain active sites in the crystal structure (the average root-mean-square deviation (rmsd) in Cα atomic positions of active-site residues is 0.83 Å). In each screen, the differences in binding energies are small, suggesting that the ranking is not discriminatory, and all top scorers should be examined. We combined the two screens, merged stereoisomers, and found 16 candidates which give promising binding models (etoposide and its phosphate counted as one) ( Table 3). One drug (dirlotapide) which is not intended for human use was excluded. All possible isomers of compounds with multiple stereoisomers are found in the full screening results of 1500, in particular: 38 of hesperidin, 34 of teniposide, 32 of etoposide and 21 of etoposide-phosphate.
Table 3. The results of virtual screening of drugs on the active sites of SARS-CoV-2 3CL pro model.
The left and right columns are the results of A and B chains, respectively. The top scorers are listed first, then the equivalent top scorers of the other chain listed at the lower half. ‘M.W.’: molecular weight in g mol -1. ‘B.E.’: AutoDock Vina binding energy in kcal mol -1. The ‘Hits’ column is the number of times a compound appears as top scorers (representing different stereoisomers) out of the total number of stereoisomers of that compound in the library; only the binding energy of the top-ranking hit was shown. Etoposide and its phosphate are listed separately in the screens. ‘n.f.’ = not found. Approved and pre-approved drugs are shown in green and orange, respectively. Except dihydroergocristine and ditercalinium, all approved drugs have undergone post-market surveillance, i.e. Phase 4. The mean score of each screen (1500 results), scores of lopinavir and ritonavir are included at the bottom for reference.
| A Chain | B Chain | ||||||
|---|---|---|---|---|---|---|---|
| A Top scorers | M.W. | B.E. | Hits | B Top scorers | M.W. | B.E. | Hits |
| diosmin | 609 | -10.1 | 1/1 | etoposide | 669 | -8.7 | 1/32 |
| hesperidin | 611 | -10.1 | 8/38 | R428 | 507 | -8.6 | 2/2 |
| MK-3207 | 558 | -10.1 | 1/4 | MK-3207 | 558 | -8.6 | 1/4 |
| venetoclax | 868 | -10.0 | 1/1 | teniposide | 657 | -8.5 | 2/34 |
| dihydroergocristine | 612 | -9.8 | 1/6 | UK-432097 | 778 | -8.5 | 1/2 |
| bolazine | -9.8 | -9.8 | 1/1 | eluxadoline | 570 | -8.4 | 1/1 |
| R428 | 507 | -9.8 | 2/2 | venetoclax | 868 | -8.4 | 1/1 |
| ditercalinium | 719 | -9.8 | 1/1 | ledipasvir | 889 | -8.4 | 1/1 |
| etoposide-phosphate | 669 | -9.8 | 1/21 | irinotecan | 587 | -8.4 | 1/1 |
| lumacaftor | 452 | -8.4 | 1/1 | ||||
| velpatasvir | 883 | -8.4 | 1/5 | ||||
| (B Top scorers) | (A Top scorers) | ||||||
| teniposide | -9.7 | hesperidin | -8.3 | ||||
| etoposide | -9.7 | etoposide-phosphate | -8.3 | ||||
| UK-432097 | -9.6 | bolazine | -8.3 | ||||
| irinotecan | -9.5 | dihydroergocristine | -8.1 | ||||
| lumacaftor | -8.9 | diosmin | -7.9 | ||||
| velpatasvir | -8.5 | ditercalinium | -7.7 | ||||
| eluxadoline | -8.0 | ||||||
| ledipasvir | n.f. | ||||||
| (Reference) | (Reference) | ||||||
| Mean of 1500 | -8.2 | Mean of 1500 | -7.1 | ||||
| lopinavir | -8.0 | lopinavir | -6.8 | ||||
| ritonavir | -7.9 | ritonavir | -6.9 |
Assessment of the candidate drugs
We checked the actions, targets and side effects of the 16 candidates. Among these, we first noticed velpatasvir ( Figure 1A, Figure 1D) and ledipasvir, which are inhibitors of the NS5A protein of the hepatitis C virus (HCV). Both are marketed as approved drugs in combination with sofosbuvir, which is a prodrug nucleotide analogue inhibitor of RNA-dependent RNA polymerase (RdRp, or NS5B). Interestingly, sofosbuvir has recently been proposed as an antiviral for the SARS-CoV-2 based on the similarity between the replication mechanisms of the HCV and the coronaviruses 17. Our results further strengthen that these dual-component HCV drugs, Epclusa (velpatasvir/sofosbuvir) and Harvoni (ledipasvir/sofosbuvir), may be attractive candidates to repurpose because they may inhibit two coronaviral enzymes. A drug that can target two viral proteins substantially reduces the ability of the virus to develop resistance. These direct-acting antiviral drugs are also associated with very minimal side effects and are conveniently orally administered ( Table 4). These computational results provide a rationale for experimental validation of inhibiting the SARS-CoV-2 with velpatasvir and ledipasvir, which is underway.
