

The ability of T. gondii to promote a STAT3- and STAT6-dependent M2 phenotype in macrophages by injecting the virulence factor ROP16 in cis and trans subsequently limits the magnitude of the parasite-specific T cell response.
Abstract
The ability of Toxoplasma gondii to inject the rhoptry kinase ROP16 into host cells results in the activation of the transcription factors STAT3 and STAT6, but it is unclear how these events impact infection. Here, parasites that inject Cre-recombinase with rhoptry proteins were used to distinguish infected macrophages from those only injected with parasite proteins. Transcriptional profiling revealed that injection of rhoptry proteins alone was sufficient to induce an M2 phenotype that is dependent on STAT3 and STAT6, but only infected cells displayed reduced expression of genes associated with antimicrobial activity and protective immunity. In vivo, the absence of STAT3 or STAT6 improved parasite control, while the loss of ROP16 resulted in a marked reduction in parasite numbers and heightened parasite-specific T cell responses. Thus, ROP16 is a virulence factor that can act in cis and trans to promote M2 programs and which limits the magnitude of parasite-specific T cell responses.
Graphical Abstract

Introduction
Toxoplasma gondii is an intracellular parasite that exhibits strain-dependent virulence in a range of warm-blooded hosts (Hunter and Sibley, 2012). As T. gondii invades host cells, there is an initial wave of proteins secreted from the microneme and rhoptry organelles that allow parasite attachment and invasion, followed by rhoptry and dense granule protein secretion into the host cell, which establishes the parasitophorous vacuole (PV; Bougdour et al., 2014; Sibley, 2011). Many of the later-stage ROP and GRA proteins are exported across the PV into the host cell cytosol where they act as virulence factors that alter host cell function to promote immune evasion and parasite replication (Clough and Frickel, 2017; El Hajj et al., 2007; Olias et al., 2016; Saeij et al., 2007; Steinfeldt et al., 2010; Taylor et al., 2006). For example, the rhoptry proteins ROP5 and ROP18 are transported to the surface of the PV where they block recruitment of host effector molecules (Behnke et al., 2012; Etheridge et al., 2014; Niedelman et al., 2012). Similarly, the T. gondii inhibitor of STAT1 transcriptional activity (TgIST) protein present in the secretory granules of T. gondii traffics to the host cell nucleus, where it recruits a repressive complex that blocks signal transducer and activator of transcription (STAT) 1–mediated transcription (Gay et al., 2016; Olias et al., 2016). Since the ability of IFN-γ to activate STAT1 is required for control of T. gondii, parasites that lack TgIST grow normally over the first 5 d of infection in vivo but thereafter have decreased virulence. These studies highlight the ability of T. gondii effectors to act in cis within the infected host cell, and polymorphisms in their genes contribute to the variance in virulence between the different lineages of the parasite, broadly referred to as type I, type II, and type III strains (Saeij et al., 2005). The effects of these virulence factors have been studied primarily in the context of cells infected with T. gondii, but rhoptry proteins can be injected into host cells without parasite invasion (Christian et al., 2014; Koshy et al., 2010, 2012), although their impact on uninfected cells is not well understood.
While many pathogens block pathways associated with innate recognition of microbial products, another evasion strategy is to activate host cell processes that promote microbial growth. Alternatively activated (M2) macrophages, normally induced by cytokines (IL-4 and IL-10) that signal through STAT3 or STAT6, have a reduced ability to kill intracellular bacteria and parasites, and the ability of several organisms to promote M2 polarization is an evolutionarily conserved strategy that provides a refuge for pathogen replication (Buchacher et al., 2015; Chaves et al., 2001; Mège et al., 2011; Price and Vance, 2014; Yao et al., 2005). ROP16 is a kinase present in the rhoptries of T. gondii that is injected into host cells, where it directly phosphorylates STAT3 and STAT6 and in macrophages induces an M2-like phenotype (Butcher et al., 2011; El Kasmi et al., 2008; Jensen et al., 2011, 2013; Ong et al., 2010). While the ability of ROP16 to activate STAT3 and STAT6 was recognized based on a genetic screen to identify the basis of T. gondii virulence (Saeij et al., 2006, 2007), the ability to define its function in vivo has been complicated by differences between parasite strains. Thus, depending on the model, it is unclear if ROP16 is a positive or negative regulator of the survival and growth of T. gondii (Butcher et al., 2011; Fox et al., 2016; Jensen et al., 2011; Murray, 2011; Saeij et al., 2007). The majority of studies on the role of ROP16 proteins have used a virulent type I strain of T. gondii, which limits the ability to dissect its impact on the host immune response. The type II strains are less virulent but contain a polymorphic form of ROP16 that is a poor activator of STAT3 and STAT6 (Jensen et al., 2013; Yamamoto et al., 2009). In contrast, the type III strains of T. gondii are considered to be the least virulent strains in mice but express a form of ROP16 that is a potent activator of STAT3 and STAT6. These characteristics make type III strains a tractable system to assess the impact of ROP16 on the host response to infection.
The capacity of diverse pathogens to inject effector molecules into uninfected host cell populations represents an important mechanism that acts in trans to modify host function (Autenrieth et al., 2010; Pechous and Goldman, 2015; Pinaud et al., 2017). However, the ability to identify and track uninfected but injected cells to determine the consequences of injection remains a technical challenge for many of these microbes. The use of parasites that express Cre recombinase in their rhoptries (Toxoplasma-Cre parasites) has revealed that T. gondii can inject rhoptry proteins into cells that are not infected (Christian et al., 2014; Koshy et al., 2010, 2012). Little is known about whether injection of rhoptry proteins alone alters host cell function, but the use of the Toxoplasma-Cre parasites provides an opportunity to compare the effects of injection versus infection on host cells. Therefore, the type III CEP strain of T. gondii was engineered to express Cre in order to compare infected and injected macrophage populations in vitro and in vivo. These approaches revealed that injection of Toxoplasma-derived rhoptry proteins is sufficient to activate STAT3 and STAT6 and induce M2 polarization. Moreover, while the loss of STAT3 or STAT6 in vivo results in improved parasite control, this effect was most prominent with parasites that lacked ROP16, which stimulated enhanced T cell responses. Thus, ROP16 is a critical virulence factor that can act in cis and trans to modulate macrophage phenotypes and which contributes to the ability of T. gondii to limit the magnitude of parasite-specific T cell responses.
Results
Injection of rhoptry proteins induces STAT6-dependent M2 polarization in vitro
To compare the impact of injection versus infection by different strains of T. gondii, bone marrow–derived macrophages (BMMøs) were generated from Ai6 reporter mice and challenged with type II (Pru) or type III (CEP) Toxoplasma-Cre strains of T. gondii that secrete the toxofilin-Cre fusion protein and express tdTomato (Pru-Cre-tdTomato or CEP-Cre-tdTomato). At 24 h postinfection (hpi), flow cytometric analysis of the cultures revealed four populations of interest, quadrants I–IV (Fig. 1 A). Quadrant I contained bystander cells that had not been infected or injected by T. gondii and that had not phagocytosed the parasite and thus did not express the reporter or contain detectable parasite-derived material. Quadrant II consisted of cells that contained detectable parasite-derived material from phagocytosed organisms or were recently infected and had not yet expressed the ZsGreen1 reporter. Quadrant III contained infected cells that were ZsGreen1+, while quadrant IV was composed of injected cells that did not contain an intact parasite or parasite debris but were ZsGreen1+. BMMøs challenged with Pru-Cre-tdTomato did not show STAT6 phosphorylation in any quadrant (Fig. 1 B). For cultures challenged with CEP-Cre-tdTomato, STAT6 activation was not detected in bystander cells (quadrant I provides an internal control), but was apparent in a portion of the cells in quadrant II and was readily detected in all cells in quadrants III and IV (Fig. 1 B). Previous studies have reported that although Pru induces STAT3 phosphorylation, a polymorphism in ROP16 results in a reduced ability to sustain that activity (Yamamoto et al., 2009). Indeed, direct comparison of Pru and CEP in these assays revealed that Pru did induce STAT3 phosphorylation in quadrant III, but the impact of CEP on the activation of STAT3 was present in quadrants II, III, and IV (Fig. S1 A). The ability of CEP to phosphorylate STAT6 and STAT3 to comparable levels in cells injected (quadrant IV) and infected (quadrant III), but not in bystander cells (quadrant I), demonstrates that injected parasite proteins are sufficient to mediate these events and that they are not a secondary consequence of soluble factors that act on bystander or injected cells.
Figure 1.

Impact of injection and infection on macrophage phenotype in vitro. (A) Primary BMMøs from Ai6 mice were challenged with CEP-Cre-tdTomato, and at 24 hpi they were analyzed by flow cytometry for expression of tdTomato and ZsGreen. Populations were divided into fractions I (uninfected), II, III (infected), and IV (injected). Representative flow plot from one of four independent experiments (n = 3 replicates/experiment). (B) BMMøs from Ai6 mice were challenged with CEP-Cre-tdTomato or Pru-Cre-tdTomato, and fractions I–IV were assessed for pSTAT6 (gray, fluorescence minus one [FMO]; blue, Pru; red, CEP). Bar graphs depict the mean fluorescence intensity (MFI) of pSTAT6 in each fraction. Summary data from one of three independent experiments (n = 3 replicates/condition/experiment). (C) Experimental plan for transcriptional profiling of Ai6 BMMøs of the WT or STAT6−/− BMMøs infected with Pru-Cre-tdTomato or CEP-Cre-tdTomato strains. After 20 hpi, fractions I, III, and IV were sorted for microarray analysis. (D) Donut charts that show the numbers of genes altered by injection or infection for Pru and CEP and the impact of STAT6 on the changes induced by CEP. The numbers in each circle indicate total genes in set (orange, injection-specific induced; blue, infection-specific induced). (E) GSEA enrichment plots of Pru and CEP infected or injected cells showing enrichment of up-regulated M2 genes (GEO accession nos. GSE32690 and GSE51466). Enrichment score refers to the degree to which the gene set is overrepresented at the top or bottom of the ranked input list of genes. NES, normalized enrichment score (adjusted for gene set size or multiple hypothesis testing). (F) BMMøs were stimulated in culture with IFN-γ (M1), IL-4 (M2a), or IL-10 (M2c) for 24 h, and RNA sequencing was performed to generate gene signatures associated with these cytokines. The datasets generated in C were then compared with the M1, M2a, and M2c signatures, and differences were expressed as a heat map. (G) Identification of the CEP injection-induced STAT6-dependent gene profile. Cluster A genes associated with the innate immune response were inhibited by injection, while cluster B (M2) and C (oxidation-reduction processes) genes were promoted by injection. Summary statistics represent mean ± SD; ****, P < 0.0001 (two-tailed unpaired Student’s t test). FDR, false discovery rate.
