

Bohannon et al. show that polyunsaturated fatty acids (PUFAs) vary in their ability to activate cardiac IKs potassium channels depending on the pKa of the PUFA head group, allowing PUFAs to be tuned to treat long QT syndrome mutants with different disease severity.
Abstract
Repolarization and termination of the ventricular cardiac action potential is highly dependent on the activation of the slow delayed-rectifier potassium IKs channel. Disruption of the IKs current leads to the most common form of congenital long QT syndrome (LQTS), a disease that predisposes patients to ventricular arrhythmias and sudden cardiac death. We previously demonstrated that polyunsaturated fatty acid (PUFA) analogues increase outward K+ current in wild type and LQTS-causing mutant IKs channels. Our group has also demonstrated the necessity of a negatively charged PUFA head group for potent activation of the IKs channel through electrostatic interactions with the voltage-sensing and pore domains. Here, we test whether the efficacy of the PUFAs can be tuned by the presence of different functional groups in the PUFA head, thereby altering the electrostatic interactions of the PUFA head group with the voltage sensor or the pore. We show that PUFA analogues with taurine and cysteic head groups produced the most potent activation of IKs channels, largely by shifting the voltage dependence of activation. In comparison, the effect on voltage dependence of PUFA analogues with glycine and aspartate head groups was half that of the taurine and cysteic head groups, whereas the effect on maximal conductance was similar. Increasing the number of potentially negatively charged moieties did not enhance the effects of the PUFA on the IKs channel. Our results show that one can tune the efficacy of PUFAs on IKs channels by altering the pKa of the PUFA head group. Different PUFAs with different efficacy on IKs channels could be developed into more personalized treatments for LQTS patients with a varying degree of IKs channel dysfunction.
Introduction
The ventricular cardiac action potential is controlled by the activation of depolarizing and repolarizing ionic currents. One of the dominant repolarizing currents during the ventricular action potential is the slow delayed-rectifier potassium current (IKs), which is critical for the timing of action potential termination (Barhanin et al., 1996; Sanguinetti et al., 1996; Salata et al., 1996). Ion channel mutations, or channelopathies, are the root of many pathological conditions, including the arrhythmogenic disorder long QT syndrome (LQTS; Bohnen et al., 2017; Alders and Christiaans, 2003; Schwartz et al., 2012). LQTS is an inherited disorder that is characterized by a prolonged QT interval—the time between ventricular depolarization and repolarization—on the electrocardiogram (Schwartz et al., 2012; Waddell-Smith and Skinner, 2016). LQTS-causing channelopathies have been discovered in many different channels, including voltage-gated Na+ channels, Ca2+ channels, and K+ channels (Alders and Christiaans, 2003; Bohnen et al., 2017; Drum et al., 2014; Harmer et al., 2010; Rivolta et al., 2002). However, the most common form of LQTS (LQT1) is caused by mutations in the voltage-gated K+ channel known as the IKs channel.
The IKs channel underlies the slow component of the delayed-rectifier K+ current and is composed of the voltage-gated K+ channel, Kv7.1 α subunit, and the KCNE1 accessory β subunit (Barhanin et al., 1996; Salata et al., 1996; Sanguinetti et al., 1996). The Kv7.1 α subunit has six transmembrane-spanning segments called S1–S6 (Peroz et al., 2008; Smith et al., 2007). Segments S1–S4 make up the voltage-sensing domain, in which S4 functions as a voltage sensor due to the presence of several positively charged arginine residues (Peroz et al., 2008). Segments S5 and S6 comprise the pore domain. When the membrane becomes depolarized, the S4 segment moves outward, leading to a conformational change that allows the pore to open and K+ ions to flow outward. Kv7.1 forms a macromolecular complex with the β subunit KCNE1, which dramatically alters the voltage dependence and kinetics of Kv7.1 channel activation, to generate the physiological IKs current (Barhanin et al., 1996; Salata et al., 1996; Sanguinetti et al., 1996; Barro-Soria et al., 2014; Osteen et al., 2010).
Loss-of-function mutations in the cardiac IKs channel that lead to LQT1 result in reduced repolarizing IKs current and, as a result, prolongation of the ventricular action potential. LQT1 is particularly dangerous because it predisposes individuals to torsades de pointes, which can lead to ventricular fibrillation and sudden cardiac death (Bohnen et al., 2017). Entry into cardiac arrhythmia in patients with LQT1 is often triggered by β-adrenergic stimulation, whether by exercise or intense emotional stress (Bohnen et al., 2017; Schwartz, et al., 2012; Wu et al., 2016). Treatment options for LQTS include pharmacological attenuation of β-adrenergic stimulation by β blockers or the implantation of a cardioverter defibrillator (Schwartz et al., 2012; Cho, 2016; Waddell-Smith and Skinner, 2016). Though these treatments help to prevent arrhythmia or stop arrhythmia, they do not work for all individuals (Chockalingam et al., 2012; Schwartz et al., 2012), and they do not directly target the underlying channelopathies that lead to LQTS (Schwartz et al., 2017). Therefore, there is a need for new therapeutics that directly target the channelopathies that lead to LQTS.
We previously showed that the activity of the IKs channel can be modified by lipids, such as polyunsaturated fatty acids (PUFAs; Liin et al., 2015, 2016). PUFAs and PUFA analogues are amphipathic molecules that have two distinct structural regions that can participate in interactions with membrane proteins: (1) a charged hydrophilic head group, and (2) a long hydrophobic tail with two or more double bonds. PUFAs and PUFA analogues influence the activation of K+ channels through a lipoelectric mechanism in which the hydrophobic tail integrates into the cell membrane near the voltage-sensing domain and electrostatically attracts the positively charged S4 through its negatively charged hydrophilic head group, thus facilitating channel activation (Fig. 1, A and B). We recently demonstrated that in PUFAs with a carboxyl head group, the position of the double bonds in the tail correlates significantly with apparent binding affinity to the IKs channel (Bohannon et al., 2019). Specifically, having the first double bond close to the carboxyl head group is important for high apparent binding affinity for the IKs channel and PUFA-induced enhancement of IKs current (Bohannon et al., 2019). It is known that a negatively charged head group is necessary for activation of voltage-gated K+ channels (Liin et al., 2015; Börjesson et al., 2008). Docosahexaenoic acid (DHA), which can bear a negative charge at its carboxyl head group, shifts the voltage dependence of Kv7.1 channel activation to more negative voltages; however, coexpression of KCNE1 abolishes DHA sensitivity (Liin et al., 2015). KCNE1 was recently shown to tune PUFA sensitivity by inducing a conformational change of the S5-P-helix loop that results in protonation of the PUFA head group (Larsson et al., 2018). This protonation can be circumvented by using PUFA analogues that are negatively charged at physiological pH (pH 7.4), such as DHA-glycine or N-arachidonoyl taurine (Liin et al., 2015, 2016). In addition to an electrostatic effect on the voltage sensor of the IKs channel, our group also recently demonstrated that PUFA analogues have an additional effect on the pore of the IKs channel (Liin et al., 2018): A lysine residue (K326) in the S6 helix of the IKs channel electrostatically interacts with the negatively charged head group of PUFA analogues, and this electrostatic interaction increases the maximal conductance (Gmax) of the cardiac IKs channel (Liin et al., 2018; Fig. 1, B–D).
Figure 1.

Illustration of the lipoelectric mechanism and measured effects on the cardiac IKs channel. (A) Schematic side view (left) of the IKs channel with S4 in green. Illustration (right) of the electrostatic interaction of a PUFA analogue (orange) with the voltage sensor (green) of the cardiac IKs channel, which leads to potentiation of upward S4 movement. (B) Schematic top view (left) of the IKs channel with Kv7.1 in blue (light blue, pore domain; dark blue, voltage-sensing domain) and KCNE1 in purple. Illustration (right) of the electrostatic interaction of a PUFA analogue (orange) with the positively charged lysine residue K326 in the S6 segment of the cardiac IKs channel, which leads to an increase in the Gmax of the IKs channel. (C) Activation protocol for the cardiac IKs channel using two-electrode voltage clamp and raw current traces in 0 µM PUFA analogue (left) and 20 µM PUFA analogue (right), with arrows indicating tail currents. Red trace occurs at 20 mV for visualization of PUFA-induced increases in current. (D) Representative current versus voltage relationship in 0 µM (black line) and 20 µM PUFA (blue line) highlighting an increase in I/I0 at 0 mV, leftward shift in the V0.5, and increase in Gmax denoted by arrows.
A thorough characterization of PUFA analogues with a wide range of effects on the cardiac IKs channel provides a means to develop novel treatments for LQT1-causing mutations of different severity. LQT1 is variable in its severity and can present with different symptoms based on the individual (Schwartz et al., 2012). For example, some patients carrying a mutation in KCNQ1 can have milder phenotypes associated with less severe prolongation of the QT interval (Schwartz et al., 2012; Wu et al., 2016; Amin et al., 2012; Chouabe et al., 2000). For example, R533W, which causes a positive shift of ∼15 mV in the voltage dependence of activation, is associated with a milder cardiac phenotype (Chouabe et al., 2000). In other cases, such as for the KCNQ1 mutation A341V, that is one of the most severe presentations of LQT1; >30% of patients experience cardiac arrest or sudden cardiac death (Crotti et al., 2007; Schwartz et al., 2012). These examples highlight extreme differences in the manifestation of LQT1 in the clinical population that occur in a mutation-specific manner. Treatment for such distinct phenotypes requires an individualized approach. For this reason, there is a need to find new ways in which the effects of PUFA analogues can be tuned, allowing for more personalized treatment options for patients with LQT1. The purpose of the present study was to evaluate different PUFA head groups to determine if the activating effects of PUFA analogues can be enhanced or attenuated through modifications to the charged PUFA head group.