Figure 1. Virtual screening results for the SARS-CoV-2 3CL pro protease.
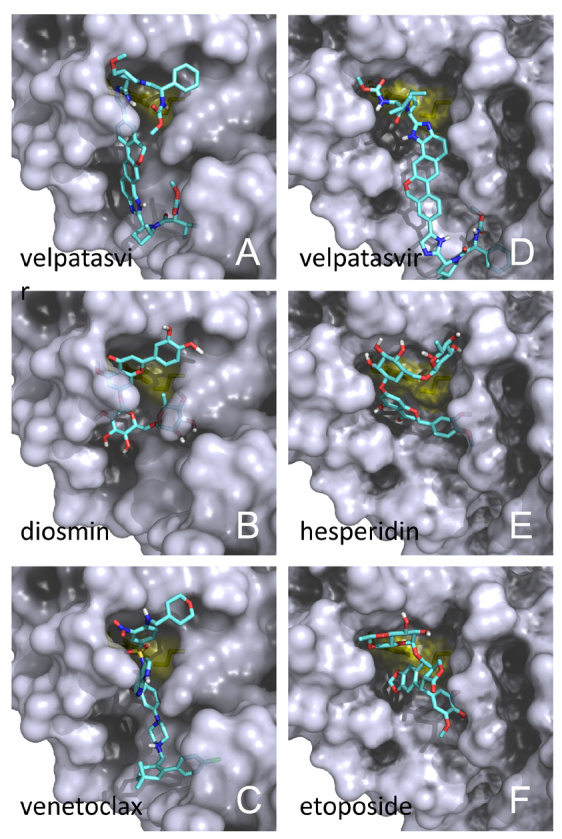
Docking of representative drugs into the active sites of A chain ( A, B, C) and that of B chain ( D, E, F). The catalytic residue surfaces are coloured in yellow. Atom colours of drug: C: cyan; O: red; N: blue; H: white; S: yellow; only polar hydrogens are shown. Prepared with PyMOL.
Table 4. Possible side effects and routes of administration of the drugs identified from virtual screening for SARS-CoV-2 3CL pro.
| Drug | Possible side effects
(adverse reactions) |
Admin. |
|---|---|---|
| Diosmin a, b | Mild gastrointestinal
disorders; skin irritations; nausea; heart arrhythmias |
Topical; oral |
| Hesperidin a, d | Stomach pain and upset;
diarrhea; headache |
Oral |
| MK-3207 c | No information | Oral |
| Venetoclax a, b | Neutropenia; nausea;
anaemia, diarrhea; upper respiratory tract infection |
Oral |
| Dihydroergocristine a | No information | Oral |
| Bolazine b | No information | Intramuscular |
| R428 b | No information | Oral |
| Ditercalinium | No information | No info |
| Etoposide a, b | Alopecia; constipation;
diarrhea; nausea; vomiting; secondary malignancies |
Intravenous |
| Teniposide a, b | Gastrointestinal toxicity;
hypersensitivity reactions; reversible alopecia |
Intravenous |
| UK-432097 c | No information | Inhaled |
| Irinotecan a, b | Gastrointestinal
complication |
Intravenous |
| Lumacaftor a | Dyspnea;
nasopharyngitis; nausea; diarrhea; upper respiratory tract infection |
Oral |
| Velpatasvir a, b | Headache; fatigue;
nausea |
Oral |
| Eluxadoline a, b | Constipation; nausea;
fatigue, bronchitis, viral gastroenteritis; pancreatitis |
Oral |
| Ledipasvir a | Fatigue; headache | Oral |
Sources of information: a DrugBank.ca (main), b Wikipedia.org, c ClinicalTrials.gov and d WebMD.com.
