

ABSTRACT
An effective prophylactic hepatitis B virus (HBV) vaccine has long been available but is ineffective for chronic infection. The primary cause of chronic hepatitis B (CHB) and greatest impediment for a therapeutic vaccine is the direct and indirect effects of immune tolerance to HBV antigens. The resulting defective CD4+/CD8+ T cell response, poor cytokine production, insufficient neutralizing antibody (nAb) and poor response to HBsAg vaccination characterize CHB infection. The objective of this study was to develop virus-like-particles (VLPs) that elicit nAb to prevent viral spread and prime CD4+/CD8+ T cells to eradicate intracellular HBV. Eight neutralizing B cell epitopes from the envelope PreS1 region were consolidated onto a species-variant of the HBV core protein, the woodchuck hepatitis core antigen (WHcAg). PreS1-specific B cell epitopes were chosen because of preferential expression on HBV virions. Because WHcAg and HBcAg are not crossreactive at the B cell level and only partially cross-reactive at the CD4+/CD8+ T cell level, CD4+ T cells specific for WHcAg-unique T cell sites can provide cognate T-B cell help for anti-PreS1 Ab production that is not curtailed by immune tolerance. Immunization of immune tolerant HBV transgenic (Tg) mice with PreS1-WHc VLPs elicited levels of high titer anti-PreS1 nAbs equivalent to wildtype mice. Passive transfer of PreS1 nAbs into human-liver chimeric mice prevented acute infection and cleared serum HBV from mice previously infected with HBV in a model of CHB. At the T cell level, PreS1-WHc VLPs and hybrid WHcAg/HBcAg DNA immunogens elicited HBcAg-specific CD4+ Th and CD8+ CTL responses.
KEYWORDS: Hbv, immune tolerance, pres1, neutralizing antibody, therapeutic vaccine
Introduction
According to the Centers for Disease Control and Prevention (CDC), there are 1.2 million carriers of HBV in the U.S. and approximately 240 million worldwide. In contrast to adult infection, neonatal HBV infection is rarely cleared and as many as 90% of perinatally infected children become chronically infected.1 Vertical transmission of HBV is the major source of chronic infection in endemic areas, creating a cycle of perinatal infection, chronicity, and late-term complications including fibrosis and hepatocellular carcinoma (HCC) with over 1 million deaths annually. The large number of HBV chronic carriers represents a significant health problem since they serve as a reservoir for further infection as well as being at risk for the development of HCC. Therefore, in addition to worldwide vaccine programs to prevent new infections, methods for treating HBV chronic carriers will be necessary to eradicate this disease. Although a number of nucleotide and nucleoside analogs are quite effective at reducing HBV viral load, treatment is associated with poor-sustained responses. Use of immunomodulatory-pegylated interferon alfa-2b, alone or in combination with antiviral drugs, is resulting in improved rates of sustained response but is still less than 30%.2 It has been hoped that antiviral treatments would permit the immune system to “reset” allowing for innate/adaptive immunity to recover. However, this approach has been only marginally successful at restoring immunity. One consequence of the standard antiviral therapies is that reduced HBV replication and antigenic loads deprive the immune system of necessary antigenic stimuli. Therefore, therapeutic vaccination would compliment antiviral therapy by providing the necessary immunogenic stimulus to drive innate/adaptive immune responses. Because resolution of CHB infection has been achieved through bone marrow transplantation, vaccine-based immunotherapy is highly promising, especially in combination with antiviral drugs.
Although a safe and efficacious preventative vaccine for HBV has been available for 30 years, it is not effective against chronic infection. The primary cause of CHB infection and the greatest impediment to developing a therapeutic vaccine is the direct and indirect effects of T and B cell immune tolerance mediated primarily by secreted HBeAg and HBsAg.3–14 The resulting defective CTL responses, poor cytokine production, insufficient nAb levels and non-response to conventional HBsAg vaccination (a direct measure of immune tolerance) characterize CHB infection. The current HBV treatments (nucleoside analogs and INFalpha) can achieve a “functional cure”, defined as sustained, undetectable HBsAg and HBV DNA in serum with or without seroconversion to anti-HBs after completion of a finite course of treatment with persistent low levels of covalently closed circular DNA (cccDNA).15 A “complete cure” will require cccDNA clearance from all hepatocytes and immune restoration to maintain it.
The objective of this study was to develop dual-purpose virus-like particles (VLPs) to elicit and enhance PreS1-specific nAb production to prevent viral spread and secondly to prime HBcAg-specific CD4+/CD8+ T cells to eradicate intracellular HBV and prevent accumulation of cccDNA.5,16 A number of therapeutic vaccine clinical trials have been conducted using the HBV envelope antigens (i.e., HBsAg, PreS2 and PreS1-containing subviral particles) singly or combined, delivered as proteins in adjuvant or as DNA constructs, all with rather disappointing results.2,17,18 In many of these studies, antiviral drugs were also used in order to inhibit viral replication. Nevertheless, the HBV envelope vaccines to date have not demonstrated clear clinical efficacy. Most, if not all, previous therapeutic vaccine strategies utilized HBV-derived antigens or DNA constructs, which are not sufficiently immunogenic in patients who have high viral and antigen loads and are immune tolerant.17,18 In our view, the primary immune defect in CHB infection is the absence and/or dysfunction of high avidity HBV-specific Th cells due to immune tolerance mechanisms including clonal deletion, clonal anergy, clonal ignorance, Treg, clonal exhaustion and upregulation of inhibitory molecules such as PD-1, CTLA-4, TIM-3, etc.3–5,8-10,19,20 In order to circumvent the direct and indirect effects of immune tolerance, chimeric VLPs were constructed consisting of eight HBV envelope, PreS1-specific, neutralizing B cell epitopes inserted onto the species-variant WHcAg, which is approximately 66-68% homologous with the HBcAg (Figure 1). PreS1-specific B cell epitopes were chosen because of the viral neutralizing capacity of PreS1 antibodies, the correlation between anti-PreS1 Ab production and viral clearance, the sequential nature of the PreS1 epitopes and the preferential expression of PreS1 on HBV virions as opposed to the much more abundant subviral particles, which can “adsorb out” anti-HBs antibodies.21–26 The WHcAg was chosen because of the capacity to enhance multivalent (i.e., 240 monomers/VLP) Ab production and demonstrated efficacy as a vaccine carrier,27,28 and importantly for this application, WHcAg is not crossreactive at the B cell level with the HBcAg and only partially cross-reactive at the CD4+/CD8+ T cell levels.29–31 Therefore, CD4+ T cells specific for foreign, WHcAg-unique T cell sites (i.e., heterospecific) can provide cognate Th-B cell help for anti-PreS1 antibody production that is not compromised by immune tolerance as opposed to the dysfunctional HBV-specific CD4+ T cells (homospecific) present in chronic HBV carriers (see Figures 1 and 2).32
Figure 1.
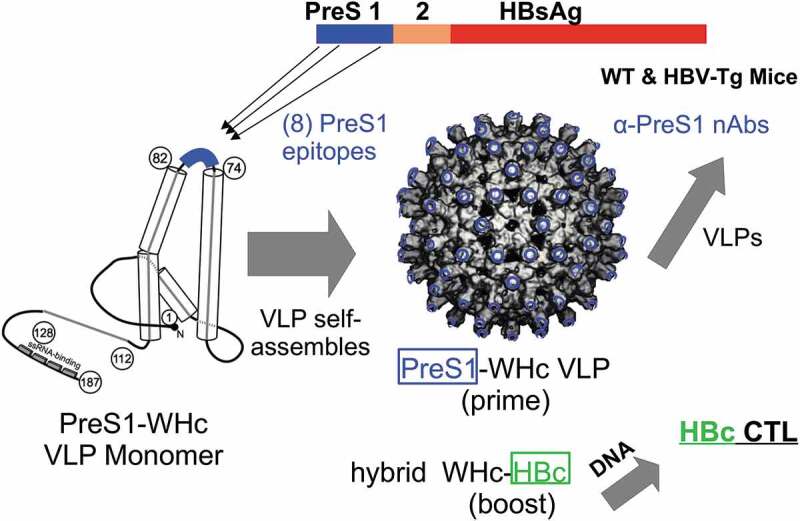
Therapeutic HBV Vaccine Design. Eight PreS1 B cell epitopes were inserted onto hybrid WHc-VLPs to elicit neutralizing antibodies and hybrid WHcAg/HBcAg VLP constructs were used to elicit HBcAg-specific CTL.
Figure 2.
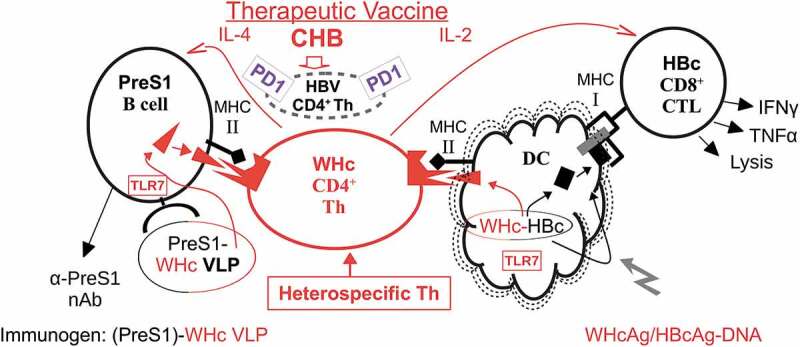
Cellular pathways for the presentation of PreS1-WHc VLPs and hybrid WHcAg/HBcAg DNA. The therapeutic vaccine utilizes the WHcAg as a carrier of HBs-PreS1 B cell and HBcAg-specific CTL epitopes, which provides “heterospecific” T cell helper function. The absent or dysfunctional HBV-specific CD4+ Th cell population is represented by the small broken circle showing over-expression of the inhibitory PD-1 molecule. A VLP prime/DNA boost strategy is envisioned.
The use of Hepatitis B immune globulin (HBIG) in preventing neonatal infection and in HBV-associated liver transplantation demonstrates the importance of anti-envelope nAbs, which act as viral entry inhibitors to prevent hepatocyte-to-hepatocyte viral infection. Other important roles for nAbs in chronic HBV infection include: (1) clearing serum HBV, which may reduce T cell exhaustion mechanisms;33 (2) immune complexes mediate ADCC and other Fc-mediated effector responses;34,35 (3) nAbs reduce the risk of viral reactivation;36 (4) nAbs reduce the accumulation of HBV cccDNA by preventing viral spread;16 (5) nAb may prevent CTL escape mutants from surviving in the periphery; and (6) may allow safe withdrawal of antiviral drugs “under the cover” of nAbs. Recent studies have demonstrated the value of anti-PreS1 antibodies in the clearance of “CHB infection” in animal models.34,37 An alternative technology, which consists of injecting a PreS1 peptide (Myrcludex B),16 also functions as a viral entry-inhibitor but is not as cost-efficient or practical as vaccination and injected peptide will not mediate many of the functions of nAbs listed above. However, Myrcludex B treatment does reduce the accumulation of HBV cccDNA in humanized mice, presumably by preventing cell-to-cell viral spread and has shown clinical efficacy against hepatitis delta virus (HDV) infection.16,38
Complete recovery from acute HBV infection requires cellular immune responses, especially multispecific, polyclonal CTL responses.39 Further, patients with chronic HBV who experience remission also demonstrate CTL and Th1 responses.40 In contrast, the CTL and Th responses are undetectable or very weak in patients with ongoing chronic infection.19,20 Therefore, we designed DNA constructs capable of circumventing immune tolerance at the level of T cell help for CTL by taking advantage of the same technology used to bypass poor T cell help for anti-PreS1 Ab production. Coexpression of the HBcAg linked with the WHcAg in DNA vectors allows the foreign Th epitopes on WHcAg to elicit “heterospecific” T cell help for HBcAg-specific CTL, which can be “helpless” in the context of chronic HBV infection (Figure 1). As shown in Figure 2, absent or defective (i.e., PD-1+) HBcAg-specific “homospecific” Th cells can be substituted for by WHcAg- “heterospecific” Th cells as long as the WHcAg-specific Th cell epitopes are physically linked to the HBcAg-specific CTL epitopes within the same VLP in order to ensure that both are taken up by the same dendritic cell (DC) or antigen-presenting cell (APC). T cell help for CTL function is not mediated directly by Th-CTL interaction as exists in the direct Th-B cell interaction. T cell help for CTLs is mediated indirectly through activation of DCs or other APC; therefore, the concept of “heterospecific” T cell help for CTL function has not been exploited to the same degree as in conjugate T-B epitope vaccines, except for a few examples primarily in tumor vaccinology. Tumor antigen ”homospecific” Th cells are often absent or dysfunctional due to tolerance mechanisms and experimental tumor vaccines that provide heterospecific Th cells have shown enhanced in vivo antitumor activity.41–44 These precedents suggest that Th cell defects in chronic viral diseases may also be circumvented by “heterospecific” Th cells.