Materials and methods
Molecular biology
Kv7.1 and KCNE1 channel complementary RNA were transcribed using the mMessage mMachine T7 kit (Ambion). 50 ng of complementary RNA was injected at a 3:1, weight/weight (Kv7.1/KCNE1) ratio into defolliculated Xenopus laevis oocytes (Ecocyte) for IKs channel expression. Site-directed mutagenesis was performed using the Quickchange II XL Mutagenesis Kit (QIAGEN Sciences) for mutations in the Kv7.1 α subunit. Injected cells were incubated for 72–96 h in standard ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM HEPES; pH 7.5) containing 1 mM pyruvate at 16°C before electrophysiological recordings.
Two-electrode voltage clamp
X. laevis oocytes were recorded in the two-electrode voltage-clamp configuration. Recording pipettes were filled with 3 M KCl. The chamber was filled with ND96 recording solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM Tricine; pH 9.0 or 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM HEPES; pH 7.5). PUFAs were obtained from Cayman Chemical or were synthesized in-house (University of Linköping) and kept at −20°C as 100-mM stock solutions in ethanol. Serial dilutions of the different PUFAs were prepared from stocks to make 0.2-, 0.7-, 2-, 7-, 20-, and 70-µM concentrations in ND96 solutions (ND96 recording solutions were made at both pH 7.5 and pH 9.0). PUFAs were perfused into the recording chamber using the Rainin Dynamax Peristaltic Pump (Model RP-1). Combinations of PUFA analogues, monounsaturated fatty acids (MUFAs), saturated fatty acids (SFAs), and albumin (A7906-10G; Sigma-Aldrich) were applied to emulate physiological circulation of fatty acids in the human body. We applied the PUFA analogue linoleoyl glycine (lin-glycine; 0.2 mM), the MUFA oleic acid (0.2 mM), and the SFA stearic acid (0.2 mM) in combination with albumin (0.1 mM) to estimate the effect of physiological levels of PUFAs on IKs channels in the presence of physiological levels of the fatty acid–binding protein albumin and other fatty acids in interstitial space (Abdelmagid et al., 2015; Tsukamoto and Sugawara, 2018).
Electrophysiological recordings were obtained using Clampex 10.3 software (Axon, pClamp; Molecular Devices). During the application of PUFAs, the membrane potential was stepped every 30 s from −80 mV to 0 mV for 5 s before stepping to −40 mV and back to −80 mV. The application protocol was used to ensure that the PUFA effects reached steady state. The PUFA effects reached steady state in 10 min when applied at the highest concentrations (7 and 20 µM). A voltage-step protocol (Fig. 1 C, inset) was used to measure the current versus voltage (I-V) relationship before PUFA application and after the PUFA effects had reached steady state for each concentration of PUFA. During the activation protocol, cells were held at −80 mV followed by a hyperpolarizing prepulse to −140 mV. The voltage was then stepped from −100 to 60 mV (Δ20 mV) followed by a voltage step to −20 mV to measure tail currents. Following the test pulse to measure tail currents (Fig. 1 C, arrows), cells were held again at −80 mV.
Data analysis
Tail currents were analyzed using Clampfit 10.3 software in order to obtain conductance versus voltage (G-V) curves to determine the voltage dependence of channel activation. The V0.5, the voltage at which half the maximal tail current occurs, was obtained by fitting the G-V curves from each concentration of PUFA with a Boltzmann equation (Fig. 1 D):
where Gmax is the maximal conductance at positive voltages and s is the slope factor in millivolts. The current values for each concentration at 0 mV were used to plot the dose–response curves for each PUFA. Dose–response curves were fit using the Hill equation in order to obtain the Km value for each PUFA:
where A is the relative increase in current (ΔI/I0) caused by the PUFA at saturating concentrations, Km is the apparent affinity of the PUFA, x is the concentration, and n is the Hill coefficient. In some cases, there was variability in the V0.5 between batches of oocytes. To correct for variability between batches of oocytes, we applied a correction to compensate for a V0.5 that differs greatly from 20 mV (the typical V0.5 for the IKs channel). This allowed us to more consistently measure PUFA-induced IKs current increases. In our correction, we subtracted the V0.5 (obtained from using the Boltzmann equation) by 20 mV and used the current measured at the resulting voltage. The Gmax for each concentration was obtained from the fitted values given by the Boltzmann fit and then normalized to the Gmax in control solution (0 µM), Gmax0. All data is given as mean ± SEM. Graphs plotting mean and SEM for I/I0, ΔV0.5, Gmax, and Km were generated using Origin 9 software. PUFA artwork was made using CorelDRAW Software. To determine if there were significant differences between PUFA-induced effects on I/I0, ΔV0.5, and Gmax, we conducted one-way ANOVA followed by Tukey’s honestly significant difference test (HSD) for multiple comparisons. For data in which dose–response curves were well fitted, we used the fitted max values and Km for the statistical tests. For data in which dose–response curves were not clearly reaching saturation, we used the values at 20 µM for the statistical tests. Significance α-level was set at P < 0.05; asterisks denote significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
Estimated pKa values
pKa, the negative log of the acid dissociation constant, values of PUFA analogues in solution were calculated using Marvin Software (ChemAxon). However, studies of PUFAs in lipid bilayers and our previous studies on PUFA–IKs channel interactions showed that there is a large difference in the pKa values in solution (calculated according to the structure) compared with the measured pKa of PUFAs in the lipid bilayer (Börjesson and Elinder, 2011; Elinder and Liin, 2017) and in close contact with the IKs channel (Elinder and Liin, 2017; Liin et al., 2015). The average difference between the calculated solution pKa values and experimental found pKa values for PUFAs associated with IKs channels is ∼3.5 (Liin et al., 2015). We therefore added this correction factor to the calculated solution pKa values to generate our estimated pKa value for PUFAs associated with the IKs channel.
Hierarchical cluster analysis
Hierarchical cluster analysis was performed using BioVinci data visualization software (BioTuring). Effects on I/I0, ΔV0.5, and Gmax at 20 µM were normalized to the PUFA analogue with the largest influence on each of the three effects so that these effects were now scaled from 0.0 to 1.0, 1.0 being the largest effect. Each parameter (I/I0, ΔV0.5, and Gmax) was then used as input for clustering to generate the dendrogram and heat map.
Online supplemental material
Specific synthesis pathways for PUFA analogues that were synthesized in-house are described in detail in the Supplemental materials and methods. Fig. S1 contains data on the current versus voltage relationship between pH 7.5 and pH 9.0. Fig. S2 shows that reduction of IKs current by the application of DHA-taurine is not intrinsically related to the taurine head group alone. Fig. S3 shows that residues in the voltage sensor and pore are important for electrostatic activation of the cardiac IKs channel. Fig. S4 contains effects of lin-AP3 on IKs activation at pH 9.0.
Figure S1.
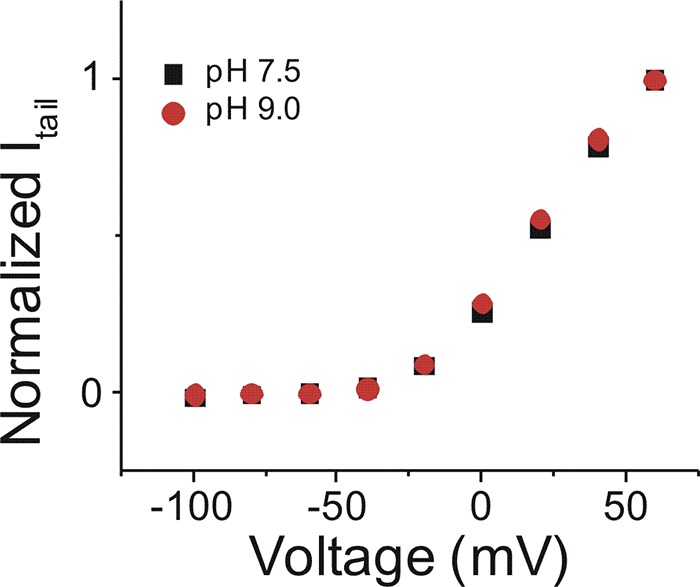
Current versus voltage relationship between pH 7.5 and pH 9.0. Current–voltage relationship of IKs channel in pH 7.5 (black squares; mean ± SEM; n = 4) and pH 9.0 (red circles; mean ± SEM; n = 4).
Figure S2.
