

The expression of expanded CUG-repeat RNAs in myotonic dystrophy type 1 patients is associated with cardiac electrophysiological defects and an elevated risk of sudden cardiac death. Tylock et al. reveal that expression of these RNAs in the murine heart results in prolonged QRS and corrected QT-intervals due to alterations in sodium and potassium channel activity.
Abstract
Myotonic dystrophy type 1 (DM1), the most common form of muscular dystrophy in adults, results from the expression of toxic gain-of-function transcripts containing expanded CUG-repeats. DM1 patients experience cardiac electrophysiological defects, including prolonged PR-, QRS-, and QT-intervals, that increase susceptibility to sudden cardiac death (SCD). However, the specific biophysical and molecular mechanisms that underlie the electrocardiograph (ECG) abnormalities and SCD in DM1 are unclear. Here, we addressed this issue using a novel transgenic mouse model that exhibits robust cardiac expression of expanded CUG-repeat RNA (LC15 mice). ECG measurements in conscious LC15 mice revealed significantly prolonged QRS- and corrected QT-intervals, but a normal PR-interval. Although spontaneous arrhythmias were not observed in conscious LC15 mice under nonchallenged conditions, acute administration of the sodium channel blocker flecainide prolonged the QRS-interval and unveiled an increased susceptibility to lethal ventricular arrhythmias. Current clamp measurements in ventricular myocytes from LC15 mice revealed significantly reduced action potential upstroke velocity at physiological pacing (9 Hz) and prolonged action potential duration at all stimulation rates (1–9 Hz). Voltage clamp experiments revealed significant rightward shifts in the voltage dependence of sodium channel activation and steady-state inactivation, as well as a marked reduction in outward potassium current density. Together, these findings indicate that expression of expanded CUG-repeat RNA in the murine heart results in reduced sodium and potassium channel activity that results in QRS- and QT-interval prolongation, respectively.
Introduction
With an estimated prevalence of 1:7,400, myotonic dystrophy type 1 (DM1) is the most common form of adult muscular dystrophy (Harper, 2001). DM1 patients carry an autosomal dominant mutation in the dystrophia myotonica protein kinase (DMPK) gene, which leads to the unstable expansion of a (CTG)n-repeat tract (n > 50) in the 3′-untranslated region (Mahadevan et al., 1992). Importantly, transcription of the mutant DMPK gene results in the expression of a messenger RNA (mRNA) containing expanded trinucleotide CUG-repeats (Brook et al., 1992; Fu et al., 1992). Toxic gain-of-function effects produced by the mRNA containing expanded CUG-repeats (or CUGexp RNA) are a major pathogenic mechanism for the phenotypes associated with DM1 (Thornton, 2014).
DM1 is characterized by abnormal regulation of alternative splicing and alternative polyadenylation in muscle (Ho et al., 2004; Batra et al., 2014). The presence of toxic CUGexp RNA leads to (1) nuclear retention and aggregation of mutant mRNA transcripts (nuclear foci; Taneja et al., 1995), (2) sequestration in nuclear foci containing splicing cofactors called muscleblind-like (Mbnl) proteins (Miller et al., 2000; Lin et al., 2006), (3) Mbnl-dependent changes of alternative polyadenylation, mRNA transport, and pre–micro-RNA 1 processing (Rau et al., 2011; Wang et al., 2012; Batra et al., 2014), and (4) altered splicing for Mbnl1-dependent transcripts that typically reflect embryonic splicing patterns (Philips et al., 1998; Ho et al., 2004; Jiang et al., 2004; Lin et al., 2006; Wang et al., 2012). Thus, adult DM1 patients express a subset of embryonic mRNA splice isoforms in tissues where the DMPK gene is expressed, which results in pathogenic outcomes when the embryonic splice isoform does not adequately support function in adult tissue (Savkur et al., 2001; Charlet-B et al., 2002; Mankodi et al., 2002; Dansithong et al., 2005). As one clear example, embryonic splicing of pre-mRNA for the skeletal muscle chloride channel (CLC1) results in a transcript that undergoes nonsense-mediated decay, a marked reduction in muscle chloride conductance, hyperexcitability, and myotonia (Mankodi et al., 2002; Lueck et al., 2007a; Lueck et al., 2007b; Wheeler et al., 2007).
Since the DMPK transcript is expressed in a wide range of tissues, DM1 patients exhibit multisystemic symptoms. In fact, DMPK expression is highest in the heart (Maeda et al., 1995; Sarkar et al., 2004). As a consequence, ≥65% of DM1 patients develop cardiac conduction abnormalities, including PR-interval and QRS-interval prolongation, as well as QT-interval prolongation as observed on surface electrocardiograms (ECGs; Phillips and Harper, 1997; Groh et al., 2008; Wahbi et al., 2012; Park et al., 2013). Clinical studies have correlated age, larger CTG expansion size, and degrees of PR- and QRS-interval prolongation with risk of developing high-grade atrioventricular (AV) block, bradycardia, and asystole. As a result, at-risk DM1 patients are often provided implanted pacemakers (Groh et al., 2002; Groh et al., 2008; Nazarian et al., 2011). DM1 patients experience an increased incidence of premature death, due in part to sudden cardiac death (SCD; de Die-Smulders et al., 1998; Mathieu et al., 1999; Groh et al., 2008; Chong-Nguyen et al., 2017), even in patients with pacemakers (Laurent et al., 2011; Chong-Nguyen et al., 2017; Wahbi et al., 2017). In spite of this, little is known regarding the precise biophysical and molecular mechanisms that underlie the ECG defects observed in DM1 patients and the specific pathogenic role of CUGexp RNA in the heart.
Pre-mRNA of the α1-subunit of the cardiac sodium channel (encoded by the SCN5A gene) undergoes a developmentally regulated splicing event that involves selection between two alternative forms of exon 6 (fetal exon 6A versus adult exon 6b; Ou et al., 2005). The two exons differ by 31 nucleotides, resulting in a 7–amino acid difference within transmembrane segment 3 and the 3–4 linker of domain I of the channel. The fetal isoform results in a loss of function due to a depolarizing shift in the voltage dependence of activation, slowed activation/inactivation kinetics, and slower recovery from inactivation (Onkal et al., 2008). The fetal isoform is increased in hearts of DM1 patients (Wahbi et al., 2013; Freyermuth et al., 2016; Pang et al., 2018), and mice with forced expression of the fetal Scn5a isoform exhibit prolonged PR- and QRS-intervals and AV conduction defects (Freyermuth et al., 2016). However, the direct cause of SCD and the potential for phenotype reversibility by correcting SCN5A splicing have yet to be demonstrated. Moreover, SCN5A mis-splicing is unlikely to account for the entirety of the electrophysiological defects observed in DM1 patients since increased expression of the fetal Scn5a isoform does not result in a prolonged QT-interval (Freyermuth et al., 2016; Pang et al., 2018). These gaps in knowledge present a significant obstacle to effective therapeutic intervention. To address these issues, we characterized the ECG abnormalities, as well as ventricular myocyte action potential (AP), sodium channel, and potassium channel properties, using a new transgenic mouse model with high expression of expanded CUG-repeat RNA in the heart (LC15 mice).
Materials and methods
All animal procedures were reviewed and approved by the University Committee on Animal Resources at the University of Rochester. Mice were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Pelleted food and standard HYDROPAC water were provided ad libitum.
Generation of LC15 mice
WT FVB/n breeders were purchased from Taconic Biosciences. Transgenic lines were derived using the construct diagrammed in Fig. 1 A, and maintained on a congenic FVB/n background. The repeat tract was generated using cell-free assembly procedures described previously (Osborne and Thornton, 2008). The uninterrupted CTG repeat was embedded in the natural context of the human DMPK 3′-UTR. Expression of the transcript is driven by a cytomegalovirus (CMV) enhancer/β-actin promoter and contains a luciferase open reading frame. Previous work has shown that the CMV enhancer/β-actin promoter drives ubiquitous expression of transgenes in mice (Hadjantonakis et al., 1998). Transgenic mice in line LC15 exhibit higher constitutive expression of CUGexp RNA in the heart than in skeletal muscle. The integration site is multicopy, such that not all copies contain the same size repeat tract. Lengths of the expanded repeat varied from 250 to 400 repeats, as determined by Southern blot analysis.
Figure 1.

LC15 transgenic mice exhibit robust CUGexp RNA expression in the heart. (A) Schematic of transgene used to generate LC15 mice. An uninterrupted expanded CTG-repeat tract generated using a cell-free assembly procedure described previously (Osborne and Thornton, 2008) was embedded in the natural context of the human Dmpk 3′-UTR. CMV-βA, CMV enhancer fused to chicken β-actin promoter. Luciferase is from Gaussia. (B) Summary of transgene mRNA expression by qRT-PCR (normalized to the housekeeping gene Gtf2b) in heart and quadriceps (Quads) muscle from five LC15 mice. The LC15 line displayed a higher transgene expression in heart (approximately fourfold) than skeletal muscle. (C) Southern blot of genomic DNA from three different LC15 mice (lanes 2–4) and one WT mouse (lane 5) used to determine CTG repeat size. Calibration controls (lane 1) indicate fragment size, expressed as number of CTG repeats. The CTG expansion ranges from 250 to 400 repeats. Note slight instability of the repeat tract in lane 3 compared with lanes 2 and 4. (D) FISH on four representative frozen sections of left ventricle from LC15 mice using a CAG8-Texas Red probe showing foci (red) of CUGexp RNA in nuclei (blue). Control section of WT muscle showed no signal (not shown). Nuclear foci are found in >80% of ventricular myocytes from LC15 mice. (E) Splicing of Sorbs1 exon 23 (top), Cacna1s exon 29 (middle), and Tnnt2 exon 5 (bottom) revert to their respective fetal patterns in hearts from five LC15 adult mice compared with that of two WT mice. Mbnl1−/− mice were used as controls for Mbnl1-dependent splicing.
Quantification of transgene expression
Human DMPK 3′UTR transgene mRNA expression was determined by quantitative (q) reverse transcription (RT)-PCR with the relative expression level in each tissue examined in triplicate and normalized to Gtf2b expression. Five hearts and quadriceps muscles from LC15 mice were examined. Transgene expression was quantified by qPCR using the following primer–probe set: Probe: 5′-/56-FAM/AAGCTTTCTTGTGCATGACGCCC/3IABkFQ/-3′; Primer1: 5′-CTATCGTTGGTTCGCAAAGTG-3′; Primer2: 5′-GCAAATTTCCCGAGTAAGCAG-3′.
A commercially available qPCR primer-probe set was used to quantify endogenous Gtf2b mRNA expression (Mm01323562_g1; Thermo Fisher Scientific) and performed according to manufacturer’s instructions.
Fluorescence in situ hybridization (FISH)
FISH was performed on frozen sections of heart tissue, as described previously (Mankodi et al., 2001). The probe used for FISH was a CAG-repeat 2-O-methyl oligoribonucleotide, 20 nucleotides in length, conjugated at the 3′ end with Texas Red.