As illustrated in Figure 2, immunization with DNA encoding hybrid WHcAg/HBcAg VLPs will allow DC to present or cross-present foreign, WHcAg Th cell epitopes in the context of MHC class II molecules to CD4+ WHcAg-heterospecific Th cells and the same, now-activated, DCs will present the linked HBcAg-specific CTL epitopes in the context of MHC class I molecules to CD8+, HBcAg-specific CTLs. Therefore, the licensing or activation of the DC can occur in the absence of homospecific T cell help. Additional advantages to the use of the WHcAg as a carrier for HBV-specific B cell and CTL epitopes are: (1) in addition to possessing foreign, WHcAg-unique CD4+/CD8+ T cell epitopes, WHcAg and HBcAg share conserved epitopes which allows priming of WHcAg/HBcAg crossreactive CD4+ and CD8+ T cells in addition to heterospecific Th cells; (2) the WHcAg encapsidates a TLR7 ligand, which serves as an additional co-stimulus for B cells and DC APC;45 and (3) the particulate structure of WHcAg enhances B cell/DC antigen uptake.
The studies described herein demonstrated that immunization with PreS1-WHc VLPs elicited levels of high titer anti-PreS1 nAbs in immune tolerant HBV-Tg mice equivalent to wildtype mice. Further, passive transfer of PreS1-specific nAbs into human-liver chimeric mice prevented acute infection and cleared serum HBV from mice previously infected with HBV (i.e., a model of CHB). At the T cell level, PreS1-WHc VLPs and hybrid WHcAg/HBcAg DNA immunogens elicited HBcAg-specific CD4+ Th and CD8+ CTL responses necessary for clearance of intracellular HBV.
Materials & methods
Mice
Inbred C57BL/6 (B6) (H-2b), C57BL/10 (B10) (H-2b), B6/BALBc (H-2bxd) mice and B10, TLR7-KO mice were obtained from the Jackson Laboratory. B10-Tg mice with intrahepatic expression of the HBcAg protein (HBc-Tg, 0.2 to 2 μg/g liver protein) or the HBeAg protein (HBe-Tg, 4 to 10 μg/ml serum) were obtained from Dr. J Ou (USC).46,47 The HBV-Tg mice (1.3.32) were obtained from F. Chisari (TSRI).48 PreS1-Tg mice (lineage 107–5) were provided by F. Chisari and L. Guidotti (TSRI).49
Recombinant WHcAg hybrid VLP construction
The WHcAg and hybrid WHcAg VLPs were expressed from the pUC-WHcAg vector expressing the full-length WHcAg protein codon optimized for expression in E. coli. The sequence for WHcAg (accession NC_004177) was cloned into the pUC19 vector. For inserting heterologous B cell epitopes, EcoRI-XhoI restriction sites were engineered into the open reading frame between amino acids 78 and 79 of the core protein gene. The engineered restriction sites add a Gly-Ile-Leu linker on the N-terminal side and a Leu linker on the C-terminal side of the inserted epitopes. For VLP-1.6 the heterologous B cell epitope was directly fused between amino acids 78 and 79 by the polymerase chain reaction using overlapping primers. The PreS1 sequences and numbering are based on HBV genotype D, serotype ayw (Accession X65259):MGQNLSTSNP LGFFPDHQLD PAFRANTANP DWDFNPNKDT WPDANKVGAG AFGLGFTPPH GGLLGWSPQA QGILHTVPAN PPPASTNRQS GRQPTPLSPP LRNTHPQA. For fusing the heterologous T950-969 T cell epitope, the AvaI restriction site was used in the HyW VLP previously described.50 Epitopes were cloned into the VLP gene using synthetic oligonucleotides comprising the desired epitope coding sequence and the appropriate engineered restriction sites or overlapping primers. All WHcAg constructs were transformed into Alpha-Select competent E. coli (Bioline USA, Inc.) and confirmed by DNA sequencing.
The VLP constructs delivered as DNA were codon optimized for mammals and cloned into pVAX1, grown in Alpha-Select cells and plasmid purified by the ZymoPure II MaxiPrep kit (D4202, Zymo Research) according to the manufacturer’s instructions. DNA was formulated in PBS and concentration was determined spectrophotometrically.
Purified proteins, synthetic peptides, and mabs
The VLP particles were expressed in Alpha-Select E. coli cells grown in Terrific Broth (Fisher BP2468). Cells were lysed by passage through an EmulsiFlex-C3 (Avestin, Ottawa, ON, Canada) and the lysate heated to 65°C for approximately 10 min, then clarified by centrifugation. The WHcAg particles were selectively precipitated by the addition of solid ammonium sulfate up to approximately 45% saturation (277 g/L) and the precipitates were collected by centrifugation. Precipitated VLPs were redissolved in minimum buffer (10 mM Tris, pH 8), dialyzed against the same buffer and applied to a Sepharose CL4B column (5x100 cm) or ultrafiltered by tangential flow using a WaterSep Discover 12, 750K molecular weight cutoff. Finally, VLPs were formulated in 20 mM Tris, pH8, 100 mM NaCl, 0.5 mM EDTA. Endotoxin was removed from the core preparations by phase separation with Triton X-114.50,51 The purified VLPs were 0.2 um sterile-filtered, characterized and aliquoted. Characterization typically includes custom ELISA, native agarose gel electrophoresis PAGE, heat stability, circular dichroism and dynamic light scattering as previously described.50
To produce rPreS1 + 2 and myr-PreS1 + 2, the gene encoding ayw preS1S2 (aa1-163) fused to a six-histidine tag was cloned alone or together with the yeast N-methyltransferase one gene into the pET Duet vector and transformed into HMS174(DE3) E coli. Bacteria were grown on LB medium supplemented with 2 g/l glucose, at 30°C, until the A600 was between 0.6 and 0.8. In the case of the dual expression cells, the medium was then supplemented with 10 ml of 5 mM myristic acid in 0.6 mM BSA in water. At this time 100 mg of IPTG was added per liter of culture, and the bacteria allowed to continue to grow for 3–4 h. Bacteria were collected by centrifugation and stored frozen until processed. Bacterial pellets were suspended in 6M urea and disrupted by a single passage through an Avestin Emulsiflex C3 operating at a pressure of 25000 psi. The use of urea was to prevent the rapid proteolysis of the soluble protein upon disruption of the bacteria. The lysate was clarified by centrifugation and applied to a nickel column (BioRad), extensively washed with 6 M urea until the absorbance at 280 nm had returned to baseline values then washed with water to remove the urea. The protein was eluted using 50 mM citric acid and dialyzed against 10 mM acetate buffer pH 5.0.
Synthetic peptides were synthesized by and purchased from Eton Biosciences (San Diego, CA) and Abclonal (Woburn, MA).
PreS1-specific Mab 18/7 was provided by W. Gerlich (Justus Liebig University Giessen). PreS1-specific Mabs AP-2 and KR-127 were purchased from Santa Cruz Biotechnology (Dallas, TX). PreS1-specific Mab Ab001 was purchased from Beacle, Inc. (Okayama, Japan).
Human-liver chimeric mice
Homozygous NRG-fumarylacetoacetate hydrolase (fah/fah) mutant mice (NRG/F) were maintained with 8 μg/ml 2-(2-nitro-4-fluromethylbenzoyl)-1,3 cyclohexanedine (NTBC).52 Anesthetized mice were injected in the spleen with 1 × 106 human primary hepatocytes (Triangle Research Laboratories, NC). After transplantation, the NRG/F-hu mice were subjected to three rounds of NTBC drug recycling to eliminate mouse hepatocytes and to provide space for human hepatocyte growth. Thereafter, mice were infected with 1 × 106 GE copies of HBV/genotype C (isolated from HBV-infected human-liver chimeric mouse serum) injected retro-orbitally. Human albumin in mouse sera was measured with a modified ELISA method (Bethyl Labs Human Albumin ELISA Quantitation Set).53
Passive transfer of antisera
Anti-PreS1-WHc VLP sera (0.2 ml) or control anti-WHc sera (0.2 ml) were injected i.v. into human-liver chimeric mice prior to infection with 1 × 106 GE copies of HBV per mouse in the control and acute groups. For the chronic group, 0.2 ml of anti-PreS1-WHc VLP sera were injected i.v. 2 and 5 weeks after HBV infection. To measure HBV DNA, sera were collected from the tail vein and HBV DNA was extracted with QIAamp MinElute Virus Spin Kit according to the manufacturer’s instructions. Primer 1 (HBV2270F: 5ʹ-GAGTGTGGATTCGCACTCC-3ʹ) and Primer 2 (HBV2392R: 5ʹ-GAGGCGAGGGAGTTCTTCT-3ʹ) were used in the Q-PCR reaction to measure HBV DNA. A human serum with known viral titer was used as an HBV DNA standard.53
Virus neutralization assay
Neutralization was assessed as previously described.54 Briefly, HDV (HBV genotype D, L/M/S -HBsAg subtype ayw) particles were derived from the culture medium from transfected cells and suspended at 1 × 109 particles per ml. One hundred microliter of HDV-HBV particles were mixed with 100 μl of sample (sera or purified Mab neat, 1/10 and 1/100 dilutions in PBS) and incubated at 37°C for 1 hr such that final dilutions of sera were 1:40, 1:400 and 1:4000 and Mab was 5, 0.5, and 0.05 μg. The mix was adjusted to 5% PEG and inoculated on HepaRG (106 cells for a multiplicity of infection of 100 viral particles per hepatocyte). After 16 hr, the inoculum was removed and replaced with fresh medium. Cells were harvested at day 7 post inoculation for the detection of intracellular genomic HDV RNA as a marker of infection by Northern Blot and quantified by densitometry.
Immunizations and serology
Groups of mice were immunized intraperitoneally (i.p.) with the PreS1-WHc VLPs (usually 10–20 μg) emulsified in incomplete Freund’s adjuvant (IFA) for both antibody production and T cell experiments. DNA immunization was performed i.m. in the tibialis cranialis muscle with 50 μg pVAX-VLP-1.6 plasmid in a volume of 50 μl. Immediately after immunization, the muscle was electroporated (EP) using Clinporator 2 device (IGEA, Carpi, Italy) with a pulse pattern of one 1 ms 600 V/cm pulse followed by a 400 mg 60 V/cm pulse. A booster dose was given 1 month later and mice were sacrificed 2 weeks thereafter. For antibody experiments, mice were bled retro-orbitally and sera pooled from each group. Periodically individual mouse sera were tested to confirm the fidelity of the pooled sera results. Anti-WHc and anti-insert IgG antibodies were measured in murine sera by an indirect solid-phase ELISA by using the homologous WHcAg (50 ng/well), HBV virions, rPreS1 + 2, or synthetic peptides (0.5 μg/well), representing the inserted PreS1 sequences, as solid-phase ligands as described previously.55 Serial dilutions of both test sera and preimmunization sera were made and the data are expressed as antibody titers representing the reciprocals of the highest dilutions of sera required to yield an optical density at 492 nm (OD 492) three times an equal dilution of preimmunization sera.
In vitro T cell cytokine assays
Spleen cells from groups of three mice each of the various lineages were harvested and pooled 4–6 weeks after immunization with the PreS1-WHc VLPs. Spleen cells (5 × 105) were cultured with varying concentrations of WHcAg, HBcAg or synthetic peptides derived from the WHcAg, HBcAg or PreS region. For cytokine assays, culture supernatants were harvested at 48 h for IL-2 determination and at 96 h for interferon-gamma (IFNγ) determination by ELISA. IFNγ production was measured by a two-site ELISA using mAb 170 and a polyclonal goat anti-mouse IFNγ (Genzyme Corp., Boston, MA).
Results
Construction of PreS1-WHc VLPs
The HBV encodes three envelope proteins termed small (HBs), middle (M) and large (L) proteins, which share the C-terminal HBsAg domain. The M and L proteins carry additional N-terminal extensions of 55aa (PreS2 region) and 108 or 119aa depending on genotype (PreS1 region). The 1-108aa (genotype D) PreS1 sequence numeration is used throughout. The stoichiometric ratio of L/M/HBs proteins in HBV virions is approximately 1:1:4, whereas the most abundant secreted small noninfectious subviral particles contain almost exclusively the small HBsAg protein, lesser amounts of PreS2 and only trace amounts of the PreS1 region.56 Therefore, for a therapeutic HBV vaccine designed to elicit HBV virus-specific antibodies, inclusion of PreS B cell epitopes is imperative. Antibodies produced to the HBsAg domain can be “adsorbed-out” by subviral HBsAg particles that circulate at levels as high as 1.0 mg/ml in chronic HBV sera. A second imperative is that the PreS B cell epitopes chosen must elicit HBV-specific nAbs. A number of PreS1-specific B cell epitopes have been identified in mice by immunization with HBsAg/L particles, PreS1-derived synthetic peptides and by serological analysis of human HBV-infected blood samples (i.e., 1–21, 21–47 and 83–106).55,57 Further, in vitro neutralization studies and in vivo immunization studies with PreS1 epitope-specific synthetic peptides demonstrated that PreS1-specific antibodies could protect chimpanzees from experimental HBV challenge in the absence of anti-HBs region antibody.21–23 Subsequently, several groups delineated the PreS1 residues (aa 9–18) and (aa28-48) involved in HBV-hepatocyte receptor recognition.58–60
Based on these earlier studies, we initially chose four PreS1 B cell epitopes (1.1, 1.2, 1.3 and 1.4; see Table 1) for insertion onto the exposed loop region of the WHcAg platform. Four PreS1-WHc VLPs were selected from a larger library based on assembly, yield and antigenicity determined by binding to a series of PreS1-specific monoclonal antibodies (Mabs). The inserted PreS1 sequences were modified in a second set of VLPs designated 1.1+, 1.3+, 1.4+ and 1.5 in order to broaden recognition by the panel of PreS1-specific Mabs. As shown in Table 1, purified HBV virions, recombinant (r) PreS1/PreS2 protein and myristoylated rPreS1/PreS2 protein were recognized by all four PreS1-specific Mabs and an anti-PreS1 peptide (aa83-106) polyclonal antisera. Note that Mab Ab001, which binds aa1-15 is not blocked by myristoylation as previously suggested, which indicates heterogenicity in the Ab response to the PreS1 amino terminus.61 The PreS1-specific Mab binding profiles for the selected PreS1-WHc VLPs and a series of synthetic peptides demonstrated that the inserted PreS1 sequences were accessible on the surface of the VLPs and appropriately antigenic. Based on antigenicity and immunogenicity data, we consolidated the inserted PreS1 sequences from VLP-1.1+ and VLP-1.4+ into the loop region of a single VLP (1.6). We also consolidated the PreS1 sequences from VLP1.3+ (inserted into the WHcAg loop) and VLP1.2 (fused to the N-terminus of the WHcAg) onto a single VLP (1.9). Note that the combination of VLPs 1.6 + 1.9 is efficiently bound by all four PreS1-specific Mabs and the anti-aa83-106 antisera in a solid-phase ELISA format (Table 1).