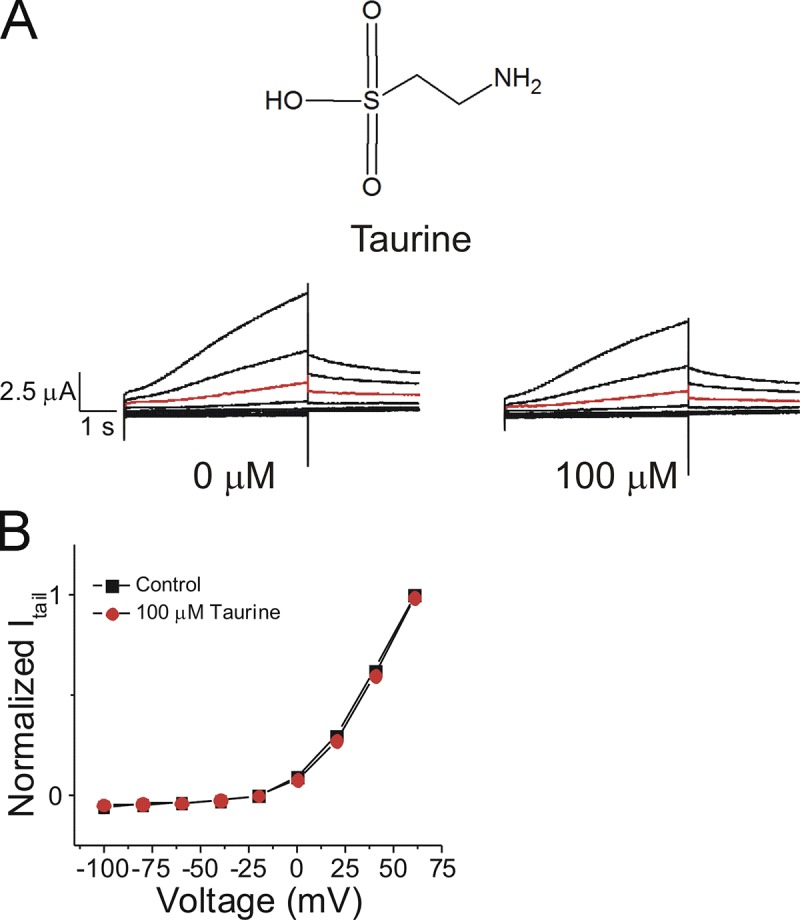
Reduction of IKs current by the application of DHA taurine is not intrinsically related to the taurine head group alone. (A) Structure of taurine and raw current traces measured in 0 µM taurine (left) and 100 µM taurine (right). Red trace shows currents at 20 mV for visualization of no effects of taurine on the voltage dependence. (B) Current versus voltage relationship in control (0 µM taurine) and following the addition of 100 µM taurine (mean ± SEM; n = 4).
Figure S3.

Residues in the voltage sensor and pore are important for electrostatic activation of the cardiac IKs channel. (A) Current–voltage relationship for Kv7.1 R231Q Q234R + KCNE1 with the application of lin-glycine (mean ± SEM; n = 4). (B-D) Dose-dependent effects of lin-glycine on Kv7.1 R231Q Q234R + KCNE1 (black squares) and the wild- type IKs channel (red dashed line) on (B) I/I0, (C) ΔV0.5, and (D) Gmax. (E) Current-voltage relationship for Kv7.1 K326Q + KCNE1 with the application of lin-glycine (mean ± SEM; n = 4). (F-H) Dose-dependent effects of lin-glycine on Kv7.1 K326Q + KCNE1 (black squares) and the wild-type IKs channel (red dashed line) on (F) I/I0, (G) ΔV0.5, (H) Gmax.
Figure S4.
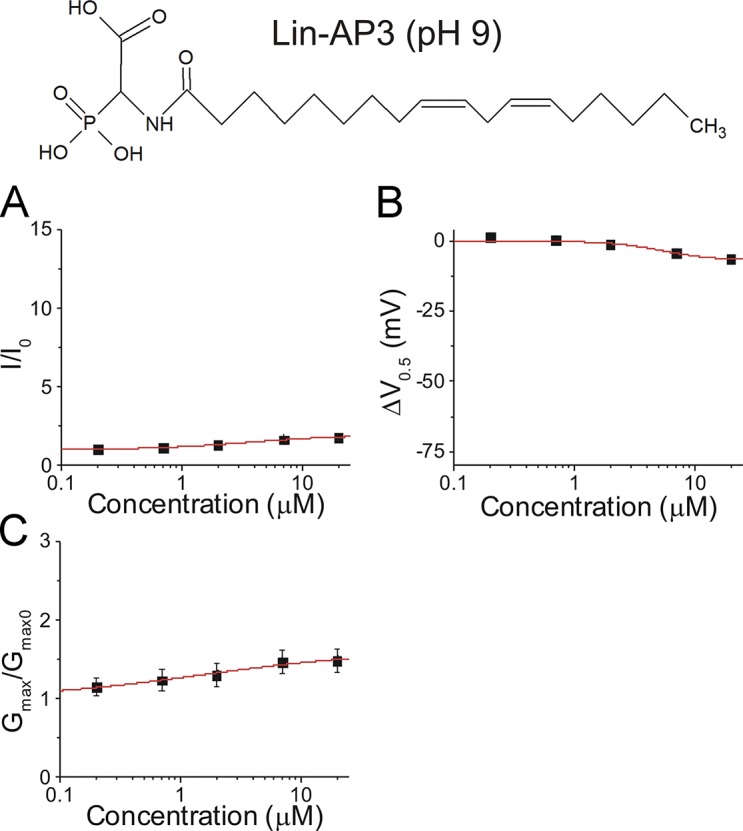
Effects of lin-AP3 on IKs activation at pH 9.0. (A-C) Dose-dependent effects of lin-AP3 on (A) IKs current (I/I0), (B) IKs voltage dependence (ΔV0.5), and (C) IKs Gmax at pH 9.0 (mean ± SEM at maximal concentration; n = 3).
Results
Linoleoyl-taurine (lin-taurine) and lin-glycine increased the IKs current by differentially affecting the V0.5 and the Gmax
We previously demonstrated that the negative charge of a PUFA with a carboxyl head group is neutralized by the presence of KCNE1 in IKs channels (Larsson et al., 2018). For this reason, PUFAs with a carboxyl head group tend to have little effect on IKs channel activation at physiological pH. In this study, we investigated the effects of other head groups that are expected to promote IKs channel activation through the lipoelectric mechanism (Fig. 1, A and B) with effects on the voltage sensor (Fig. 1 A) and the pore (Fig. 1 B). We compared the effects between PUFAs with varying functional groups of the hydrophilic PUFA head but with the same hydrocarbon tail. To do this, we used a two-electrode voltage clamp and a series of depolarizing voltage steps to measure the effects of PUFAs on IKs current (Fig. 1 C). This allowed us to measure the effects on the normalized current at 0 mV (I/I0), the shift in voltage dependence of IKs channel activation (ΔV0.5), and the Gmax (Fig. 1 D).
We first compared three PUFAs and PUFA analogues that have a linoleic acid tail: linoleic acid, lin-glycine, and lin-taurine (Fig. 2, A–C). Application of 20 µM linoleic acid (Fig. 2 A), which has a carboxyl head group, did not increase in I/I0 (0.5 ± 0.1; Fig. 2 D), did not left-shift the V0.5 of IKs channel activation (4.7 ± 0.9 mV; Fig. 2 F), and did not increase the Gmax (0.7 ± 0.1; Fig. 2 H). Lin-glycine, when applied at 20 µM (Fig. 2 B), produced a moderate increase in I/I0 (5.3 ± 0.5; Fig. 2 D) and a moderate shift in the V0.5 (−26.4 ± 4.4 mV; Fig. 2 F) and produced the largest increase in the Gmax (2.4 ± 0.2; Fig. 2 H). Lin-taurine, when applied at 20 µM (Fig. 2 C), produced the largest increase in I/I0 (10.4 ± 4.0; Fig. 2 D) and the largest left-shift in the V0.5 (−73.1 ± 2.6 mV; Fig. 2 F) and increased the Gmax (2.0 ± 0.6; Fig. 2 H). Statistical analysis of the fitted parameters of the dose–response curves show that lin-taurine had the biggest increase in I/I0 (Fig. 2 E) and V0.5 (Fig. 2 G), whereas lin-glycine had the biggest increase in Gmax (Fig. 2 I). The size of the voltage shifts caused by the three PUFAs correlates with the predicted protonation (i.e., charge) of the different head groups (from the pKa values estimated for carboxyl, glycine, and taurine head groups in the lipid bilayer) at physiological pH (Table 1). In contrast, the effects on Gmax did not correlate with the predicted charge of the PUFA head groups.
Figure 2.

Lin-taurine produces the most potent activation of the IKs channel compared with lin-glycine and linoleic acid. (A–C) Structure of and raw current traces measured in 0 µM (left) and 20 µM (right) (A) linoleic acid, (B) lin-glycine, and (C) lin-taurine. Red trace shows currents at 20 mV for visualization of PUFA-induced effects on current. (D) Dose-dependent effects of linoleic acid (n = 5), lin-glycine (n = 4), and lin-taurine (n = 3) on IKs current (I/I0; mean ± SEM at maximal concentration). (E) Statistical differences on I/I0 effects (I/I0 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (F) Dose-dependent effects of linoleic acid, lin-glycine, and lin-taurine on IKs voltage dependence (ΔV0.5). (G) Statistical differences on ΔV0.5 effects (ΔV0.5 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (H) Dose-dependent effects of linoleic acid, lin-glycine, and lin-taurine on IKs Gmax. (I) Statistical differences on Gmax effects (Gmax fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Table 1. PUFAs/PUFA analogues and their estimated pKa values.
| PUFA | pKa1 | pKa2 | pKa3 |
|---|---|---|---|
| Linoleic acid | 8.5 | NA | NA |
| Lin-glycine | 7.6 | NA | NA |
| Lin-taurine | 2.7 | NA | NA |
| Lin-glycine+1C | 8.0 | NA | NA |
| Lin-glycine+2C | 8.0 | NA | NA |
| Lin-aspartate | 7.6 | 9.1 | NA |
| Lin-cysteic acid | 2.6 | 7.1 | NA |
| Lin-AP3 | 5.0 | 7.7 | 11.8 |
| DHA | 8.3 | NA | NA |
| DHA-glycine | 7.5 | NA | NA |
| DHA-taurine | 2.8 | NA | NA |
| Pinolenic acid | 8.4 | NA | NA |
| Pin-glycine | 7.5 | NA | NA |
| Pin-taurine | 2.8 | NA | NA |
Estimated pKa values for PUFAs and PUFA analogues associated with the cardiac IKs channel were calculated by adding a factor of 3.5 to the starting pKa value calculated in solution.