RT-PCR splicing assays
Total RNA was isolated from frozen ventricular tissue according to the manufacturer’s instructions (TriZol; Molecular Research Center). Complementary DNA was synthesized using SuperScript II RT (Invitrogen) with oligo(dt) primers. Splicing of Sorbs1, Tnnt2, Scn5a, and Cacna1s were analyzed by RT-PCR using the following primers flanking the alternatively spliced exon of interest: Sorbs1: (forward [fwd]) 5′-FAM-ACCTTCCAGCTCAGCTTCCAC-3′ and (reverse [rev]) 5′-ACTTCTCATCCACCTGTCGGAG-3′;Tnnt2: (fwd) 5′-AGCCGAGAGCATGTCTGACG-3′ and (rev) 5′-FAM-TCTGTCTCAGCCTCACCCTCAG-3′;Scn5a: (fwd) 5′-FAM-CTTCTGCCTGCATGCGTTCAC-3′ and (rev) 5′-GGCCTCCACGGAACCATT-3′;Cacna1s: (fwd) 5′-GCTATTTTGGAGATCCTTGGAA-3′ and (rev) 5′-FAM-AGCAGGGTGCGCACACCCTC-3′.
RT-PCR products were resolved on agarose gels and analyzed using an Amersham Typhoon Gel and Bolt Imaging system. Relative band intensities were measured by densitometry analysis using ImageQuant TL. To measure the relative level of exon 6a inclusion in Scn5a, amplified PCR products were visualized on a 2% agarose TBE gel alongside an aliquot of PCR product digested to completion with restriction enzyme Apa1 (16 h at 37°C) in order to cleave any product containing exon 6B.
Echocardiography
Echocardiography was performed by the Mouse Surgical Core Facility at the University of Rochester Aab Cardiovascular Research Institute. Following removal of chest hair from mice anesthetized with isoflurane (2% mixed with oxygen), 2-D guided M-mode echocardiography was performed at the mid-papillary level (Visualsonics Vevo 2100). The heart rate and internal diameters of the left ventricle (LV) during systole and diastole were measured. These values were used to calculate LV volume, stroke volume, ejection fraction, fractional shortening, cardiac output, and LV mass.
Anesthetized ECG acquisition
Mice were anesthetized with isoflurane as described above and were provided with supplemental heat to maintain a rectal temperature between 36°C and 38°C. Subcutaneous pin electrodes were placed in a lead II configuration and connected to a DP-301 differential amplifier (Warner Instruments). ECGs were recorded continuously for 3–5 min using Axoscope software (pCLAMP 10.0; Molecular Devices). A continuous 3-s stable, baseline recording was taken from the final minute of each recording of each mouse and used to calculate the standard ECG parameters (RR-, PR-, QRS-, and QT-intervals). For experiments assessing the effect of acute administration of flecainide on ECG parameters, 18-mo-old WT and LC15 mice were anesthetized with 2% isoflurane as described above. After obtaining control baseline ECG recordings for 3–5 min, flecainide acetate was injected intraperitoneally (20 mg/kg body weight) while continuously recording ECGs for an additional 5–20 min following drug administration. The relative sensitivity of each mouse for exhibiting an arrhythmia 5 min following flecainide administration was grouped into one of three general categories (mild, moderate, and severe). Mice were described as exhibiting “no or mild sensitivity” if they did not develop a detectable change to ECG waveform morphology that obstructed standard analysis of ECG parameters. Mice were characterized as exhibiting “moderate sensitivity” if they maintained sinus rhythm but showed ECG waveform changes that exhibited overlapping S- and T-waves to yield unreliable determinations of QRS- and/or corrected QT (QTc)-interval duration. Finally, mice were determined to exhibit “severe sensitivity” to flecainide when ECGs revealed dropped ventricular beats, an irregular rhythm, or a heart rate that decreased to <350 beats per minute (bpm).
Surgical implantation of Data Sciences International ETA-F10 telemeters
Radiotelemetry devices (model ETA-F10; Data Sciences International) were implanted subcutaneously in anesthetized mice. To provide adequate pain management, mice were administered analgesics (0.1 mg/kg body weight buprenorphine and 2.5 mg/kg body weight flunazine) 1 hr before surgery and then one to two additional times per day for the next 3–4 d. Following surgery, implanted mice were housed singly for 7 d within the ECG recording room (with a 6 AM to 6 PM light/dark cycle) to allow for full recovery from surgery and acclimation to the recording environment. Mice were checked regularly after surgery and euthanized if they experienced pain/discomfort that could not be managed with analgesics. After mice had recovered from surgery and were off analgesics for 4–5 d, radiotelemeters were turned on using magnets, and conscious ECG data were continuously acquired for 60 h using Ponemah (Data Sciences International) hardware/software.
Calculation of ECG parameters
Blinded analyses of ECG parameters were performed using homemade offline software (previously described; Auerbach et al., 2013) that identifies and aligns all R-peaks (or, alternatively, S-anti-peaks) within a provided range of ECG signals and then averages the signals to generate a representative ECG recording from P- to T-wave. From this signal-averaged trace, PR-, QRS-, and QT-intervals were measured via manual identification of wave markers. QT-intervals were measured from the start of the Q-wave to the T-anti-peak since this is the most reliable way to measure the QT-interval within genetic mouse models (Zhang et al., 2014). To correct for variability in heart rate, QTc-intervals were calculated using the approach of Mitchell et al. (1998): QTc = QT/(RR/100)1/2.
Isolation of single mouse cardiac ventricular myocytes
Cardiac ventricular myocytes were isolated from adult (6–8-mo-old) WT and LC15 transgenic littermate mice on a congenic FVB background. Mice were anesthetized via intraperitoneal injection of 100 mg/kg ketamine, 10 mg/kg xylazine, and 2 mg/kg acepromazine. Anesthetized mice were administered 20 ml of heparin (1,000 USP units per 1 ml; intraperitoneal injection) to prevent blood clotting. Five minutes after heparin administration, the chest cavity was opened by cutting across the abdomen and up both sides. The rib cage and sternum were retracted to expose the heart. The aorta was carefully isolated from the surrounding vasculature with a hooked instrument, and a small incision was made in the ascending aorta. The aorta was cannulated quickly with a blunted 22-gauge needle and anchored in place by a silk suture knot. The heart was then excised from the chest cavity and attached to a Langendorff perfusion system.
The cannulated heart was initially perfused for 5 min at 6 ml/min with 37°C isolation buffer (IB) consisting of 120 mM NaCl, 15 mM KCl, 0.6 mM Na2HPO4, 0.6 mM KH2PO4, 1.2 mM MgSO4, 10 mM HEPES, 4.6 mM NaHCO3, 30 mM taurine, 5.5 mM glucose, and 10 mM 2,3-butanedione monoxime (pH 7.4) at 37°C in order to remove residual blood from the coronary vessels. The heart was then perfused at 4 ml/min with IB supplemented with 12.5 µM CaCl2, 0.025% trypsin, 0.17 U/ml collagenase A, and 0.42 U/ml collagenase D at 37°C for 7–10 min. Following digestion, the atria and aorta were removed, and the ventricular tissue was minced using fine tweezers. For voltage clamp experiments, the ventricular septum was removed, and only the ventricular free wall and apex tissues were used to isolate single myocytes. Myocytes were dissociated from tissue via gentle trituration using a plastic transfer pipette in stop buffer (IB with 10% FBS and 12.5 µM CaCl2), filtered through a 200-µm mesh, and then allowed to equilibrate in stop buffer for 10 min. The resulting pellet was washed once in stop buffer, and then Ca2+ was added back to the solution gradually to a final concentration of 1 mM. The myocytes were then sedimented by gravity for a final 10 min. The supernatant was removed and the myocyte pellet resuspended in MEM culture media supplemented with 10% FBS, 1% penicillin and streptomycin, and 10 µM blebbistatin to inhibit spontaneous contractions. Myocytes were plated on laminin-treated coverslips and allowed to equilibrate for ≥1 h in an incubator at 37°C with 5% CO2.
Current and voltage clamp protocols and solutions
Electrical properties were measured in whole-cell current and voltage clamp conditions in myocytes using glass micropipettes (1–3 MΩ resistance), an Axopatch 200B amplifier, Digidata 1320A digitizer, and pCLAMP 8.2 data acquisition/analysis software (Molecular Devices). Current clamp data were acquired at 20 kHz and filtered at 10 kHz; voltage clamp data were acquired at 10 kHz and filtered at 5 kHz (K currents) or 2 kHz (Na currents). The internal (pipette) and extracellular solutions are provided below. After gigaohm seal formation, the patch was ruptured with mild suction, and the myocyte was allowed to equilibrate with the pipette internal solution before initiating data acquisition.
For current clamp measurement of APs, the internal solution included 148 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM HEPES, 2 mM creatine, 5 mM K2-ATP, and 5 mM phosphocreatine (pH 7.2) with KOH. The extracellular recording solution for these experiments included 148 mM NaCl, 5.4 mM KCl, 0.4 mM NaH2PO4, 1 mM CaCl2, 1 mM MgCl2, 5.5 mM glucose, and 15 mM HEPES (pH 7.35) with NaOH.
For voltage clamp measurements of inward sodium currents, the internal solution included 5 mM NaCl, 135 mM CsF, 10 mM EGTA, 5 mM MgATP, and 5 mM HEPES (pH 7.2) at RT. The extracellular recording solution for these experiments included 5 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 0.1 mM CdCl2, 11 mM glucose, 132.5 mM CsCl, and 20 mM HEPES (pH 7.35).
For voltage clamp measurements of outward potassium currents, the internal solution included 135 mM KCl, 1 mM MgCl2, 10 mM EGTA, 10 mM HEPES, and 4 mM K2-ATP (pH 7.2) with KOH. The extracellular recording solution for these experiments included 136 mM NMDG, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, and 5 mM CoCl2 (pH 7.4) with HCl.
Collection and analysis of AP recordings
APs were recorded at 35°C–37°C from ventricular myocytes with membrane capacitances ranging between 100 and 200 pF. No holding current was applied to alter the resting membrane potential. Under current clamp, increasing levels of current were injected starting at 2 nA (Δ 0.1 nA steps, 0.3 ms) until an AP threshold was reached. Once the threshold was determined, 110% of the current necessary to elicit an AP was delivered 20 consecutive times at pacing cycle lengths (PCLs) ranging from 111 (9 Hz) to 1,000 ms (1 Hz). Only myocytes that exhibited no more than two dropped beats for every PCL and a change in voltage >90 mV from the holding potential to the peak of the AP were included for subsequent analyses. In addition, all myocytes exhibiting low excitability (i.e., injected current >7.5 nA) were discarded from further analysis. AP parameters, including AP duration (APD; APD-25%, APD-75%, and APD-90% repolarization), upstroke velocity (dV/dt), resting membrane potential, and peak change in voltage were quantified using custom AP analysis software (National Instruments LabView; Auerbach et al., 2013). The software analyzed all 20 APs during a train, and average values for a given PCL were calculated in Microsoft Excel. The first AP of each train was discarded from analysis as the PCL for that AP was technically undefined.
