

Abstract
Background & Aims:
Mutations in the tetratricopeptide repeat domain 7A gene (TTC7A) cause intestinal epithelial and immune defects. Patients can become immune deficient and develop apoptotic enterocolitis, multiple intestinal atresia, and recurrent intestinal stenosis. The intestinal disease in patients with TTC7A deficiency is severe, untreatable, and recurs despite resection or allogeneic hematopoietic stem cell transplant. We screened drugs for those that prevent apoptosis of in cells with TTC7A deficiency and tested their effects in an animal model of the disease.
Methods:
We developed a high-throughput screen to identify compounds approved by the Food and Drug Administration that reduce activity of caspases 3 and 7 in TTC7A-knockout HAP1 (human haploid) cells and reduce the susceptibility to apoptosis. We validated the effects of identified agents in HeLa cells that stably express TTC7A with point mutations found in patients. Signaling pathways in cells were analyzed by immunoblots. We tested the effects of identified agents in zebrafish with disruption of ttc7a, which develop intestinal defects, and colonoids derived from biopsies of patients with and without mutations in TTC7A. We performed real-time imaging of intestinal peristalsis in zebrafish and histologic analyses of intestinal tissues from patients and zebrafish. Colonoids were analyzed by immunofluorescence and for ion transport.
Results:
TTC7A-knockout HAP1 cells have abnormal morphology and undergo apoptosis, due to increased levels of active caspases 3 and 7. We identified drugs that increased cell viability; leflunomide (used to treat patients with inflammatory conditions) reduced caspase 3 and 7 activity in cells by 96%. TTC7A-knockout cells contained cleaved caspase 3 and had reduced levels of phosphorylated AKT and XIAP; incubation of these cells with leflunomide increased levels of phosphorylated AKT and XIAP and reduced levels of cleaved caspase 3. Administration of leflunomide to ttc7a−/− zebrafish increased gut motility, reduced intestinal tract narrowing, increased intestinal cell survival, increased sizes of intestinal luminal spaces, and restored villi and goblet cell morphology. Exposure of patient-derived colonoids to leflunomide increased cell survival, polarity, and transport function.
Conclusions:
In a drug screen, we identified leflunomide as an agent that reduces apoptosis and levels of caspase 3 and activates AKT signaling and in TTC7A-knockout cells. In zebrafish with disruption of ttc7a, leflunomide restores gut motility, reduces intestinal tract narrowing, and increases intestinal cell survival. This drug might be repurposed for treatment of TTC7A deficiency.
Keywords: genetic, monogenic IBD, animal model, cell death
Graphical Abstract
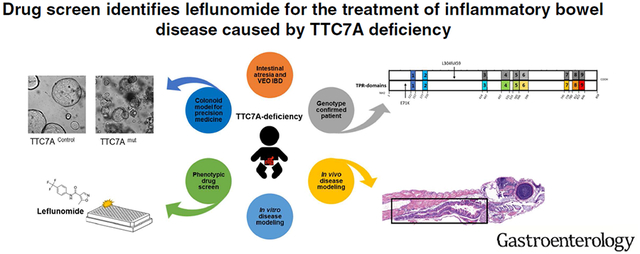
Lay Summary:
In a screen of drugs, leflunomide was identified as an agent that might be used to treat patients with intestinal defects caused by mutations in the TTC7A gene.
Introduction
Biallelic mutations in tetratricopeptide repeat domain 7A (TTC7A) result in a combined primary intestinal epithelial and immune defect, that presents clinically as immunodeficiency, very early onset inflammatory bowel disease (VEOIBD) and/or multiple intestinal atresia (MIA).1–3 Since 2013, there have been > 50 reported cases of TTC7A-deficiency with two-thirds of affected children dying within the first 12 months of life.3–5 Children with TTC7A-deficiency present with severe multisystemic disease including apoptotic enterocolitis (i.e., intestinal inflammation arising from elevated intestinal epithelial cell apoptosis), friable and/or exfoliative mucosa, congenital intestinal atresia and recurrent stenosis, lymphocytopenia, and combined immunodeficiency (CID). Poor response to standard treatment regimens including steroids, surgery and allogeneic hematopoietic stem cell transplant (HSCT), as well as the recurrence of bowel disease and the high mortality rate in these patients1 motivated our expedited search for effective therapies to treat TTC7A-deficiency.