The different effects of lin-glycine and lin-taurine on the V0.5 were not due to differences in the lengths of the PUFA head groups
One structural difference between the head groups of lin-glycine and lin-taurine was that the glycine group was shorter in length than the taurine group (Fig. 2, B and C). Therefore, we explored whether the different lengths of the head groups could explain the different activating effects of the two PUFAs on the IKs channel. To do so, we inserted additional carbons into the head group of lin-glycine to elongate the glycine head group and then compared the effects of lin-glycine, lin-glycine+1C (Fig. 3 A), and lin-glycine+2C (Fig. 3 B). With the insertion of one additional carbon in the glycine head group, lin-glycine+1C had a similar length as lin-taurine. Application of lin-glycine produced an increase in I/I0 (5.3 ± 0.5), whereas lin-glycine+1C and +2C surprisingly produced a smaller increase in I/I0 (1.7 ± 0.1 and 1.6 ± 0.1, respectively; Fig. 3, C and I). In addition, lin-glycine produced the largest shift in the V0.5 (−26.4 ± 4.4 mV) compared with lin-glycine+1C (−7.2 ± 2.5 mV) and lin-glycine+2C (−8.7 ± 0.5 mV; Fig. 3, D and J). Lin-glycine increased the Gmax of the IKs channel (2.4 ± 0.2), whereas lin-glycine+1C and lin-glycine+2C produced no change in the Gmax (1 ± 0.1 and 0.9 ± 0.1, respectively; Fig. 3, E and K).
Figure 3.

Increasing the length of the lin-glycine head group alters the pKa and reduces the activating effect on the IKs channel. (A and B) Structure of (A) lin-glycine with the addition of one carbon in the head group (lin-glycine+1C) and (B) lin-glycine with the addition of two carbons in the head group (lin-glycine+2C). (C–H) Dose-dependent effects of lin-glycine (n = 4), lin-glycine+1C (n = 3), and lin-glycine+2C (n = 3) on (C) IKs current (I/I0) at pH 7.5, (D) IKs voltage dependence (ΔV0.5) at pH 7.5, (E) IKs Gmax at pH 7.5, (F) I/I0 at pH 9.0, (G) ΔV0.5 at pH 9.0, and (H) Gmax at pH 9.0 (mean ± SEM at maximal concentration). (I–K) Statistical differences at 20 µM on (I) I/I0 effect, (J) ΔV0.5 effect, and (K) Gmax effect measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant.
One possible mechanism behind the decreased effects of lin-glycine+1C and lin-glycine+2C compared with lin-glycine is that the addition of carbons in the glycine head group shifts the pKa of the head group, which thereby promotes protonation and loss of the negative charge in the head group. We therefore repeated the experiments with lin-glycine, lin-glycine+1C, and lin-glycine+2C at pH 9.0. Using PUFAs with a carboxyl head group, we previously demonstrated that conducting experiments at pH 9.0 can deprotonate the head group to restore its negative charge and allow PUFAs to activate the IKs channel (Bohannon et al., 2018; Liin et al., 2015). Changing the solution from pH 7.5 to pH 9.0 did not alter the normal activation of the IKs channel (Fig. S1). At pH 9.0, lin-glycine, lin-glycine+1C, and lin-glycine+2C all produced a similar left-shift in the voltage dependence of IKs channel activation at 20 µM (−43.6 ± 1.6 mV, −41.7 ± 1.7 mV, and −47.9 ± 2.4 mV, respectively; Fig. 3, G and J). Note that at pH 9.0, application of lin-glycine resulted in a larger left-shift in the voltage dependence of the IKs channel compared with the left-shifting effect of lin-glycine at pH 7.5 (Fig. 3 J). This is consistent with our estimated pKa = 7.6 for lin-glycine: At pH 7.5, 50% of lin-glycine was negatively charged, whereas at pH 9.0, lin-glycine was almost fully in its deprotonated and negatively charged form. Lin-glycine displayed higher apparent affinity and began to shift the V0.5 at lower concentrations compared with lin-glycine+1C and lin-glycine+2C (Fig. 3 G). Although the left-shifting effects of lin-glycine, lin-glycine+1C, and lin-glycine+2C were improved at pH 9.0 (Fig. 3, G and J), all three PUFA analogues decreased the Gmax of the channel (0.6 ± 0.1, 0.9 ± 0.1, and 0.5 ± 0.1, respectively) at pH 9.0 (Fig. 3, H and K). The reason for this decrease in Gmax is unclear. At pH 9.0, lin-glycine, lin-glycine+1C, and lin-glycine+2C all increased I/I0 (2.3 ± 0.1, 2.7 ± 0.3, and 1.8 ± 0.04, respectively; Fig. 3, F and I).
The finding that the voltage-shifting effects of lin-glycine+1C and lin-glycine+2C are similar to that of lin-glycine at pH 9.0 but smaller at pH 7.5 suggests that the addition of one and two additional carbons in the glycine head group shifts the pKa of the glycine head group and reduces the likelihood that the glycine head will be deprotonated and negatively charged at pH 7.5. The size of the voltage shifts for lin-glycine+1C and lin-glycine+2C at pH 7.5 (50% smaller than for lin-glycine) is consistent with our estimated pKa values of lin-glycine+1C and lin-glycine+2C, which are both ∼8.0 compared with 7.6 for lin-glycine (Table 1). That the voltage-shifting effects of lin-glycine, lin-glycine+1C, and lin-glycine+2C at pH 9.0 are all similar suggests that is it not the length of the head group that renders lin-taurine more effective that lin-glycine but mainly the protonation state of the PUFA head groups.
Increasing the number of potentially charged moieties on the PUFA head group did not further promote IKs channel activation
Because we previously found that the charge of the head group is important for activating the cardiac IKs channel, we tested whether it is possible to further improve the activating effects of PUFA analogues by increasing the charge available on the PUFA head group. To do so, we compared PUFA analogues that have one possible charge (lin-taurine and lin-glycine), two possible charges (linoleoyl-aspartate [lin-aspartate] and linoleoyl-cysteic acid [lin-cysteic acid]; Fig. 4, A and B), and three possible charges (lin-AP3; Fig. 4 C). Interestingly, increasing the number of potentially negatively charged groups on the PUFA head group did not further improve the effects on I/I0, V0.5, or Gmax. Lin-aspartate, which has two potentially charged moieties, moderately increased I/I0 (4.0 ± 0.1; Fig. 4 D), moderately left-shifted the V0.5 (−34.5 ± 2.3 mV; Fig. 4 E), and moderately increased Gmax (1.4 ± 0.1; Fig. 4 F). The effects of lin-aspartate were similar to the effects of lin-glycine, which has only one potentially charged moiety (Fig. 4, G–I). Lin-cysteic acid (Fig. 4 C), which also possesses two potentially charged moieties, substantially increased I/I0 (9.2 ± 0.4; Fig. 4 D), substantially left-shifted the V0.5 of IKs channel activation (−58.4 ± 2.8 mV; Fig. 4 E), and substantially increased the Gmax (2.0 ± 0.2; Fig. 4 F). The effects of lin-cysteic acid were similar to the effects of lin-taurine, which has only one potentially charged moiety (Fig. 4, G–I). Lastly, lin-AP3, which has three potential negative charges, produced the smallest increase in I/I0 (1.8 ± 0.3; Fig. 4, D and G) and the smallest left-shift in the V0.5 (−5.7 ± 1.3 mV; Fig. 4, E and H) and produced no change in the Gmax (1.1 ± 0.1; Fig. 4, F and I). Together, these data show that having more than one potentially charged moiety of the head group does not necessarily improve the efficacy of PUFA analogues, leading us to concentrate on glycine and taurine head groups as potential therapeutics for LQTS.
Figure 4.

Increasing the number of potentially charged moieties of the PUFA head group does not improve PUFA-induced IKs activation. (A–C) Structure of and raw current traces measured in 0 µM (left) and 20 µM (right) (A) lin-aspartate, (B) lin-cysteic acid, and (C) lin-AP3. Red trace shows currents at 20 mV for visualization of PUFA-induced effects on current. (D–F) Dose-dependent effects of lin-glycine (n = 4), lin-taurine (n = 3), lin-aspartate (n = 4), lin-cysteic acid (n = 5), and lin-AP3 (n = 3) on (D) IKs current (I/I0), (E) IKs voltage dependence (ΔV0.5), and (F) IKs Gmax (mean ± SEM at maximal concentration). (G-I) Statistical differences at 20 µM on (G) I/I0 effect, (H) ΔV0.5 effect, and (I) Gmax effect measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant.
Taurine compounds had the largest current increase and left-shifting effect on the IKs channel
We next compared PUFAs and PUFA analogues that have a DHA or pinolenic acid tail group to determine if the efficacy of glycine and taurine head groups are consistent across PUFA tail groups. DHA, which has a carboxyl head group, produced little change in IKs current at 20 µM (Fig. 5 A) and produced a slight increase in I/I0 (2.0 ± 0.6; Fig. 5 D). DHA-glycine, which has a glycine head group, produced a larger increase in I/I0 (4.7 ± 1.3 at 20 µM) relative to DHA. DHA-taurine produced the most robust increases in IKs current at 7 µM compared with PUFA analogues with a DHA tail, increasing I/I0 by 5.1 ± 0.7 at 7 µM (Fig. 5 D). Surprisingly, at concentrations >7 µM (20 µM), DHA-taurine decreased the current for reasons that are unclear. For this reason, we report the effects observed at 7 µM. When the effects on the V0.5 of the IKs channel were measured, DHA did not left-shift the V0.5 (0.1 ± 1.4 mV; Fig. 5 F), DHA-glycine had a moderate left-shifting effect (−16.5 ± 1.3 mV at 20 µM), and DHA-taurine had a more robust left-shifting effect (−45.3 ± 2.9 mV at 7 µM; Fig. 5 F). DHA, DHA-glycine, and DHA-taurine all increased the Gmax (1.7 ± 0.3 at 20 µM, 2.0 ± 0.2 at 20 µM, and 1.7 ± 0.1 at 7 µM, respectively; Fig. 5 H). Statistical analysis of the fitted parameters of the dose–response curves show that DHA-taurine had the biggest increase in the V0.5 (Fig. 5 G), whereas DHA-glycine had the biggest increases in I/I0 (Fig. 5 E) and Gmax (Fig. 5 I).