Collection and analysis of Na+ currents
Voltage clamp recordings of inward sodium currents were performed at room temperature with 5 mM [Na+] in the external recording solution. Voltage clamp data were acquired at 10 kHz and filtered at 2 kHz. Upon establishing whole-cell configuration, the capacitance and series resistance compensation were optimized (>70% compensation) while the cell was held at −120 mV. Current amplitudes were normalized to cell capacitance (pA/pF). P/N leak subtraction (eight subpulses of opposite polarity to waveform) was executed before each sweep.
To measure sodium current amplitude and voltage dependence of activation, myocytes were stepped for 300 ms from a holding potential of −120 mV to test potentials ranging from −150 mV to +60 mV (in 5-mV increments). Steady-state inactivation was measured using a brief (30-ms) step to −40 mV immediately following a 300-ms depolarization to potentials ranging from −150 mV to +60 mV (in 5-mV increments). In all cases, individual voltage steps were delivered at 5.5-s intervals to allow for complete recovery from inactivation. The normalized voltage dependence of channel conductance and availability were fit to modified Boltzmann functions.
Collection and analysis of K+ currents
Total outward voltage-gated K+ currents were measured at 35°–37°C in whole-cell voltage clamp experiments. Cell capacitance and series resistance compensation were optimized (>80% compensation) while the cell was depolarized in 10-mV pulses from a −70-mV holding potential. Depolarizing voltage steps used to elicit outward K+ currents consisted of 4.5-s depolarizing test pulses to potentials from −40 to +50 mV in 10-mV increments delivered every 10 s from a holding potential of −70 mV. Peak and steady-state outward currents at each test potential were normalized to cell capacitance (pA/pF) analyzed using Clampfit 10.4 software.
Kinetic analysis of outward K+ current decay
K+ current traces elicited during depolarization to +40 mV as described above were decimated 40-fold and then the decay phase of the current was fitted according to the following double exponential function (Chebyshev method):
where (t) is the current at time t after the peak of the current during depolarization, A1 and A2 are amplitudes for fast and slow decay components with their respective time constants (τ1 and τ1, respectively), and C represents the steady-state current level at the end of the test pulse.
4-Aminopyridine (4-AP)–sensitive K+ currents
To determine the sensitivity of the different kinetic components of the outward K+ current to block by 4-AP, recording solutions containing low (50-µM) and high (500-µM) concentrations of 4-AP were sequentially added to the bath. After recording baseline outward K+ currents, an external solution containing 50 µM 4-AP was perfused into the chamber for 2 min, after which the voltage clamp protocol was completed a second time. The external solution was subsequently replaced with a solution containing 500 µM 4-AP, and the voltage clamp protocol was completed a third time. K+ current traces recorded in the presence of 500 µM 4-AP were digitally subtracted from traces recorded in the presence of 50 µM 4-AP in order to generate currents sensitive to a high concentration of 4-AP. Peak 4-AP–sensitive K+ currents were then quantified as described above. In addition, the integral, or area under the curve, of 4-AP-sensitive K+ currents was quantified for the 200 ms following the peak of the current.
Statistical analyses
All results are reported as mean ± SEM. Statistical significance was determined using Student’s t test when comparing two groups, one-way ANOVA followed by post hoc Tukey test when making multiple comparisons, or the χ2 test when analyzing categorical data. In all cases, differences were considered to be statistically significant at P < 0.05.
Online supplemental material
Fig. S1 presents Mbnl1 and Mbnl2 expression levels in hearts of WT and LC15 mice. Fig. S2 shows levels of Scn5a exon 6a (fetal) and 6b (adult) mRNA splice isoforms in hearts of WT, Mbnl1−/−, and LC15 mice. Fig. S3 compares Kv1.4 protein expression levels in ventricular myocytes from WT and LC15 mice.
Figure S1.

Mbnl1 and Mbnl2 expression levels in hearts from LC15 mice. (A) Representative Western blot of Mbnl1 (top) and gapdh (bottom) in lysates from ventricular myocytes from WT and LC15 mice. (B) Relative Mbnl1 band intensity (normalized to gapdh) for lysates from ventricular myocytes isolated from WT (n = 4; closed circles) or LC15 (n = 3; open circles) mice. (C) Representative Western blot of Mbnl2 (top) and gapdh (bottom) in lysates from ventricular myocytes isolated from WT and LC15 mice. The blot was stripped and then probed a second time with anti-gapdh. (D) Relative Mbnl2 band intensity (normalized to gapdh) for lysates from ventricular myocytes isolated from WT (n = 4; closed circles) or LC15 (n = 4; open circles) mice. Error bars represent SEM. *, P < 0.05.
Figure S2.
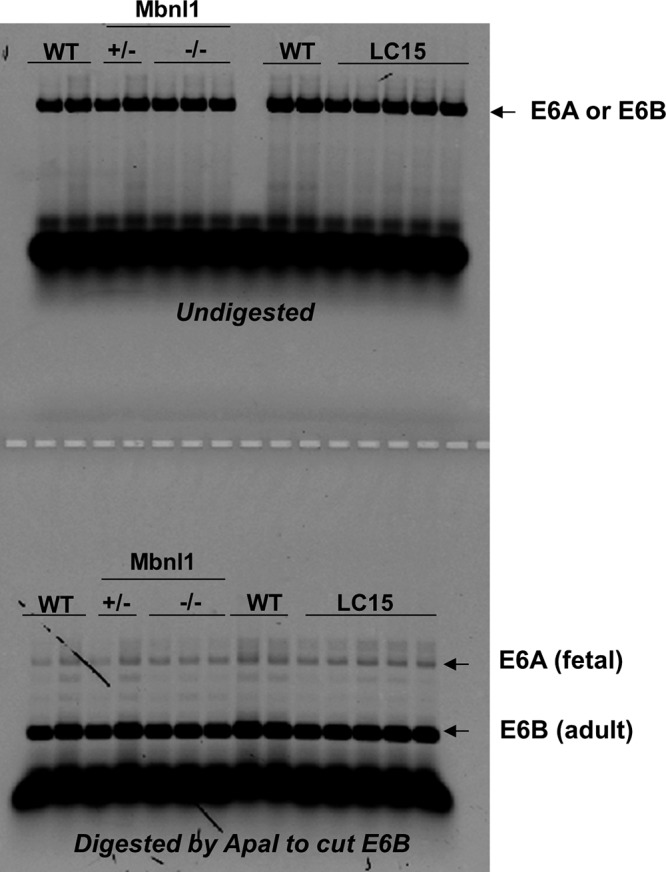
Splicing of fetal Scn5a exon 6A is not increased in heart tissue from LC15 mice. Mutually exclusive Scn5a exon 6A/B expression was measured by PCR using complementary DNA made from total mRNA isolated from adult mouse heart tissue. PCR product containing the exon 6B splice variant was sensitive to restriction enzyme cleavage. Thus, the upper band remaining after digestion reflects the fraction of Scn5a exon 6A transcript, corresponding to the fetal isoform of NaV1.5. Densitometric analysis of the exon 6A and 6b bands revealed a similarly modest, but statistically significant (P < 0.05), increase in exon 6A inclusion in hearts from LC15 (1.2% ± 0.1%; n = 5) and Mbnl1−/− (1.2% ± 0.1%; n = 3) mice compared with that of WT (0.6% ± 0.1%; n = 3) mice.
Figure S3.

Kv1.4 protein expression is reduced in ventricular myocytes from LC15 mice. (A) Representative Western blot of Kv1.4 (APC-007 antibody, Alomone) in lysates from ventricular myocytes isolated from WT and LC15 mice (top). A separate Western blot was performed using anti-gapdh on the lower molecular weight half of the blotting membrane (bottom). (B) Relative Kv1.4 band intensity (normalized to gapdh) in myocytes adult WT (n = 3 mice; closed circle) and LC15 (n = 4 mice; open circle) mice. Error bars represent SEM. *, P < 0.05.
Results
Expression of CUGexp RNA in the heart results in formation of nuclear foci, RNA mis-splicing, and upregulation of Mbnl2
To model DM1-associated RNA toxicity in the heart, mice expressing CUGexp RNA were generated using the transgene diagrammed in Fig. 1 A. We selected a transgenic line, LC15, that exhibits high CUGexp RNA expression in cardiac tissue, as evidenced by qRT-PCR. Transgene expression was approximately fourfold higher in the heart compared with that of the quadriceps muscle (Fig. 1 B). Although the construct used to generate these mice contained ~400 uninterrupted CTG-repeats, LC15 mice harbored a multicopy integration (estimated at four copies) having ~250–400 repeats per copy, as determined by Southern blot analysis of genomic DNA (Fig. 1 C).
LC15 mice did not exhibit detectable myotonia or splicing misregulation in skeletal muscle (data not shown), consistent with transgene expression in muscle being below the threshold for inducing strong sequestration of Mbnl1 protein. FISH using a Texas Red-tagged CAG8-repeat probe revealed widespread formation of RNA aggregates (foci) within nuclei of ventricular cardiomyocytes (Fig. 1 D). The nuclear foci observed in this transgenic line were intense, similar to those observed in human DM1 tissue (Taneja et al., 1995), and were detected in >80% of cardiac nuclei. Ventricular RNA was isolated from cardiac tissue and assessed for splicing of three alternative exons (Sorbs1 exon 23, Cacna1s exon 29, and Tnnt2 exon 5), known to be mis-spliced in the hearts of DM1 patients and mice with Mbnl gene deletion. As expected, LC15 hearts exhibited DM1-like mis-splicing of RNAs for all three transcripts (Fig. 1 E). In addition, Western blot analysis revealed a significant compensatory increased expression (~50%) of Mbnl2 relative to Gapdh in the absence of a significant change in the level of Mbnl1 expression in ventricular myocytes from LC15 mice compared with that observed for myocytes from WT littermates (Fig. S1).
Anesthetized LC15 mice exhibit cardiac repolarization defects with normal LV function
Given the findings above, LC15 mice provide a unique opportunity to characterize the impact of expression of CUGexp RNA on the electrical and contractile properties of the murine heart. Since DM1 patients exhibit multiple ECG defects, including prolongation of PR-, QRS-, and QT-intervals, we compared the ECGs of anesthetized adult WT and LC15 mice. As the transgene in LC15 mice is X-linked, transgene expression in female mice might be regulated by X inactivation. This possibility, coupled with significant sex- and age-dependent differences in ECG parameters in mice (Rijnbeek et al., 2014), led us to perform all electrophysiology analyses in age-matched male WT and LC15 mice.