Figure S1.

Impact of injection and infection on BMMø phenotype. (A) BMMøs from Ai6 mice were challenged with CEP-Cre-tdTomato (CEP) or Pru-Cre-tdTomato (Pru), and fractions I–IV were assessed for pSTAT3. Bar graphs depict the MFI of pSTAT3 of cells from indicated fraction. Summary data from one of three independent experiments (n = 3 replicates/condition). (B) Hierarchically clustered samples with dendrograms from infected Ai6 BMMøs. (C and D) Ai6 BMMøs were infected with Pru or CEP, and at 1 dpi the expression of CD206 and CD301b or Arg1 was assessed by flow cytometry, and the percentage that were CD301b+CD206+ was quantified (C), while the MFI for CD206, CD301b, and Arg1 for cells in fraction I, III, or IV were then quantified (D). Flow plots and summary data are representative of one of two independent experiments (n = 3 replicates/condition). Summary statistics represent mean ± SD; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (two-tailed unpaired Student’s t test). Fig. S1 is related to Fig. 1.
Because the ability of IL-4 and IL-13 to activate STAT6 promotes an M2 transcriptional program, experiments were performed to compare the impact of injection alone versus infection on the induction of STAT6-dependent M2 genes. BMMøs from Ai6 or Ai6/STAT6−/− mice were challenged with either Pru-Cre-tdTomato or CEP-Cre-tdTomato, and the populations in quadrants I, III, and IV were sorted for transcriptional profiling (Fig. 1 C). Hierarchical cluster analysis of gene expression in these three populations showed that the highest-order clustering was determined by the STAT6-dependent changes induced by infection or injection by CEP (Fig. S1 B). BMMøs lacking STAT6 and infected or injected by CEP were more closely clustered to BMMøs infected or injected by the Pru parasites that do not induce sustained phosphorylation of STAT6. To understand the effect of injection alone on BMMøs, the fraction of genes altered by injection (quadrant IV) was compared with global changes induced by infection (quadrant III; Fig. 1 D). Compared with naive BMMøs, infection with Pru resulted in differential expression of ∼900 genes, and ∼10% of these changes can be attributed to injection alone. For CEP, infection altered expression of ∼1,800 genes, and almost one third of these alterations occur with injection alone. Analysis of Ai6/STAT6−/− BMMøs infected with CEP showed that 40% of the changes in gene expression in injected cells was STAT6 dependent compared with only 2% of the genes differentially expressed in the infected cells (Fig. 1 D).
To compare infected and injected BMMø populations to established macrophage phenotypes, the transcriptomes following Pru or CEP infection were compared with a set of M2 genes curated from accession nos. GSE32690 (Riquelme et al., 2013) and GSE51466 (Taguchi et al., 2014; Fig. 1 E and Table S1). Gene set enrichment analysis (GSEA) revealed that both infection and injection of BMMøs by CEP induced a transcriptional program enriched for M2 phenotype genes, while infection or injection by Pru did not (Fig. 1 E). Next, we generated de novo gene expression signatures for BMMøs that are induced by STAT6 (M2a, IL-4) or STAT3 (M2c, IL-10) pathways, as well as a gene expression signature of M1 macrophages that result from STAT1 activation (M1, IFN-γ). These signatures were then compared with the transcriptional profile of BMMøs injected or infected by Pru or CEP, and the degree of similarity is represented as a heat map. No BMMø infected by either strain of T. gondii exhibited a gene expression profile indicative of STAT1 activation, and cells exposed to Pru did not exhibit a signature characteristic of STAT6 or STAT3 activation (Fig. 1 F). In contrast, BMMøs exposed to CEP showed expression of an IL-4/STAT6 transcriptional profile that was present in infected and injected cells and was clearly impacted by the loss of STAT6. BMMøs injected and infected with CEP also displayed a less prominent IL-10/STAT3 gene expression profile (Fig. 1 F). Phenotypic analysis for canonical M2a markers, mannose receptor C type 1 (Mrc1, CD206), macrophage galactose N-acetylgalactosamine–specific lectin 2 (Mgl2, CD301b), and arginase 1 (Arg1) by flow cytometry confirmed that BMMøs infected or injected by CEP expressed these M2-associated proteins (Fig. S1, C and D).
Overall, ∼150 STAT6-dependent genes were identified in CEP-injected cells, and three functional clusters based on gene ontology (GO) terms could be identified (Fig. 1 G). Cluster A contains genes down-regulated by parasite injection and includes several (Tlr2, Tlr5, Tlr7, and Nod1) that are broadly linked to innate recognition of pathogens, including Tlr2, which has been implicated in resistance to T. gondii (Mun et al., 2003). Within this cluster, the down-regulation of IFN-γ–induced GTPase, which is essential for PV disruption and resistance to T. gondii (Melzer et al., 2008), highlights the ability of T. gondii to interfere with critical innate immune pathways. Conversely, gene clusters B and C contain genes up-regulated in injected BMMøs and include a cassette of M2-associated genes in cluster B and genes enriched in GO terms related to the oxidation-reduction processes in cluster C. These data show that infection or injection by the CEP strain of T. gondii readily induces a STAT6-dependent gene signature associated with M2 macrophages, and injection alone of rhoptry proteins, including ROP16, is sufficient to account for ∼30% of the global changes in the gene expression program observed in BMMøs infected by CEP.
Effects of injection versus infection on macrophage populations in vivo
To determine if injection or infection by CEP can promote an M2-like phenotype in vivo, Ai6 mice were infected i.p. with CEP-Cre-tdTomato parasites. At 1 d postinfection (dpi), 89 ± 4% of infected cells (ZsGreen+Toxo-tdTomato+) in the peritoneal cavity were large peritoneal macrophages (LPM; CD11b+CD64+CD102+), and a small proportion of LPM expressed ZsGreen but did not contain parasites (ZsGreen+Toxo-tdTomato−; Fig. 2, A and B). The injected cells were not restricted to the peritoneum and were readily detected in the omentum (site of drainage from the peritoneum) at 3 dpi (Fig. 2 C), where they were frequently present in clusters associated with the presence of infected cells.
Figure 2.

Identification and analysis of macrophage populations from CEP-infected mice. (A) Ai6 mice were infected i.p. with 106 CEP-Cre-tdTomato, and at 1 dpi the PECs were analyzed by flow cytometry to identify resident LPM. The interactions of these subsets with CEP were analyzed by plotting Toxo-tdTomato versus ZsGreen fluorescence. Average frequency of each quadrant is shown. (B) The number of infected (Toxo+ZsGreen+) or injected (Toxo−ZsGreen+) LPM at the site of infection. Summary data are from one of two representative experiments (n = 4 mice). (C) Imaging of the omentum at 3 dpi revealed large numbers of green cells that contained parasites (red) as well as green-only populations typically present in foci of parasite replication. Representative micrograph from one of two independent experiments. Scale bar, 25 µm. (D) Ai6 mice were infected with CEP-Cre-tdTomato, and at 1 dpi, three populations (uninfected, infected, and injected) of LPM were sorted and used for RNA sequencing. PC analysis of sorted populations uninfected (gray circles), infected (red triangles), or injected (green squares) for all replicates is shown (n = 4 replicate groups of four mice). (E) GSEA enrichment plots of CEP infected or injected cells, showing M2 enrichment of up-regulated genes (GEO accession no. GSE51466). Enrichment score refers to the degree to which the gene set is overrepresented at the top or bottom of the ranked input list of genes. NES is adjusted for gene set size or multiple hypothesis testing. (F) Comparison of LFC (infected versus uninfected) and LFC (injected versus uninfected) is shown together with peritoneal M2 genes highlighted (red). Genes up-regulated (red area) and down-regulated (blue area) by both injected and infected cells are highlighted in shaded regions. The top 10 up-regulated M2 genes and selected immune-related genes (blue) are labeled. (G) A volcano plot of the DEGs between injected and uninfected LPM. The significance cutoff was set to a FDR of 0.05 (−log(adjusted P value ≥1.3)), and the biological cutoff was set to a fold change of ±2 (−1 ≥ LFC ≥ 1). The red (IL-4), green (IL-10), or blue (IL-4 and IL-10) dots represent the transcripts induced by the indicated cytokines. Select M2 genes are labeled. (H) GSEA enrichment plots for cell cycle and IFN-γ signaling pathways from the KEGG database. Enrichment curves for injected (IV versus I) and infected (III versus I) cells are shown in blue and green, respectively. NES and FDR are shown. Summary statistics represent mean ± SD.
To assess the impact of injection and infection by CEP on LPM, Ai6 Cre reporter mice were infected i.p. with CEP-Cre-tdTomato parasites, and at 1 dpi, the bystander, infected, and injected populations of LPM were sort-purified and transcriptionally profiled. Consistent with the sort-purification strategy, parasite-derived transcripts were only detected in the infected LPM population (Fig. S2 A). The use of a Pearson correlation associated with hierarchical clustering showed that the uninfected samples clustered together, while samples that corresponded to infected or injected populations were more closely related and also clustered according to the quadrant of the sort-purified cells (Fig. S2 B). Principal component (PC) analysis identified three groups in which the bystander cells clearly separated from the infected and injected populations along PC1 (71.2%), while infected and injected cells separated along PC2 (10.6%; Fig. 2 D). Thus, injection alone can alter LPM gene expression similar to infection, but does not fully recapitulate the transcriptional profile of infected cells.