TTC7A contains 9 tetratricopeptide repeat (TPR) domains which are structural motifs found in a range of proteins typically associated with trafficking and scaffolding.6 Our group previously reported that TTC7A acts as a scaffolding protein by shuttling phosphatidylinositol 4-kinase-three alpha (PI4KIIIα) to the plasma membrane, where PI4KIIIα catalyzes the synthesis of phosphatidylinositol 4-phosphate (PI4P) from phosphatidylinositol (PI) membrane lipids.1 Homeostatic levels of anionic PI4P are important for plasma membrane identity, apicobasal polarity, cell survival, and production of poly-phosphorylated phosphatidylinositol phosphate lipids PI (4,5)P2/ PI(3,4,5)P3.7. Plasma membrane PI4P synthesis is exclusively dependent on the function and localization of PI4KIIIα. Mouse and zebrafish models have provided strong evidence for a relationship between PI homeostasis, PI4KIIIα-function and intestinal health.8, 9
To accelerate viable treatment options for TTC7A-deficiency patients, we carried out a phenotypic high-throughput drug screen using a FDA-approved drug library and identified leflunomide, a disease modifying anti-rheumatic drug (DMARD)10, as a candidate therapeutic. In TTC7A-knockout (KO) cells, ttc7a-mutant zebrafish and TTC7A-deficient patient-derived colonoids, we demonstrate that leflunomide rescues defective AKT signaling and improves multiple intestinal phenotypes. Our findings therefore support leflunomide as a candidate drug to move forward for further clinical studies.
Materials and methods
Cells and drug treatment
Generating an intestinal TTC7A knockout (TTC7A-KO) stable cell line was unsuccessful; thus, HAP1 (human haploid) cells were selected as our in vitro model because they were commercially available via Horizon Discovery (Cambridge, UK) and engineered using CRISPR-Cas9 genome editing. Drugs and concentrations included: Dimethylsulfoxide (DMSO) (0.5%vol/vol), cyanocobalamin (CYANO 10 μM) (Selleckchem, USA), leflunomide (LEF 4 μM) (Selleckchem, USA), tiaprofenic acid (TIA 4 μM) (Prestwick Chemical Library, USA), fenbufen (FEN 10 μM) (Selleckchem, USA) fasudil (FAS 5 μM) (Selleckchem, USA), Y27632 (10 μM) (Selleckchem, USA), and 4-PBA (5 mM) (Sigma, USA).
Western blot
Western blotting was completed as per standard protocols. Primary antibodies included: anti-Caspase 3 Rabbit, anti-p-AKT (S473) Rabbit anti-p-AKT (T308) Rabbit mab, anti-AKT Rabbit, anti-XIAP (D2Z8W) Rabbit mab (all from Cell Signaling, USA).
Caspase activity assay and high-throughput drug screen
TTC7A-KO cells were screened with Prestwick, TOCRIS, and LOPAC drug libraries at 8 μM, 8 μM, and 5 μM concentrations, respectively, at the SMART laboratory for high-throughput programs (Toronto, ON). Mean Caspase activity of controls were plotted and compounds that reduced Caspase activity below 3 standard deviations of the WT control cells (hit threshold= μWT - 3σ), providing a confidence limit of 99.73%, were selected as hits.11 Concentration-response curves (40 to 0.04 μM) and IC50 values were generated with Graphpad software.
Zebrafish ttc7a−/− model, maintenance, husbandry, and drug treatment
All protocols and procedures involving zebrafish were performed in accordance with Canadian Council on Animal Care (CCAC) guidelines. Mutant ttc7a strains were generated and maintained by the Zebrafish Core Facility at Sickkids’ Peter Gilgan Centre for Research and Learning using CRISPR/Cas9 mutagenesis following previously described protocols.12 Heterozygous (ttc7a+/−) and homozygous (ttc7a+/+) fish displayed normal and indistinguishable intestinal phenotypes (i.e., size, structure and motility); thus, both served as healthy controls for comparison to ttc7a−/− fish. Zebrafish were treated during the larval stage (3 to 7) days post fertilization (dpf). Drugs were dissolved directly into the water resulting in final concentrations corresponding to those used in in vitro experiments: DMSO (0.5%vol/vol), cyanocobalamin (CYANO-10 μM), leflunomide (LEF-4 μM), tiaprofenic acid (TIA-4 μM), and fenbufen (FEN-10 μM).