Table 1.
Antigenicity of PreS1-WHc VLPs.
| PreS1-Specific Mabs (Min binding conc. [ng/ml]) |
|||||
|---|---|---|---|---|---|
| Ab001 | 18/7 | AP-2 | KR-127 | α-p(83–106) | |
| Antigens | aa(1–15) | aa(20–23) | aa(23–34) | aa(26–34) | aa(83–106) |
| HBV (virions) | 0.5 | 2.0 | 25.0 | 75.0 | + |
| rPreS1 + 2 | 0.3 | 0.3 | 1.6 | 1.6 | + |
| myr-rPreS1 + 2 | 0.3 | 0.3 | 1.6 | 1.6 | + |
| PreS1-VLPs | |||||
| VLP-1.1 (21–43) | 0 | 0 | 1.6 | 1.6 | 0 |
| VLP1.1+ (19–43) | 0 | 0.3 | 40 | 1,000 | 0 |
| VLP-1.2 (83–106) | 0 | 0 | 0 | 0 | + |
| VLP-1.3 (1–21) | 0.3 | 0 | 0 | 0 | 0 |
| VLP-1.3+ (1–25) | 0.3 | 1.6 | 0 | 0 | 0 |
| VLP-1.4 (15–31) | 0 | 0.3 | 0 | 0 | 0 |
| VLP-1.4+ (15–34) | 0 | 8.0 | 8.0 | 8.0 | 0 |
| VLP-1.5 (1–34) | 0.3 | 0.3 | 0 | 0 | 0 |
| VLP-1.6 (15–40) | 0 | 0.3 | 1.6 | 1.6 | 0 |
| VLP-1.9 (83-106-NH2/ 1-25-loop) | 0.3 | 1.6 | 0 | 0 | + |
| VLPs 1.6 + 1.9 | 0.3 | 0.3 | 1.6 | 1.6 | + |
| PreS1-Peptides | |||||
| p(1–15) | 20 | 0 | 0 | 0 | 0 |
| p(5–16) | 1.6 | 0 | 0 | 0 | 0 |
| p(11–21) | 0 | 0 | 0 | 0 | 0 |
| p(18–25) | 0 | 8.0 | 0 | 0 | 0 |
| p(1–36) | 0.3 | 1.6 | 8.0 | 1.6 | 0 |
| p(21–42) | 0 | 0 | 8.0 | 1.6 | 0 |
| p(83–106) | 0 | 0 | 0 | 0 | + |
The depicted panel of antigens (virions, PreS1 + 2 proteins, PreS1-WHc VLPs, and synthetic peptide) were coated on ELISA plates as solid-phase ligands. Dilutions of PreS1-specific Mabs or polyclonal murine anti-p83-106 were incubated with the solid-phase antigens to determine binding by ELISA. Binding is expressed as the minimum concentration of mAb to yield 3x the O.D. 492 of an irrelevant Mab. Binding by the polyclonal anti-p83-106 sera is depicted as titers plus (+) or minus (0) binding. myr-rPreS1 + 2, myristoylated rPreS1 + 2.
Immunogenicity of PreS1-WHc VLPs
Each PreS1-WHc VLP was analyzed for immunogenicity and anti-PreS1 Ab fine specificity by injecting groups of B10 mice each with 20 μg and boosting with 10 μg of VLP formulated in incomplete Freund’s adjuvant (IFA) (Table 2). All VLPs elicited anti-PreS1 IgG endpoint titers of at least 1:625,000 after a boost measured on rPreS1/2 protein and the titers ranged between 1:125,000 and 1:6x106 (i.e., VLP1.6) as measured on purified HBV virions demonstrating the relevance of the anti-PreS1 Ab response to the virus. The fine specificity of the anti-PreS1 Abs correlated well with the PreS1 sequences inserted on the VLPs. For example, VLP-1.2 elicited Abs specific for only aa83-106, the inserted sequence; and VLP-1.3 elicited Abs specific for the aa1-15 insert. Further, fine specificity mapping of the anti-VLP antisera on a large panel of PreS1 peptides (see Table 3) revealed that the PreS1 B cell epitopes segregated into an N-terminal domain (aa1-21: containing three overlapping epitopes, aa1-15, aa5-16 and aa11-21); a central domain (aa20-47: containing three overlapping epitopes, aa20-23, aa22-32, and aa30-42); and a C-terminal domain (aa83-106: containing two epitopes, aa83-94 and aa95-106) revealing at least eight PreS1 epitopes identified by antisera raised against this panel of 10 PreS1-WHc VLPs. Note that VLPs 1.6 and 1.9 elicited a complementary set of PreS1-specific Abs and combining these two VLPs for immunization yielded the full spectrum of anti-PreS1 Ab specificities (Table 3).
Table 2.
Immunogenicity and Fine Specificity of Anti-PreS-WHc VLP Sera.
| Antibody titer (1/dilution) vs PreS1 Antigens |
|||||||
|---|---|---|---|---|---|---|---|
| anti-VLP Sera | HBV1 | rPreS1± 2 | p(1–15) | p(18–25) | p(1–36) | p(21–42) | p(83–106) |
| VLP-1.1 | 125K2 | 625K | 0 | 0 | 625K | 3x106 | 0 |
| VLP-1.1+ | 125K | 3x106 | 0 | 125K | 6x106 | 125K | 0 |
| VLP-1.2 | 625K | 625K | 0 | 0 | 0 | 0 | 625K |
| VLP-1.3 | 125K | 625K | 3x106 | 0 | 3x106 | 0 | 0 |
| VLP-1.3+ | 625K | 3x106 | 625K | 5K | 3x106 | 0 | 0 |
| VLP-1.5 | 125K | 625K | 50K | 10K | 2.1x106 | 50K | 0 |
| VLP-1.4 | 625K | 625K | 0 | 100K | 1.2x106 | 5K | 0 |
| VLP-1.4+ | 3x106 | 3x106 | 0 | 50K | 6.4x106 | 625K | 0 |
| Hybrid VLPs | |||||||
| VLP-1.6 | 6x106 | 6x106 | 0 | 625K | 6.4x106 | 3x106 | 0 |
| VLP-1.9 | 125K | 1.2x106 | 125K | 5K | 625K | 0 | 125K |
Groups of three B10 mice each were immunized i.p. (20 µg) and boosted (10 µg) with the indicated PreS1-WHc VLPs (emulsified in IFA). Four weeks post-boost sera were collected, pooled and binding to a panel of PreS1-specific antigens was determined by solid-phase ELISA. Endpoint titers (1/dilution) are depicted as the highest serum dilution to yield 3x the OD 492 of equivalent dilutions of normal mouse serum (NMS). 1 HBV = purified virions, 2 K = thousands.
Table 3.
Immunogenicity and fine specificity of the combination of VLP-1.6 and VLP-1.9.
| Antibody Titer (1/dilution) vs. PreS1 Antigens |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immunogen | WHc | rPreS1± 2 | p(1–15) | p(5–16) | p(11–21) | p(18–25) | p(1–36) | p(22–32) | p(30–42) | p(21–42) | p(83–106) | HBV (virions) |
| VLP-1.6 + VLP-1.9 |
250K | 2x106 | 57K | 34K | 32K | 0.5K | 625K | 0.8K | 6K | 250K | 23K | 37K |
Groups of five mice were immunized once with the combined VLP-1.6 + VLP-1.9 (20 µg each, i.p. in IFA). Three weeks post-immunization sera were collected, pooled and anti-PreS1 fine specificities determined by ELISA using the panel of antigens depicted as solid-phase ligands. Endpoint Ab titer (1/dilution) was determined by the highest dilution of sera to yield 3x OD492 of NMS. K = thousands
Important characteristics of the PreS1-specific Abs elicited by PreS1-WHc VLPs include the ability to bind native PreS1 epitopes expressed on L/M/S-HBsAg particles and to recognize both major serotypes (i.e., ay and ad). Although the PreS1 region is highly conserved, especially the receptor binding N-terminal domain, there are genotype-specific differences. Therefore, anti-PreS1-WHc VLP murine sera were tested by ELISA for binding to a panel of solid-phase L/M/S-HBsAg particles purified from infected patients, representing the two major serotypes (Figure 3). Although the amount of PreS1 sequence among the four L/M/S-HBsAg preparations varied, Mab 18/7 served as a reference antibody. Anti-VLPs 1.2, 1.5 and 1.6 Abs each recognized all four L/M/S-HBsAg particles at varying dilutions, indicating a high degree of crossreactivity for the major ay and ad serotypes, especially because all of the inserted PreS1 sequences were derived from the ay serotype. Although the results indicate it is probably not necessary to insert genotype-specific PreS1 sequences, if required it can be easily accomplished with this technology.
Figure 3.
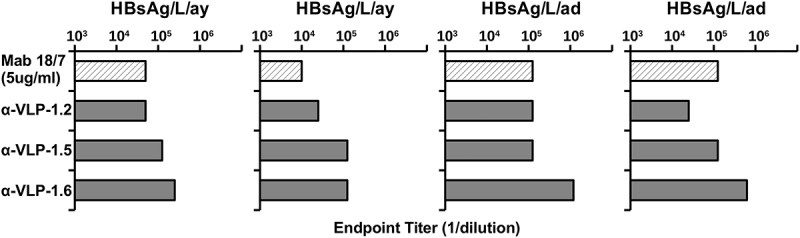
Anti-PreS1-WHc VLP Abs bind multiple L/M/S-HBsAg particles of both major serotypes (ad/ay). Four L/M/S-HBsAg preparations, either ay or ad, derived from HBV-infected sera were used as solid-phase ligands to measure anti-PreS1 Ab binding by ELISA. Groups of three mice were immunized (i.p.) with 20 µg and boosted with 10 µg of the indicated PreS1-WHc VLPs and pooled sera were tested for binding to the panel of L/M/S-HBsAg antigens by ELISA. Endpoint titers (1/dilution) are shown. The Mab 18/7 was included for reference.
One characteristic of hepadnavirus core proteins that contributes to enhanced immunogenicity is the presence of a domain at the C-terminus that incorporates a TLR7 ligand into the assembled particles.45 To determine the influence of TLR7 signaling on the immunogenicity of PreS1-WHc-VLPs, TLR7-knock out (KO) mice and wildtype mice were immunized with VLP-1.6 and anti-PreS1 Ab titers were determined three and 5 weeks after a single injection (Figure 4). Wildtype mice produced significantly higher endpoint titers of anti-PreS1 Ab at week 3 (1:1.2x106) and at week 5 (1:15x106) as compared to TLR7-KO mice at week 3 (1:50,000) and week 5 (1:250,000). This result indicates that innate TLR7 signaling is operative at least during the primary humoral response to PreS1-WHc VLP immunization.
Figure 4.
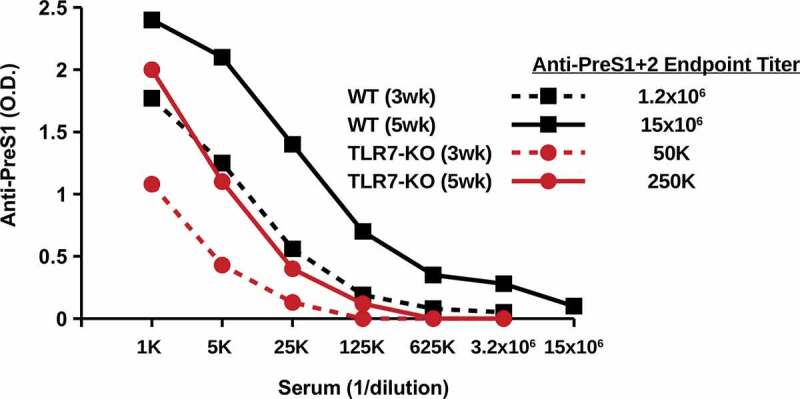
Immunogenicity of PreS1-WHc VLP-1.6 in WT and TLR7-KO Mice. Groups of three WT or TLR7-KO mice were immunized (i.p.) with a single dose of 20ug of PreS1-WHc VLP-1.6 emulsified in IFA and 3 and 5 weeks later IgG anti-PreS1 endpoint titers were determined on pooled sera by ELISA.