Figure 5.

DHA-taurine at 7 µM produces the most potent activation of the IKs channel compared with DHA-glycine and DHA at 20 µM. (A–C) Structure of and raw current traces measured in 0 µM (left) and 20 µM (right) (A) DHA, (B) DHA-glycine, and (C) DHA-taurine, 0 µM (left) and 7 µM (right). We report effects of DHA-taurine at 7 µM due to an unclear reduction in current caused by the application of 20 µM. Red trace shows currents at 20 mV for visualization of PUFA-induced effects on current. (D) Dose-dependent effects of DHA (n = 4), DHA-glycine (n = 4), and DHA-taurine (n = 3) on IKs current (I/I0; mean ± SEM at maximal concentration). (E) Statistical differences on I/I0 effects (I/I0 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (F) Dose-dependent effects of DHA, DHA-glycine, and DHA-taurine on IKs voltage dependence (ΔV0.5). (G) Statistical differences on ΔV0.5 effects (ΔV0.5 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (H) Dose-dependent effects of DHA, DHA-glycine, and DHA-taurine on IKs Gmax. (I) Statistical differences on Gmax effects (Gmax fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. *, P < 0.05; ****, P < 0.0001. ns, not significant.
Application of 20 µM pinolenic acid (Fig. 6 A), which has a carboxyl head group, increased I/I0 slightly (1.5 ± 0.3; Fig. 6 D), had little left-shifting effect on the V0.5 of IKs channel activation (−6 ± 1.8 mV; Fig. 6 F), and produced a slight increase in the Gmax (1.4 ± 0.2; Fig. 6 H). Application of 20 µM pinoleoyl-glycine (pin-glycine; Fig. 6 B) produced a moderate increase in I/I0 (3.8 ± 0.2; Fig. 6 D), had a moderate left-shifting effect on the V0.5 of IKs channel activation (−21.1 ± 2.5 mV; Fig. 6 F), and increased the Gmax (1.8 ± 0.1; Fig. 6 H). Application of 20 µM pinoleoyl-taurine (pin-taurine; Fig. 6 C) produced a robust increase in I/I0 (9.0 ± 1.4; Fig. 6 D), potently left-shifted the V0.5 of IKs channel activation (−51.6 ± 3.5 mV; Fig. 6 F), and increased the Gmax (1.9 ± 0.3; Fig. 6 H) relative to other PUFA analogues with a pinolenic acid tail. Statistical analysis of the fitted parameters of the dose–response curves show that pin-taurine had the biggest increase in I/I0 (Fig. 6 E) and the V0.5 (Fig. 6 G), whereas there were no significant differences in Gmax among the three compounds (Fig. 6 I).
Figure 6.

Pin-taurine produces the most potent activation of the IKs channel compared with pin-glycine and pinolenic acid. (A–C) Structure of and raw current traces measured in 0 µM (left) and 20 µM (right) (A) pinolenic acid, (B) pin-glycine, and (C) pin-taurine. Red trace shows currents at 20 mV for visualization of PUFA-induced effects on current. (D) Dose-dependent effects of pinolenic acid (n = 3), pin-glycine (n = 3), and pin-taurine (n = 4) on IKs current (I/I0; mean ± SEM at maximal concentration). (E) Statistical differences on I/I0 effects (I/I0 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (F) Dose-dependent effects of pinolenic acid, pin-glycine, and pin-taurine on IKs voltage dependence (ΔV0.5). (G) Statistical differences on ΔV0.5 effects (ΔV0.5 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (H) Dose-dependent effects of pinolenic acid, pin-glycine, and pin-taurine on IKs Gmax. (I) Statistical differences on Gmax effects (Gmax fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant.
As previously mentioned, DHA-taurine produced an unexpected decrease in I/I0 and Gmax at 20 µM. The largest effect on Gmax induced by DHA-taurine on the IKs channel occurred at 7 µM, followed by a drastic decrease in Gmax at 20 µM. We also observed a similar decrease in Gmax at 20 µM with pin-taurine; however, this decrease in Gmax was not as pronounced as we saw with 20 µM of DHA-taurine. The source of the reduction in Gmax with the application of some taurine compounds is not known. One possibility is that it is caused by a steric effect of the longer taurine head group, resulting in obstruction of the IKs channel pore. To determine whether the reduction in Gmax is intrinsic to the taurine head group, we applied 100 µM taurine to the IKs channel. However, 100 µM taurine alone did not change I/I0, ΔV0.5, or Gmax (Fig. S2), suggesting that the taurine head group alone is not responsible for the reduction in Gmax. Therefore, PUFA-induced decreases in Gmax at concentrations ≥20 µM must be due to a different mechanism that occurs through the combination of the taurine head group and the PUFA tail.
We directly compared the effects of PUFA analogue head groups across different PUFA tails to see if there were any differences in apparent binding affinity or effects on I/I0, ΔV0.5, or Gmax depending on the tail. Our previous data and the data in this study suggest that PUFA analogues with glycine head groups have a pKa of ∼7.5–7.6 when associated with IKs channels, suggesting that half the PUFA molecules with a glycine head group will be deprotonated and negatively charged at pH 7.5. PUFA analogues with a glycine head group produced similar max effects on I/I0 (Fig. 7 A) and ΔV0.5 (Fig. 7 B), whereas Gmax was more varied (Fig. 7 C). PUFA analogues with taurine head groups had an estimated pKa of ∼2.6, suggesting that all of the PUFA molecules with a taurine head group will be deprotonated and negatively charged at pH 7.5. PUFA analogues with a taurine head group all produced much larger effects on ΔV0.5 than those with glycine head groups (Fig. 7 B), whereas the effects on Gmax were all in a relatively similar range (Fig. 7 C). Lin-taurine and pin-taurine produced much larger effects on I/I0 than those with glycine head groups, whereas DHA-taurine produced a similar effect on I/I0 as those with glycine head groups (Fig. 7 A). In summary, the major difference between PUFAs with taurine and glycine head groups is in the effects on ΔV0.5. This difference is mainly due to the pKa (i.e., the charge) of the PUFA head group, with little influence from the hydrophobic PUFA tail groups.
Figure 7.

Comparison of effects by glycine head groups and taurine head groups on IKs current (I/I0), voltage dependence (ΔV0.5), and Gmax. (A–C) Dose-dependent effects of DHA-glycine (n = 4), lin-glycine (n = 4), pin-glycine (n = 3), DHA-taurine (n = 3), lin-taurine (n = 3), and pin-taurine (n = 4) on (A) IKs current (I/I0), (B) IKs voltage dependence (ΔV0.5), and (C) IKs Gmax (mean ± SEM at maximal concentration). Gray open square is for DHA-taurine at 20 μM (not included in fit).
Hierarchical cluster analysis grouped PUFA analogues that have similar functional effects
We used hierarchical cluster analysis as an unbiased method to group PUFAs and PUFA analogues according to similarity of their effects on I/I0, ΔV0.5, and Gmax at 20 µM (Fig. 8 and Table 2). The hierarchical cluster analysis resulted in three distinct clusters of PUFAs and PUFA analogues. The first branch point results in the most distinct cluster (cluster 1) of PUFA analogues that include lin-taurine, lin-cysteic acid, pin-taurine, and DHA-taurine, which had the largest effects on the V0.5. The second branch point divides clusters 2 and 3. Cluster 2 includes lin-aspartate, pin-glycine, DHA-glycine, and lin-glycine, which had intermediate effects on I/I0 and Gmax. Cluster 3 includes linoleic acid, DHA, lin-AP3, and pinolenic acid, which had the smallest effects on IKs channel activation. The results of the hierarchical cluster analysis suggest that PUFA analogues with a glycine head group have the most consistent effects on increasing Gmax and that PUFA analogues with a taurine head group are most consistent in left-shifting the voltage dependence of IKs channel activation.
Figure 8.

Hierarchical cluster analysis and heat map demonstrate that taurine head groups are most similar in their voltage-shifting effects and glycine head groups are most similar in their effects on Gmax. The dendrogram displays groupings of PUFAs and PUFA analogues according to similarity of their effects. The heat map displays the magnitude of the effects, with warmer colors representing PUFAs and PUFA analogues that have larger relative effects (closer to 1.0) on I/I0, Gmax, and ΔV0.5 and cooler colors representing PUFAs and PUFA analogues with smaller relative effects (closer to 0.0).