The flavonoid glycosides diosmin ( Figure 1B) and hesperidin ( Figure 1E), obtained from citrus fruits, fit very well into and block the substrate binding site. Yet, these compounds cause mild adverse reactions ( Table 4). Hesperidin has 38 stereoisomeric forms and several of these showed up among the top scorers ( Table 3; Figure 1E). It has been reported to be a good inhibitor of the SARS-CoV 3CL pro with an IC 50 of 8.3 µM in a cell-based assay 18.
Teniposide and etoposide (and its phosphate) are chemically related and exhibited good binding models ( Figure 1F). However, these chemotherapy drugs have a lot of strong side effects and need intravenous administration ( Table 4). The approved drug venetoclax ( Figure 1C) and investigational drugs MK-3207 and R428 scored well in both screens. Venetoclax is another chemotherapy drug that is burdened by side effects including upper respiratory tract infection ( Table 4). Not much has been disclosed about MK-3207 and R428.
We subjected the crystal structure to the same virtual screening procedures. A very similar list of candidates showed up consistently ( Extended data 15, Table S2) with high scores although ledipasvir was not found.
We noticed that most of the compounds on the list have molecular weights (MW) over 500 ( Table 3), except lumacaftor (MW=452). The largest one is ledipasvir (MW=889). This is because the size of the peptide substrate and the deeply buried protease active site demand a large molecule that has many rotatable dynamics to fit into it.
Discussion
We identified five trials on ClinicalTrials.gov involving antiviral and immunomodulatory drug treatments for SARS ( Table 5), all without reported results; i.e., at present, there are no safe and effective drug candidates against SARS-CoV. This is because once the epidemic is over, there are no patients to recruit for clinical trials. Only the study with streptokinase succeeded in completion of phase 3. It is disappointing that little progress in SARS drug development has been made in the past 17 years. After the 2003 outbreak, numerous inhibitors for the 3CL pro enzyme have been proposed 19, 20, yet no new drug candidates have succeeded to enter the clinical phase 1.
Table 5. Drugs targeting SARS that are registered for the U.S. Food and Drug Administration (USFDA) clinical trials (as of mid-February 2020).
‘n.i.’ = no information.
| Drug | Condition | Phase | Status | From | To | Location |
|---|---|---|---|---|---|---|
| Lopinavir / Ritonavir +
Ribavirin |
SARS | Unknown | Unknown | n.i. | n.i. | Hong Kong |
| Alferon LDO | SARS | Phase 2 | Completed | Nov 04 | Apr 06 | Hong Kong |
| Poly-ICLC | Respiratory
viruses a |
Phase 1 | Completed | Mar 08 | Dec 09 | USA |
| Streptokinase vs.
Heparin |
SARS, ARDS | Phase 3 | Completed | Feb 16 | Jan 18 | n.i. |
| Glucocorticoid
(methylprednisolone) therapy |
Coronavirus
infections b |
Phase 2,
Phase 3 |
Unknown | Jan 20 | Dec 20
(Est.) |
China |
a This covers unknown respiratory viruses. b This includes the COVID-19. ‘Est.’ = estimated. ‘ARDS’ = acute respiratory distress syndrome.
One record which receives a lot of attention amid the current outbreak is the lopinavir/ritonavir combination 21. They are protease inhibitors originally developed against HIV. During the 2003 SARS outbreak, despite lacking a clinical trial, they were tried as an emergency measure and found to offer improved clinical outcome 21. However, some scientists did express scepticism 22. By analogy, these compounds were speculated to act on SARS-CoV 3CL pro specifically, but there is as yet no crystal structure to support that, although docking studies were carried out to propose various binding modes 23– 26. The IC 50 value of lopinavir is 50 μM ( K i = 14 μM) and that for ritonavir cannot be established 27. These two compounds turned up in our virtual screening results, with scores slightly lower than the mean scores ( Table 3). Based on our results that the two CoV 3CL pro enzymes are identical as far as active sites and substrate specificities are concerned, we were of the opinion that it was still one of the recommended routes for immediate treatment at the time of writing the first version (mid-February 2020). Disappointedly, the latest trial of lopinavir/ritonavir on COVID-19 showed no clinical benefit 28.
If we look beyond the 3CL pro, an earlier screen produced 27 candidates that could be repurposed against both SARS-CoV and MERS-CoV 29. In addition, the other coronaviral proteins could be targeted for screening. Treatment of the COVID-19 with remdesivir (a repurposed drug in development targeting the RdRp) showing improved clinical outcome has earlier been reported and clinical trial is now underway 30.