Ability of PreS1-WHc VLPs to circumvent immune tolerance
Several investigators have suggested using the HBcAg as a carrier for PreS protective B cell epitopes in a possible therapeutic vaccine, which is problematic given immune tolerance to the HBc/HBeAgs in CHB infection, especially during the immune tolerant (IT) phase.61,62 As a means of circumventing immune tolerance, we have utilized the WHcAg as a carrier of eight PreS1 B cell epitopes. Immunization of HBeAg-MUP-Tg mice and HBeAg x HBcAg-MUP double-Tg mice, which are extremely tolerant to the HBc/HBeAgs at the Th/CTL levels,63 with the HBcAg (20ug, IFA) demonstrated a high degree of Th cell tolerance as reflected by a 900-fold reduction in anti-HBc antibody as compared to wildtype B10 mice (Figure 5(a)) and a 20-fold reduction in crossreactive anti-WHc Abs. The low-level anti-HBc Abs in the HBe/HBcAg-Tg mice reflects the contribution of T cell-independent antibody production.64 In contrast, immunization of HBeAg-MUP-Tg or HBe/HBcAg-MUP-Tg mice with a PreS1-WHc VLP (VLP-1.1) yielded equivalent high titer anti-WHc and, more importantly, anti-PreS1 Abs compared to wildtype mice, whereas, crossreactive anti-HBc Ab production was significantly reduced (25–12.5-fold) (Figure 5(b)). Similarly, immunization with PreS1-WHc VLPs 1.1+ and 1.2 elicited equivalent high titer anti-PreS1 Abs in HBeAg-MUP-Tg mice and B10 wild-type mice (data not shown).
Figure 5.
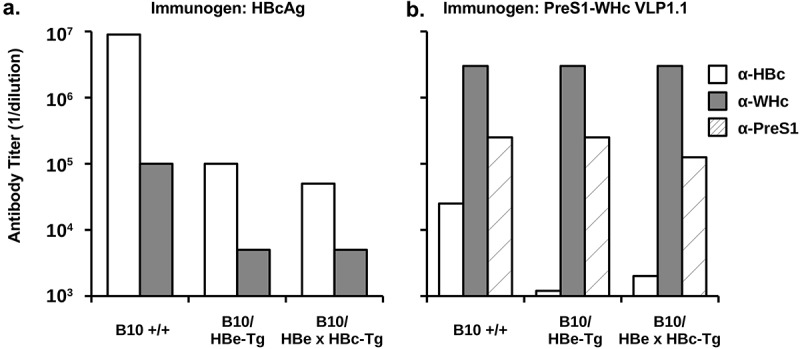
Immunization with PreS1-WHc VLP1.1 circumvents immune tolerance in HBe/HBcAg-Tg mice. Groups of three B10 wildtype, B10 HBeAg-Tg, and B10 HBe/HBcAg double-Tg mice were immunized (i.p.) with a single 20 µg dose of HBcAg (a) or PreS1-WHc VLP1.1 (b) emulsified in IFA. Four weeks after immunization sera were collected, pooled and tested by ELISA for IgG anti-HBc, anti-WHc, and anti-PreS1 Abs expressed as endpoint (1/dilution) titers.
Furthermore, HBV-Tg mice, which express the HBsAg S/M/L envelope antigens as well as the HBe/HBcAgs and are immune tolerant to the HBV structural antigens,65 were immunized and boosted with a mixture of 20ug each of PreS1-WHc VLPs (1.2, 1.3 and 1.6) and anti-PreS1 humoral responses compared to wildtype mice (Table 4). Anti-PreS1 antibody production in HBV-Tg mice detected by binding to the rPreS1/2 protein, HBV virions and 3 of 5 PreS1 peptides was equivalent to or higher than in wildtype mice and lower against two peptides (aa18-25 and aa83-106), possibly due to greater adsorption of these anti-PreS1-specific Abs by circulating PreS1 antigen-bearing particles. Similarly, PreS1-Tg mice immunized and boosted with a combination of VLPs 1.6 and 1.9 produced equivalent or higher titer anti-PreS1 Abs as compared to wildtype mice with the same exception of anti-aa18-25 and anti-aa83-106 Abs, again suggesting these two specificities may be preferentially adsorbed by circulating PreS1 antigen (Table 4). It is important to note that anti-PreS1 Ab production did not result in liver injury in either HBV-Tg or PreS1-Tg mice determined by the lack of serum ALT elevation (data not shown).
Table 4.
Comparative immunogenicity of PreS1-WHc VLPs in HBV-Tg, PreS1-Tg and Wildtype Mice.
| Antibody Titers (1/dilution) vs. PreS1 Antigens |
||||||||
|---|---|---|---|---|---|---|---|---|
| n | HBV | rPreS1± 2 | p(1–15) | p(1–36) | p(18–25) | p(21–42) | p(83–106) | |
| Wildtype (B6/BALB/c) |
5 | 50K | 125K | 125K | 3x106 | 625K | 625K | 125K |
| HBV-Tg (B6/BALB/c) |
5 | 72K | 190K | 625K | 6x106 | 50K | 875K | 0.5K |
| PreS1-Tg (B6) |
6 | 110K | 2x106 | 70K | 3x106 | 70K | 900K | 14K |
Groups of five wild-type or HBV-Tg mice were immunized i.p. and boosted with a mix of PreS1-WHc VLPs-1.2, −1.3 and −1.6 (20ug each in IFA). A group of six PreS1-Tg mice was immunized i.p. and boosted with a mix of PreS1-WHc VLPs-1.6 and −1.9. Five weeks after the boost, sera was collected, pooled and binding to a panel of PreS1-specific antigens was determined by solid-phase ELISA. Endpoint titers (1/dilution) are depicted as the highest serum dliution to yield 3x the OD492 of NMS. K = thousands.
To analyze the Th cell response to PreS1-WHc VLPs, HBV-Tg and wildtype mice were immunized with either VLP-1.1+ or VLP-1.3 and splenic IL-2 and IFNγ cytokine production in response to culture with a panel of recall antigens was determined (Figure 6). As expected, native WHcAg was the dominant source of Th cell cytokine production in response to PreS1-WHc VLP immunization as well as the constituent WHcAg-derived peptides W60-80 and W120-140 in both HBV-Tg and wildtype mice. Note that Th cells crossreactive for the HBcAg were also primed by immunization with PreS1-WHc VLPs 1.1+ and 1.3 but produced IL-2 and IFNγ to a lesser degree than in response to the WHcAg, especially in HBV-Tg mice. However, the fine specificity of the crossreactive HBcAg-specific T cells could not be determined in HBV-Tg mice because HBV-Tg mice on a B6/BALBc background are tolerant to the H120-140 dominant HBcAg-specific T cell site, unlike wild-type mice, in which H120-140-specific Th cells were detected. The W120-140 and H120-140 sequences differ by only two amino acids. Therefore, WHcAg-specific Th cells are dominant in PreS1-WHc VLP immunized mice, although low-level HBcAg-crossreactive Th cells are also primed even in HBV-Tg mice; however, the lack of a H120-140-specific Th cell response is evidence of HBcAg-specific Th cell tolerance in HBV-Tg mice.
Figure 6.
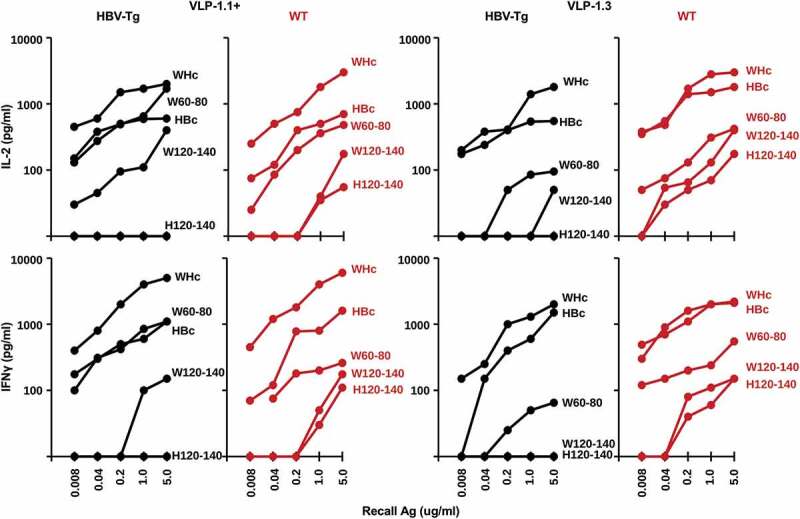
Analysis of CD4+ Th cell response to PreS1-WHc VLP immunization in HBV-Tg mice. Groups of three HBV-Tg or wildtype (B6/BALBc), mice were immunized (s.c.) with 20ug of either PreS1-WHc VLP-1.1+ or VLP-1.3 emulsified in IFA. Four weeks later spleen cells were harvested and cultured (5x105) with varying concentrations of the indicated WHcAg, HBcAg or WHc(W)- or HBc(H)-derived synthetic peptides. Culture supernatants were collected at 48 h for IL-2 determination and at 96 h for IFNγ determination by 2-site ELISA. The results represent single mice but are representative of three mice/group.
In summary, the results from immunization studies in HBe/HBcAg-MUP-Tg mice, HBV-Tg mice, and PreS1-Tg mice reveal that use of the WHcAg platform to carry PreS1 B cell epitopes is capable of circumventing HBe/HBcAg-specific and L/M/S-HBsAg-specific Th cell tolerance, which characterizes CHB infection. It is also notable that the HBV-Tg and PreS1-Tg lineages are not tolerant at the B cell level to PreS1 B cell epitopes. The presence of PreS1/2-specific as well as HBsAg-specific IgG in immune complexes in CAH patients is also consistent with a lack of B cell tolerance during the immune clearance phases of chronic HBV infection.66
Immunization with Pres1-WHc VLPs elicits HBV neutralizing antibodies
The previous results indicate that PreS1-WHc VLPs are capable of circumventing Th cell immune tolerance and can elicit high titer anti-PreS1-specific Abs of at least eight different specificities. However, to be relevant, the anti-PreS1-specific Abs produced must be virus-neutralizing (i.e., nAb). As shown in Figure 7, the anti-PreS1 antibodies elicited by immunization with six PreS1-WHc VLPs efficiently neutralized/prevented infection of a HepaRG human hepatocyte cell line by a hepatitis delta virus (HDV) coated with HBV envelope proteins (i.e., L/M/S HBsAg/ay) in an infection assay developed by Sureau, et al.54 Note that antisera to PreS1-WHc VLPs 1.4+ and 1.1+ were capable of completely preventing HDV-HBV infection of HepaRG cells even at final dilutions of 1:4000 as did 0.05 µg/ml of Mab 18/7 (a standard anti-PreS1 neutralizing Mab). In an attempt to find an endpoint dilution for viral neutralization, a higher stringency infection was performed (lower panel, Figure 7). In this high stringency assay Mab 18/7 even at 5.0 μg/ml was not able to totally neutralize infection; however, the three polyclonal anti-PreS1-WHc VLP antisera completely neutralized infection at final dilutions of 1:400 (VLPs 1.1+ and 1.4+) and 1:40 (VLP 1.5). Similarly, anti-PreS1-WHc VLP 1.6 antibody completely prevented infection at a final dilution of 1:400 (data not shown).
Figure 7.
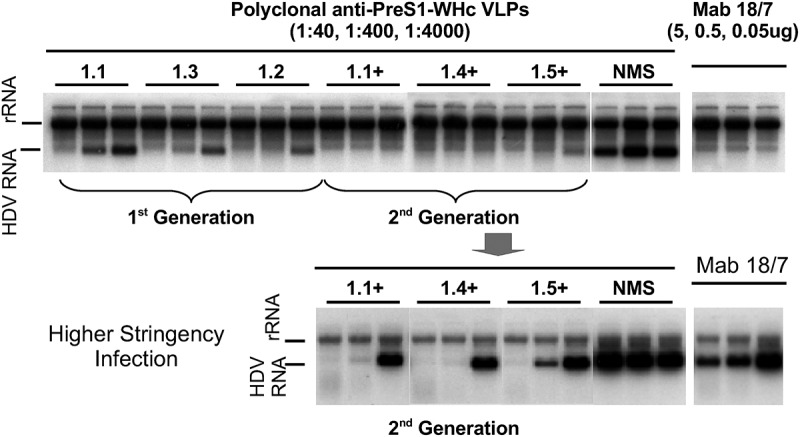
Neutralization of HBV Infection by PreS1-WHc VLP Antisera. Groups of three mice were immunized with the six depicted PreS1-WHc VLPs containing separate neutralizing B cell epitopes. Mice received a primary (20 μg) and a single booster (10 μg) immunization (i.p., IFA). Antisera were collected after the boost, pooled and the neutralization activity was determined in an in vitro infection assay using a modified hepatocyte cell line (HepaRG) infected with HDV particles coated with HBV envelope proteins.54 The bottom panel represents a higher stringency neutralization assay.