Three-month-old male WT (n = 11) and LC15 (n = 14) mice were lightly anesthetized using inhaled isoflurane (2% mixed with oxygen) and were provided with supplemental heat to maintain a rectal temperature between 36°C and 38°C. ECGs (lead II configuration) were recorded for 3–5 min, and 3 s of continuous recording were extracted from the final minute to generate an average ECG trace for analysis (Fig. 2 A). Anesthetized 3-mo-old LC15 mice exhibited a modestly higher heart rate (P < 0.01) compared with that of the WT littermate mice (510 ± 11 vs. 442 ± 14 bpm, respectively), despite being administered an equal level of inhaled anesthetic agent. Consistent with the changes observed in DM1 patients, LC15 mice exhibited a significant prolongation in the QTc-interval duration compared with age-matched WT mice (QTc, 14.4 ± 0.3 vs. 12.1 ± 0.3 ms, respectively; Fig. 2, A and B). However, unlike DM1 patients, anesthetized adult LC15 mice exhibited PR- and QRS-interval durations that were not significantly different from that of the WT mice.
Figure 2.

ECG analyses in anesthetized WT and LC15 mice. (A) Representative signal-averaged ECG traces aligned to the start of the Q-wave from 3-mo-old WT (n = 11 mice; black) and LC15 (n = 14 mice; red) littermate mice. The traces reflect the average of 3 s of continuous ECG recording from each mouse with beat-to-beat overlay aligned to S-wave anti-peaks. (B) Average ECG parameters calculated from the signal-averaged traces obtained from anesthetized WT (closed circles) and LC15 (open circles) mice. A subset of these mice was reanalyzed at 12 mo old (WT, n = 5 mice; LC15, n = 7 mice). (C) Representative 2-D guided M-mode echocardiography performed in 12-mo-old WT (top) and LC15 (bottom) mice. (D) Calculated average echocardiography parameters, including ejection fraction, fractional shortening, and LV mass, from WT and LC15 mice. (E) LV mass normalized to body weight. Error bars represent SEM. *, P < 0.05.
Anesthetized ECGs were conducted on a subset of these mice at 12 mo of age in order to investigate potential progression of age-dependent ECG phenotypes (WT, n = 5; LC15, n = 7 mice). The average heart rate was not significantly different between the two groups (WT, 495 ± 12 bpm; LC15, 517 ± 14 bpm), most likely because the heart rate of the anesthetized 12-mo-old WT mice was higher than that observed at 3 mo. Nevertheless, similar to that observed at a younger age, the 12-mo-old LC15 mice also exhibited significant QTc-interval prolongation compared with that of the WT littermate mice (16.6 ± 0.6 vs. 13.2 ± 0.4 ms, respectively) in the absence of a change in the PR- or QRS-interval duration (Fig. 2, A and B). We also observed a modest, but significant, age-dependent increase in QTc in both WT and LC15 mice (WT, 12.1 ± 0.3 ms vs. 13.2 ± 0.4 ms, P < 0.05; LC15, 14.4 ± 0.3 ms vs. 16.6 ± 0.6 ms, P < 0.01). However, the average increase in the QTc-interval duration observed in LC15 mice compared with that of the WT mice was not different between the two ages.
To determine whether the LC15 mice developed significant ventricular dysfunction, hypertrophy, or dilated cardiomyopathy, in vivo 2-D guided M-mode echocardiography was performed in 12-mo-old WT and LC15 mice. Hearts from LC15 mice exhibited normal function and LV wall dimensions (Fig. 2 C). All calculated parameters, including ejection fraction, fractional shortening, LV mass, and LV mass/body weight were comparable in the 12-mo-old WT and LC15 mice (Fig. 2, D and E).
Conscious LC15 mice exhibit QRS- and QTc-interval prolongation
To eliminate any potential masking effects of anesthesia on ECG parameters, a cohort of 12-mo-old WT and LC15 mice were implanted with radiotelemetry devices capable of transmitting continuous ECGs (Fig. 3). During a 3-d period, mouse heart rates cycled between distinct 12-h periods of relatively slow (day) and fast (night) average heart rates (Fig. 3 A). These periods coincided with the room’s timed lighting cycle, consistent with the nocturnal nature of mice when left undisturbed. A blinded analysis of 72 h of continuous ECG recordings failed to reveal arrhythmias in any WT or LC15 mouse. Representative ECG segments were analyzed from three different periods of slow and fast heart rates. The three periods of “fast heart rate” ECG traces analyzed separately and averaged for each mouse were reported as a single data point (Fig. 3 B). An identical analysis was completed during periods of slow heart rates. The average heart rates in the slow and fast heart rate groups differed by ~140 bpm (WT, 499 ± 22 bpm vs. 638 ± 14 bpm; LC15, 485 ± 13 bpm vs. 628 ± 13 bpm) but were comparable between WT and LC15 mice (Fig. 3 C). Compared with WT mice, ECG recordings from conscious LC15 mice exhibited prolonged QTc-intervals (slow heart rate, QTc, 31.9 ± 0.7 ms vs. 25.8 ± 0.9 ms, P < 0.01; fast heart rate, QTc, 32.7 ± 0.4 ms vs. 27.3 ± 0.9 ms, P < 0.01) and QRS-intervals (slow heart rate, QRS, 10.3 ± 0.4 ms vs. 8.9 ± 0.3 ms, P = 0.02; fast heart rate, QRS, 10.1 ± 0.6 ms vs. 7.9 ± 0.4 ms, P = 0.02) at both slow and fast heart rates. However, unlike DM1 patients, the PR-interval was not significantly prolonged in ECG recordings from conscious LC15 mice at either fast or slow heart rates (Fig. 3 C).
Figure 3.

ECG analyses of conscious WT and LC15 mice using implanted radiotelemeters. ECGs were recorded continuously for 60 h in mice surgically implanted with radiotelemetry devices (ETA-F10). (A) Plot of average heart rate (HR) over 5-min intervals during the 60 h of recording for a cohort of representative WT and LC15 mice (n = 2 mice for each genotype) that received implants on the same day and were recorded simultaneously. HRs cycled together during daily periods of faster and slower HRs, coinciding with the dark or light room environment, respectively. Each mouse was analyzed at three different periods of fast and slow HRs (i.e., six analysis periods per mouse), and data shown represent averaged values of the three analyses. Expanded view: Representative ECG signals recorded during a period of fast HR showing high lead II ECG signal/noise. (B) Representative signal-averaged ECG traces from a period of fast HR aligned to the start of the Q-wave from WT (n = 5 mice; black) and LC15 (n = 5 mice; red) littermate mice. (C) Average ECG parameters from conscious WT (closed circles) and LC15 (open circles) mice during periods of slow (left) and fast (right) HR. Error bars represent SEM. *, P < 0.05.
LC15 mice exhibit increased susceptibility to arrhythmia during flecainide administration
Since we found that LC15 mice exhibit a significantly prolonged QRS-interval, we tested the effect of acute administration of low-dose flecainide, a Na channel blocker and class Ic anti-arrhythmic agent, on arrhythmia susceptibility in anesthetized WT and LC15 mice. Three continuous seconds of ECG recording were analyzed during the final minute of the baseline recording and again 2 and 5 min after drug administration (Fig. 4 A). The average heart rate decreased significantly (P < 0.05) in LC15 mice after flecainide administration (561 ± 14 bpm vs. 529 ± 15 bpm; Fig. 4 B). In contrast, the heart rate was not significantly different (P = 0.43) in WT mice 2 min after flecainide administration (545 ± 14 bpm vs. 531 ± 17 bpm). In addition, ECG intervals were significantly (P < 0.05) prolonged in LC15 mice 2 min after flecainide administration, including PR-intervals (29.6 ± 0.8 ms vs. 37.4 ± 1.0 ms), QRS-intervals (8.1 ± 0.5 ms vs. 10.0 ± 0.9 ms), and QTc-intervals (16.3 ± 0.5 ms vs. 20.9 ± 1.6 ms; Fig. 4 B). As observed in younger mice, LC15 mice exhibited significantly (P < 0.05) prolonged QTc-intervals compared with those of WT mice at baseline (13.8 ± 0.5 ms vs. 16.3 ± 0.5), and this difference was even greater 2 min after flecainide injection (15.5 ± 1.3 ms vs. 20.9 ± 1.6 ms). The relative sensitivity of each mouse in exhibiting a ventricular arrhythmia 5 min following flecainide administration was grouped into one of three general categories: mild, moderate, or severe (see Materials and methods for details). The relative distribution across these groups was significantly (P < 0.05; χ2 analysis) shifted to the more severe categories for the LC15 mice compared with the WT mice (Fig. 4 C). In total, five of seven LC15 mice exhibited “severe sensitivity” to flecainide administration, whereas no WT mice were classified into this group. These results indicate that that the cardiac conduction in LC15 mice exhibits greater sensitivity to flecainide.
Figure 4.

LC15 mice exhibited increased sensitivity for arrhythmias following acute flecainide administration. (A) Representative 1-s ECG recordings obtained from WT and LC15 mice immediately before (left), 2 min after (middle), and 5 min after (right) flecainide administration (20 mg/kg intraperitoneally) in anesthetized 18-mo-old WT and LC15 mice. (B) Average ECG parameters (PR-interval, QRS-interval, QTc-interval, and heart rate) calculated from the signal-averaged traces obtained from anesthetized WT (closed circles; n = 5) and LC15 (open circles; n = 7) mice before and 2 min after flecainide administration. A PR-interval could not be determined for one of the seven LC15 mice. (C) Pie charts summarizing the fraction of mice exhibiting altered ECG morphology 5 min after flecainide administration classified into three categories: mild, moderate, and severe sensitivity (see Materials and methods for details). Error bars represent SEM. *, P < 0.05 compared with WT. #, P < 0.05 compared with baseline.
Maximal AP upstroke velocity in ventricular myocytes from LC15 mice is reduced at fast pacing frequencies
We found significant ventricular depolarization (QRS-interval prolongation) and repolarization (QTc-interval prolongation) defects in conscious LC15 mice that exhibited high levels of CUGexp RNA expression in the heart. As the echocardiographic analyses indicated normal contractile function in 1-yr-old LC15 mice, the observed ECG defects were unrelated to major structural changes in the heart. Thus, we hypothesized that the ECG changes in LC15 mice reflect differences in ionic currents that comprise the ventricular AP.
As an initial test of this hypothesis, AP properties were measured at 37°C in whole-cell current clamp experiments of acutely dissociated ventricular myocytes from WT and LC15 mice. Following dialysis with internal solution, neither average resting membrane potential (−75.2 ± 2.3 mV and −75.7 ± 1.1 mV) nor total cell capacitance (137 ± 8 pF and 122 ± 7 pF) were significantly different in myocytes from WT and LC15 mice. Using a brief current injection (110% of the current required to elicit an AP), a train of 20 consecutive APs were initiated at different PCLs ranging from 1,000 ms (1 Hz) to 111 ms (9 Hz). Average (n = 20) AP waveforms elicited at 1 and 9 Hz in myocytes from WT and LC15 mice are shown in Fig. 5 A. The average current injection required to evoke an AP in myocytes from LC15 and WT mice was not significantly different (5.5 ± 0.4 nA versus 4.5 ± 0.4 nA, respectively; P = 0.1; Fig. 5 B). As the PCL decreased (i.e., as the pacing frequency increased), maximal dV/dt of the phase-0 AP upstroke was progressively reduced in myocytes from LC15 mice. Specifically, at the shortest PCL (9 Hz), maximal dV/dt was significantly reduced (P < 0.05) in myocytes from LC15 mice (Fig. 5, C and D). Consistent with this, although the maximal dV/dt at 9 Hz was not significantly different statistically (P = 0.14) from that observed at 1 Hz in myocytes from WT mice (415.3 ± 19.5 V/s vs. 474.4 ± 30.0 V/s at 9 Hz and 1 Hz, respectively), this comparison was statistically significant (P < 0.05) in myocytes from LC15 mice (294.6 ± 30.4 V/s vs. 441.8 ± 23.9 V/s at 9 Hz and 1 Hz, respectively; Fig. 5 D).