We consider this work part of the global efforts responding in a timely fashion to fight this deadly communicable disease. We are aware that there are similar modelling, screening and repurposing exercises targeting 3CL pro reported or announced 23, 31– 37 (up to mid-February 2020). Our methods did not overlap, and we share no common results with these studies. During revision, another crystal structure paper was published 38.
Data availability
Source data
The 11 SARS-CoV-2 polyprotein PP1AB and 3CL pro sequences used in this study were obtained from NCBI GenBank, accession numbers MN908947 , MN938384, MN975262, MN985325, MN988668, MN988669, MN988713, MN994467, MN994468, MN996527 and MN996528, available on 1 February 2020.
The SARS-CoV PP1AB sequence was obtained from NCBI Protein, accession number ABI96956.
The two coronavirus protease structures used were obtained from Protein Data Bank, ID 2DUC and 6LU7.
Extended data
Open Science Framework: SARS-CoV-2 (2019-nCoV) 3CLpro Model and Screening. https://doi.org/10.17605/OSF.IO/FD243 15.
The “Virtual Screening” folder contains the following extended data:
2019-nCoV-3CLpro.pdb. (3D model of the 3CL pro: A and B chains.)
A-screen4500.pdbqt, B-screen4500.pdbqt, X-screen4500.pdbqt. (Virtual screening 3D results of Model A chain, Model B chain and the crystal-structure (A chain) in PDBQT format (can be viewed by any text editor). Use the software PyMOL to open these files. Each result file contains 4500 drug-to-protein docking hits ranked by AutoDock Vina binding energies in kcal mol -1.)
A-screen1500.table.csv, B-screen1500.table.csv, X-screen1500.table.csv. (Virtual screening results (names only) of Model A chain, Model B chain and the crystal-structure (A chain) in CSV format (can be opened by Excel or any text editor). This is a summary of the top 1500 drug-to-protein docking hits ranked by AutoDock Vina binding energies in kcal mol -1.)
The “Extended Results” folder contains the following extended data:
Tab S1.docx (Sequence homology of the 3CL pro cleavage junctions of PP1AB between SARS-CoV-2 and SARS-CoV).
Tab S2-v2.docx (The results of virtual screening of drugs on the active site of SARS-CoV-2 3CL pro crystal structure).
Fig S1.pptx (The structural model of the SARS-CoV-2 3CL pro protease).
Compare Crystal.docx (A comparison, with Figure S2, of the active sites of model chains A, B and the crystal structure).
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Funding Statement
We acknowledge support from the Innovation and Technology Commission of Hong Kong, the Hong Kong Polytechnic University and the Life Science Area of Strategic Fund 1-ZVH9.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 2; peer review: 3 approved]
References
- 1. Chan JF, Yuan S, Kok KH, et al. : A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muramatsu T, Takemoto C, Kim YT, et al. : SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci U S A. 2016;113(46):12997–13002. 10.1073/pnas.1601327113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krivov GG, Shapovalov MV, Dunbrack RL, Jr: Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77(4):778–795. 10.1002/prot.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Labbé CM, Rey J, Lagorce D, et al. : MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 2015;43(W1):W448–W454. 10.1093/nar/gkv306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trott O, Olson AJ: AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang C, Wei P, Fan K, et al. : 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43(15):4568–4574. 10.1021/bi036022q [DOI] [PubMed] [Google Scholar]
- 7. Hsu MF, Kuo CJ, Chang KT, et al. : Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280(35):31257–31266. 10.1074/jbc.M502577200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barrila J, Bacha U, Freire E: Long-range cooperative interactions modulate dimerization in SARS 3CL pro. Biochemistry. 2006;45(50):14908–14916. 10.1021/bi0616302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barrila J, Gabelli SB, Bacha U, et al. : Mutation of Asn28 disrupts the dimerization and enzymatic activity of SARS 3CL pro. Biochemistry. 2010;49(20):4308–4317. 10.1021/bi1002585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu T, Zhang Y, Li L, et al. : Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology. 2009;388:324–334. 10.1016/j.virol.2009.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen S, Zhang J, Hu T, et al. : Residues on the dimer interface of SARS coronavirus 3C-like protease: dimer stability characterization and enzyme catalytic activity analysis. J Biochem. 2008;143(4):525–536. 10.1093/jb/mvm246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng SC, Chang GG, Chou CY: Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J. 2010;98(7):1327–1336. 10.1016/j.bpj.2009.12.4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lovell SC, Word JM, Richardson JS, et al. : The penultimate rotamer library. Proteins. 2000;40(3):389–408. [DOI] [PubMed] [Google Scholar]