Anti-PreS1-WHc Abs prevent acute infection and clear serum HBV in previously infected mice in vivo in human-liver chimeric mice
In addition to the ability of PreS1-specific Abs to neutralize HBV infection of a human hepatocyte cell line in vitro (Figure 7), to determine the efficacy of PreS1-specific nAbs in an infectious in vivo system we utilized mice made chimeric with human-liver cells.67,68 Human-liver chimeric mice are immune compromised, so first wildtype mice were immunized with a combination of VLP-1.6, VLP-1.2 and VLP-1.3 and 0.2 ml of secondary anti-PreS1 antisera or control anti-WHc sera was adoptively transferred into human-liver chimeric mice: (1) prior to HBV infection (acute infection and controls); or (2) 2 weeks after HBV infection (“chronic infection”) with 1 × 106 HBV GE copies/mouse in each challenge. HBV DNA in the serum was monitored every 2 weeks for 8 weeks and HBV DNA in the liver was determined at termination at 8 weeks post-infection (Figure 8). Control mice demonstrated escalating serum HBV DNA levels that peaked at 6–8 weeks post-infection. The four mice adoptively transferred with anti-PreS1 Abs prior (day −1) to HBV infection were protected against acute infection with the exception of one “breakthrough” at 8 weeks post-infection, as nAb levels waned. The acute group only received a single injection of 0.2 ml of anti-PreS1 sera, while the chronic group received adoptive transfer of 0.2 ml of anti-PreS1 sera at 2 and 5 weeks after the HBV infection. All chronically infected mice cleared serum HBV DNA by week 6 post-infection and remained negative for serum HBV DNA at the termination of the experiment (Figure 8(a)). At termination, liver HBV DNA levels were determined and no virus was detected in the livers of the acute group. Although not statistically significant, the HBV DNA levels in the livers of the chronic group were somewhat lower than those in the control group (Figure 8(b)). Anti-PreS1 nAb were not expected to clear the preexisting infection in the liver and the reduced HBV DNA liver load compared to controls most likely represents the ability of circulating anti-PreS1 nAbs to prevent viral spreading to uninfected hepatocytes since HBV infection requires secretion of cell-free virus. Immunohistology staining for HBsAg detected significant HBsAg in three of the four control livers, no HBsAg in the acute livers and HBsAg staining in only one of six “chronic” livers (Figure 8(c)).
Figure 8.
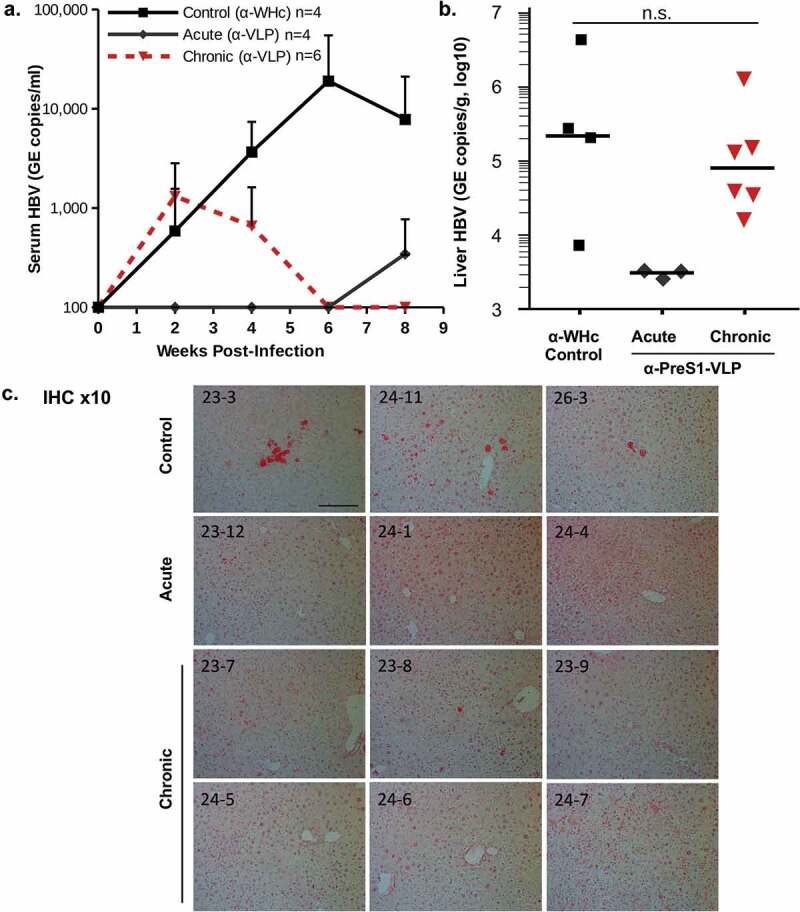
Anti-PreS1 Abs prevent acute infection and clear serum HBV from chronically infected human-liver chimeric mice. Wildtype B10 mice were immunized and boosted with a mixture of PreS1-WHc VLPs-1.2, −1.3 and −1.6 (20 μg each) and 5 weeks after the boost sera was collected, pooled and used for the adoptive transfer. 0.2 ml of anti-PreS1-WHc sera or control anti-WHc sera were transferred into human-liver chimeric mice prior to infection with 1 × 106 GE copies of HBV/mouse in the control and acute groups. For the chronic group, 0.2 ml of anti-VLP sera was transferred 2 and 5 weeks after the HBV infection. (a). Serum HBV-DNA was determined at the indicated time points post-infection, (b). Liver HBV-DNA was determined at termination. The difference between the Control and Chronic groups were not statistically significant at α = 0.05 (Wilcoxon Two-Sample Test). (c). Liver immunohistology staining for HBsAg. The scale bar is 200um.
These results provide a proof-of-concept that the PreS1-WHc VLPs can elicit nAbs capable of preventing an acute HBV infection into human-liver cells and are sufficient to clear serum HBV from “chronically infected” mice and may prevent spreading of HBV amongst human-liver cells in vivo as evidenced by the decreased HBsAg staining in the chronic group.
WHcAg-based DNA constructs designed to circumvent immune tolerance and to elicit HBcAg-specific CTL
We demonstrated that immunization with PreS1-WHc VLPs could elicit noncrossreactive WHcAg-specific heterospecific CD4+ T cells and to a lesser degree WHcAg/HBcAg crossreactive CD4+ T cells that may mediate viral clearance via cytokine production. For example, immunization with PreS1-WHc VLP 1.1+ primed HBcAg-crossreactive CD4+ T cells in both wildtype and HBV-Tg mice (Figure 6). To determine the ability of the PreS1-WHc VLPs to elicit a CD8+ CTL response in either a DNA or VLP format, we immunized groups of mice with either VLP-1.6 protein or DNA encoding VLP-1.6. As shown in Figure 9(a), the DNA version elicited superior CD4+ T cell responses to VLP-1.6 and its constituent three WHc-specific CD4+ T cell epitopes (WHc60-80, WHc 80–95 and WHc120-140) as compared to the protein version of VLP-1.6. Moreover, the DNA immunogen elicited a strong CD8+ CTL response to WHc10-25 and a cross-reactive CTL response to the HBcAg-specific HBc93-100 CTL epitope, whereas, protein VLP-1.6 did not prime CTL responses efficiently. Therefore, just as crossreactive HBcAg-specific CD4+ T cells can be primed by PreS1-WHc VLPs (Figure 6), crossreactive HBcAg-specific CD8 + T cells can be primed by DNA encoding PreS1-WHc VLPs (Figure 9(a)). It is also notable that, despite the superior priming of WHcAg-specific CD4+ T cells by the DNA vaccine compared to the VLP protein, the VLP-1.6 protein immunization elicited far superior (at least 10x higher) anti-WHc and anti-PreS1 Ab responses (Figure 9(b)). This is a dramatic demonstration of the complementarity between the two forms of immunization and that the best method to ensure high titer nAb production as well as effective CTL responses is to use a VLP prime/DNA boost strategy. These WHcAg/HBcAg crossreactive CD4+/CD8+ T cells represent only one method of eliciting HBcAg-specific CD4+/CD8+ T cells. It was anticipated that the WHcAg-“heterospecific” Th cells primed by a hybrid WHcAg/HBcAg DNA vaccine would enable the direct priming of a significantly stronger HBcAg-specific CD8+ CTL response.
Figure 9.
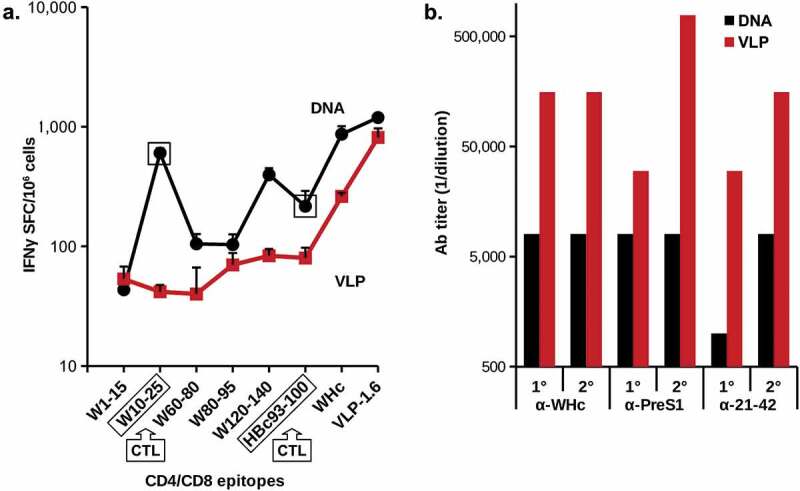
Comparison of delivery of PreS1-WHc VLP-1.6 as a DNA plasmid or as a protein. Groups of five wild-type B6 mice were immunized i.m. with 50 µg of pVAX-VLP-1.6 DNA by electroporation (EP) using the Clinporator device (IGEA, Italy) and boosted 1 month later or mice were immunized (i.m. with 20ug) of VLP-1.6 in IFA and boosted with 10 g one month later. A. Detection of IFNγ-specific spot-forming cells (SFC)/106 spleen cells were determined with a commercial ELISPOT assay. Error bars indicate standard deviation of five pooled spleens measured in triplicate. B. Ab production after the primary (1°) and the boost (2°) was determined by ELISA of pooled sera on the panel of antigens shown. The MHC class I-restricted CD8+CTL epitopes on WHcAg (W10-25) and on HBcAg (H93-100) for B6 mice are indicated. The other peptides are MHC class II-restricted CD4+Th cell epitopes.
Therefore, the goal was to co-express the WHcAg with the HBcAg in the same VLP in order to allow WHcAg-specific (heterospecific) Th cells to provide T cell help for the priming and maintenance of HBcAg-specific CD8+ CTLs (see Figures 1, 2). Because the WHcAg and the HBcAg are 68% homologous and structurally very similar it was possible to obtain hybrid WHcAg/HBcAg assembled VLPs in E. coli using two different strategies. First, full-length WHcAg188 and truncated HBcAg149 genes were co-expressed in E. coli to form hybrid WHcAg/HBcAg VLPs. The subunit for assembly of this VLP is a dimer and biochemical analysis of VLPs from E. coli co-expressing WHcAg and HBcAg showed that, in addition to homodimers, there was a significant fraction of mixed WHcAg/HBcAg dimers, indicating the presence of hybrid WHcAg/HBcAg VLPs (Figure 10(a)). The presence of hybrid WHcAg/HBcAg VLPs was confirmed by ELISA analysis that utilized a WHcAg-specific mAb that did not cross-react with HBcAg and reciprocally an HBcAg-specific mAb that did not cross-react with WHcAg to capture and/or detect hybrid VLPs in solution (Figure 10, lower panels). These hybrid-specific ELISAs do not detect either WHcAg or HBcAg homogeneous particles.
Figure 10.
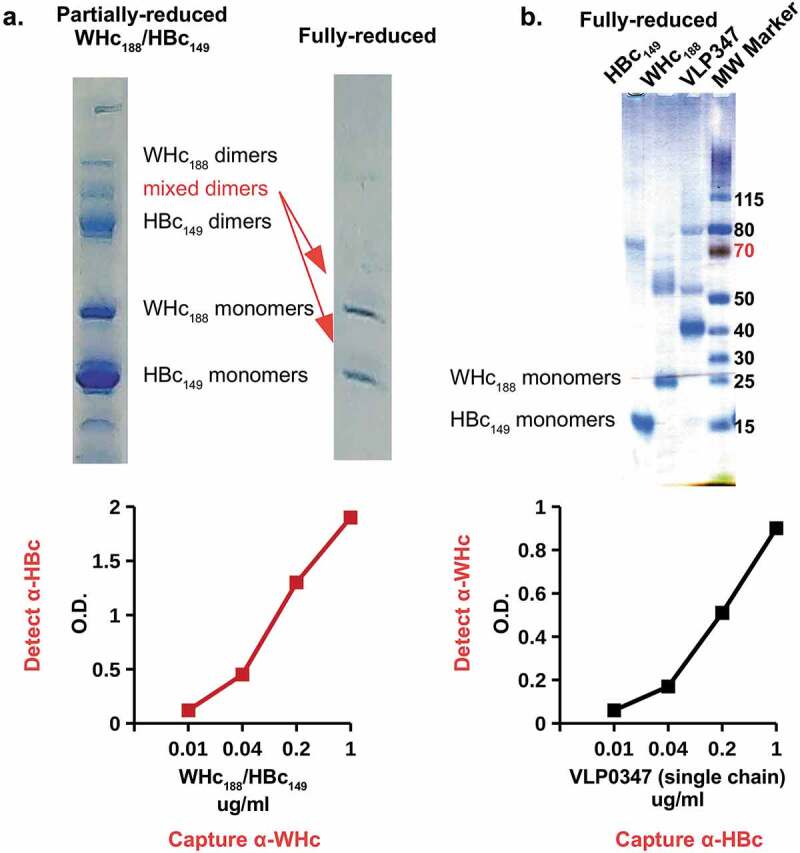
Production of hybrid WHcAg/HBcAg VLPs. (a). Full-length WHcAg188 and truncated HBcAg149 genes were co-expressed in E. coli. The mixed dimer band was excised from the gel which was run under partially-reducing conditions and then fully reduced and run on a second gel (arrows). (b). The HBcAg149 gene was fused to the WHcAg188 gene with a flexible linker to form a “single-chain dimer” expressed as a single open reading frame in E. coli. Purified VLPs were analyzed in capture ELISAs specific for hybrid WHcAg/HBcAg VLPs and incapable of detecting homologous WHcAg or HBcAg VLPs. Molecular Weight Marker (MW Marker) is Thermo Scientific PAGE Ruler with manufacturer’s kilodalton size estimates for this gel chemistry.