Table 2. Summary of effects of PUFA analogues on the cardiac IKs channel.
| PUFA name | I/I0 | ΔV0.5 (mV) | Gmax/Gmax0 | n |
|---|---|---|---|---|
| Linoleic acid | 0.5 ± 0.04 | 4.7 ± 0.9 | 0.7 ± 0.04 | 5 |
| Lin-glycine | 5.3 ± 0.5 | −26.4 ± 4.4 | 2.4 ± 0.2 | 4 |
| Lin-taurine | 10.4 ± 4.0 | −73.1 ± 2.6 | 2.0 ± 0.6 | 3 |
| Lin-glycine+1C | 1.7 ± 0.2 | −7.2 ± 2.5 | 1.3 ± 0.02 | 3 |
| Lin-glycine+2C | 1.9 ± 0.4 | −8.7 ± 0.5 | 1.1 ± 0.2 | 3 |
| Lin-cysteic acid | 9.2 ± 0.4 | −58.4 ± 2.8 | 2.0 ± 0.2 | 5 |
| Lin-aspartate | 4.0 ± 0.1 | −34.5 ± 2.3 | 1.4 ± 0.1 | 4 |
| Lin-AP3 | 1.8 ± 0.3 | −5.7 ± 1.3 | 1.1 ± 0.1 | 3 |
| DHA | 2.0 ± 0.6 | 0.1 ± 1.4 | 1.7 ± 0.3 | 4 |
| DHA-glycine | 4.7 ± 1.3 | −16.5 ± 1.3 | 2.0 ± 0.2 | 4 |
| DHA-taurine | 5.1 ± 0.7 | −45.3 ± 2.9 | 1.7 ± 0.1 | 3 |
| Pinolenic acid | 1.5 ± 0.3 | −6 ± 1.8 | 1.4 ± 0.2 | 3 |
| Pin-glycine | 3.8 ± 0.2 | −21.1 ± 2.5 | 1.8 ± 0.1 | 3 |
| Pin-taurine | 9.0 ± 1.4 | 51.6 ± 3.5 | 1.9 ± 0.3 | 4 |
Summary of the effects of PUFAs on IKs I/I0, ΔV0.5 (mV), and Gmax/Gmax0 with the number of experiments (n). Data are represented as mean ± SEM at the maximum concentration used (effects of DHA-taurine are reported at 7 µM due to a decrease in current observed at 20 µM).
Circulating concentrations of lin-glycine with albumin and other fatty acids promoted the activation of the cardiac IKs channel by left-shifting the voltage dependence of activation
In the body, PUFAs circulate in complex with serum albumin but interact with channel proteins in the free fatty acid form. In addition, PUFAs in the bloodstream are in circulation with other types of fatty acids, including MUFAs and SFAs. To emulate the effects of PUFAs under physiological conditions, we applied lin-glycine in combination with the MUFA oleic acid, the SFA stearic acid, and albumin (Tsukamoto and Sugawara, 2018; Abdelmagid et al., 2015). We applied 0.1 mM albumin/0.2 mM lin-glycine/0.2 mM oleic acid/0.2 mM stearic acid, which we refer to as albumin + fatty acids (Abdelmagid et al., 2015; Tsukamoto and Sugawara, 2018). Following the application of albumin + fatty acids, we saw an increase in IKs current (Fig. 9 A). In the current versus voltage relationship, we observed that the application of albumin + fatty acids increased Gmax and caused a leftward shift in the voltage dependence of IKs activation (Fig. 9 B). Lin-glycine in combination with MUFAs, SFAs, and albumin produced a significant increase in IKs current (2.0 ± 0.1) compared with control (P = 0.003; Fig. 9 C). In addition, we observed a leftward shift in the voltage dependence of IKs activation (−11.4 ± 1.1 mV) compared with control (P = 0.0004) and a significant increase in the Gmax of the IKs channel (1.15 ± 0.04; P = 0.007; Fig. 9, D and E). These data together suggest that there is still a substantial concentration of lin-glycine in the free fatty acid form that is available to promote the activation of the cardiac IKs channel by left-shifting the voltage dependence of activation and increasing Gmax.
Figure 9.
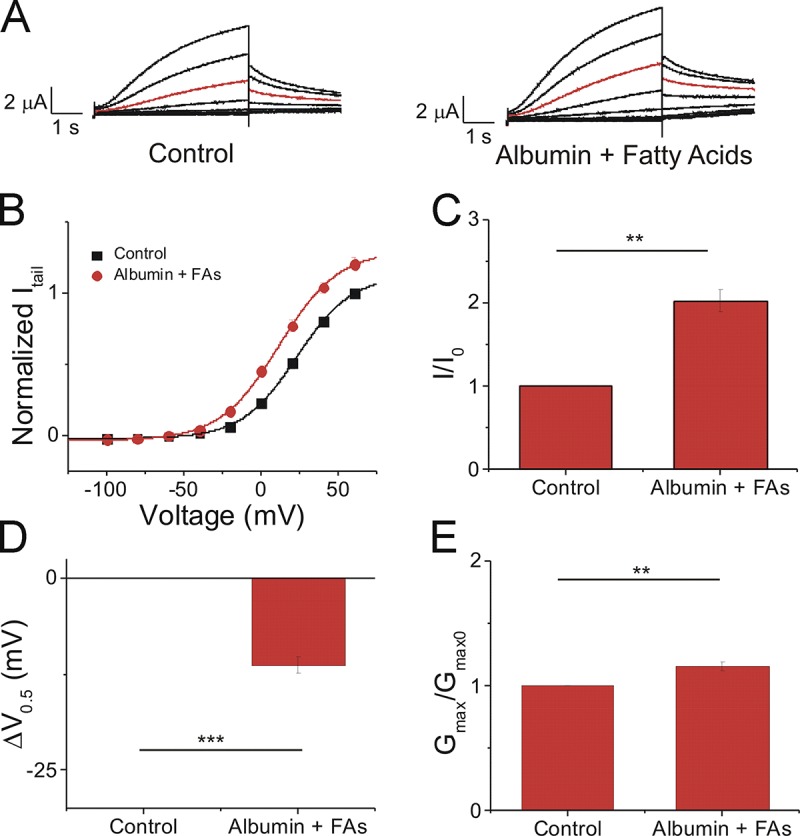
Lin-glycine in combination with physiological concentrations of monounsaturated and saturated fatty acids and albumin promotes the activation of the IKs channel. (A) Raw current traces measured in control ND96 (left) and in the presence of 0.1 mM albumin + 0.2 mM lin-glycine/0.2 mM oleic acid/0.2 mM stearic acid (Fatty Acids; right). Red trace shows currents at 20 mV for visualization of PUFA-induced effects on current. (B) Current-voltage relationship of cells in control ND96 (black squares) and in 0.1 mM albumin + 0.2 mM lin-glycine/0.2 mM oleic acid/0.2 mM stearic acid (fatty acids [FAs]; red circles; mean ± SEM; n = 4). (C) Statistical differences on I/I0 effects (I/I0 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (D) Statistical differences on ΔV0.5 effects (ΔV0.5 fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. (E) Statistical differences on Gmax effects (Gmax fitted from the dose–response curve) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. **, P < 0.01; ***, P < 0.001. ns, not significant.
PUFA analogues rescued LQT1-associated loss-of-function mutation Kv7.1 V215M + KCNE1 by left-shifting voltage dependence of IKs activation
To evaluate the therapeutic potential of the PUFA analogues as potential treatments for LQTS, we expressed the IKs channel bearing a mutation that causes LQT1 (V215M). V215M (in which a valine residue is replaced with methionine) is a loss-of-function mutation located in the S3 segment of the Kv7.1 α subunit of the cardiac IKs channel (Eldstrom et al., 2010; Fig. 10 A). The V215M mutation causes a rightward shift in the voltage dependence of channel activation and alters the activation and deactivation kinetics compared with the wild-type channel (Eldstrom et al., 2010). To determine the ability of PUFA analogues to restore IKs channel loss of function, we applied both lin-glycine and lin-taurine to the IKs channel bearing the V215M mutation. From the current versus voltage relationship, we found that the V215M mutation resulted in a significant rightward shift in the voltage dependence of IKs channel activation relative to the wild-type channel (V215M V0.5 = 40.2 ± 0.2 mV; wild type V0.5 = 16.6 ± 0.4 mV; and ΔV0.5 = +24.2 ± 2.3 mV; Fig. 10, B and C). However, the application of lin-glycine (at 10 µM) and lin-taurine (at 5 µM) both strongly left-shifted the voltage dependence of activation compared with the V0.5 of activation of V215M mutant channels (ΔV0.5 = −23.8 ± 3.3 mV and −29.7 ± 0.8 mV, respectively), fully restoring the wild-type voltage dependence of the IKs channel (Fig. 10, B and C). These data demonstrating PUFA-induced effects on LQT1-causing mutations suggest that PUFA analogues are potent enough activators of the IKs channel that they are capable of restoring the normal voltage dependence of LQT1 mutation–bearing IKs channels.
Figure 10.

PUFA analogues lin-glycine and lin-taurine rescue LQT1-associated loss-of-function mutation, V215M. (A) Topology of Kv7.1 and KCNE1 with location of V215M indicated. (B) Current–voltage relationship of the wild-type IKs channel (black squares; mean ± SEM; n = 4), Kv7.1 V215M + KCNE1 (red circles; mean ± SEM; n = 3), Kv7.1 V215M + KCNE1 with lin-glycine (green triangles; mean ± SEM; n = 3), and Kv7.1 V215M + KCNE1 with lin-taurine (blue triangles; mean ± SEM; n = 3). (C) Statistical differences on the voltage dependence (V0.5) effects (V0.5 fitted using the Boltzmann equation) measured by one-way ANOVA followed by Tukey’s HSD post hoc analysis. ****, P < 0.0001. Error bars represent SEM.