- 14. The PyMOL molecular graphics system (Schrödinger, LLC). Reference Source [Google Scholar]
- 15. Chen YW: SARS-CoV-2 (2019-nCoV) 3CLpro Model & Screening. 2020. 10.17605/OSF.IO/FD243 [DOI]
- 16. Jin Z, Du X, Xu Y, et al. : Structure-based drug design, virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. bioRxiv. 2020. 10.1101/2020.02.26.964882 [DOI] [Google Scholar]
- 17. Ju J, Kumara S, Li X, et al. : Nucleotide analogues as inhibitors of viral polymerases. bioRxiv. 2020. 10.1101/2020.01.30.927574 [DOI] [Google Scholar]
- 18. Lin CW, Tsai FJ, Tsai CH, et al. : Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antiviral Res. 2005;68(1):36–42. 10.1016/j.antiviral.2005.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pillaiyar T, Manickam M, Namasivayam V, et al. : An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595–6628. 10.1021/acs.jmedchem.5b01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuo CJ, Liang PH: Characterization and inhibition of the main protease of severe acute respiratory syndrome coronavirus. ChemBioEng Reviews. 2015;2:118–132. 10.1002/cben.201400031 [DOI] [Google Scholar]
- 21. Chu CM, Cheng VC, Hung IF, et al. : Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59(3):252–256. 10.1136/thorax.2003.012658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stockman LJ, Bellamy R, Garner P: SARS: systematic review of treatment effects. PLoS Med. 2006;3(9):e343. 10.1371/journal.pmed.0030343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gruber C, Steinkellner G: Coronavirus COVID-19 (formerly known as Wuhan coronavirus and 2019-nCoV) - what we can find out on a structural bioinformatics level. 2020. Reference Source [Google Scholar]
- 24. Dayer MR, Taleb-Gassabi S, Dayer MS: Lopinavir; a potent drug against coronavirus infection: insight from molecular docking study. Arch Clin Infect Dis. 2017;12(4):e13823 10.5812/archcid.13823 [DOI] [Google Scholar]
- 25. Nukoolkarn V, Lee VS, Malaisree M, et al. : Molecular dynamic simulations analysis of ritonavir and lopinavir as SARS-CoV 3CL pro inhibitors. J Theor Biol. 2008;254(4):861–867. 10.1016/j.jtbi.2008.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang XW, Yap YL: Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg Med Chem. 2004;12(10):2517–2521. 10.1016/j.bmc.2004.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu CY, Jan JT, Ma SH, et al. : Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci U S A. 2004;101(27):10012–10017. 10.1073/pnas.0403596101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cao B, Yeming Y, Wang D, et al. : A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dyall J, Coleman CM, Hart BJ, et al. : Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58(8):4885–4893. 10.1128/AAC.03036-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holshue ML, DeBolt C, Lindquist S, et al. : First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020;382(10):929–936. 10.1056/NEJMoa2001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu Z, Peng C, Shi Y, et al. : Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. 2020. 10.1101/2020.01.27.921627 [DOI] [Google Scholar]
- 32. Liu X, Wang Xj: Potential inhibitors for 2019-nCoV coronavirus M protease from clinically approved medicines. bioRxiv. 2020. 10.1101/2020.01.29.924100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stoermer MJ: Homology models of Wuhan coronavirus 3CL pro protease. ChemRxiv. 2020. 10.26434/chemrxiv.11637294.v1 [DOI] [Google Scholar]
- 34. Global Health Drug Discovery Institute: Targeting 2019-nCoV Portal.2020. Reference Source [Google Scholar]
- 35. Beck BR, Shin B, Choi Y, et al. : Predicting commercially available antiviral drugs that may act on the novel coronavirus (2019-nCoV), Wuhan, China through a drug-target interaction deep learning model. bioRxiv. 2020. 10.1101/2020.01.31.929547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao K, Nguyen DD, Wang R, et al. : Machine intelligence design of 2019-nCoV drugs. bioRxiv. 2020. 10.1101/2020.01.30.927889 [DOI] [Google Scholar]
- 37. Li Y, Zhang J, Wang N, et al. : Therapeutic drugs targeting 2019-nCoV main protease by high-throughput screening. bioRxiv. 2020. 10.1101/2020.01.28.922922 [DOI] [Google Scholar]
- 38. Zhang L, Lin D, Sun X, et al. : Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020; pii: eabb3405. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]