Figure S2.

Gene expression analysis of LPM from CEP-infected mice. (A) Reads that map to parasite transcriptome from sorted populations of uninfected (I), infected (III), and injected (IV) at 1 dpi with CEP. (B) Hierarchically clustered sorted samples from four replicated (rep) samples with dendrograms from infected Ai6 mice. (C) Numbers of DEGs induced by infection or injection. (D) Quantitative radar plot representation of the degree of enrichment of GO terms for major immune cell subsets using the DAVID online tool. Numbers represent the number of enrichment genes and provide an overall comparison of the infected versus injected populations. Fig. S2 is related to Fig. 2.
Further analysis of the bystander, infected, and injected cell populations identified ∼7,118 differentially expressed genes (DEGs; log fold change [LFC] >1; P < 0.05) between all of the populations. Specifically, there were 1,670 DEGs between bystander and infected LPM and 381 DEGs between bystander and injected LPM (Fig. S2 C). Comparison of DEGs between bystander and infected LPM to bystander and injected LPM revealed a subset (324) of genes that are regulated by both infection and injection. A comparison of the up-regulated genes shared by injected and infected LPM with genes associated with M2 macrophage polarization (Fleming et al., 2015) confirmed that many of the most highly up-regulated genes are part of an M2 signature (Fig. 2 E). Further comparison with the M2a and M2c profiles generated in Fig. 1 revealed that injected cells showed the most significant and highest-magnitude changes in M2a (STAT6) signature genes, such as Mgl2 (CD301b), a hallmark of M2a-activated macrophages, with M2c (STAT3) signature genes largely unaffected (Fig. 2, F and G). Thus, those genes most significantly altered by injection are related to M2a polarization, which indicates that rhoptry proteins can act in trans to alter macrophage function.
The ability to induce an M2 signature is present in both the infected and injected LPM, but these datasets also identify changes that are specific to infection. The use of GSEA to examine clusters of genes regulated in these pathways illustrates the positive impact of infection on cell cycle, while the elevated IFN-γ signaling signature seen in bystander LPM is absent from the infected LPM (Fig. 2 H). Further, the use of GO analysis, presented as radar plots for GO terms (Fig. S2 D), affirms that infection or injection in vivo is sufficient to generate an M2 population, and that CEP acts in cis to up-regulate cell processes associated with cell division and DNA repair but limit macrophage functions associated with protective immunity.
Impact of STAT3 and STAT6 on macrophage responses and control of CEP in vivo
The transcriptional profiling analyses described above highlight the potential impact of injection of parasite effectors on the proliferation and polarization of LPM. To assess the extent of these changes at the protein level, Ai6 mice were infected with CEP-Cre-tdTomato parasites and at 1 dpi stained for surface expression of CD301b, a canonical marker of M2 macrophages, and the intracellular molecule Ki67 as an indicator of cellular proliferation (Fig. 3 A). Bystander LPM did not express CD301b or Ki67, but the majority of LPM that had been infected or injected up-regulated CD301b, and a significant proportion of the CD301b+ population also expressed Ki67 (Fig. 3, A–C). Analysis of infected and injected LPM from the Ai6 and Ai6/STAT6−/− mice at 1 dpi showed that while STAT6 was required for maximal CD301b and Ki67 expression in both the injected and infected cells, the injected cells were more impacted by the loss of STAT6 (Fig. 3, D and E). These data demonstrate that injection of rhoptries alone induces a STAT6-dependent M2 phenotype in vivo.
Figure 3.

Response of STAT6−/− and LysM-STAT3 mice to CEP infection. (A) To assess the impact of infection on macrophage proliferation, Ai6 mice were injected i.p. with 106 CEP-Cre-tdTomato. At 1 dpi, LPM fractions I–IV were assessed by flow cytometry for expression of CD301b and Ki67. Representative flow plots from one of two independent experiments (n = 4 mice). (B and C) The percentage of Ki67+ or CD301b+ LPM for fractions I, III, and IV. Summary data are from one of two independent experiments (n = 4 mice). (D and E) Ai6 (open bars) and Ai6/STAT6−/− (filled bars) mice were infected i.p. with 106 CEP-Cre-tdTomato, and at 1 dpi, LPM fractions I, III, and IV expression of CD301b and Ki67 were compared. Summary data are from one of two independent experiments (n = 4 mice/genotype). (F and G) Stat3flox, LysM-STAT3, or STAT6−/− mice were infected i.p. with 106 CEP-Cre-tdTomato, and at 1 dpi, LPM were examined for expression of CD301b and CD206 in uninfected (CEP−) and infected (CEP+) populations. Flow plots and summary data from one of two representative experiments (n = 4–5 mice/genotype). (H) C57BL/6 or STAT6−/− mice were infected i.p. with 105 CEP, and on 5 dpi, PECs were used to prepare cytospins, and the percentage of cells infected was determined. Summary data presented are the means from four individual experiments compared by two-tailed paired t test (n = 4–5 mice/genotype/experiment). (I) Stat3flox or LysM-STAT3 mice were infected i.p. with 105 CEP-Cre-tdTomato, and on 5 dpi, PECs were used to prepare cytospins, and the percentage of cells infected was determined. Summary data presented are the means from three individual experiments compared by two-tailed paired t test (n = 4–5 mice/genotype/experiment). (J) Stat3flox, LysM-STAT3, or STAT6−/− mice were infected i.p. with 105 CEP-Cre-tdTomato, and at 10 dpi, the levels of IFN-γ in the serum were measured by ELISA. Summary data from one of two independent experiments (n = 4–5 mice/genotype). Summary statistics represent mean ± SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed unpaired Student’s t test).
To test the role of STAT3 in parasite-induced M2 polarization, LysMCre mice were crossed with Stat3flox mice to generate progeny (LysM-STAT3) in which myeloid cells lacked STAT3. The presence of the LysMCre precludes tracking of injected cells with the Ai6 system, but the polarization of LPM infected with fluorescent CEP could be compared between Stat3flox, LysM-STAT3, and STAT6−/− mice. At 1 dpi, the uninfected LPM in the Stat3flox, LysM-STAT3, and STAT6−/− mice did not express CD301b or CD206 at high levels, whereas infected LPM from control Stat3flox mice were positive for CD301b and CD206 (Fig. 3, F and G). As expected, the infected STAT6−/− LPM were impaired in levels of CD206 and CD301b, whereas infected STAT3-deficient LPM up-regulated CD301b while expression of CD206 was reduced (Fig. 3, F and G). Thus, while STAT6 has a prominent role in the ability of infection to promote an M2 phenotype, STAT3 also contributes to these events.
The roles of STAT3 and STAT6 in the immune response to the parasite were also examined by infecting WT and STAT6−/− mice as well as Stat3flox and LysM-STAT3 mice with CEP. At 5 dpi, across multiple experiments in the STAT6−/− and LysM-STAT3 mice, there was a modest decrease in parasite burden compared with relevant controls in the peritoneal exudate cells (PECs) and peripheral tissues (Fig. 3, H and I; and Fig. S3 A). Despite this lower parasite burden at 5 dpi, both the LysM-STAT3 and STAT6−/− mice had a significantly higher fraction of activated T cells (CD11aHICD62LLO) in the spleen than the Stat3flox mice at 10 dpi (Fig. S3 B), and restimulation of splenocytes with soluble Toxoplasma antigen (STAg) resulted in increased production of IFN-γ (Fig. S3 C). Interestingly, at 10 dpi, the LysM-STAT3 mice had the highest levels of IFN-γ in the serum (Fig. 3 J), and their macrophages, monocytes, and type 2 conventional dendritic cells (cDC2s) were more activated on the basis of expression of B7 molecules (CD80 and CD86) compared with the Stat3flox and STAT6−/− mice (Fig. S3 D). At this time point, these populations are not infected, and this phenotype likely results from the absence of IL-10–mediated activation of STAT3 in LysM+ cells that limits inflammation during infection (Gao et al., 2018; Gazzinelli et al., 1996; Neyer et al., 1997). These data suggest that the ability of ROP16 to phosphorylate STAT6 and STAT3 results in suppression of early parasite control mechanisms, but the interpretation of these datasets is complicated by the involvement of these transcription factors in cytokine signaling pathways that affect inflammation.
Figure S3.

Analysis of STAT6−/− and LysM-STAT3 mice during CEP infection. (A) Stat3flox or LysM-STAT3 mice were infected i.p. with 105 CEP-Cre-tdTomato, and on 5 dpi parasite burden from the liver, lung, and PECs was measured by quantitative PCR. The amount of parasite DNA measured per 200 ng of host DNA in each tissue was averaged for all the mice of each genotype in each experiment and plotted pairwise across the Stat3flox and LysM-STAT3 genotypes. Summary data of all the tissues measured in three independent experiments were compared between Stat3flox and LysM-STAT3 groups by Wilcoxon test (n = 4–5 mice/genotype/experiment). (B–D) Stat3flox, LysM-STAT3, or STAT6−/− mice were infected i.p. with 105 CEP-Cre-tdTomato, and at 10 dpi, the adaptive and innate responses were measured. (B) Numbers of activated (CD11aHICD62LLO) CD8+ and CD4+ T cells in the spleen were measured by flow cytometry (mean ± SD). (C) Splenocytes were stimulated with STAg for 72 h, and IFN-γ production was measured by ELISA (mean ± SD). (D) The fraction of cDC2 in the peritoneum and monocytes and cDC in the spleen that were activated (CD80+CD86+) was measured by flow cytometry (mean ± SD). Summary data from one of two independent experiments (n = 4–5 mice/genotype). Summary statistics represent mean ± SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed unpaired Student’s t test). Fig. S3 is related to Fig. 3.