The second method used was to fuse the HBcAg149 gene to the N-terminus of the WHcAg188 gene with a flexible linker to form a “single-chain dimer” and to express the one contiguous open reading frame in E. coli. Again, the biochemical and ELISA analysis indicated that hybrid WHcAg/HBcAg VLPs were produced and designated as VLP-347 (Figure 10(b)). The advantage of the single polypeptide method is that only hybrid VLPs are possible, whereas, separate expression of the two gene constructs could theoretically produce a mixture of homologous and hybrid assembled VLPs, although our results indicate this is not likely in E. coli, at least.
To establish the ability of hybrid WHcAg/HBcAg VLP DNA constructs to elicit HBcAg-specific CTL, B6 mice were immunized intramuscularly (i.m.) with DNA (100 g, 2 times) encoding VLP-347, hybrid WHcAg188/HBcAg149 or HBc alone (Figure 11). The hybrid WHcAg/HBcAg DNA construct elicited equal or superior CTL responses compared to HBcAg-DNA to the dominant CTL epitope on the HBcAg in B6 mice, namely HBc93-100. CTL specific for HBc13-23, presumably due to crossreactivity with the WHcAg-specific CTL epitope at the WHc13-23 site, were also produced. Because a dominant CD4+ Th cell site in B6 mice (aa120-140) is highly conserved (19 of 21aa) between WHcAg and HBcAg, it was not surprising that the WHcAg and HBcAg 120–140 peptides recalled a strong IFNγ response after immunization with all three DNA constructs. However, the H120-140 Th epitope is not functional in HBV-Tg mice due to immune tolerance (see Figure 6). Note that IFNγ responses to the WHcAg-specific Th epitopes WHc60-80 and WHc80-95 were primed by the VLP-347 and hybrid WHc188/HBc149 DNAs, but not by HBc DNA immunization. Therefore, in HBV-Tg mice, it is anticipated that these “heterospecific”, WHcAg-unique T cells will be able to replace the defective homospecific H120-140-specific Th cells and become the dominant source of Th cells for the induction of HBcAg-specific CTL (see Figure 2).
Figure 11.
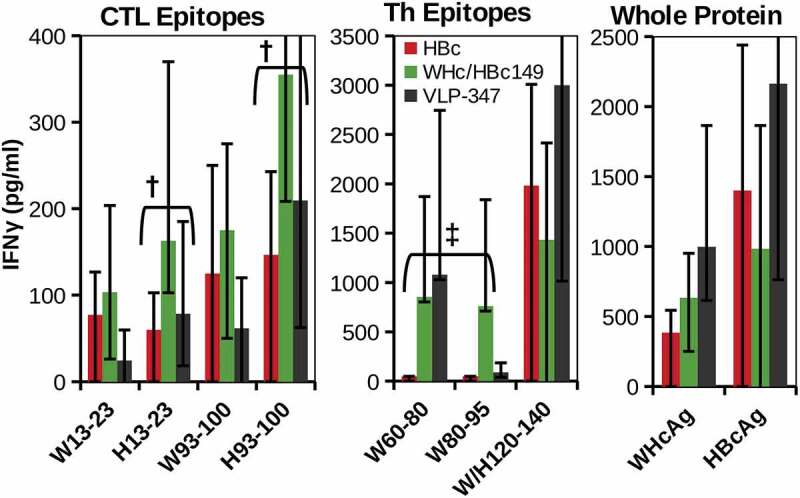
Hybrid WHcAg/HBcAg VLP DNA constructs can prime efficient HBcAg-specific CTL. Groups of three wild-type B6 mice were immunized with DNA constructs (100 µg, two doses, s.c.) encoding VLP-347, WHcAg188/HBcAg149 or HBcAg alone. Splenic CTL or Th cell (5x105) IFNγ responses recalled by the panel of peptides corresponding to CTL or Th epitopes and whole protein antigens are shown. HBcAg-specific CTL (†) and WHcAg-”heterospecific” Th cell (‡) responses are highlighted. IFNγ was measured in 4-day culture supernatants by two-site ELISA. Data represent the mean ± standard deviation of 3 mice/group. Control spleen cells from unimmunized mice cultured with antigens or DNA immunized mice cultured with media alone served as negative controls.
It is also possible to insert so-called “universal” Th cell epitopes derived from tetanus (TT) and diphtheria (DT) toxoid proteins into the PreS1-WHc VLPs and the hybrid WHcAg/HBcAg DNA constructs in order to elicit additional heterospecific CD4+ Th cells. This would be most relevant in HBV/HIV co-infection as TT/DT-specific memory Th cells most likely were primed prior to HIV/HBV infections and remain viable alternatives to provide “heterospecific” Th cell function for HBV-specific B cells and CTL. As shown in Figure 12, TT-immune mice (a model for TT-immunized humans) produced early and enhanced anti-WHc Ab responses when injected with a single dose of WHcAg-TT VLPs via the action of TT-heterospecific Th cells.
Figure 12.
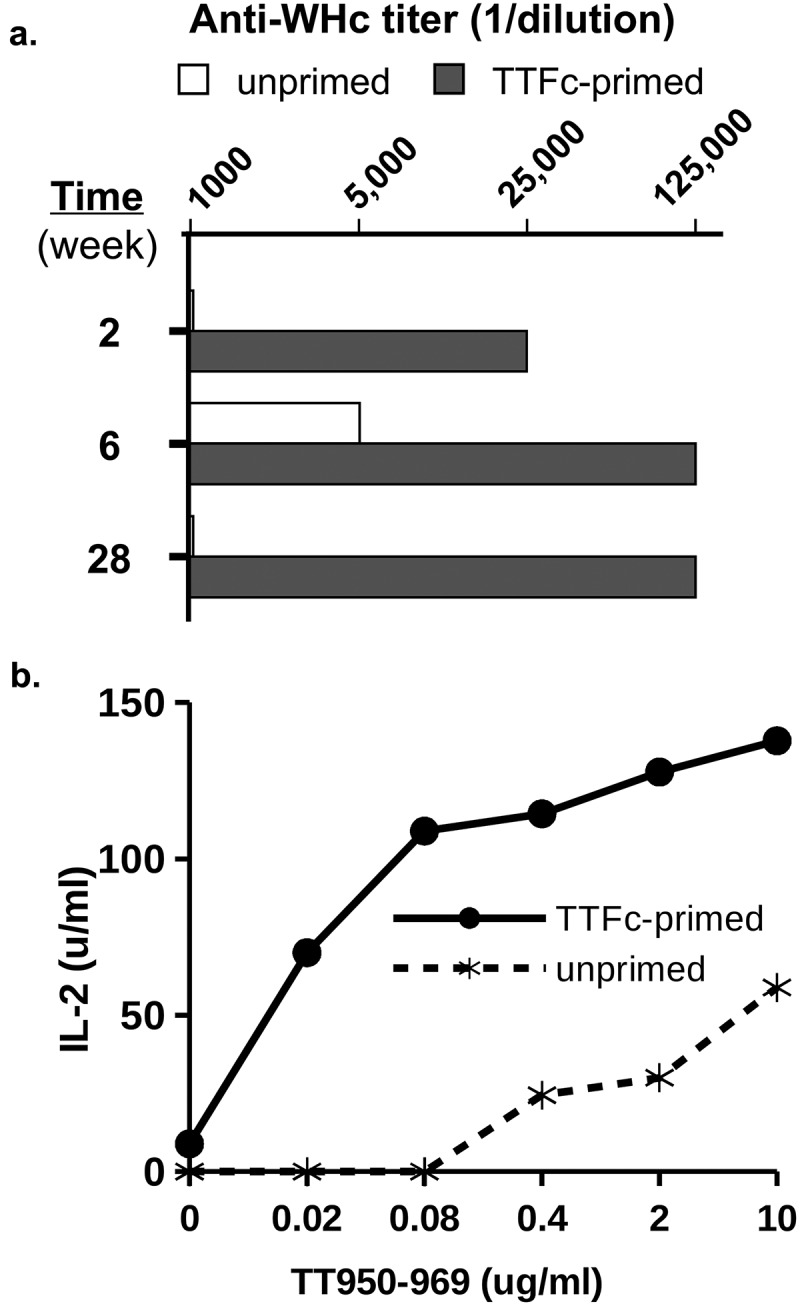
Tetanus toxoid (TT) priming provides heterospecific T cell help for VLPs carrying a TT epitope. Groups of three mice (B10 strain) were first primed with tetanus toxin fragment C (TTFc, 20 µgin IFA) to mimic TT immunization in humans, or unprimed. Two months later TTFc-primed and unprimed mice were injected with hybrid WHc-TT950-969 VLPs (10 g in saline). (a). At 2, 6, and 28 weeks sera were pooled and anti-WHc antibody levels were determined by ELISA. (b). Splenic T cell recognition of the TT950-969 peptide was measured by harvesting spleen cells and culturing with the TT950-969 peptide, followed by IL-2 determination by ELISA.
Discussion
Immune tolerance to an infectious agent such as the HBV is not a binary event (tolerance vs no tolerance), but rather represents complex host-viral interactions. Because immune tolerance is clonal, all viral antigens and their constituent epitopes are not equally tolerogenic nor are all cell types equally susceptible to tolerance induction. For example, the secreted HBeAg is more tolerogenic than the cellular HBcAg,69 and one HBeAg-specific epitope is more tolerogenic than another epitope on the same HBeAg.70,71 Similarly, one would expect different degrees of T cell tolerance to epitopes on the PreS1, PreS2, and HBsAg envelope antigens because T cell recognition is distinct between these envelope regions.55 At the cellular level, Th cells are often more susceptible to tolerance induction than B cells or CTL.72 These phenomenon represent forms of “split tolerance”. Immune tolerance preferentially occurs in high avidity clones and is mediated by deletional and non-deletional mechanisms. Therefore, low avidity T cells that have escaped deletion most likely constitute the bulk of the T cell repertoire during chronic HBV infection and low avidity T cells are more prone to negative regulation (i.e., checkpoint inhibitors, regulatory T cells, metabolic dysfunction, and clonal exhaustion). Other hosts and viral factors such as age of infection, immune status, antigen load, secreted or cytosolic antigen and phase of infection affect immune tolerance as well.9,10 For vaccine immunotherapy to be effective it must target either non-tolerant clones or non-deleted clones in which tolerance can be reversed or circumvented. Therefore, we targeted PreS1-specific B cells and HBcAg-specific CTL effector cells and bypassed the more tolerant prone Th cells by substituting heterospecific, WHcAg-specific Th cells for HBV-specific Th cells. Although B cell defects have been defined during chronic HBV infection,6 low-level antibodies to most viral antigens are present during the active phases of infection often masked in immune complexes.66 Further, HBc/HBeAg-specific CTL can be detected in the periphery and the liver during chronic infection,73,74 however, similar to B cell antibody production, CTL function is suboptimal and not sufficient to mediate viral clearance. It is anticipated that both of these nascent effector pathways can be enhanced by providing more efficient Th cell function using the WHcAg platform to present neutralizing, PreS1-specific B cell epitopes and HBcAg-specific CTL epitopes.
Accomplishments of the current study include:
Defined eight PreS1 B cell epitopes (highly conserved amongst HBV genotypes).
Inserted each of the eight PreS1 B cell epitopes onto WHcAg carrier VLPs.
Consolidated all eight PreS1 epitopes onto two PreS1-WHc VLPs (VLP-1.6 and VLP-1.9).
Demonstrated that PreS1-WHc VLPs elicited PreS1-specific Ab that recognizes native HBV virions and multiple L/M/S-HBsAg particles of both major serotypes (ad/ay).
Demonstrated that six PreS1-WHc VLPs elicited nAbs specific for N-terminal, central and C-terminal B cell domains of the PreS1 region, however, the central B cell domain elicited superior neutralizing Ab. A previous study of PreS1-HBc VLPs concluded that anti-PreS1 Abs specific for the N- and C-terminus were not viral neutralizing.61 This suggests that the fine specificity of the B cell response to the PreS1 region is variable and can be influenced by the choice of HBcAg or WHcAg carrier.