Discussion
We have characterized several different head groups of PUFA analogues in order to determine the range of their effects of PUFA analogues on the current, voltage dependence, and Gmax of the cardiac IKs channel. Our findings demonstrate that PUFA analogues with a glycine head group consistently produced moderate activation of the cardiac IKs channel. In addition, we demonstrated that PUFA analogues with a taurine or cysteic acid head group produced the most potent activation of the cardiac IKs channel. Lastly, we showed that increasing the number of potentially charged moieties did not necessarily improve PUFA-induced activation of the cardiac IKs channel. This is most likely due to the pKa of the additional potentially charged moieties as well as potential steric hindrance of PUFAs with multiple potentially charged groups.
We previously presented evidence that the charged head group of PUFAs electrostatically interacts with the arginines in S4 or K326 in S6 of the IKs channel (Liin et al., 2018). We assume that the PUFAs and PUFA analogues tested here also interact by similar mechanisms with the IKs channel. As an example, we showed here that neutralization mutations of charges in S4 or S6 decrease the effects of lin-glycine on the voltage shift and Gmax (Fig. S3), as if lin-glycine also interacts with the S4 arginines and K326 in the pore. Most of the variability in the effects of the different PUFA head groups on IKs channels can be explained by the predicted pKa of the different head groups, which determines their protonation state in the membrane bound to the IKs channel. The experimentally determined pH dependence of the effect on IKs channels of a PUFA with a carboxyl head group was consistent with a pKa of ∼8.5, suggesting that PUFAs with a simple carboxyl head group are protonated and neutral at pH 7.5. We therefore propose that PUFAs with a simple carboxyl head group are unable to participate in an electrostatic interaction with S4 arginines or K326 in S6 the IKs channel. The experimental determined pKa value of PUFA analogues with a glycine head group associated with the IKs channel is ∼7.6 (Börjesson and Elinder, 2011; Elinder and Liin, 2017; Liin et al., 2015). Therefore, half of the PUFA molecules with a glycine head group will be deprotonated and able to participate in an electrostatic interaction with S4 arginines or K326 in S6 (Liin et al., 2015). Our data comparing the effects of lin-glycine at pH 7.5 and pH 9.0 support the idea that half of the PUFA molecules with a glycine head group are able to have an electrostatic interaction with the S4 segment of IKs channels. Notably, the left-shift in the V0.5 of lin-glycine at pH 9.0 (Fig. 3 H) was approximately doubled compared with the left-shift at pH 7.5 (Fig. 3 E), consistent with our estimate of a pKa of ∼7.5–7.6 for PUFA analogues with glycine head groups (Table 1). At pH 9.0, all of the lin-glycine molecules will be deprotonated and able to participate in an electrostatic interaction with the S4 segment, leading to a larger left-shift in the voltage dependence of IKs channel activation.
Finally, the predicted pKa for PUFA analogues with a taurine head group associated with IKs channels was ∼2.6, so all taurine head groups were able to participate in an electrostatic interaction with the S4 arginines or K326 in S6 even at pH 7.5. The predicted pKas for taurine and glycine compounds are consistent with the approximate half size of the voltage-shifting effect of glycine compounds compared with taurine compounds. Why lin-glycine gives a larger increase in Gmax than lin-taurine is not clear, but may be due to different access for lin-glycine than for lin-taurine to the PUFA binding site that promotes an increase in Gmax. We estimated pKa1 and pKa2 values of lin-aspartate and lin-cysteic acid, as well as the pKa3 of lin-AP3, to compare them with estimated pKa values of lin-glycine and lin-taurine (Table 1). In lin-aspartate, the pKa1 was ∼7.6, which is similar to the pKa value of lin-glycine, suggesting that the first potentially charged group is likely to reside in its deprotonated form 50% of the time at pH 7.5. The pKa2 of lin-aspartate was ∼9.1, which means that the second potentially charged moiety would be protonated and uncharged at pH 7.5. Therefore, lin-aspartate has approximately the same functional charge on the hydrophilic head group as lin-glycine. Indeed, the overall effect of lin-aspartate on I/I0 is not significantly different than the I/I0 effect of lin-glycine. In lin-cysteic acid, the pKa1 was ∼2.4, which is very similar to the pKa value of lin-taurine, meaning lin-cysteic acid should be at least as potent as lin-taurine. The pKa2 of lin-cysteic acid was ∼7.1, meaning that the second group is likely to reside in its deprotonated form >50% of the time at physiological pH. However, lin-cysteic acid did not have a larger effect than lin-taurine, suggesting that the second charge group was not interacting with the channel, or it is possible that nearby residues in the IKs channel protein modified the pKa2 so that this group remained protonated at pH 7.5. Lastly, in lin-AP3, the pKa1 and pKa2 were ∼5.0 and ∼7.7 while the pKa3 was ∼11.8, which suggests that the first site would be deprotonated and the second site would be deprotonated 50% of the time while the third group was protonated and uncharged at pH 7.5. However, lin-AP3 has little to no effect on I/I0, ΔV0.5, or Gmax, suggesting that lin-AP3 does not effectively interact with the voltage sensor/pore or that it is not effectively deprotonated/negatively charged. We observed small effects of lin-AP3 when applied at pH 9.0 (in an attempt to help unmask potentially charged groups; Fig. S4), suggesting that there may be steric hindrance preventing the bulky AP3 head group from interacting favorably with the IKs channel.
Similar to our findings on the importance of the pKa of the PUFAs for shifting the voltage dependence of IKs channels, Ottosson et al. (2015) found that lowering the pKa of resin acid molecules resulted in greater left shift in the voltage dependence of the Shaker potassium channel. This further shows the importance of a deprotonated and charged compound for a strong activating effect on voltage-gated K+ channels by the lipoelectric mechanism. In addition, they noted that with some substitutions wherein a bulky group was added to the scaffold, the efficacy of these resin acid compounds was reduced (Ottosson et al., 2015). They suggested that adding a bulky group may impede the ability of the small molecule to interact with the voltage sensor of the Shaker K+ channel (Ottosson et al., 2015). This is similar to our data using the more bulky PUFA analogue lin-AP3.
The pH dependence of PUFA head group ionization was also shown in the Slo1 BK channel by Tian et al. (2016). They found that DHA produces potent activation of Slo1 BK channels and that this effect can be reduced when the pH is decreased, leading to protonation of the PUFA head group, and that the effect can be potentiated when the pH is increased, leading to deprotonation of the PUFA head group (Tian et al., 2016). Similar to our results, Tian et al. (2016) found that the addition of a phosphate head group leads to an attenuated effect on BK channel activation compared with DHA and DHA-glycine, which is similar to the effects we saw when applying lin-AP3.
In addition to the charge of the PUFA head group, the degree of unsaturation in the PUFA tail also plays an important role in PUFA-induced activation of the IKs channel. We and others have found that the PUFA-induced activation of IKs channels and Shaker K channels requires that the PUFA tail structure has at least two double bonds in cis-configuration in the tail (Liin et al., 2015, 2016; Börjesson et al., 2008). We recently conducted a systematic analysis of the PUFA tail (Bohannon et al., 2019) and found that neither the length of the carbon tail nor the number of double bonds in the tail correlated significantly with effects on or apparent binding affinity for the cardiac IKs channel. However, the position of the double bonds in the tail was strongly correlated with stronger activation of and better apparent affinity for the cardiac IKs channel (Bohannon et al., 2019).
Lipophilic compounds have the ability to form micelles. The concentration at which micelle formation takes place is called the critical micellar concentration. If the critical micellar concentration for our compounds was reached, micelle formation had the potential to interfere with the efficacy of the PUFAs and PUFA analogues being applied. However, the critical micellar concentration that is estimated for the majority of PUFAs and other unsaturated fatty acids is between 60 and 150 µM (Serth et al., 1991; Richieri et al., 1992; Mukerjee and Mysels, 1971). The experiments reported here were done at concentrations between 0.2 and 20 µM, which is well under the expected critical micellar concentration reported for unsaturated fatty acids. For this reason, we expect that the PUFAs applied in our preparation remained in the free fatty acid form, meaning that it is unlikely that any lack of effect from a PUFA could be attributed to the formation of micelles.
A range of effective compounds for activating the cardiac IKs channel is useful in the design of personalized therapeutics for LQT1. Patients with different LQT1 mutations have IKs channels with different degrees of channel malfunction (e.g., different-sized voltage shifts in their voltage dependence of activation) and present symptoms of varying severity. For this reason, individual LQT1 patients will not benefit from a one-size-fits-all treatment, producing a need for more personalized treatments. The findings presented here suggest that patients with more severe loss-of-function mutations of the cardiac IKs channel would most likely benefit from PUFA analogues with a taurine head group. In particular, PUFA analogues with a taurine or cysteic acid head group would be the most effective to rescue loss-of-function mutations in the IKs channel that lead to large shifts of the voltage dependence of IKs activation because these head groups produce the most robust effects on the V0.5. Patients with a milder LQT1 phenotype, however, may benefit more from treatment with a glycine PUFA analogue that has more moderate effects on IKs channel activation, and especially loss-of-function mutations that alter the Gmax of the IKs channel. Effective PUFA analogues can thus be selected for specific patients according to the severity of LQT1 pathology.
Acknowledgments
Sharona E. Gordon served as editor.
We thank Thea Wennman, Amanda Dahl, Frida Marshagen, Fiola Beqiri, Levi Lindroos, and Sankhero Gewarges for their contributions to experiments during their time as visiting scholars at the University of Miami. We thank Dr. Peter Konradsson at Linköping University for valuable discussions regarding synthesis strategies.
This work was supported by National Institutes of Health R01-HL131461 (to H.P. Larsson) and by the Swedish Society for Medical Research and the Swedish Research Council (2017-02040 to S.I. Liin).