Impact of ROP16 deletion on host response to infection
To directly assess the contribution of ROP16 to the development of an M2-like phenotype in macrophages and its impact on parasite survival and growth, ROP16-deficient parasites (CEPΔrop16) were generated from CEP-Cre-tdTomato parasites. While WT CEP parasites induced phosphorylation of STAT6 and STAT3 and in injected and infected populations polarized BMMøs toward an M2 phenotype (CD301b+CD206+), the CEPΔrop16 parasite failed to do so (Fig. 4, A and B). The CEPΔrop16 parasites complemented with ROPIII16 restored the ability to activate STAT6 and generate an M2 phenotype (Fig. 4, A and B). To determine whether ROP16 impacted parasite replication, BMMøs were infected with either CEP or CEPΔrop16. In resting BMMøs, CEP and CEPΔrop16 parasites showed similar levels of parasite growth (Fig. S4). Further, when BMMøs were activated with IFN-γ and TNF-α before infection, the ability of these stimuli to limit the growth of CEP and CEPΔrop16 was similar. Thus, loss of ROP16 does not impact baseline parasite replication or alter the capacity of M1 macrophages to control parasite growth.
Figure 4.

Effect of depletion of ROP16 in CEP on macrophage polarization in vitro and in vivo. (A) Primary Ai6 BMMøs were infected with CEP-Cre-tdTomato (CEP), CEP-Cre-tdTomato-Δrop16 (Δrop16), or CEP-Cre-tdTomato-Δrop16:ropIII16 (Δrop16:ropIII16) parasites, and at 24 hpi, the ability of the parasite to phosphorylate STAT6 was assessed by flow cytometry (gray, FMO [fluorescence minus one]; red, CEP; blue, Δrop16; green, Δrop16:ropIII16). Bar graphs depict the MFI of pSTAT6 in each fraction (I–IV). Flow plots and summary data are representative of one of two independent experiments (n = 3 replicates/condition). (B) After infection with CEP, Δrop16, or Δrop16:ropIII16 strain, the frequency of uninfected (I), infected (III), or injected (IV) cells that expressed CD301b and CD206 was quantified by flow cytometry. Summary data are representative of one of two independent experiments (n = 3 replicates/condition). (C and D) Ai6 mice were infected i.p. with 106 CEP or Δrop16, and at 24 hpi, LPM in fractions I, III, and IV were analyzed by flow cytometry for the expression of CD301b versus CD206 (C) and CD301b versus NOS2 (D). The percentage of each population is quantified and presented as a bar graph. Summary data are representative of one of two independent experiments (n = 4 mice/condition). (E) Ai6 mice were infected i.p. with 106 CEP, Δrop16, or Δrop16:ropIII16, and at 24 hpi, LPM in fractions I, III, and IV were analyzed by flow cytometry for the expression of CD301b. MFI is quantified and presented as a bar graph. Flow plots and summary data are representative of one of two independent experiments (n = 4 mice/condition). Summary statistics represent mean ± SD; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; *****, P < 0.00001; ******, P < 0.000001 (two-tailed unpaired Student’s t test).
Figure S4.
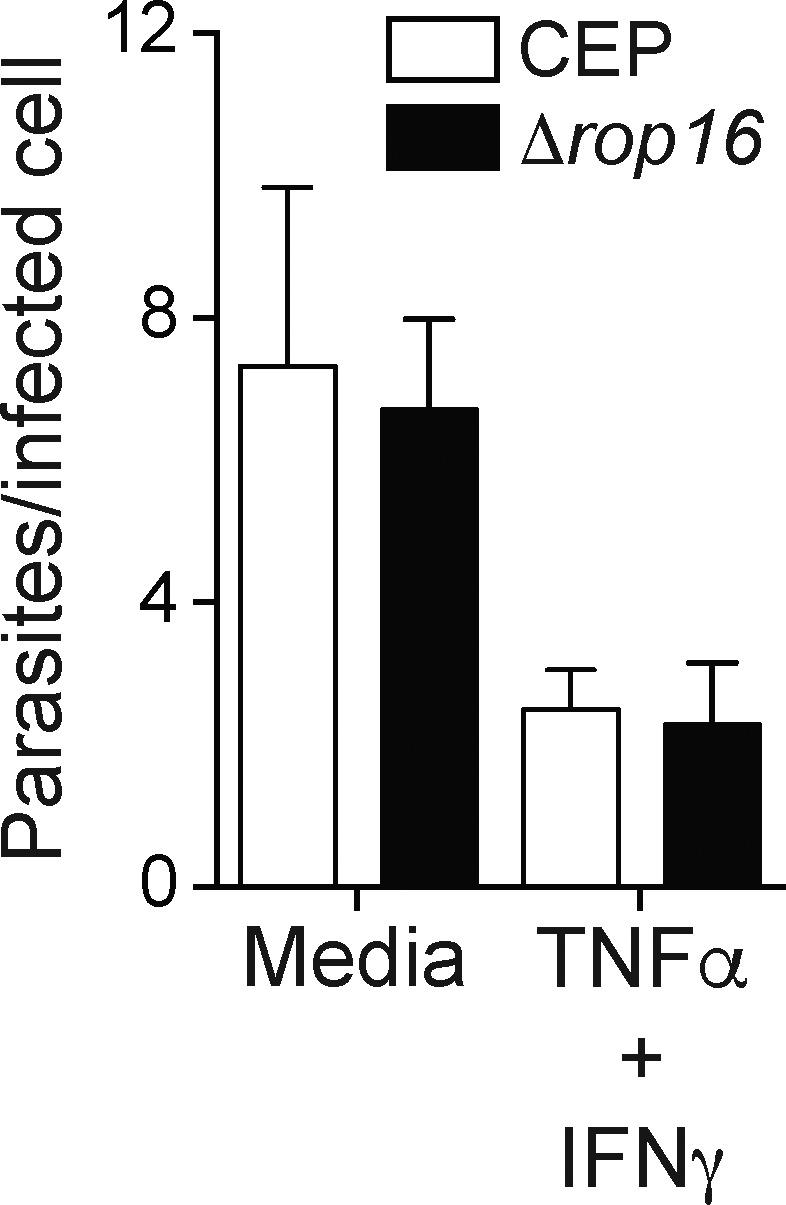
Growth and control of CEP in BMMø is ROP16 independent. Resting BMMøs or BMMøs stimulated with IFN-γ plus TNF for 10 h were infected with CEP or Δrop16, and 20–24 h later cytospins were used to calculate the percentage of cells infected and the numbers of parasites per infected cell. Summary data from one of two independent experiments (n = 3 replicates/condition). Summary statistics represent mean ± SD. Fig. S4 is related to Fig. 4.
To assess the impact of ROP16 on the phenotype of macrophage populations in vivo, Ai6 mice were infected with CEP or CEPΔrop16. While LPM infected or injected by CEP were polarized toward an M2-like phenotype, LPM infected or injected by CEPΔrop16 did not up-regulate these M2 markers (Fig. 4 C). The injected population was most affected by the loss of ROP16, while the majority of infected cells were still CD206+ but the levels of CD301b were reduced. Similarly, the loss of ROP16 also resulted in a reduced percentage of injected or infected cells that were Ki67+ (Fig. S4 C). Along with the induction of an M2 phenotype, the impact of ROP16 loss in injected and infected LPM on the induction of an M1 phenotype was assessed by examining inducible nitric oxide synthase (iNOS), a functional marker of M1 polarization, versus CD301b expression. At 1 dpi, a small fraction of bystander LPM produce iNOS, and infection with CEP and CEPΔrop16 resulted in induction of CD301b expression and the absence of iNOS expression (Fig. 4 D). Interestingly, LPM injected by CEP were mostly CD301b+ and no cells expressed iNOS, but LPM injected by CEPΔrop16 showed a significant increase in the numbers of iNOS+ cells and a reduction in the proportion of CD301b+ LPM (Fig. 4 D). It should be noted that infected or injected LPM from mice infected with the complemented Δrop16:ropIII16 CEP strain recapitulated the up-regulation of CD301b observed with the WT strain (Fig. 4 E).
To determine if the loss of ROP16 impacted parasite replication at early time points in vivo, mice were challenged with CEP or CEPΔrop16 and parasite burden was measured. By 4 dpi, the loss of ROP16 resulted in a 40–50% reduction of infected cells in the peritoneum (Fig. 5 A), and at 10 dpi the CEP parasites had disseminated to the lung, brain, heart, and liver, and the overall levels of the CEPΔrop16 were markedly reduced (Fig. 5 B). This effect was not a consequence of a reduced ability of the mutant parasite to replicate, because infection of mice treated with IFN-γ–blocking antibodies or that lacked IL-12p40 (Il12b−/− mice) resulted in high parasite burdens that were comparable between CEP and CEPΔrop16 infection (Fig. 5, C and D). To assess how ROP16 affects the parasite-specific immune response, WT mice were challenged with CEP or CEPΔrop16 and assessed at 10 dpi, the peak of infection-induced T cell responses. Systemic cytokine levels and T cell responses are typically proportional to parasite burden during T. gondii infection (Gavrilescu and Denkers, 2001; Mordue et al., 2001), but despite the reduced parasite burden, mice infected with CEPΔrop16 had elevated serum concentrations of IL-12p40 and IFN-γ (Fig. 5 E). Moreover, after stimulation of splenocytes with STAg, those isolated from mice challenged with CEPΔrop16 produced the highest levels of IFN-γ (Fig. 5 F). The use of parasite-specific MHC class I and II tetramers revealed that both CEP and CEPΔrop16 induced parasite-specific CD4+ and CD8+ T cells, but the mutant induced the highest levels (Fig. 5 G). These parasite-specific T cells can be divided into three subsets based on expression of C-X-C motif chemokine receptor 3 (CXCR3) and killer cell lectin-like receptor G1 (KLRG1) (Chu et al., 2016). In the absence of ROP16, there was an increase in the proportion of terminally differentiated (KLRG1+CXCR3−) effector T cells (Fig. S5, A and B). To determine the ability of parasite-specific CD8+ T cells to lyse target cells, an in vivo CTL assay was performed using a parasite-specific peptide (SVLAFRRL). Mice infected with CEP or CEPΔrop16 showed specific loss of peptide pulsed targets, with the highest levels of cytotoxicity observed in mice infected with CEPΔrop16 (Fig. 5 H). However, there was a linear correlation between the lysis of transferred target cells and the number of parasite-specific CD8+ T cells in individual mice (Fig. 5 I). These data indicate that during CEP infection, the presence of ROP16 limits the magnitude, but not the functionality, of the parasite-specific T cell response.