Demonstrated that PreS1-WHc VLPs circumvented immune tolerance and elicited equivalent levels of nAb to the PreS1 region in wildtype, HBV-Tg, HBc/HBeAg-Tg, and PreS1-Tg mice lineages.
Demonstrated that PreS1-WHc VLPs primed crossreactive, HBcAg-specific CD4+/CD8+ T cells as well as WHcAg-heterospecific CD4+/CD8+ T cells.
Demonstrated that in human-liver chimeric mice passive transfer of PreS1 nAbs prevented an acute HBV infection.
Demonstrated that in human-liver chimeric mice previously infected with HBV (model of chronic infection), PreS1 nAbs cleared serum HBV and may arrest HBV spread in the liver possibly reducing cccDNA levels.16
Produced hybrid WHcAg/HBcAg particles as DNA constructs, which elicited HBcAg-specific CD8+ CTL.
The relative scarcity of PreS1 antigen relative to the major HBsAg is a limiting factor for anti-PreS1 nAb production during a natural HBV infection. The capacity of the highly immunogenic WHcAg carrier to display multiple PreS1 neutralizing B cell epitopes overcomes this limitation. For example, 240 copies of each of the eight PreS1 B cell epitopes are displayed per PreS1-WHc VLP. A combined PreS1-WHc VLP-1.6/VLP-1.9 vaccine formulated in an adjuvant suitable for human use given in a prime/boost protocol with an optimized WHcAg/HBcAg DNA construct would represent a strong candidate therapeutic HBV vaccine. This vaccine would be capable of circumventing immune tolerance and eliciting multiple PreS1 nAb specificities as well as HBcAg-specific CD8+ CTL to target intracellular HBV DNA including cccDNA. Although a PreS1-WHc VLP prime – hybrid WHcAg/HBcAg DNA boost regimen could be given as a monotherapy, combination with an antiviral agent would enhance efficacy by reducing viral load. Inserting multiple neutralizing B cell PreS1 epitopes will mitigate the possibility of nAb escape mutants, which may be problematic when treating an established HBV infection. Bacterial production of PreS1-WHc VLPs together with a WHcAg/HBcAg DNA immunogen would be cost-efficient and compatible with any antiviral treatment for maximum efficacy. The ultimate goal is to produce both PreS1-WHc VLPs and WHcAg-HBcAg hybrid VLP DNA constructs for the dual purposes of eliciting PreS1-specific nAbs and HBcAg-specific CTL. The current study was limited primarily to evaluating the PreS1-specific humoral response in immune tolerant HBV-Tg mice and in human-liver chimeric mice infected with HBV. Future studies will be necessary to evaluate the ability of the WHcAg-HBcAg hybrid DNA constructs produced herein to circumvent HBcAg-specific CTL immune tolerance.
Although therapeutic HBV vaccines have not been successful to date, a number of experimental vaccines show promise in animal models or in the clinic,74,75 especially PreS-containing candidates.76,77 However, most experimental vaccines rely exclusively on the use of HBV-derived antigens, subject to the direct and indirect effects of immune tolerance, unlike the current PreS1-WHc VLP technology. An alternative approach for bypassing immune tolerant, dysfunctional endogenous T cells is the adoptive transfer of “engineered” HBV-specific T cells, which are demonstrating efficacy in animal models and in one clinical trial.78–80 In a recent study, a single adoptive transfer of transfected HBV-specific T cells demonstrated effective reduction in HBV DNA without severe liver disease in an animal model. However, achieving long-term control of HBV infection required the combined use of the PreS1 peptide viral entry-inhibitor Myrcludex B.16,78 We argue that regardless of the efficacy of any and all antiviral therapies, the induction of nAbs should be included to prevent the spread of HBV to uninfected hepatocytes, which may indeed be a requirement for the complete cure of CHB infection.
In addition to use as a therapeutic HBV vaccine, other possible applications for PreS1-WHc VLPs include: Use as a preventative vaccine in low-to-nonresponders to the conventional HBsAg vaccine; vaccination of pregnant HBV+ carrier mothers in order to provide passive transfer of PreS1-specific neutralizing Abs to block transmission during and after birth; an immunotherapy for chronic HDV infection; and prior to immunosuppressive therapy, vaccination of HBV+ liver transplant recipients in order to prevent infection of the new liver.
Funding Statement
This work was supported by the Agence Nationale de Recherches sur le Sida et les Hépatites Virales;National Institute of Allergy and Infectious Diseases [R01 AI049730];National Institute of Allergy and Infectious Diseases [R44 AI08819];Vardalinstitutet (SE);Stockholms Läns Landsting.
Acknowledgments
The authors are grateful to Drs. Luca Guidotti, Frank Chisari and James Ou for access to the HBV-Tg lineages; Jean-Noel Billaud for the WHc-TT construct; John Gerin for the purified HBV preparation; Wolfram Gerlich for providing the 18/7 Mab; and Paul Pockros for helpful discussion of clinical applications for the PreS1-WHc VLPs. This work was supported by NIH grants R01 AI049730 and R44 AI08819 to DM; grants from Swedish Science Council, Cancer Foundation, and ALF grant from Stockholm County Council to MS and LF; and a grant from ANRS (French National Agency for Research on AIDS and Viral Hepatitis) to CS.
Disclosure of potential conflicts of interest
No potential conflict of interest.
References
- 1.Stevens CE, Beasley RP, Tsui J, Lee WC.. Vertical transmission of hepatitis B antigen in Taiwan. N Engl J Med. 1975;292:771–74. doi: 10.1056/NEJM197504102921503. [DOI] [PubMed] [Google Scholar]
- 2.Lok AS-F. The maze of treatments for hepatitis B. N Engl J Med. 2005;352:2743–46. doi: 10.1056/NEJMe058119. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Dong Q, Li Q, Li Y, Zhao D, Sun J, Fu J, Meng F, Lin H, Luan J, et al. Dysregulated response of follicular helper T cells to hepatitis B surface antigen promotes HBV persistence in mice and associates with outcomes of patients. Gastroenterology. 2018;154:2222–36. doi: 10.1053/j.gastro.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Zhang M, Li H, Chen Z, Luo A, Liu B, Chen M, Peng M, Ren H, Hu P. Tfh cell-mediated humoral immune response and HBsAg level can predict HBeAg seroconversion in chronic hepatitis B patients receiving peginterferon-α therapy. Mol Immunol. 2016;73:37–45. doi: 10.1016/j.molimm.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Park -J-J, Wong DK, Wahed AS, Lee WM, Feld JJ, Terrault N, Khalili M, Sterling RK, Kowdley KV, Bzowej N, et al. Hepatitis B virus–specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology. 2016;150:684–695.e5. doi: 10.1053/j.gastro.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, Alberts E, Davidson BR, Kennedy PT, Gill US, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest. 2018;128:4588–603. doi: 10.1172/JCI121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salimzadeh L, Le Bert N, Dutertre C-A, Gill US, Newell EW, Frey C, Hung M, Novikov N, Fletcher S, Kennedy PT, et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J Clin Invest. 2018;128:4573–87. doi: 10.1172/JCI121957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian Y, Kuo C-F, Akbari O, Ou J-HJ. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity. 2016;44:1204–14. doi: 10.1016/j.immuni.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milich DR. The concept of immune tolerance in chronic hepatitis B virus infection is alive and well. Gastroenterology. 2016;151:801–04. doi: 10.1053/j.gastro.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Protzer U, Knolle P. “To Be or Not to Be”: immune tolerance in chronic hepatitis B. Gastroenterology. 2016;151:805–06. doi: 10.1053/j.gastro.2016.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Cote PJ, Korba BE, Miller RH, Jacob JR, Baldwin BH, Hornbuckle WE, Purcell RH, Tennant BC, Gerin JL. Effects of age and viral determinants on chronicity as an outcome of experimental woodchuck hepatitis virus infection. Hepatol Baltim Md. 2000;31:190–200. doi: 10.1002/(ISSN)1527-3350. [DOI] [PubMed] [Google Scholar]
- 12.Ma Z, Zhang E, Gao S, Xiong Y, Lu M. Toward a functional cure for hepatitis B: the rationale and challenges for therapeutic targeting of the B cell immune response. Front Immunol. 2019;10:2308. doi: 10.3389/fimmu.2019.02308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Bert N, Salimzadeh L, Gill US, Dutertre C-A, Facchetti F, Tan A, Hung M, Novikov N, Lampertico P, Fletcher SP, et al. Comparative characterization of B cells specific for HBV nucleocapsid and envelope proteins in patients with chronic hepatitis B. J Hepatol. 2019. doi: 10.1016/j.jhep.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Poonia B, Ayithan N, Nandi M, Masur H, Kottilil S. HBV induces inhibitory FcRL receptor on B cells and dysregulates B cell-T follicular helper cell axis. Sci Rep. 2018;8:15296. doi: 10.1038/s41598-018-33719-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: from discovery to regulatory approval. Hepatol Baltim Md. 2017;66:1296–313. doi: 10.1002/hep.29323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, Alexandrov A, Urban S, Petersen J, Lütgehetmann M, et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58:861–67. doi: 10.1016/j.jhep.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Michel M-L, Deng Q, Mancini-Bourgine M. Therapeutic vaccines and immune-based therapies for the treatment of chronic hepatitis B: perspectives and challenges. J Hepatol. 2011;54:1286–96. doi: 10.1016/j.jhep.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 18.Lim SG, Agcaoili J, De Souza NNA, Chan E. Therapeutic vaccination for chronic hepatitis B: A systematic review and meta-analysis. J Viral Hepat. 2019;26:803–17. doi: 10.1111/jvh.13085. [DOI] [PubMed] [Google Scholar]
- 19.Webster GJM, Reignat S, Brown D, Ogg GS, Jones L, Seneviratne SL, Williams R, Dusheiko G, Bertoletti A. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol. 2004;78:5707–19. doi: 10.1128/JVI.78.11.5707-5719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivino L, Le Bert N, Gill US, Kunasegaran K, Cheng Y, Tan DZ, Becht E, Hansi NK, Foster GR, Su T-H, et al. Hepatitis B virus-specific T cells associate with viral control upon nucleos(t)ide-analogue therapy discontinuation. J Clin Invest. 2018;128:668–81. doi: 10.1172/JCI92812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neurath AR, Seto B, Strick N. Antibodies to synthetic peptides from the preS1 region of the hepatitis B virus (HBV) envelope (env) protein are virus-neutralizing and protective. Vaccine. 1989;7:234–36. doi: 10.1016/0264-410X(89)90235-1. [DOI] [PubMed] [Google Scholar]
- 22.Neurath AR, Kent SB, Strick N, Parker K, Courouce AM, Riottot MM, Petit MA, Budkowska A, Girard M, Pillot J. Antibodies to synthetic peptides from the pre-S1 and pre-S2 regions of one subtype of the hepatitis B virus (HBV) envelope protein recognize all HBV subtypes. Mol Immunol. 1987;24:975–80. doi: 10.1016/0161-5890(87)90009-5. [DOI] [PubMed] [Google Scholar]
- 23.Thornton GB, Moriarty AM, Milich DR, Eichberg JW, Purcell RH, Gerin JL. Protection of chimpanzees from hepatitis-B virus infection after immunization with synthetic peptides: identification of protective epitopes in the pre-S region. In: Lerner R, Ginsberg H, Chanock R, Brown F editors. Modern approaches to new vaccines including prevention of AIDS. New York: Cold Spring Harbor Laboratory Publications; 1989. p. 467–71. [Google Scholar]
- 24.Niedre-Otomere B, Bogdanova A, Skrastina D, Zajakina A, Bruvere R, Ose V, Gerlich WH, Garoff H, Pumpens P, Glebe D, et al. Recombinant Semliki Forest virus vectors encoding hepatitis B virus small surface and pre-S1 antigens induce broadly reactive neutralizing antibodies. J Viral Hepat. 2012;19:664–73. doi: 10.1111/jvh.2012.19.issue-9. [DOI] [PubMed] [Google Scholar]
- 25.Milich DR, Thornton GB, Neurath AR, Kent SB, Michel ML, Tiollais P, Chisari FV. Enhanced immunogenicity of the pre-S region of hepatitis B surface antigen. Science. 1985;228:1195–99. doi: 10.1126/science.2408336. [DOI] [PubMed] [Google Scholar]
- 26.Budkowska A, Dubreuil P, Poynard T, Marcellin P, Loriot MA, Maillard P, Pillot J. Anti-pre-S responses and viral clearance in chronic hepatitis B virus infection. Hepatology. 1992;15:26–31. doi: 10.1002/hep.1840150106. [DOI] [PubMed] [Google Scholar]