Figure 5.

AP recordings from isolated cardiac ventricular myocytes. (A) Representative group-averaged AP traces from current clamp recordings of single ventricular myocytes from WT (n = 5; black) and LC15 (n = 8; red) mice. AP traces reflect the aligned signal average of 20 consecutive APs (excluding the first AP) elicited by injecting 110% of the threshold current at PCLs ranging from 1,000 ms (left) to 111 ms (right). Dotted line represents 0 mV. (B) The minimum amount of injected current required to evoke a single AP in myocytes from WT (closed circles) and LC15 (open circles) mice. (C) Maximal phase 0 AP upstroke velocity (dV/dt) at different PCLs in myocytes from WT (closed circles) and LC15 (open circles) mice. (D) Cumulative difference (from 1 Hz) in maximal dV/dt for different stimulation frequencies. (E) Average APDs measured by determining the time required for 25% (APD-25%; triangles), 75% (APD-75%; inverted triangles), and 90% (APD-90%; squares) repolarization from the peak of the AP to the resting membrane potential. For these data, all statistical symbols shown comparing WT and LC15 values apply to every PCL. (F) Average peak change in voltage during the AP upstroke (top), maximal voltage at the peak of the AP (middle), and resting membrane potential before the AP upstroke (bottom) in ventricular myocytes from WT (closed circles) and LC15 (open circles) mice recorded at different PCLs. Error bars represent SEM. *, P < 0.05.
APD is prolonged in ventricular myocytes from LC15 mice in the absence of a change in resting membrane potential or peak change in voltage during the AP
The APD was measured at three distinct points during repolarization: early/phase 1 (APD-25%), plateau/phase 2 (APD-75%), and late/phase 3 (APD-90%). A significant prolongation of the ventricular AP was observed in myocytes from LC15 mice for each of these repolarization intervals at every stimulation frequency compared with that of myocytes from WT mice (Fig. 5 E). However, unlike the frequency dependence observed for the reduction in maximal AP upstroke velocity (Fig. 5, C and D), the prolonged APD observed in myocytes from LC15 mice (Fig. 5 E) did not depend on stimulation frequency. In spite of these changes in APD, no differences in either resting membrane potential or peak change in voltage at the peak of the AP were observed at any PCL in myocytes from WT and LC15 mice (Fig. 5 F).
Voltage dependence of sodium current activation and steady-state inactivation are right shifted, but peak current density is unaltered in ventricular myocytes from LC15 mice
The increased QRS-interval duration in conscious LC15 mice (Fig. 3 C) and reduced maximal AP upstroke velocity in myocytes from LC15 mice (Fig. 5, C and D) could be explained in part by a reduction in peak sodium current density and/or changes in channel activation/availability. To test this idea, we performed whole-cell voltage clamp measurements of sodium currents in ventricular myocytes from WT and LC15 mice. The average cell capacitance of myocytes used in these studies was not significantly different (P = 0.80) between LC15 (181 ± 19 pF) and WT mice (188 ± 12 pF).
From a holding potential of −120 mV, sodium currents were elicited under low sodium conditions using test potentials ranging from −150 mV to +60 mV (Fig. 6 A). The average voltage dependence of peak inward sodium current density and relative sodium channel conductance (G/Gmax) are shown in Fig. 6, B and C, respectively. Neither peak inward sodium current density (Fig. 6 B) nor maximal channel conductance (Fig. 6 D) were significantly different in myocytes from LC15 and WT mice. However, the voltage dependence of sodium channel activation was shifted significantly (P < 0.05) to more positive potentials in ventricular myocytes from LC15 mice compared with that of WT mice (−56.9 ± 1.0 mV vs. −63.0 ± 0.4 mV, respectively; Fig. 6 E). In a separate set of measurements, steady-state inactivation was assessed from the relative current measured during a brief (30-ms) test depolarization to −40 mV immediately following 300-ms preconditioning test pulses ranging from −150 mV to +60 mV (Fig. 7 A). Similar to that observed for channel activation, the voltage dependence of sodium channel availability (I/Imax) was also shifted significantly (P < 0.05) to more positive potentials in ventricular myocytes from LC15 mice compared with that of WT mice (−91.1 ± 1.6 mV vs. −97.7 ± 0.9 mV, respectively; Fig. 7, B and C).
Figure 6.

Peak inward sodium current density is unaltered and the voltage dependence of activation is right-shifted in ventricular myocytes from LC15 mice. (A) Representative superimposed inward sodium current traces measured during 300-ms voltage steps from −150 mV to +60 mV in 5-mV increments. Steps were delivered at 5.5-s intervals to provide sufficient time for recovery from inactivation. (B) Average peak sodium current density versus voltage relationship for myocytes from WT (n = 3; closed circles) and LC15 (n = 5; open circles) mice. (C) Normalized conductance (G/Gmax) versus voltage relationships. (D and E) Maximum channel conductance (Gmax; D) and the membrane potential for half-maximal activation (E) in ventricular myocytes from WT (closed circles) and LC15 (open circles) mice. Error bars represent SEM. *, P < 0.05.
Figure 7.

Voltage dependence of steady-state sodium channel inactivation is right-shifted in ventricular myocytes from LC15 mice. (A) Representative inward sodium currents measured during 30-ms voltage steps to −40 mV (indicated by arrow) following 300-ms inactivating prepulses of varied amplitude in a ventricular myocyte from a WT mouse. Prepulses were sequentially increased by 5 mV, from −150 mV to +60 mV, and delivered at 5.5-s intervals in order to provide sufficient time for full recovery from inactivation. (B) Voltage dependence of steady-state inactivation of relative peak sodium current amplitude (I/Imax) in myocytes from WT (n = 3; closed circles) and LC15 (n = 5; open circles) mice. (C) Average voltage for half-maximal steady-state inactivation (SSV50) measured in myocytes from WT (closed circles) and LC15 (open circles) mice. Error bars represent SEM. *, P < 0.05.
Peak outward potassium current density is reduced without a change in steady-state current density in ventricular myocytes from LC15 mice
The results presented in Fig. 5 E demonstrate that prolongation of the ventricular myocyte APD occurs throughout all phases of membrane repolarization from APD-25% through APD-90%, which could be explained by a reduction in peak outward potassium current density. To test this hypothesis, we performed whole-cell voltage clamp measurements of outward potassium currents in ventricular myocytes from WT and LC15 mice. To measure both peak and steady-state potassium current components, myocytes were depolarized from the holding potential (−70 mV) to test potentials ranging from −40 mV to +50 mV for 4.5 s. Test pulses were delivered at 10-s intervals in order to allow the K+ channels to fully recover from inactivation. Average cell capacitance in these experiments was not significantly different (P = 0.42) between myocytes recorded from WT (154.5 ± 6.3 pF) and LC15 (147.4 ± 6.1 pF) mice. Peak outward potassium current density was significantly reduced in ventricular myocytes from LC15 mice compared with WT mice (at +40 mV, 32.5 ± 1.9 pA/pF vs. 41.6 ± 2.6 pA/pF; P = 0.006; Fig. 8, A and B). However, the steady-state, or “plateau,” current, measured during the final 200 ms of each 4.5-s voltage step, was not significantly (P = 0.93) different between myocytes from WT (6.2 ± 0.3 pA/pF) and LC15 mice (6.1 ± 0.3 pA/pF).
Figure 8.

Peak outward potassium current density is reduced in ventricular myocytes from LC15 mice. (A) Representative outward potassium currents measured during 4.5-s voltage steps from a holding potential of −70 mV in myocytes from WT (left; black) and LC15 (right; red) mice. Voltage steps from −40 mV to +50 mV were sequentially increased in 10-mV increments and delivered at 10-s intersweep intervals. Insets: Currents during the initial 125 ms of depolarization on an expanded scale. (B) Average voltage dependence of peak (circles) and steady-state (diamonds) potassium current density in myocytes from WT (n = 24; closed symbols) and LC15 (n = 25; open symbols) mice. Steady-state currents were measured during the final 200 ms of each voltage step. (C) Representative potassium current decay during depolarization to +40 mV in myocytes from WT (black) and LC15 (red) mice fitted according to a second order exponential equation (blue solid curves). Error bars represent SEM. *, P < 0.05.
Amplitudes of the two kinetic components of potassium current decay are both reduced in ventricular myocytes from LC15 mice
The decay phase of the potassium current in murine ventricular myocytes contains multiple kinetic components (Xu et al., 1999a; Xu et al., 1999b). Specifically, the outward potassium current elicited at +40 mV consists of three distinct kinetic components: a fast-inactivating transient outward current (Ito), a slower inactivating current (IK,slow), and a steady-state current component (Iss). To assess the potential changes in these three kinetic components of the potassium current, the decay phase of the current elicited at +40 mV was fitted according to a second order exponential function plus a steady-state component (Fig. 8 C). In all cases, the potassium current decay was well-described by this fitting procedure (correlation coefficient, 0.980 and 0.978 for myocytes from WT and LC15 mice, respectively). The results indicated that although the time constants of the two kinetic components of decay and the amplitude of the steady-state current (Iss) were not significantly different between myocytes from WT and LC15 mice, the amplitudes of the Ito and IK,slow components of decay were both significantly reduced (Table 1).
Table 1. Two-exponential curve fitting results.
| Ipeak | Ito | IK,slow | Iss | |
|---|---|---|---|---|
| WT (n = 24) | ||||
| Τ decay (ms) | – | 185.3 ± 11.0 | 1,115.2 ± 30.5 | – |
| Density (pA/pF) | 41.9 ± 2.9 | 17.0 ± 1.5 | 18.9 ± 1.4 | 5.9 ± 0.3 |
| Percent Ipeak | – | 41 | 45 | 14 |
| LC15 (n = 25) | ||||
| Τ decay (ms) | – | 166.3 ± 6.5 | 1051.8 ± 35.3 | – |
| Density (pA/pF) | 31.5 ± 1.8* | 12.3 ± 1.1* | 13.2 ± 0.9* | 5.9 ± 0.3 |
| Percent Ipeak | – | 39 | 42 | 19 |
Results from fitting a second order exponential equation to the entire decay phase of the potassium current elicited by a 4.5-s depolarization to +40 mV from a holding potential of −70 mV. *, statistically significant.