A patent application (62/032,739) has been submitted by the University of Miami with S.I. Liin and H.P. Larsson as inventors. The other authors declare no competing financial interests.
Author Contributions: B.M. Bohannon, acquisition of data, analysis and interpretation of data, and drafting or revising the article; H.P. Larsson, conception and design, analysis and interpretation of data, and drafting or revising the article; S.I. Liin, conception and design, acquisition of data, analysis and interpretation of data, and drafting or revising the article; M.E. Perez, acquisition of data; X. Wu, contributing new compounds and acquisition of data.
References
- Abdelmagid S.A., Clarke S.E., Nielsen D.E., Badawi A., El-Sohemy A., Mutch D.M., Ma D.W.L., and Staff P.L.O.S.O.N.E.. PLOS ONE Staff . 2015. Correction: Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS One. 10:e0128167 10.1371/journal.pone.0116195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alders M., and Christiaans I.. 2003. Long QT syndrome. In GeneReviews. Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., and Amemiya A., editors. [Internet] University of Washington, Seattle: 1993–2019. [PubMed] [Google Scholar]
- Amin A.S., Giudicessi J.R., Tijsen A.J., Spanjaart A.M., Reckman Y.J., Klemens C.A., Tanck M.W., Kapplinger J.D., Hofman N., Sinner M.F., et al. 2012. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 33:714–723. 10.1093/eurheartj/ehr473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., and Romey G.. 1996. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 384:78–80. 10.1038/384078a0 [DOI] [PubMed] [Google Scholar]
- Barro-Soria R., Rebolledo S., Liin S.I., Perez M.E., Sampson K.J., Kass R.S., and Larsson H.P.. 2014. KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat. Commun. 5:3750–3761. 10.1038/ncomms4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohannon B.M., Perez M.E., Liin S.I., and Larsson H.P.. 2019. ω-6 and ω-9 polyunsaturated fatty acids with double bonds near the carboxyl head have the highest affinity and largest effects on the cardiac IKs potassium channel. Acta Physiol. (Oxf.). 225:e13186 10.1111/apha.13186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen M.S., Peng G., Robey S.H., Terrenoire C., Iyer V., Sampson K.J., and Kass R.S.. 2017. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 97:89–134. 10.1152/physrev.00008.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson S.I., and Elinder F.. 2011. An electrostatic potassium channel opener targeting the final voltage sensor transition. J. Gen. Physiol. 137:563–577. 10.1085/jgp.201110599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson S.I., Hammarström S., and Elinder F.. 2008. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 95:2242–2253. 10.1529/biophysj.108.130757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y. 2016. Management of Patients with Long QT Syndrome. Korean Circ. J. 46:747–752. 10.4070/kcj.2016.46.6.747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chockalingam P., Crotti L., Girardengo G., Johnson J.N., Harris K.M., van der Heijden J.F., Hauer R.N.W., Beckmann B.M., Spazzolini C., Rordorf R., et al. 2012. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J. Am. Coll. Cardiol. 60:2092–2099. 10.1016/j.jacc.2012.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C., Neyroud N., Richard P., Denjoy I., Hainque B., Romey G., Drici M.D., Guicheney P., and Barhanin J.. 2000. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc. Res. 45:971–980. 10.1016/S0008-6363(99)00411-3 [DOI] [PubMed] [Google Scholar]
- Crotti L., Spazzolini C., Schwartz P.J., Shimizu W., Denjoy I., Schulze-Bahr E., Zaklyazminskaya E.V., Swan H., Ackerman M.J., Moss A.J., et al. 2007. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 116:2366–2375. 10.1161/CIRCULATIONAHA.107.726950 [DOI] [PubMed] [Google Scholar]
- Drum B.M.L., Dixon R.E., Yuan C., Cheng E.P., and Santana L.F.. 2014. Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J. Mol. Cell. Cardiol. 66:63–71. 10.1016/j.yjmcc.2013.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldstrom J., Xu H., Werry D., Kang C., Loewen M.E., Degenhardt A., Sanatani S., Tibbits G.F., Sanders C., and Fedida D.. 2010. Mechanistic basis for LQT1 caused by S3 mutations in the KCNQ1 subunit of IKs J. Gen. Physiol. 135:433–448. 10.1085/jgp.200910351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinder F., and Liin S.I.. 2017. Actions and mechanisms of polyunsaturated fatty acids on voltage-gated ion channels. Front. Physiol. 8:43 10.3389/fphys.2017.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer S.C., Wilson A.J., Aldridge R., and Tinker A.. 2010. Mechanisms of disease pathogenesis in long QT syndrome type 5. Am. J. Physiol. Cell Physiol. 298:C263–C273. 10.1152/ajpcell.00308.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J.E., Larsson H.P., and Liin S.I.. 2018. KCNE1 tunes the sensitivity of KV7.1 to polyunsaturated fatty acids by moving turret residues close to the binding site. eLife. 7:e37257 10.7554/eLife.37257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S.I., Silverå Ejneby M., Barro-Soria R., Skarsfeldt M.A., Larsson J.E., Starck Härlin F., Parkkari T., Bentzen B.H., Schmitt N., Larsson H.P., and Elinder F.. 2015. Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. USA. 112:5714–5719. 10.1073/pnas.1503488112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S.I., Larsson J.E., Barro-Soria R., Bentzen B.H., and Larsson H.P.. 2016. Fatty acid analogue N-arachidonoyl taurine restores function of IKs channels with diverse long QT mutations. eLife. 5:e20272 10.7554/eLife.20272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S.I., Yazdi S., Ramentol R., Barro-Soria R., and Larsson H.P.. 2018. Mechanisms Underlying the Dual Effect of Polyunsaturated Fatty Acid Analogs on Kv7.1. Cell Reports. 24:2908–2918. 10.1016/j.celrep.2018.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerjee P., and Mysels K.J.. 1971. National Standards Reference Data Service. National Bureau of Standards, Washington, D.C. [Google Scholar]
- Osteen J.D., Gonzalez C., Sampson K.J., Iyer V., Rebolledo S., Larsson H.P., and Kass R.S.. 2010. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. USA. 107:22710–22715. 10.1073/pnas.1016300108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosson N.E., Wu X., Nolting A., Karlsson U., Lund P.E., Ruda K., Svensson S., Konradsson P., and Elinder F.. 2015. Resin-acid derivatives as potent electrostatic openers of voltage-gated K channels and suppressors of neuronal excitability. Sci. Rep. 5:13278 10.1038/srep13278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroz D., Rodriguez N., Choveau F., Baró I., Mérot J., and Loussouarn G.. 2008. Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 586:1785–1789. 10.1113/jphysiol.2007.148254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richieri G.V., Ogata R.T., and Kleinfeld A.M.. 1992. A fluorescently labeled intestinal fatty acid binding protein. Interactions with fatty acids and its use in monitoring free fatty acids. J. Biol. Chem. 267:23495–23501. [PubMed] [Google Scholar]
- Rivolta I., Clancy C.E., Tateyama M., Liu H., Priori S.G., and Kass R.S.. 2002. A novel SCN5A mutation associated with long QT-3: altered inactivation kinetics and channel dysfunction. Physiol. Genomics. 10:191–197. 10.1152/physiolgenomics.00039.2002 [DOI] [PubMed] [Google Scholar]
- Salata J.J., Jurkiewicz N.K., Jow B., Folander K., Guinosso P.J. Jr., Raynor B., Swanson R., and Fermini B.. 1996. IK of rabbit ventricle is composed of two currents: evidence for IKs Am. J. Physiol. 271:H2477–H2489. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M.C., Curran M.E., Zou A., Shen J., Spector P.S., Atkinson D.L., and Keating M.T.. 1996. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 384:80–83. 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- Schwartz P.J., Crotti L., and Insolia R.. 2012. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 5:868–877. 10.1161/CIRCEP.111.962019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P.J., Ackerman M.J., and Wilde A.A.M.. 2017. Channelopathies as Causes of Sudden Cardiac Death. Card. Electrophysiol. Clin. 9:537–549. 10.1016/j.ccep.2017.07.005 [DOI] [PubMed] [Google Scholar]
- Serth J., Lautwein A., Frech M., Wittinghofer A., and Pingoud A.. 1991. The inhibition of the GTPase activating protein-Ha-ras interaction by acidic lipids is due to physical association of the C-terminal domain of the GTPase activating protein with micellar structures. EMBO J. 10:1325–1330. 10.1002/j.1460-2075.1991.tb07651.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A., Vanoye C.G., George A.L. Jr., Meiler J., and Sanders C.R.. 2007. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry. 46:14141–14152. 10.1021/bi701597s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y., Aursnes M., Hansen T.V., Tungen J.E., Galpin J.D., Leisle L., Ahern C.A., Xu R., Heinemann S.H., and Hoshi T.. 2016. Atomic determinants of BK channel activation by polyunsaturated fatty acids. Proc. Natl. Acad. Sci. USA. 113:13905–13910. 10.1073/pnas.1615562113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto I., and Sugawara S.. 2018. Low levels of linoleic acid and α-linolenic acid and high levels of arachidonic acid in plasma phospholipids are associated with hypertension. Biomed. Rep. 8:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell-Smith K.E., and Skinner J.R.. members of the CSANZ Genetics Council Writing Group . 2016. Update on the Diagnosis and Management of Familial Long QT Syndrome. Heart Lung Circ. 25:769–776. 10.1016/j.hlc.2016.01.020 [DOI] [PubMed] [Google Scholar]
- Wu J., Ding W.G., and Horie M.. 2016. Molecular pathogenesis of long QT syndrome type 1. J. Arrhythm. 32:381–388. 10.1016/j.joa.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]