Figure 5.

Loss of ROP16 results in improved parasite control and enhanced T cell responses. (A) C57BL/6 mice were infected i.p. with 105 tachyzoites of CEP or Δrop16, and at 5 dpi, the percentage of infected PECs was determined by cytospin analysis. Summary data are representative of one of two independent experiments (n = 4 or 5 mice/condition). (B) C57BL/6 mice were infected i.p. with CEP or Δrop16 and sacrificed at 10 dpi. DNA was extracted from liver, lung, heart, and brain, and levels of parasite DNA were assessed using quantitative PCR. Summary data from one of two independent experiments (n = 5 mice/condition). (C and D) C57BL/6 mice treated with an isotype control antibody or α-IFN-γ or IL-12p40−/− mice were infected i.p. with 105 tachyzoites of CEP or Δrop16. On day 5, PECs were collected and the percentage of cells infected was calculated. Summary data are representative of two experiments (n = 5 mice/condition). Scale bars, 50 µm. (E and F) C57BL/6 mice were infected with CEP or Δrop16, and at 10 dpi, serum was used to determine levels of IL-12 and IFN-γ by ELISA (E), while splenocytes were stimulated with STAg for 72 h, and IFN-γ production was measured by ELISA (F). Summary data from one of two independent experiments (n = 5 mice/condition). (G) Parasite-specific T cells in the spleen were measured using a combination of LFA1 and tetramers specific for MHC I (tetramer H2Kb) or II (tetramer_I-Ab) parasite-derived peptides. Flow plots and summary data from one of two independent experiments (n = 5 mice/condition). (H) In vivo CTL assay of mice infected with CEP or Δrop16 for 10 d. Representative histograms of target cells recovered from indicated mice. Frequency of control target cells (CellTrace Violet low, upper left) and those pulsed with the SVLAFRRL T. gondii peptide (CellTrace Violet high, upper right) are indicated. Populations of transferred cells in the spleen were analyzed at 10 h after transfer. Target cells transferred into naive mice were analyzed as a negative control. Flow plots are representative of one of two independent experiments (n = 5 mice/condition). (I) The frequency of tetramer-positive CD8+ T cells specific for SVLAFRRL from mice infected with CEP or Δrop16 is plotted against the percentage of target cell lysis. Linear regression was performed and plotted. R2 = 0.71; P < 0.01. Summary data are representative of one of two independent experiments (n = 5 mice/condition). Summary statistics represent mean ± SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (two-tailed unpaired Student’s t test).
Figure S5.

ROP16-dependent T cell response to CEP infection. (A and B) To characterize the T cell response, splenocytes from CEP or Δrop16 infected mice (10 dpi) were analyzed by flow cytometry for KLRG1 and CXCR3 on LFA1hi or parasite-specific CD4+ (A) and CD8+ (B) T cells. Flow plots and summary data are representative of one of two independent experiments (n = 5 mice/group). Summary statistics represent mean ± SD; *, P < 0.05; **, P < 0.01 (two-tailed unpaired Student’s t test). Fig. S5 is related to Fig. 5.
Discussion
The ability of T. gondii to attach to, invade, and establish the PV within host cells is a complex process that involves proteins secreted from the micronemes, rhoptries, and dense granules. Effector proteins from micronemes and rhoptries that are required for parasite attachment and invasion are termed “early” effectors, while those from rhoptries and dense granules that aid in establishing the PV and nutrient acquisition are considered “late” effectors that include ROP5, ROP18, and TgIST. Consequently, the ability to isolate the impact of early effectors in isolation has been a challenge, an issue relevant to pathogens that introduce microbial effectors into uninfected host cells. The Toxoplasma-Cre system has been critical to be able to demonstrate that cells injected by the parasite can be differentiated from infected and bystander populations and subsequently tracked to understand how early virulence factors impact host cells. The studies here illustrate the strain-dependent impact of these early effector proteins, as injected proteins were responsible for approximately one third of the transcriptional changes associated with a type III strain, but significantly reduced in cells infected with a type II strain (Pru). Indeed, ~40% of the changes that resulted from injection by CEP were STAT6 dependent. While the ability of certain strains of T. gondii to promote an M2 phenotype are dependent on ROP16 (Jensen et al., 2011), the ability to compare infected and injected cells provided the ability to distinguish the role of early- and late-stage parasite effectors on macrophage function in vivo.
One of the overarching themes that has emerged in the biology of T. gondii is its ability to use diverse mechanisms to target the STAT family of transcription factors (STAT1, 3, 5, and 6; Denkers et al., 2012). Recent work has shown that all strains of T. gondii use the effector protein TgIST to block STAT1 signaling and thus inhibit IFN-γ–associated antimicrobial mechanisms after day 5 of infection (Gay et al., 2016; Olias et al., 2016). Interestingly, the ability to use the CEP strain shows that the loss of ROP16 results in a marked reduction in parasite levels as early as 4–5 dpi, which is even more significant by 10 dpi and has been extended to the chronic phase of infection (Tuladhar et al., 2019). Here, transcriptional profiling of macrophages infected and injected by CEP in vivo showed that suppression of IFN-γ–dependent innate immune mechanisms was only observed in infected cells, consistent with the role of TgIST as a late effector protein. Perhaps the most profound effect of CEP injection or infection of macrophages in vivo is the ability of ROP16 to induce an M2 phenotype, a process that requires both STAT3 and STAT6. Infection with CEPΔrop16 revealed that ROP16 is needed for optimal M2 polarization in infected and injected cells, but in vivo other parasite effector proteins may contribute to the M2 phenotype of infected cells but not injected cells (Fig. 4 C). It has been suggested that the ROP16-induced STAT3 induction engages anti-inflammatory signaling, while STAT6 activity may allow increased parasite replication (Butcher et al., 2005, 2014). The studies presented here do not distinguish between these possibilities but could explain why the reduced parasite burden observed with the CEPΔrop16 parasites was more striking than that observed with CEP in the STAT6−/− or LysM-STAT3 mice. This result may indicate an interaction between STAT3 and STAT6, perhaps as combinations of homo- and heterodimers, to enforce the maximal M2 phenotype in affected macrophages.
M2 macrophages have an important role in wound healing and tumor progression, and there is broad interest in dissecting the pathways that promote the generation of these cells by pathogens such as Mycobacterium tuberculosis, Brucella abortus, and Listeria monocytogenes (Abdullah et al., 2012; Mahajan et al., 2012; Price and Vance, 2014; Rajaram et al., 2010). It has been proposed that the altered metabolic state of M2 macrophages supports the persistence of Salmonella typhimurium, B. abortus, and Chlamydia pneumoniae (Buchacher et al., 2015; Eisele et al., 2013; Xavier et al., 2013), while CD206hi macrophages are permissive for the growth of Leishmania major (Lee et al., 2018). Thus, the ability of a range of intracellular pathogens to either generate or target M2-like macrophage populations is evolutionarily conserved. Currently, the fate and impact of the injection-induced M2 populations on the outcome of infection is uncertain and will require novel approaches to target this population. M2 cells have reduced antimicrobial activities and may provide a refuge for parasite replication, or their ability to resolve inflammation or inhibit T cell responses may be another strategy to promote microbial growth (Gordon and Martinez, 2010; Huber et al., 2010; Osborne et al., 2014; Sutherland et al., 2014). In support of this idea, the marked increase in the magnitude of the parasite-specific T cells observed with CEPΔrop16 infection despite a marked reduction in parasite burden indicates a previously unappreciated role for ROP16 to mitigate T cell responses and correlates with a significant decrease in the M2 populations. There are a number of potential mechanisms whereby M2 macrophages can antagonize the development of cell-mediated immunity, and ongoing studies are focused on understanding the basis for this ROP16-mediated suppression of T cell responses.
Materials and methods
Mice and parasites
All procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania (Animal Welfare Assurance reference number A3079-01) and were carried out in accordance with the guidelines set forth in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). C57BL/6 mice were obtained from Taconic. Ai6 mice (stock 007906), a Cre reporter strain that expresses ZsGreen1 downstream of a CAG promoter in the Rosa26 locus (Madisen et al., 2010), STAT6−/− (stock 005977), LysM-Cre (stock 004781), Stat3flox (stock 016923), and Il12b−/− (stock 002693) mice were originally obtained from The Jackson Laboratory and bred at the University of Pennsylvania. Littermate controls were used for experiments with Stat3flox and LysM-STAT3 mice, C57BL/6 mice were used as controls in experiments with STAT6−/− mice, and WT Ai6 mice were used as controls in experiments with Ai6/STAT6−/− mice.