- 27.Schickli JH, Whitacre DC, Tang RS, Kaur J, Lawlor H, Peters CJ, Jones JE, Peterson DL, McCarthy MP, Van Nest G, et al. Palivizumab epitope-displaying virus-like particles protect rodents from RSV challenge. J Clin Invest. 2015;125:1637–47. doi: 10.1172/JCI78450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitacre DC, Espinosa DA, Peters CJ, Jones JE, Tucker AE, Peterson DL, Zavala FP, Milich DR. P. falciparum and P. vivax epitope-focused VLPs elicit sterile immunity to blood stage infections. PLoS One. 2015;10:e0124856. doi: 10.1371/journal.pone.0124856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Billaud J-N, Peterson D, Lee BO, Maruyama T, Chen A, Sallberg M, Garduño F, Goldstein P, Hughes J, Jones J, et al. Advantages to the use of rodent hepadnavirus core proteins as vaccine platforms. Vaccine. 2007;25:1593–606. doi: 10.1016/j.vaccine.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Billaud J-N, Peterson D, Schödel F, Chen A, Sallberg M, Garduno F, Goldstein P, McDowell W, Hughes J, Jones J, et al. Comparative antigenicity and immunogenicity of hepadnavirus core proteins. J Virol. 2005;79:13641–55. doi: 10.1128/JVI.79.21.13641-13655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ameiss K, Ashraf S, Kong W, Pekosz A, Wu W-H, Milich D, Billaud J-N, Curtiss R. Delivery of woodchuck hepatitis virus-like particle presented influenza M2e by recombinant attenuated Salmonella displaying a delayed lysis phenotype. Vaccine. 2010;28:6704–13. doi: 10.1016/j.vaccine.2010.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milich DR, McLachlan A, Thornton GB, Hughes JL. Antibody production to the nucleocapsid and envelope of the hepatitis B virus primed by a single synthetic T cell site. Nature. 1987;329:547–49. doi: 10.1038/329547a0. [DOI] [PubMed] [Google Scholar]
- 33.Bachmann MF, Hunziker L, Zinkernagel RM, Storni T, Kopf M. Maintenance of memory CTL responses by T helper cells and CD40-CD40 ligand: antibodies provide the key. Eur J Immunol. 2004;34:317–26. doi: 10.1002/(ISSN)1521-4141. [DOI] [PubMed] [Google Scholar]
- 34.Li D, He W, Liu X, Zheng S, Qi Y, Li H, Mao F, Liu J, Sun Y, Pan L, et al. A potent human neutralizing antibody Fc-dependently reduces established HBV infections. eLife. 2017;6. doi: 10.7554/eLife.26738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halper-Stromberg A, Lu C-L, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158:989–99. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul S, Dickstein A, Saxena A, Terrin N, Viveiros K, Balk EM, Wong JB. Role of surface antibody in hepatitis B reactivation in patients with resolved infection and hematologic malignancy: A meta-analysis. Hepatology. 2017;66:379–88. doi: 10.1002/hep.v66.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bian Y, Zhang Z, Sun Z, Zhao J, Zhu D, Wang Y, Fu S, Guo J, Liu L, Su L, et al. Vaccines targeting preS1 domain overcome immune tolerance in hepatitis B virus carrier mice. Hepatology. 2017;66:1067–82. doi: 10.1002/hep.29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, Lehr T, Lempp FA, Wedemeyer H, Haag M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. 2016;65:490–98. doi: 10.1016/j.jhep.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 39.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 40.Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, Herberg J, Gilson R, Alisa A, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–80. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinaa L, Rasmussen PB, Rygaard J, Mouritsen S, Gautam A. Generation of autoreactive CTL by tumour vaccines containing foreign T helper epitopes. Scand J Immunol. 2007;65:240–48. doi: 10.1111/sji.2007.65.issue-3. [DOI] [PubMed] [Google Scholar]
- 42.Shimizu K, Thomas EK, Giedlin M, Mulé JJ. Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res. 2001;61:2618–24. [PubMed] [Google Scholar]
- 43.de Goër de Herve M-G, Cariou A, Simonetta F, Taoufik Y. Heterospecific CD4 help to rescue CD8 T cell killers. J Immunol Baltim Md 1950. 2008;181:5974–80. [DOI] [PubMed] [Google Scholar]
- 44.Steinaa L, Rasmussen PB, Gautam A, Mouritsen S. Breaking B-cell tolerance and CTL tolerance in three OVA-transgenic mouse strains expressing different levels of OVA. Scand J Immunol. 2008;67:113–20. doi: 10.1111/sji.2008.67.issue-2. [DOI] [PubMed] [Google Scholar]
- 45.Lee BO, Tucker A, Frelin L, Sallberg M, Jones J, Peters C, Hughes J, Whitacre D, Darsow B, Peterson DL, et al. Interaction of the hepatitis B core antigen and the innate immune system. J Immunol. 2009;182:6670–81. doi: 10.4049/jimmunol.0803683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen MT, Billaud J-N, Sällberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A. 2004;101:14913–18. doi: 10.1073/pnas.0406282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guidotti LG, Matzke B, Pasquinelli C, Shoenberger JM, Rogler CE, Chisari FV. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. J Virol. 1996;70:7056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med. 1998;188:341–50. doi: 10.1084/jem.188.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Billaud J-N, Peterson D, Barr M, Chen A, Sallberg M, Garduno F, Goldstein P, McDowell W, Hughes J, Jones J, et al. Combinatorial approach to hepadnavirus-like particle vaccine design. J Virol. 2005;79:13656–66. doi: 10.1128/JVI.79.21.13656-13666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aida Y, Pabst MJ. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J Immunol Methods. 1990;132:191–95. doi: 10.1016/0022-1759(90)90029-U. [DOI] [PubMed] [Google Scholar]
- 52.Li F, Cowley DO, Banner D, Holle E, Zhang L, Su L. Efficient genetic manipulation of the NOD-Rag1-/-IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep. 2014;4:5290. doi: 10.1038/srep05290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li F, Nio K, Yasui F, Murphy CM, Su L. Studying HBV infection and therapy in immune-deficient NOD-Rag1-/-IL2RgammaC-null (NRG) Fumarylacetoacetate Hydrolase (Fah) knockout mice transplanted with human hepatocytes. Methods Mol Biol Clifton NJ. 2017;1540:267–76. [DOI] [PubMed] [Google Scholar]
- 54.Blanchet M, Sureau C. Analysis of the cytosolic domains of the hepatitis B virus envelope proteins for their function in viral particle assembly and infectivity. J Virol. 2006;80:11935–45. doi: 10.1128/JVI.00621-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milich DR, McLachlan A, Chisari FV, Kent SB, Thorton GB. Immune response to the pre-S(1) region of the hepatitis B surface antigen (HBsAg): a pre-S(1)-specific T cell response can bypass nonresponsiveness to the pre-S(2) and S regions of HBsAg. J Immunol. 1986;137:315–22. [PubMed] [Google Scholar]
- 56.Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large surface proteins of hepatitis B virus containing the pre-s sequence. J Virol. 1984;52:396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alberti A, Cavalletto D, Chemello L, Belussi F, Fattovich G, Pontisso P, Milanesi G, Ruol A. Fine specificity of human antibody response to the PreS1 domain of hepatitis B virus. Hepatology. 1990;12:199–203. doi: 10.1002/(ISSN)1527-3350. [DOI] [PubMed] [Google Scholar]
- 58.Barrera A, Guerra B, Notvall L, Lanford RE. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J Virol. 2005;79:9786–98. doi: 10.1128/JVI.79.15.9786-9798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glebe D, Urban S, Knoop EV, Cag N, Krass P, Grün S, Bulavaite A, Sasnauskas K, Gerlich WH. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology. 2005;129:234–45. doi: 10.1053/j.gastro.2005.03.090. [DOI] [PubMed] [Google Scholar]
- 60.Neurath AR, Kent SB, Strick N, Parker K. Identification and chemical synthesis of a host cell receptor binding site on hepatitis B virus. Cell. 1986;46:429–36. doi: 10.1016/0092-8674(86)90663-X. [DOI] [PubMed] [Google Scholar]
- 61.Bremer CM, Sominskaya I, Skrastina D, Pumpens P, El Wahed AA, Beutling U, Frank R, Fritz H-J, Hunsmann G, Gerlich WH, et al. N-terminal myristoylation-dependent masking of neutralizing epitopes in the preS1 attachment site of hepatitis B virus. J Hepatol. 2011;55:29–37. doi: 10.1016/j.jhep.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 62.Chen X, Li M, Le X, Ma W, Zhou B. Recombinant hepatitis B core antigen carrying preS1 epitopes induce immune response against chronic HBV infection. Vaccine. 2004;22:439–46. doi: 10.1016/j.vaccine.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 63.Frelin L, Wahlström T, Tucker AE, Jones J, Hughes J, Lee BO, Billaud J-N, Peters C, Whitacre D, Peterson D, et al. A mechanism to explain the selection of the hepatitis e antigen-negative mutant during chronic hepatitis B virus infection. J Virol. 2009;83:1379–92. doi: 10.1128/JVI.01902-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milich DR, McLachlan A. The nucleocapsid of hepatitis B virus is both a T-cell-independent and a T-cell-dependent antigen. Science. 1986;234:1398–401. doi: 10.1126/science.3491425. [DOI] [PubMed] [Google Scholar]
- 65.Kakimi K, Isogawa M, Chung J, Sette A, Chisari FV. Immunogenicity and tolerogenicity of hepatitis B virus structural and nonstructural proteins: implications for immunotherapy of persistent viral infections. J Virol. 2002;76:8609–20. doi: 10.1128/JVI.76.17.8609-8620.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maruyama T, McLachlan A, Iino S, Koike K, Kurokawa K, Milich DR. The serology of chronic hepatitis B infection revisited. J Clin Invest. 1993;91:2586–95. doi: 10.1172/JCI116497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bility MT, Zhang L, Washburn ML, Curtis TA, Kovalev GI, Su L. Generation of a humanized mouse model with both human immune system and liver cells to model hepatitis C virus infection and liver immunopathogenesis. Nat Protoc. 2012;7:1608–17. doi: 10.1038/nprot.2012.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, Zhang L, Tu Z, Gao Y, Fu Y, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014;10:e1004032. doi: 10.1371/journal.ppat.1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen M, Sällberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud J-N, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol. 2005;79:3016–27. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milich DR, Jones JE, McLachlan A, Houghten R, Thornton GB, Hughes JL. Distinction between immunogenicity and tolerogenicity among HBcAg T cell determinants. Influence of peptide-MHC interaction. J Immunol Baltim Md 1950. 1989;143:3148–56. [PubMed] [Google Scholar]
- 71.Milich DR, McLachlan A, Raney AK, Houghten R, Thornton GB, Maruyama T, Hughes JL, Jones JE. Autoantibody production in hepatitis B e antigen transgenic mice elicited with a self T-cell peptide and inhibited with nonself peptides. Proc Natl Acad Sci U S A. 1991;88:4348–52. doi: 10.1073/pnas.88.10.4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zinkernagel RM, Pircher HP, Ohashi P, Oehen S, Odermatt B, Mak T, Arnheiter H, Bürki K, Hengartner H. T and B cell tolerance and responses to viral antigens in transgenic mice: implications for the pathogenesis of autoimmune versus immunopathological disease. Immunol Rev. 1991;122:133–71. doi: 10.1111/imr.1991.122.issue-1. [DOI] [PubMed] [Google Scholar]
- 73.Schuch A, Salimi Alizei E, Heim K, Wieland D, Kiraithe MM, Kemming J, Llewellyn-Lacey S, Sogukpinar Ö, Ni Y, Urban S, et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut. 2019;68:905–15. doi: 10.1136/gutjnl-2018-316641. [DOI] [PubMed] [Google Scholar]
- 74.Kosinska AD, Moeed A, Kallin N, Festag J, Su J, Steiger K, Michel M-L, Protzer U, Knolle PA. Synergy of therapeutic heterologous prime-boost hepatitis B vaccination with CpG-application to improve immune control of persistent HBV infection. Sci Rep. 2019;9:10808. doi: 10.1038/s41598-019-47149-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo J-T, Locarnini S, Zoulim F, Chang K-M, Lok AS. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62:1893–908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shouval D, Roggendorf H, Roggendorf M. Enhanced immune response to hepatitis B vaccination through immunization with a Pre-S1/Pre-S2/S vaccine. Med Microbiol Immunol (Berl). 2015;204:57–68. doi: 10.1007/s00430-014-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoa PTL, Huy NT, Thu LT, Nga CN, Nakao K, Eguchi K, Chi NH, Hoang BH, Hirayama K. Randomized controlled study investigating viral suppression and serological response following pre-S1/pre-S2/S vaccine therapy combined with lamivudine treatment in HBeAg-positive patients with chronic hepatitis B. Antimicrob Agents Chemother. 2009;53:5134–40. doi: 10.1128/AAC.00276-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wisskirchen K, Kah J, Malo A, Asen T, Volz T, Allweiss L, Wettengel JM, Lütgehetmann M, Urban S, Bauer T, et al. T cell receptor grafting allows virological control of Hepatitis B virus infection. J Clin Invest. 2019;130:2932–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koh S, Kah J, Tham CYL, Yang N, Ceccarello E, Chia A, Chen M, Khakpoor A, Pavesi A, Tan AT, et al. Nonlytic lymphocytes engineered to express virus-specific T-cell receptors limit HBV infection by activating APOBEC3. Gastroenterology. 2018;155:180–193.e6. doi: 10.1053/j.gastro.2018.03.027. [DOI] [PubMed] [Google Scholar]
- 80.Qasim W, Brunetto M, Gehring AJ, Xue S-A, Schurich A, Khakpoor A, Zhan H, Ciccorossi P, Gilmour K, Cavallone D, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol. 2015;62:486–91. doi: 10.1016/j.jhep.2014.10.001. [DOI] [PubMed] [Google Scholar]