4-AP–sensitive potassium current is reduced in ventricular myocytes from LC15 mice
The exponential curve-fitting analysis indicated that the reduction in potassium current observed in ventricular myocytes from LC15 mice might reflect a decrease in Ito and IK,slow activity. Interestingly, subcomponents of these two kinetic components to the total potassium current exhibit varying sensitivity to block by 4-AP (Xu et al., 1999a). Therefore, we compared 4-AP–sensitive outward potassium current densities in ventricular myocytes at two different concentrations of 4-AP. Outward potassium currents were first measured as described above in the absence of the drug and then followed by sequential exposure to low (50 µM) and then high (500 µM) concentrations of 4-AP (Fig. 9 A). To quantify potassium currents with low 4-AP sensitivity, current traces measured in the presence of 500 µM 4-AP were subtracted from those recorded in the presence of 50 µM 4-AP (Fig. 9 B). Consistent with the kinetic analysis, both the peak current density and integral of the low 4-AP–sensitive potassium current were reduced significantly in myocytes from LC15 mice (Fig. 9, C and D).
Figure 9.

Low 4-AP–sensitive potassium current density is reduced in myocytes from LC15 mice. (A) Representative outward potassium current traces in ventricular myocytes from WT (left; black) and LC15 (right; red) mice. Following recording in control solution, the bath solution was exchanged for one containing a low concentration (50 µM) of 4-AP and then a high concentration (500 µM) of 4-AP. (B) The low 4-AP–sensitive current component (4-AP Diff.) was determined by subtracting current traces recorded in the presence of 500 µM 4-AP from that obtained in the presence of 50 µM 4-AP. (C) Average voltage dependence of peak 4-AP Diff. current density in myocytes from WT (n = 8; closed circles) and LC15 (n = 6; open circles) mice. (D) Average voltage dependence of the integral of the 4-AP Diff. current density. Error bars represent SEM. *, P < 0.05.
Discussion
DM1 patients often exhibit a variety of different ECG abnormalities, including AV block (PR-interval prolongation), slowed propagation of ventricular depolarization (QRS-interval prolongation), and delayed ventricular repolarization (QT-interval prolongation; Phillips and Harper, 1997; Groh et al., 2008; Wahbi et al., 2012; Park et al., 2013). Since these ECG defects result in a heightened risk of SCD, it is important to establish animal models that reproduce DM1-like ECG defects in order to elucidate the underlying biophysical mechanisms and to develop effective interventions. Although there are significant limitations in using genetically engineered mice in attempts to model the cardiac phenotype observed in DM1 patients, we found that cardiomyocytes from LC15 mice exhibited nuclear foci of CUGexp RNA (Taneja et al., 1995) and altered splicing of Sorbs1, Tnnt2, and Cacna1s transcripts, presumably due to sequestration of Mbnl proteins (Mankodi et al., 2005). However, the precise degree by which cardiac CUGexp RNA expression in LC15 mice leads to loss of Mbnl function is unclear.
Mbnl2 upregulation limits the ECG phenotype of LC15 mice
Mbnl proteins are an important family of RNA binding proteins that regulate alternative splicing transitions executed during postnatal development (Pascual et al., 2006). The two Mbnl isoforms expressed in the heart (Mbnl1 and Mbnl2) regulate overlapping splicing events, thus reflecting a functional redundancy (Pascual et al., 2006; Fernandez-Costa et al., 2011). Consistent with this, complete knockout of Mbnl1 expression causes cardiac conduction defects that only partially model the ECG phenotype of DM1 patients (Dixon et al., 2015), whereas compound knockout of Mbnl1 and Mbnl2 in mice more accurately models PR-, QRS-, and QTc-interval prolongation and premature death (Lee et al., 2013). Thus, development of PR-interval prolongation and SCD in mice apparently requires loss of both Mbnl1 and Mbnl2 function. Based on the observation that Mbnl1 knockout mice exhibit increased expression of Mbnl2 protein, Lee et al. (2013) proposed a titration model for DM1, whereby Mbnl2 expression is upregulated when Mbnl1 is significantly sequestered by CUGexp RNA (Lee et al., 2013). This compensatory mechanism effectively limits the scope and severity of the cardiac phenotype of Mbnl1 knockout mice. Consistent with this model, we found that Mbnl2 expression was increased in the hearts of LC15 mice.
Since expanded CUG-repeat RNA binds Mbnl1 and Mbnl2 with high affinity (Sznajder et al., 2016), we initially expected LC15 mice to exhibit an ECG phenotype similar to that of compound-Mbnl1/2 knockout mice. However, the cardiac phenotype of LC15 mice is more comparable to that of Mbnl1 knockout mice (Dixon et al., 2015). Specifically, both Mbnl1 knockout (Dixon et al., 2015) and LC15 mice exhibit significant QRS- and QTc-interval prolongation, but lack PR-interval prolongation. Based on these observations, we propose that CUGexp expression in the hearts of LC15 mice impacts Mbnl1 and Mbnl2 function but not to the extent of combinatorial gene deletion, thus permitting sufficient residual Mbnl function to protect against AV conduction block and SCD.
A large study of DM1 mis-splicing by Freyermuth et al. (2016) identified mis-splicing of SCN5A (the gene encoding the cardiac voltage-gated sodium channel or NaV1.5) in the human DM1 heart. The fetal isoform of SCN5A, which includes exon 6A, is variably increased in human DM1 hearts (to ~60%) and mis-spliced in compound-Mbnl1/2 knockout hearts (to ~15–20%; Freyermuth et al., 2016). Interestingly, mice with forced expression of the “fetal” SCN5A isoform exhibit PR-interval prolongation (Freyermuth et al., 2016; Pang et al., 2018). Using CRISPR-mediated expression of “fetal” SCN5A, Pang et al. (2018) found that increasing exon 6A expression to close to 100% in homozygous Scn5aΔe6B/Δe6B mice, but not to ~70% in heterozygous Scn5aΔe6B/+ mice, is required to increase PR-interval prolongation (Pang et al., 2018). The absence of PR-interval prolongation in LC15 mice is consistent with the finding that fetal exon 6A is not increased in the hearts of these mice (Fig. S2), possibly due to lower expression or greater tolerance for CUG-repeat RNA.
Frequency-dependent decrease in AP upstroke velocity underlies QRS-interval prolongation in LC15 mice
We found that the maximal rate of depolarization during the AP upstroke (dV/dt) was decreased as the pacing frequency increased from 1 to 9 Hz in myocytes from LC15 mice, but not WT mice. A similar reduction in maximal dV/dt with normal sodium current density was previously reported in ventricular myocytes from DMSXL mice, although that study did not assess the effects at different pacing frequencies (Algalarrondo et al., 2015). We found that the maximal sodium current density was not significantly different in myocytes from LC15 mice. However, the voltage dependence of sodium channel activation was right shifted significantly, which could contribute to a reduction in sodium channel activation during depolarization. However, given the observed frequency dependence of the reduction in dV/dt in myocytes from LC15 mice, a defect in Na channel recovery from inactivation could also contribute to reduced channel availability at fast pacing frequencies. Importantly, although we did not detect spontaneous arrhythmias under nonchallenged conditions, LC15 mice exhibited increased arrhythmia susceptibility in the setting of acute sodium channel inhibition with flecainide.
The reduction in maximal AP upstroke velocity in ventricular myocytes from LC15 mice observed at high pacing frequencies is consistent with the prolonged QRS-interval observed in conscious LC15 mice. Specifically, the slow (~500 bpm) and fast (~600 bpm) heart rates measured with telemeters in conscious mice were close to the 9-Hz pacing frequency in which dV/dt was reduced in myocytes from LC15 mice. The absence of a statistically significant increase in QRS-interval duration in anesthetized LC15 mice could be due to the somewhat lower heart rates of anesthetized mice. Although it is also possible that the anesthesia could have directly affected the ECG parameters, inhaled isoflurane has less effects on cardiac electrophysiology compared with injectable anesthetic mixtures (Chaves et al., 2003; Appleton et al., 2004). Thus, our findings suggest that an increased heart rate (or pacing) is an important factor in the reduced myocyte upstroke velocity and increased QRS-interval prolongation observed in LC15 mice. This idea is supported by other DM1 studies linking the heart rate to serious ventricular conduction defects. For example, compound-Mbnl1/2 knockout mice exhibit a frequency-dependent loss in ventricular cardiac conduction velocity (Chou et al., 2017). Using an optical mapping approach, the development of functional conduction block in compound-Mbnl1/2 mice was observed at ~14 Hz, but not at slower pacing frequencies (Chou et al., 2017). Separately, a clinical study of adolescent DM1 patients with severe cardiac involvement found that physical exercise often precipitated major cardiac rhythm disturbances (Bassez et al., 2004). Given these studies, it would be interesting to determine whether LC15 mice also exhibit an enhanced susceptibility to arrhythmias during acute exercise, isoproterenol injection, or pacing-induced ventricular tachyarrhythmias.
Reduction in 4-AP–sensitive outward potassium current underlies prolongation of ventricular myocyte APD and QTc-interval of LC15 mice
Ventricular myocytes from LC15 mice exhibited a significant prolongation of the APD. Specifically, the APD was prolonged across all intervals evaluated (APD-25%, APD-75%, and APD-90%) in a manner that did not depend on the pacing frequency (Fig. 5 C). These findings in single myocytes suggest that QTc-interval prolongation in LC15 mice is a direct consequence of delayed ventricular repolarization rather than secondary to structural changes of the heart.
Voltage clamp studies were conducted in single myocytes to provide evidence for the biophysical mechanism that underlies the observed prolongation of the APD and QTc-interval in LC15 mice. Since APD prolongation was observed throughout the entire AP, beginning as early as APD-25%, the magnitude of the rapidly activating outward potassium currents were compared in ventricular myocytes from age-matched WT and LC15 mice. We found that peak outward potassium current density was reduced significantly in ventricular myocytes from LC15 mice. To assess potential effects on the different kinetic components of the current, we employed a double-exponential curve-fitting analysis of the decay phase of the current, as described previously (Xu et al., 1999a; Xu et al., 1999b). A significant reduction in the current density was observed for the fast (Ito,fast/Ito,slow) and slow (IK,slow1/IK,slow2) components of potassium current decay without a change in either their respective time constants or the noninactivating steady-state component of the current. The kinetic components of potassium current decay can be subdivided into distinct molecular components: Ito,fast (Kv4.2/Kv4.3), Ito,slow (Kv1.4), IK,slow1 (Kv1.5), and IK,slow2 (Kv2.1). Xu et al. (1999a) characterized the sensitivity of these different potassium current components to low (50 µM) and high concentrations of 4-AP (500 µM). Although IK,slow2 is insensitive to 4-AP (London et al., 2001), the other components exhibit differential sensitivity to the two concentrations of 4-AP. Of the currents blocked by 500 µM 4-AP, only Ito,slow was insensitive to 50 µM 4-AP. Thus, we also used sensitivity to blockage by low and high concentrations of 4-AP to dissect the component of the potassium current that is reduced in myocytes from LC15 mice (Fig. 9). Interestingly, ventricular myocytes from LC15 mice exhibited significantly less potassium current that was sensitive to 500 µM 4-AP but insensitive to 50 µM 4-AP, consistent with the previously reported pharmacological profile of Ito,slow. Thus, the prolongation in ventricular AP duration and QTc-interval observed in LC15 mice likely results, at least in part, from a reduction in Ito,slow current density.