Transgenic Toxoplasma-Cre strains of T. gondii (Pru-Cre-tdTomato) were generated as previously described (Koshy et al., 2010). The engineering of the CEPΔROP16III strain is fully described in Tuladhar et al. (2019). In brief, CEPΔhpt was cotransfected with four plasmids: two engineered with guide RNAs to upstream or downstream 21-mers for the rop16 locus, the pTKO plasmid engineered to contain 500-bp ROP16 homology regions surrounding the hypoxanthine phosphoribosyltransferase hxgprt cassette and a toxofilin-Cre cassette, and one plasmid containing the coding sequence for tdTomato under the GRA2 promoter. The plasmids bearing regions of homology to the ROP16 locus and encoding tdTomato were linearized by restriction digest before transfection. Stable transfected parasites were selected using hxgprt and single-cell clones that were HPT+ and tdTomato+ were confirmed by PCR to have a disrupted ROP16 locus and no longer cause phosphorylation of STAT6 in human foreskin fibroblasts. A clone that was >85% efficacious at causing Cre-mediated recombination was selected for use. To complement the Cre-mediated recombination, parasites were transfected with linearized plasmid containing a FLAG-tagged ROP16III gene cassette driven by the endogenous promoter and harboring a bleomycin resistance cassette. Stable transfected parasites were selected using bleomycin (Messina et al., 1995). Single-cell clones were screened by PCR for rop16 integration.
Stable transgenic parasite lines were maintained in vitro by serial passage through human fibroblasts in parasite culture medium (DMEM [Invitrogen], 20% media M199 [Invitrogen], 10% FBS [Serum Source International], 1% penicillin-streptomycin [Invitrogen], and 25 µg/ml gentamicin [Gibco]). Tachyzoites of each strain were prepared for infection by serial needle passage and filtered through a 5-µm pore filter. Mice were routinely infected i.p. with 105 parasites, a dose that is readily controlled by an immune-competent host. In some experiments, a dose of 106 was used in order to increase the frequency of infected or injected populations required for sorting and for transcriptional profiling.
Generation of BMMøs
C57BL/6, Ai6, and Ai6/STAT6−/− mice were used to generate BMMøs. RBCs were lysed with 0.86% ammonium chloride. Bone marrow cells were then plated at a concentration of 2 × 106 cells per 10 ml of media in 100-mm plates. For BMMøs, macrophage medium was composed of DMEM supplemented with 10% serum, 25 mM Hepes, 1% penicillin-streptomycin, 1 mM sodium pyruvate [Gibco], 0.1% β-mercaptoethanol, and 30% L929 supernatant. Media was replenished on days 3 and 6. Between days 7 and 9, cells were harvested using ice-cold PBS with 5 µM EDTA. To induce M1, M2a, and M2c macrophage subtypes, BMMøs were stimulated for 24 h with IFN-γ (10 ng/ml), IL-4 (10 ng/ml), or IL-10 (10 ng/ml), respectively.
Cell sorting
BMMøs or PECs were sorted using a FACSAria III cell sorter (BD Biosciences; device: 5-ml tubes; precision: single-cell; nozzle: 100 µm). Forward-scatter area versus side-scatter area was used to exclude damaged cells and dead cells were stained with LIVE/DEAD Fixable Aqua. 50,000 cells were sorted into different 5-ml tubes filled with 700 μl of cell lysis buffer (Buffer RLT, Qiagen) for RNA extraction and stored at −80°C.
Transcriptional profiling
For RNA-sequencing analysis, cDNA was prepared from sorted macrophages using the SMART V4 Ultra Low RNA kit (Clontech), followed by library construction with Nextera XT. A Tapestation (Agilent Technologies) and Qubit (Thermo Fisher Scientific) were used for library sizing and quantification, respectively. Finally, libraries were pooled and sequenced on an Illumina NextSeq 500 sequencer. The Kallisto pseudoaligner (Bray et al., 2016) was used to map reads to the reference transcriptomes from mouse (Ensembl, GRCm38.p6) and T. gondii (ToxoDB). Analysis was performed using statistical computing environment R, RStudio software, and Bioconductor suite of packages (Huber et al., 2015). Differential gene expression analysis with a Bonferroni-Hochberg statistical analysis for multiple tests was performed to identify DEGs in group comparisons. GO analysis was performed with the DAVID (Database for Annotation, Visualization and Integrated Discovery) online tool (Huang et al., 2009). GSEA was conducted using GSEA software (version 3.0) and the MSigDB investigating “C2: Canonical Pathways” and “C7: immunologic signatures” collections from MSigDB (Subramanian et al., 2005).
Microarrays and data analyses were performed as previously described (Beiting et al., 2015). Briefly, total RNA was isolated from sorted BMMøs using the Qiagen RNeasy kit. Biotin-labeled complementary RNA was generated using the Illumina TotalPrep RNA amplification kit (Ambion) and was used to hybridize Illumina MouseRef-8 v2.0 expression BeadArrays. Scanned images were converted to raw expression using GenomeStudio v1.8 software (Illumina). Data analysis was performed using the R (v3.0.2) statistical computing environment. Raw data were background subtracted, variance stabilized, and normalized by robust spline normalization using the lumi package (Du et al., 2008). DEGs were identified by linear modeling and Bayesian statistics using the limma package (Ritchie et al., 2015). GSEA was conducted using GSEA software from the Broad Institute (Subramanian et al., 2005). Enrichment score refers to the degree to which the gene set is overrepresented at the top or bottom of the ranked input list of genes.
Analysis of macrophage and T cell responses
Peritoneal lavage with 8 ml of ice-cold PBS was used to obtain single-cell suspensions that were resuspended in RPMI 1640 supplemented with 10% fetal bovine serum. Single-cell suspensions were prepared from spleens. RBCs were lysed using 0.86% ammonium chloride. Samples for flow cytometric analysis (2 × 106) were prepared as previously described (Christian et al., 2014). The induction and detection of intracellular cytokines was performed after incubation with 5 µg/ml brefeldin A at 37°C for 6 h. Harvested cells were surface stained as described above, fixed using 2% paraformaldehyde at room temperature for 10 min, and then washed with flow cytometry buffer. Detection of phosphorylated STAT proteins was performed by harvesting BMMø in ice-cold PBS + EDTA with subsequent fixation in 2% paraformaldehyde on ice for 20 min. Cells were then incubated in 90:10 methanol:PBS at −20°C overnight. Cells were stained for pSTAT3 and pSTAT6 for 2 h at 4°C, rinsed in PBS, and analyzed by flow cytometry. Cells were then stained for intracellular cytokines in 50 µl of 0.5% saponin (Sigma-Aldrich) in flow cytometry buffer at 4°C for 1 h. Antibodies used were pSTAT6 (Tyr641)–APC; Thermo Fisher Scientific: 17-9013-41; lot: 4331158; clone: CHI254N, pSTAT3 (Tyr705)–ef450; Thermo Fisher Scientific: 48-9033-42; lot: 4294783; clone: LUVNKLA, CD301b–AF647; BioLegend: 146806; lot: B181103; clone: URA-1, CD206–BV650; BioLegend: 141723; lot: B241225; clone: C068C2, ARG1–APC; R&D Systems: IC5868A; lot: ADBC0216041, CD11b–BV605; BioLegend: 301332; lot: B245640; clone: M1/70, CD102–ef450; Thermo Fisher Scientific: 48-1021-82; lot: E09976-1631; clone: 3C4 (MIC2/4), MHCII (I-A/I-E)–BV711; BioLegend: 107643; lot: B240003; clone: M5/114.15.2, Ly6C–BV785; BioLegend: 128041; lot: B258005; clone: HK1.4, Ki67–AF700; BD Biosciences: 561277; lot: 7154974; clone: B56, CD11c–APC-Fire 750; BioLegend: 117352; lot: B231341; clone: N418, CD64–PE-Cy7; BioLegend: 139314; lot: 8257960; clone: X54-5/7.1, iNOS–PerCp-ef710; Thermo Fisher Scientific: 46-5920-80; lot: 4342191; clone: CXNFT, CD3–PE-CF594; BD Biosciences: 562332; lot: 62399771; clone: 145-2C11, CD4–BV650; BioLegend: 100555; lot: B244331; clone: RM4-5, CD8–BV605; BioLegend: 100744; lot: B246676; clone: 53–6.7, LFA1–PerCp-Cy5.5; BioLegend: 141008; lot: B249460; clone: H155-78, CD19–PerCp-Cy5.5; BioLegend: 152406; lot: B241294; clone: 1D3/CD19, B220–PerCp-Cy5.5; BioLegend: 103236; lot: B239431; clone: RA3-6B2, NK1.1–PerCp-Cy5.5; BioLegend: 108728; lot: B241349; clone: PK136, CD3–PerCp-Cy5.5; Thermo Fisher Scientific: 45-0031-82; lot: 4341614; clone: 145-2C11, KLRG1–FITC; Thermo Fisher Scientific: 11-5893-82; lot: 1995313; clone: 2F1, CXCR3–PE-Cy7; BioLegend: 126516; lot: B208132; clone: CXCR3-173, CD206–AF647; BioLegend: 141712; lot: B182140, CD301b–PE-Cy7; BioLegend: 146808; lot: B199663, F4/80–APC-eFluor780; Thermo Fisher Scientific: 47-4801-82; lot: E10234-1640, CD80–FITC; Thermo Fisher Scientific: 11-0801-85; lot: E00406-1630, CD86–AF700; BioLegend: 105024; lot: B245102, KLRG1–FITC; Thermo Fisher Scientific: 11-5893-82; lot: 1995313, CD11a–PerCy-Cy5.5; BioLegend: 101124; lot: B243970, CD62L–BV711; BioLegend: 104445; lot: B248100, CD3–APC-eFluor780; Thermo Fisher Scientific: 47-0032-82; lot: 1960148, CD102–FITC; BioLegend: 105606; lot: B179122, tetramer-MHCI–PE; NIH Tetramer Core; peptide: SVLAFRRL, tetramer-MHCII–PE; NIH Tetramer Core; peptide: AVEIHRPVPGTAPPS, tetramer-MHCI–APC; NIH Tetramer Core; peptide: SVLAFRRL, tetramer-MHCII–APC; NIH Tetramer Core; peptide: AVEIHRPVPGTAPPS.