Consistent with a reduction in Ito,slow current density in myocytes for LC15 mice, Western blot analysis revealed a significant reduction in Kv1.4 protein expression compared with WT (Fig. S3). The mechanism (e.g., altered transcription, altered mRNA transport or turnover, protein degradation) that underlies this decrease in Kv1.4 expression is unclear. Alternatively, altered regulation of Kv1.4 activity by PKC, AMPK, and/or Nedd4-2/x could also contribute to the observed reduction in outward potassium current density (Andersen et al., 2018). Thus, future work is needed to identify the specific molecular mechanism(s) responsible for the observed reduction in Ito,slow and Kv1.4 expression in LC15 mice and the degree to which this also applies to DM1 patients.
Potential molecular mechanisms
The precise molecular mechanism by which expanded CUG-repeat RNA expression in LC15 mice results in a rate-dependent reduction in maximal AP dV/dt and QRS prolongation is unclear. These rate-dependent effects are consistent with a slowing in Na channel recovery from inactivation. Although the fetal Scn5a splice variant (containing exon 6A) results in a slowing of Na channel recovery from inactivation (Onkal et al., 2008) and increased cardiac expression of this splice isoform results in QRS prolongation (Freyermuth et al., 2016; Pang et al., 2018), this cannot explain the changes in dV/dt and QRS prolongation in LC15 mice since no change in Scn5a exon 6 splicing was observed. Altered expression of other Scn5a splice variants is also possible (Schroeter et al., 2010). As one example, alternative splicing of Scn5a exon 17 results in an in-frame deletion of 120 nucleotides (amino acids 961–1,000 in the intracellular II-III loop). The deletion causes a depolarizing shift in both steady-state activation and inactivation (Camacho et al., 2006) similar to that observed for myocytes from LC15 mice and a slowing of Na channel recovery from inactivation (Walzik et al., 2011). Interestingly, inclusion of adjacent Scn5a exon 18 is developmentally regulated and mis-spliced and maximal dV/dt is reduced in DMSXL mice (Algalarrondo et al., 2015). However, baseline QRS duration is not prolonged and Na channel recovery from inactivation is accelerated in DMSXL mice.
Altered modulation of Na channel inactivation by fibroblast growth factor (FGF) homologous factors represent an intriguing alternative potential mechanism. FGF proteins interact with the C-terminus of the cardiac Na channel to regulate channel inactivation (Pitt and Lee, 2016). Interestingly, FGF13 (or fibroblast growth factor homologous factor-2) splicing is developmentally regulated (Munoz-Sanjuan et al., 2000), and FGF13 transcripts represent the major FGF isoforms expressed in rodent heart (Wang et al., 2011). In particular, interaction of the FGF13S splice isoform with the cardiac Na channel shifts steady-state inactivation to more depolarized potentials and slows both fast inactivation and channel recovery from inactivation (Yang et al., 2016). Importantly, conditional Fgf13 knockout in the heart, which eliminates all FGF13 splice isoforms, reduces maximal AP dV/dt, delays Na channel recovery from fast inactivation, and prolongs the ventricular AP duration (via effects on Ito and IK,slow, but not Iss; Wang et al., 2017). Moreover, cardiac-specific Fgf13-deficient mice exhibit a prolonged QRS interval and increased arrhythmia susceptibility in response to acute flecainide administration (Wang et al., 2017). These effects are remarkably similar to those reported here for LC15 mice. While admittedly speculative at this time, a more detailed exploration of these potential molecular mechanisms for the ECG, AP, and ion current changes observed in LC15 mice merits future investigation.
Acknowledgments
Eduardo Ríos served as editor.
We thank Deanne Mickelsen, Linda Richardson, and Sarah Leistman for technical support. We also thank Anatoli N. Lopatin (University of Michigan, Ann Arbor, MI) for kindly providing ECG analysis software and Krzysztof R. Grzeda (University of Michigan, Ann Arbor, MI) for the APD software.
This work was supported by the National Institutes of Health (grants U54NS048843 and T90DE21985), the John R. Murlin Memorial Fund, and the Howard Hughes Medical Institute “Med Into Grad” Fellowship (grant HHMI 56006775) to K.M. Tylock.
The authors declare no competing financial interests.
Author contributions: K.M. Tylock designed and performed the experiments, analyzed the data, and wrote the manuscript. C.A. Thornton generated the LC15 mouse colony, provided the data in Fig. 1, and edited the manuscript. R.T. Dirksen designed the experiments, interpreted data, and wrote and edited the manuscript. D.S. Auerbach designed the experiments, interpreted data, and edited the manuscript. Z.Z. Tang provided data for Scn5a exon 6A and Cacna1s exon 29 splicing and edited the manuscript.
References
- Algalarrondo V., Wahbi K., Sebag F., Gourdon G., Beldjord C., Azibi K., Balse E., Coulombe A., Fischmeister R., Eymard B., et al. . 2015. Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul. Disord. 25:308–320. 10.1016/j.nmd.2014.11.018 [DOI] [PubMed] [Google Scholar]
- Andersen M.N., Skibsbye L., Saljic A., Larsen M.Z., Rasmussen H.B., and Jespersen T.. 2018. Regulation of Kv1.4 potassium channels by PKC and AMPK kinases. Channels (Austin). 12:34–44. 10.1080/19336950.2017.1405196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton G.O., Li Y., Taffet G.E., Hartley C.J., Michael L.H., Entman M.L., Roberts R., and Khoury D.S.. 2004. Determinants of cardiac electrophysiological properties in mice. J. Interv. Card. Electrophysiol. 11:5–14. 10.1023/B:JICE.0000035922.14870.56 [DOI] [PubMed] [Google Scholar]
- Auerbach D.S., Jones J., Clawson B.C., Offord J., Lenk G.M., Ogiwara I., Yamakawa K., Meisler M.H., Parent J.M., and Isom L.L.. 2013. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. 8:e77843 10.1371/journal.pone.0077843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassez G., Lazarus A., Desguerre I., Varin J., Laforêt P., Bécane H.M., Meune C., Arne-Bes M.C., Ounnoughene Z., Radvanyi H., et al. . 2004. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology. 63:1939–1941. 10.1212/01.WNL.0000144343.91136.CF [DOI] [PubMed] [Google Scholar]
- Batra R., Charizanis K., Manchanda M., Mohan A., Li M., Finn D.J., Goodwin M., Zhang C., Sobczak K., Thornton C.A., and Swanson M.S.. 2014. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell. 56:311–322. 10.1016/j.molcel.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T., et al. . 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 68:799–808. 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- Camacho J.A., Hensellek S., Rougier J.S., Blechschmidt S., Abriel H., Benndorf K., and Zimmer T.. 2006. Modulation of Nav1.5 channel function by an alternatively spliced sequence in the DII/DIII linker region. J. Biol. Chem. 281:9498–9506. 10.1074/jbc.M509716200 [DOI] [PubMed] [Google Scholar]
- Charlet-B N., Savkur R.S., Singh G., Philips A.V., Grice E.A., and Cooper T.A.. 2002. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 10:45–53. 10.1016/S1097-2765(02)00572-5 [DOI] [PubMed] [Google Scholar]
- Chaves A.A., Dech S.J., Nakayama T., Hamlin R.L., Bauer J.A., and Carnes C.A.. 2003. Age and anesthetic effects on murine electrocardiography. Life Sci. 72:2401–2412. 10.1016/S0024-3205(03)00137-1 [DOI] [PubMed] [Google Scholar]
- Chong-Nguyen C., Wahbi K., Algalarrondo V., Bécane H.M., Radvanyi-Hoffman H., Arnaud P., Furling D., Lazarus A., Bassez G., Béhin A., et al. . 2017. Association between mutation size and cardiac involvement in myotonic dystrophy type 1: an analysis of the DM1-Heart Registry. Circ Cardiovasc Genet. 10:10 10.1161/CIRCGENETICS.116.001526 [DOI] [PubMed] [Google Scholar]
- Chou C.C., Chang P.C., Wei Y.C., and Lee K.Y.. 2017. Optical mapping approaches on muscleblind-like compound knockout mice for understanding mechanistic insights into ventricular arrhythmias in myotonic dystrophy. J. Am. Heart Assoc. 6 10.1161/JAHA.116.005191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansithong W., Paul S., Comai L., and Reddy S.. 2005. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 280:5773–5780. 10.1074/jbc.M410781200 [DOI] [PubMed] [Google Scholar]
- de Die-Smulders C.E., Höweler C.J., Thijs C., Mirandolle J.F., Anten H.B., Smeets H.J., Chandler K.E., and Geraedts J.P.. 1998. Age and causes of death in adult-onset myotonic dystrophy. Brain. 121:1557–1563. 10.1093/brain/121.8.1557 [DOI] [PubMed] [Google Scholar]
- Dixon D.M., Choi J., El-Ghazali A., Park S.Y., Roos K.P., Jordan M.C., Fishbein M.C., Comai L., and Reddy S.. 2015. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 5:9042 10.1038/srep09042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Costa J.M., Llamusi M.B., Garcia-Lopez A., and Artero R.. 2011. Alternative splicing regulation by muscleblind proteins: from development to disease. Biol. Rev. Camb. Philos. Soc. 86:947–958. 10.1111/j.1469-185X.2011.00180.x [DOI] [PubMed] [Google Scholar]
- Freyermuth F., Rau F., Kokunai Y., Linke T., Sellier C., Nakamori M., Kino Y., Arandel L., Jollet A., Thibault C., et al. . 2016. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 7:11067 10.1038/ncomms11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.H., Pizzuti A., Fenwick R.G. Jr., King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., de Jong P., et al. . 1992. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 255:1256–1258. 10.1126/science.1546326 [DOI] [PubMed] [Google Scholar]
- Groh W.J., Lowe M.R., and Zipes D.P.. 2002. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J. Cardiovasc. Electrophysiol. 13:444–448. 10.1046/j.1540-8167.2002.00444.x [DOI] [PubMed] [Google Scholar]
- Groh W.J., Groh M.R., Saha C., Kincaid J.C., Simmons Z., Ciafaloni E., Pourmand R., Otten R.F., Bhakta D., Nair G.V., et al. . 2008. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N. Engl. J. Med. 358:2688–2697. 10.1056/NEJMoa062800 [DOI] [PubMed] [Google Scholar]
- Hadjantonakis A.K., Gertsenstein M., Ikawa M., Okabe M., and Nagy A.. 1998. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech. Dev. 76:79–90. 10.1016/S0925-4773(98)00093-8 [DOI] [PubMed] [Google Scholar]
- Harper P.S. 2001. Myotonic Dystrophy. W.B. Saunders Company, London. [Google Scholar]
- Ho T.H., Charlet-B N., Poulos M.G., Singh G., Swanson M.S., and Cooper T.A.. 2004. Muscleblind proteins regulate alternative splicing. EMBO J. 23:3103–3112. 10.1038/sj.emboj.7600300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Mankodi A., Swanson M.S., Moxley R.T., and Thornton C.A.. 2004. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13:3079–3088. 10.1093/hmg/ddh327 [DOI] [PubMed] [Google Scholar]