Results were analyzed using FlowJo 9.9 software (Tree Star). APC-H2-Kb-SVLAFRRL and APC-I-Ab–AVEIHRPVPGTAPPSFSS tetramers were provided by the NIH Tetramer Core Facility. Cytolytic activity of antigen-specific CD8+ T cells was assessed by in vivo CTL assay, adapted from Jordan et al. (2009). Splenocytes isolated from C57BL/6 mice were pooled and half of the isolated splenocytes were labeled with 2.5 µM CellTrace Violet (pulsed) and subsequently pulsed with 1 µg/ml SVLAFRRL peptide (MBL International Corporation) for 1 h at 37°C. The remaining splenocytes were labeled with 0.125 µM CellTrace Violet (unpulsed) and then incubated for 1 h at 37°C. Pulsed and unpulsed cells were washed extensively in PBS, counted, and mixed at a 1:1 ratio. A total of 5 × 106 cells per mouse were injected i.v. into recipient mice infected 10 d prior with the indicated strain of T. gondii. Mice were sacrificed 10 h later, and spleens were analyzed for lysis of the peptide-pulsed population by gating on donor cells labeled with CellTrace Violet. Specific lysis was calculated as described previously (Jordan et al., 2009). The proportion and number of TGD057 tetramer–specific CD8+ T cells was also analyzed from spleens to normalize comparisons.
Data availability
Microarray data and RNA-sequencing data that support the findings of this study (Figs. 1 and 2) have been deposited in the Gene Expression Omnibus database for public access (accession nos. GSE55892 and GSE140774). All other data that support the findings of this study are available from the corresponding author upon reasonable request.
Online supplemental material
Supplemental information includes five figures and one table. Fig. S1 shows further differences between BMMøs infected or injected by type II or type III parasites in vitro. Fig. S2 shows further analysis of RNA sequencing of LPM infected or injected by CEP-Cre-tdTomato. Fig. S3 shows further analysis of LysM-STAT3 mice infected with CEP-Cre-tdTomato. Fig. S4 shows ROP16-dependent in vitro killing assays using BMMøs infected with either CEP or CEPΔrop16 parasites. Fig. S5 shows further characterization of T cell responses in WT mice at 10 dpi with either CEP or CEPΔrop16 parasites. Table S1 lists preranked genes for M2 macrophages.
Supplementary Material
lists preranked genes for M2 macrophages.
Acknowledgments
We thank the members of the Hunter laboratory for intellectual and experimental contributions to this study. We thank Deborah Argento for assistance with graphics.
This work was supported by grants from the National Institutes of Health (1R21AI126042-01 and R01AI125563 to C.A. Hunter, R01NS095994 and supplement 02S1 to A.A. Koshy), the Commonwealth of Pennsylvania (C.A. Hunter), the University of Arizona and the BIO5 Institute (A.A. Koshy), the Cancer Research Institute and the Robertson Foundation (A.T. Phan), and the National Natural Science Foundation of China (31030066 to X. Chen). C.A. Hunter is the Mindy Halikman Heyer President’s Distinguished Chair.
Author contributions: L. Chen, D.A. Christian, A.T. Phan, J. Oh, J.T. Clark, X. Chen, D.S. Roos, D.P. Beiting, and C.A. Hunter designed and performed experiments. L. Chen, D.A. Christian, A.T. Phan, S. Wang, C. Berry, and D.P. Beiting performed analysis. J.A. Kochanowsky and A.A. Koshy generated transgenic parasites. L. Chen, D.A. Christian, A.T. Phan, D.P. Beiting, A.A. Koshy, and C.A. Hunter wrote and reviewed the manuscript.
References
- Abdullah Z., Geiger S., Nino-Castro A., Böttcher J.P., Muraliv E., Gaidt M., Schildberg F.A., Riethausen K., Flossdorf J., Krebs W., et al. 2012. Lack of PPARγ in myeloid cells confers resistance to Listeria monocytogenes infection. PLoS One. 7:e37349 10.1371/journal.pone.0037349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autenrieth S.E., Linzer T.R., Hiller C., Keller B., Warnke P., Köberle M., Bohn E., Biedermann T., Bühring H.J., Hämmerling G.J., et al. 2010. Immune evasion by Yersinia enterocolitica: differential targeting of dendritic cell subpopulations in vivo. PLoS Pathog. 6:e1001212 10.1371/journal.ppat.1001212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke M.S., Fentress S.J., Mashayekhi M., Li L.X., Taylor G.A., and Sibley L.D.. 2012. The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Pathog. 8:e1002992 10.1371/journal.ppat.1002992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiting D.P., Hidano S., Baggs J.E., Geskes J.M., Fang Q., Wherry E.J., Hunter C.A., Roos D.S., and Cherry S.. 2015. The orphan nuclear receptor TLX is an enhancer of STAT1-mediated transcription and immunity to Toxoplasma gondii. PLoS Biol. 13:e1002200 10.1371/journal.pbio.1002200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougdour A., Tardieux I., and Hakimi M.A.. 2014. Toxoplasma exports dense granule proteins beyond the vacuole to the host cell nucleus and rewires the host genome expression. Cell. Microbiol. 16:334–343. 10.1111/cmi.12255 [DOI] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., and Pachter L.. 2016. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34:525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Buchacher T., Ohradanova-Repic A., Stockinger H., Fischer M.B., and Weber V.. 2015. M2 polarization of human macrophages favors survival of the intracellular pathogen Chlamydia pneumoniae. PLoS One. 10:e0143593 10.1371/journal.pone.0143593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher B.A., Kim L., Panopoulos A.D., Watowich S.S., Murray P.J., and Denkers E.Y.. 2005. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J. Immunol. 174:3148–3152. 10.4049/jimmunol.174.6.3148 [DOI] [PubMed] [Google Scholar]
- Butcher B.A., Fox B.A., Rommereim L.M., Kim S.G., Maurer K.J., Yarovinsky F., Herbert D.R., Bzik D.J., and Denkers E.Y.. 2011. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 7:e1002236 10.1371/journal.ppat.1002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher B.A., Reese M.L., Boothroyd J.C., and Denkers E.Y.. 2014. Interactions between Toxoplasma effectors and host immune responses. In Toxoplasma gondii. Second edition Weiss L.M. and Kim K., editors. Academic Press, Boston: pp. 505–519. 10.1016/B978-0-12-396481-6.00014-3 [DOI] [Google Scholar]
- Chaves A.C., Cerávolo I.P., Gomes J.A., Zani C.L., Romanha A.J., and Gazzinelli R.T.. 2001. IL-4 and IL-13 regulate the induction of indoleamine 2,3-dioxygenase activity and the control of Toxoplasma gondii replication in human fibroblasts activated with IFN-γ. Eur. J. Immunol. 31:333–344. [DOI] [PubMed] [Google Scholar]
- Christian D.A., Koshy A.A., Reuter M.A., Betts M.R., Boothroyd J.C., and Hunter C.A.. 2014. Use of transgenic parasites and host reporters to dissect events that promote interleukin-12 production during toxoplasmosis. Infect. Immun. 82:4056–4067. 10.1128/IAI.01643-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.H., Chan S.W., Gosling J.P., Blanchard N., Tsitsiklis A., Lythe G., Shastri N., Molina-París C., and Robey E.A.. 2016. Continuous effector CD8(+) T cell production in a controlled persistent infection is sustained by a proliferative intermediate population. Immunity. 45:159–171. 10.1016/j.immuni.2016.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough B., and Frickel E.M.. 2017. The Toxoplasma parasitophorous vacuole: an evolving host-parasite frontier. Trends Parasitol. 33:473–488. 10.1016/j.pt.2017.02.007 [DOI] [PubMed] [Google Scholar]
- Denkers E.Y., Bzik D.J., Fox B.A., and Butcher B.A.. 2012. An inside job: hacking into Janus kinase/signal transducer and activator of transcription signaling cascades by the intracellular protozoan Toxoplasma gondii. Infect. Immun. 80:476–482. 10.1128/IAI.05974-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P., Kibbe W.A., and Lin S.M.. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 24:1547–1548. 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- Eisele N.A., Ruby T., Jacobson A., Manzanillo P.S., Cox J.S., Lam L., Mukundan L., Chawla A., and Monack D.M.. 2013. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe. 14:171–182. 10.1016/j.chom.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj H., Lebrun M., Arold S.T., Vial H., Labesse G., and Dubremetz J.F.. 2007. ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog. 3:e14 10.1371/journal.ppat.0030014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kasmi K.C., Qualls J.E., Pesce J.T., Smith A.M., Thompson R.W., Henao-Tamayo M., Basaraba R.J., König T., Schleicher U., Koo M.S., et al. 2008. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9:1399–1406. 10.1038/ni.1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge R.D., Alaganan A., Tang K., Lou H.J., Turk B.E., and Sibley L.D.. 2014. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe. 15:537–550. 10.1016/j.chom.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming B.D., Chandrasekaran P., Dillon L.A., Dalby E., Suresh R., Sarkar A., El-Sayed N.M., and Mosser D.M.. 2015. The generation of macrophages with anti-inflammatory activity in the absence of STAT6 signaling. J. Leukoc. Biol. 98:395–407. 10.1189/jlb.2A1114-560R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox B.A., Rommereim L.M., Guevara R.B., Falla A., Hortua Triana M.A., Sun Y., and Bzik D.J.. 2016. The Toxoplasma gondii rhoptry kinome is essential for chronic infection. MBio. 7:e00193-16 10.1128/mBio.00193-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Basile J.I., Classon C., Gavier-Widen D., Yoshimura A., Carow B., and Rottenberg M.E.. 2018. STAT3 expression by myeloid cells is detrimental for the T- cell-mediated control of infection with Mycobacterium tuberculosis. PLoS Pathog. 14:e1006809 10.1371/journal.ppat.1006809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilescu L.C., and Denkers E.Y.. 2001. IFN-γ overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J. Immunol. 167:902–909. 10.4049/jimmunol.167.2.902 [DOI] [PubMed] [Google Scholar]
- Gay G., Braun L., Brenier-Pinchart M.-P., Vollaire J., Josserand V., Bertini R.-L., Varesano A., Touquet B., De Bock P.-J., Coute Y., et al. 2016. Toxoplasma gondii TgIST co-opts host chromatin repressors dampening STAT1-dependent gene regulation and IFN-γ-mediated host defenses. J. Exp. Med. 213:1779–1798. 10.1084/jem.20160340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzinelli R.T., Wysocka M., Hieny S., Scharton-Kersten T., Cheever A., Kühn R., Müller W., Trinchieri G., and Sher A.. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ and TNF-α. J. Immunol. 157:798–805. [PubMed] [Google Scholar]
- Gordon S., and Martinez F.O.. 2010. Alternative activation of macrophages: mechanism and functions. Immunity. 32:593–604. 10.1016/j.immuni.2010.05.007 [DOI] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., and Lempicki R.A.. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S., Hoffmann R., Muskens F., and Voehringer D.. 2010. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood. 116:3311–3320. 10.1182/blood-2010-02-271981 [DOI] [PubMed] [Google Scholar]
- Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. 2015. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 12:115–121. 10.1038/nmeth.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C.A., and Sibley L.D.. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10:766–778. 10.1038/nrmicro2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.D.C., Wang Y., Wojno E.D.T., Shastri A.J., Hu K., Cornel L., Boedec E., Ong Y.-C., Chien Y.H., Hunter C.A., et al. 2011. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe. 9:472–483. 10.1016/j.chom.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.D.C., Hu K., Whitmarsh R.J., Hassan M.A., Julien L., Lu D., Chen L., Hunter C.A., and Saeij J.P.J.. 2013. Toxoplasma gondii rhoptry 16 kinase promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect. Immun. 81:2156–2167. 10.1128/IAI.01185-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K.A., Wilson E.H., Tait E.D., Fox B.A., Roos D.S., Bzik D.J., Dzierszinski F., and Hunter C.A.. 2009. Kinetics and phenotype of vaccine-induced CD8+ T-cell responses to Toxoplasma gondii. Infect. Immun. 77:3894–3901. 10.1128/IAI.00024-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy A.A., Fouts A.E., Lodoen M.B., Alkan O., Blau H.M., and Boothroyd J.C.. 2010. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods. 7:307–309. 10.1038/nmeth.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy A.A., Dietrich H.K., Christian D.A., Melehani J.H., Shastri A.J., Hunter C.A., and Boothroyd J.C.. 2012. Toxoplasma co-opts host cells it does not invade. PLoS Pathog. 8:e1002825 10.1371/journal.ppat.1002825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Charmoy M., Romano A., Paun A., Chaves M.M., Cope F.O., Ralph D.A., and Sacks D.L.. 2018. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J. Exp. Med. 215:357–375. 10.1084/jem.20171389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S., Dkhar H.K., Chandra V., Dave S., Nanduri R., Janmeja A.K., Agrewala J.N., and Gupta P.. 2012. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J. Immunol. 188:5593–5603. 10.4049/jimmunol.1103038 [DOI] [PubMed] [Google Scholar]
- Mège J.L., Mehraj V., and Capo C.. 2011. Macrophage polarization and bacterial infections. Curr. Opin. Infect. Dis. 24:230–234. 10.1097/QCO.0b013e328344b73e [DOI] [PubMed] [Google Scholar]
- Melzer T., Duffy A., Weiss L.M., and Halonen S.K.. 2008. The γ interferon (IFN-γ)-inducible GTP-binding protein IGTP is necessary for toxoplasma vacuolar disruption and induces parasite egression in IFN-γ-stimulated astrocytes. Infect. Immun. 76:4883–4894. 10.1128/IAI.01288-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina M., Niesman I., Mercier C., and Sibley L.D.. 1995. Stable DNA transformation of Toxoplasma gondii using phleomycin selection. Gene. 165:213–217. 10.1016/0378-1119(95)00548-K [DOI] [PubMed] [Google Scholar]
- Mordue D.G., Monroy F., La Regina M., Dinarello C.A., and Sibley L.D.. 2001. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 167:4574–4584. 10.4049/jimmunol.167.8.4574 [DOI] [PubMed] [Google Scholar]
- Mun H.S., Aosai F., Norose K., Chen M., Piao L.X., Takeuchi O., Akira S., Ishikura H., and Yano A.. 2003. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int. Immunol. 15:1081–1087. 10.1093/intimm/dxg108 [DOI] [PubMed] [Google Scholar]
- Murray P.J. 2011. Macrophages as a battleground for toxoplasma pathogenesis. Cell Host Microbe. 9:445–447. 10.1016/j.chom.2011.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyer L.E., Grunig G., Fort M., Remington J.S., Rennick D., and Hunter C.A.. 1997. Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect. Immun. 65:1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedelman W., Gold D.A., Rosowski E.E., Sprokholt J.K., Lim D., Farid Arenas A., Melo M.B., Spooner E., Yaffe M.B., and Saeij J.P.. 2012. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-γ response. PLoS Pathog. 8:e1002784 10.1371/journal.ppat.1002784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olias P., Etheridge R.D., Zhang Y., Holtzman M.J., and Sibley L.D.. 2016. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe. 20:72–82. 10.1016/j.chom.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong Y.C., Reese M.L., and Boothroyd J.C.. 2010. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 285:28731–28740. 10.1074/jbc.M110.112359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne L.C., Monticelli L.A., Nice T.J., Sutherland T.E., Siracusa M.C., Hepworth M.R., Tomov V.T., Kobuley D., Tran S.V., Bittinger K., et al. 2014. Coinfection. Virus-helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science. 345:578–582. 10.1126/science.1256942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechous R.D., and Goldman W.E.. 2015. Illuminating targets of bacterial secretion. PLoS Pathog. 11:e1004981 10.1371/journal.ppat.1004981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaud L., Samassa F., Porat Z., Ferrari M.L., Belotserkovsky I., Parsot C., Sansonetti P.J., Campbell-Valois F.X., and Phalipon A.. 2017. Injection of T3SS effectors not resulting in invasion is the main targeting mechanism of Shigella toward human lymphocytes. Proc. Natl. Acad. Sci. USA. 114:9954–9959. 10.1073/pnas.1707098114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J.V., and Vance R.E.. 2014. The macrophage paradox. Immunity. 41:685–693. 10.1016/j.immuni.2014.10.015 [DOI] [PubMed] [Google Scholar]
- Rajaram M.V., Brooks M.N., Morris J.D., Torrelles J.B., Azad A.K., and Schlesinger L.S.. 2010. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor γ linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185:929–942. 10.4049/jimmunol.1000866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme P., Tomiuk S., Kammler A., Fändrich F., Schlitt H.J., Geissler E.K., and Hutchinson J.A.. 2013. IFN-γ-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol. Ther. 21:409–422. 10.1038/mt.2012.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., and Smyth G.K.. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij J.P., Boyle J.P., and Boothroyd J.C.. 2005. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 21:476–481. 10.1016/j.pt.2005.08.001 [DOI] [PubMed] [Google Scholar]
- Saeij J.P., Boyle J.P., Coller S., Taylor S., Sibley L.D., Brooke-Powell E.T., Ajioka J.W., and Boothroyd J.C.. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 314:1780–1783. 10.1126/science.1133690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij J.P., Coller S., Boyle J.P., Jerome M.E., White M.W., and Boothroyd J.C.. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 445:324–327. 10.1038/nature05395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley L.D. 2011. Invasion and intracellular survival by protozoan parasites. Immunol. Rev. 240:72–91. 10.1111/j.1600-065X.2010.00990.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeldt T., Könen-Waisman S., Tong L., Pawlowski N., Lamkemeyer T., Sibley L.D., Hunn J.P., and Howard J.C.. 2010. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 8:e1000576 10.1371/journal.pbio.1000576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., and Mesirov J.P.. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland T.E., Logan N., Rückerl D., Humbles A.A., Allan S.M., Papayannopoulos V., Stockinger B., Maizels R.M., and Allen J.E.. 2014. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat. Immunol. 15:1116–1125. 10.1038/ni.3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., Okada A., Kitamura H., Yasui T., Naiki T., Hamamoto S., Ando R., Mizuno K., Kawai N., Tozawa K., et al. 2014. Colony-stimulating factor-1 signaling suppresses renal crystal formation. J. Am. Soc. Nephrol. 25:1680–1697. 10.1681/ASN.2013060675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S., Barragan A., Su C., Fux B., Fentress S.J., Tang K., Beatty W.L., Hajj H.E., Jerome M., Behnke M.S., et al. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science. 314:1776–1780. 10.1126/science.1133643 [DOI] [PubMed] [Google Scholar]
- Tuladhar S., Kochanowsky J.A., Bhaskara A., Ghotmi Y., Chandrasekaran S., and Koshy A.A.. 2019. The ROP16III-dependent early immune response determines the subacute CNS immune response and type III Toxoplasma gondii survival. PLoS Pathog. 15(10):e1007856 10.1371/journal.ppat.1007856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier M.N., Winter M.G., Spees A.M., den Hartigh A.B., Nguyen K., Roux C.M., Silva T.M., Atluri V.L., Kerrinnes T., Keestra A.M., et al. 2013. PPARγ-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe. 14:159–170. 10.1016/j.chom.2013.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Standley D.M., Takashima S., Saiga H., Okuyama M., Kayama H., Kubo E., Ito H., Takaura M., Matsuda T., et al. 2009. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 206:2747–2760. 10.1084/jem.20091703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Li W., Kaplan M.H., and Chang C.H.. 2005. Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by dendritic cells. J. Exp. Med. 201:1899–1903. 10.1084/jem.20050324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
lists preranked genes for M2 macrophages.
Data Availability Statement
Microarray data and RNA-sequencing data that support the findings of this study (Figs. 1 and 2) have been deposited in the Gene Expression Omnibus database for public access (accession nos. GSE55892 and GSE140774). All other data that support the findings of this study are available from the corresponding author upon reasonable request.