- Laurent V., Pellieux S., Corcia P., Magro P., Pierre B., Fauchier L., Raynaud M., and Babuty D.. 2011. Mortality in myotonic dystrophy patients in the area of prophylactic pacing devices. Int. J. Cardiol. 150:54–58. 10.1016/j.ijcard.2010.02.029 [DOI] [PubMed] [Google Scholar]
- Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H., et al. . 2013. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 5:1887–1900. 10.1002/emmm.201303275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., and Thornton C.A.. 2006. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15:2087–2097. 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- London B., Guo W., Pan Xh J.S., Lee V., Shusterman C.J., Rocco D.A., Logothetis J.M., Nerbonne, and Hill J.A.. 2001. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced QT prolongation. Circ. Res. 88:940–946. 10.1161/hh0901.090929 [DOI] [PubMed] [Google Scholar]
- Lueck J.D., Lungu C., Mankodi A., Osborne R.J., Welle S.L., Dirksen R.T., and Thornton C.A.. 2007a Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am. J. Physiol. Cell Physiol. 292:C1291–C1297. 10.1152/ajpcell.00336.2006 [DOI] [PubMed] [Google Scholar]
- Lueck J.D., Mankodi A., Swanson M.S., Thornton C.A., and Dirksen R.T.. 2007b Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J. Gen. Physiol. 129:79–94. 10.1085/jgp.200609635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M., Taft C.S., Bush E.W., Holder E., Bailey W.M., Neville H., Perryman M.B., and Bies R.D.. 1995. Identification, tissue-specific expression, and subcellular localization of the 80- and 71-kDa forms of myotonic dystrophy kinase protein. J. Biol. Chem. 270:20246–20249. 10.1074/jbc.270.35.20246 [DOI] [PubMed] [Google Scholar]
- Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barceló J., O’Hoy K., et al. . 1992. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 255:1253–1255. 10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., and Thornton C.A.. 2001. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10:2165–2170. 10.1093/hmg/10.19.2165 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., and Thornton C.A.. 2002. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 10:35–44. 10.1016/S1097-2765(02)00563-4 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Lin X., Blaxall B.C., Swanson M.S., and Thornton C.A.. 2005. Nuclear RNA foci in the heart in myotonic dystrophy. Circ. Res. 97:1152–1155. 10.1161/01.RES.0000193598.89753.e3 [DOI] [PubMed] [Google Scholar]
- Mathieu J., Allard P., Potvin L., Prévost C., and Bégin P.. 1999. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 52:1658–1662. 10.1212/WNL.52.8.1658 [DOI] [PubMed] [Google Scholar]
- Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., and Swanson M.S.. 2000. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19:4439–4448. 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell G.F., Jeron A., and Koren G.. 1998. Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274:H747–H751. [DOI] [PubMed] [Google Scholar]
- Munoz-Sanjuan I., Smallwood P.M., and Nathans J.. 2000. Isoform diversity among fibroblast growth factor homologous factors is generated by alternative promoter usage and differential splicing. J. Biol. Chem. 275:2589–2597. 10.1074/jbc.275.4.2589 [DOI] [PubMed] [Google Scholar]
- Nazarian S., Wagner K.R., Caffo B.S., and Tomaselli G.F.. 2011. Clinical predictors of conduction disease progression in type I myotonic muscular dystrophy. Pacing Clin. Electrophysiol. 34:171–176. 10.1111/j.1540-8159.2010.02905.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onkal R., Mattis J.H., Fraser S.P., Diss J.K., Shao D., Okuse K., and Djamgoz M.B.. 2008. Alternative splicing of Nav1.5: an electrophysiological comparison of “neonatal” and “adult” isoforms and critical involvement of a lysine residue. J. Cell. Physiol. 216:716–726. 10.1002/jcp.21451 [DOI] [PubMed] [Google Scholar]
- Osborne R.J., and Thornton C.A.. 2008. Cell-free cloning of highly expanded CTG repeats by amplification of dimerized expanded repeats. Nucleic Acids Res. 36:e24 10.1093/nar/gkn025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S.W., Kameyama A., Hao L.Y., Horiuchi M., Minobe E., Wang W.Y., Makita N., and Kameyama M.. 2005. Tetrodotoxin-resistant Na+ channels in human neuroblastoma cells are encoded by new variants of Nav1.5/SCN5A. Eur. J. Neurosci. 22:793–801. 10.1111/j.1460-9568.2005.04280.x [DOI] [PubMed] [Google Scholar]
- Pang P.D., Alsina K.M., Cao S., Koushik A.B., Wehrens X.H.T., and Cooper T.A.. 2018. CRISPR-mediated expression of the fetal Scn5a isoform in adult mice causes conduction defects and arrhythmias. J. Am. Heart Assoc. 7:e010393 10.1161/JAHA.118.010393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.M., Shin K.J., Kim S.E., Park J., Ha S.Y., and Kim B.J.. 2013. Prolonged corrected QT interval in patients with myotonic dystrophy type 1. J. Clin. Neurol. 9:186–191. 10.3988/jcn.2013.9.3.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M., Vicente M., Monferrer L., and Artero R.. 2006. The muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 74:65–80. 10.1111/j.1432-0436.2006.00060.x [DOI] [PubMed] [Google Scholar]
- Philips A.V., Timchenko L.T., and Cooper T.A.. 1998. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 280:737–741. 10.1126/science.280.5364.737 [DOI] [PubMed] [Google Scholar]
- Phillips M.F., and Harper P.S.. 1997. Cardiac disease in myotonic dystrophy. Cardiovasc. Res. 33:13–22. 10.1016/S0008-6363(96)00163-0 [DOI] [PubMed] [Google Scholar]
- Pitt G.S., and Lee S.Y.. 2016. Current view on regulation of voltage-gated sodium channels by calcium and auxiliary proteins. Protein Sci. 25:1573–1584. 10.1002/pro.2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F., Freyermuth F., Fugier C., Villemin J.P., Fischer M.C., Jost B., Dembele D., Gourdon G., Nicole A., Duboc D., et al. . 2011. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18:840–845. 10.1038/nsmb.2067 [DOI] [PubMed] [Google Scholar]
- Rijnbeek P.R., van Herpen G., Bots M.L., Man S., Verweij N., Hofman A., Hillege H., Numans M.E., Swenne C.A., Witteman J.C., and Kors J.A.. 2014. Normal values of the electrocardiogram for ages 16-90 years. J. Electrocardiol. 47:914–921. 10.1016/j.jelectrocard.2014.07.022 [DOI] [PubMed] [Google Scholar]
- Sarkar P.S., Han J., and Reddy S.. 2004. In situ hybridization analysis of Dmpk mRNA in adult mouse tissues. Neuromuscul. Disord. 14:497–506. 10.1016/j.nmd.2004.03.012 [DOI] [PubMed] [Google Scholar]
- Savkur R.S., Philips A.V., and Cooper T.A.. 2001. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29:40–47. 10.1038/ng704 [DOI] [PubMed] [Google Scholar]
- Schroeter A., Walzik S., Blechschmidt S., Haufe V., Benndorf K., and Zimmer T.. 2010. Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J. Mol. Cell. Cardiol. 49:16–24. 10.1016/j.yjmcc.2010.04.004 [DOI] [PubMed] [Google Scholar]
- Sznajder L.J., Michalak M., Taylor K., Cywoniuk P., Kabza M., Wojtkowiak-Szlachcic A., Matłoka M., Konieczny P., and Sobczak K.. 2016. Mechanistic determinants of MBNL activity. Nucleic Acids Res. 44:10326–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja K.L., McCurrach M., Schalling M., Housman D., and Singer R.H.. 1995. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 128:995–1002. 10.1083/jcb.128.6.995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C.A. 2014. Myotonic dystrophy. Neurol. Clin. 32:705–719: viii. 10.1016/j.ncl.2014.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahbi K., Meune C., Porcher R., Bécane H.M., Lazarus A., Laforêt P., Stojkovic T., Béhin A., Radvanyi-Hoffmann H., Eymard B., and Duboc D.. 2012. Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. JAMA. 307:1292–1301. 10.1001/jama.2012.346 [DOI] [PubMed] [Google Scholar]
- Wahbi K., Algalarrondo V., Bécane H.M., Fressart V., Beldjord C., Azibi K., Lazarus A., Berber N., Radvanyi-Hoffman H., Stojkovic T., et al. . 2013. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch. Cardiovasc. Dis. 106:635–643. 10.1016/j.acvd.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Wahbi K., Babuty D., Probst V., Wissocque L., Labombarda F., Porcher R., Bécane H.M., Lazarus A., Béhin A., Laforêt P., et al. . 2017. Incidence and predictors of sudden death, major conduction defects and sustained ventricular tachyarrhythmias in 1388 patients with myotonic dystrophy type 1. Eur. Heart J. 38:751–758. [DOI] [PubMed] [Google Scholar]
- Walzik S., Schroeter A., Benndorf K., and Zimmer T.. 2011. Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS One. 6:e19188 10.1371/journal.pone.0019188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Hennessey J.A., Kirkton R.D., Wang C., Graham V., Puranam R.S., Rosenberg P.B., Bursac N., and Pitt G.S.. 2011. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109:775–782. 10.1161/CIRCRESAHA.111.247957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. . 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 150:710–724. 10.1016/j.cell.2012.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Tang H., Wei E.Q., Wang Z., Yang J., Yang R., Wang S., Zhang Y., Pitt G.S., Zhang H., and Wang C.. 2017. Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J. Mol. Cell. Cardiol. 104:63–74. 10.1016/j.yjmcc.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T.M., Lueck J.D., Swanson M.S., Dirksen R.T., and Thornton C.A.. 2007. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 117:3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Guo W., and Nerbonne J.M.. 1999a Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J. Gen. Physiol. 113:661–678. 10.1085/jgp.113.5.661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Li H., and Nerbonne J.M.. 1999b Elimination of the transient outward current and action potential prolongation in mouse atrial myocytes expressing a dominant negative Kv4 alpha subunit. J. Physiol. 519:11–21. 10.1111/j.1469-7793.1999.0011o.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Wang Z., Sinden D.S., Wang X., Shan B., Yu X., Zhang H., Pitt G.S., and Wang C.. 2016. FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific manner. Channels (Austin). 10:410–420. 10.1080/19336950.2016.1190055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wu J., King J.H., Huang C.L., and Fraser J.A.. 2014. Measurement and interpretation of electrocardiographic QT intervals in murine hearts. Am. J. Physiol. Heart Circ. Physiol. 306:H1553–H1557. 10.1152/ajpheart.00459.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]