Peristalsis assays
Peristalsis assays were adapted from Shi et al13. See Supplementary Methods for more detail.
Immunofluorescence histochemical staining on Formalin-Fixed, Paraffin-Embedded (FFPE) Sections
Human tissues were fixed in 10% neutral buffered formalin, without methanol and embedded in paraffin using routine protocols. The use of human tissue samples was approved by the Research Ethics Board (Hospital for Sick Children) and comprehensive consent was obtained. Informed consent to participate in research was obtained and a copy of the consent is available on the NEOPICS website, http://www.neopics.org/study-documents.html. Zebrafish samples were fixed at 7 dpf by zinc formalin and embedded with paraffin. See Supplementary Methods for more detail.
Consent and TTC7A patient genotyping and biopsy immunofluorescence
Human subject research was carried out under a study protocol approved by the Boston Children’s Hospital Institutional Review Board under Protocol IRB-P00000529. Targeted gene panel sequencing was carried out at Boston Children’s Hospital where a patient was identified with deleterious biallelic mutation in TTC7A (211G>A→Glu71Lys and 911delT → Leu304Arg). Mutations were validated using CLIA approved Sanger sequencing. Formalin-fixed stomach, duodenum and colon biopsies were processed for standard H&E and immunofluorescence.
Patient-derived intestinal colonoid culture
Colonic biopsies were obtained and cultured using methods modified from Sato et al.14 Briefly, crypts were dissociated from colonic biopsies obtained from a patient with TTC7A mutation or from a healthy control patient. Isolated crypts were suspended in Growth Factor Reduced Phenol Red Free Matrigel (Corning, NY) and plated as 50 μl domes in a tissue culture-treated 24-well plate (Thermofisher) with growth factor (Wnt, R-spondin, Noggin) supplemented media (See Supplemental Methods for media composition). Colonoid cultures were passaged by removal of Matrigel with Cell Revovery Solution (Corning, NY), mechanical dissociation of colonoids, and replating in Matrigel every 4 days.
Colonoid survival assay
TTC7A deficient and healthy control colonoids were plated in Matrigel with human colonoid media containing Rho-kinase inhibitor Y27632. Following establishment of colonoids, Y27632 was removed from the media and the colonoids were treated with leflunomide (10 μM and 2.5 μM in DMSO), or vehicle control (DMSO). See Supplementary Methods for more detail.
Colonoid polarity
TTC7A deficient and healthy control colonoids were cultured with human colonoid media without Rho-kinase inhibitor Y27632 and +/− leflunomide (10 μM). 48 hours after plating, colonoids (50 per group) were visually assessed by two blinded investigators and counted for the presence of multiple lumens. See Supplementary Methods for colonoid immunocytochemistry and histology.
Colonoid swelling assay
Colonoid swelling after leflunomide (10 μM), Rho-kinase inhibitor, or DMSO treatment was performed as previously described.15 Measurements of cell diameter and subsequent calculation of volume change (assuming a sphere) was facilitated by Image J.
Statistical Analysis
Data are presented as mean ±SD/SEM. Statistical significance was calculated by GraphPad Prism software version 6.0 (GraphPad, San Diego, CA) as a two-tailed 1-way or 2-way ANOVA, or unpaired Student’s t-test. Statistical significance was established at P values <0.05.
Results
Disease modeling in TTC7A-KO cells
To develop an assay for the phenotypic correction of TTC7A, we obtained human haploid (HAP1) TTC7A-KO cells, confirmed loss of TTC7A expression (Supplementary Figure 1), and characterized cellular phenotypes associated with the loss of TTC7A (Figures 1A–D, Supplementary Figure 2). Live-cell imaging revealed that TTC7A-KO cells have abnormal morphology, including active membrane blebbing (Figure 1A, Supplementary Video 1). WT cells displayed normal spreading and colony formation whereas TTC7A-KO cells were rounded with multiple membrane blebs and protrusions, a morphology that is associated with the early stages of apoptosis16 (Figure 1A).
Figure 1. TTC7A-KO cells have an increased susceptibility for apoptosis.

(A) Morphology of WT and TTC7A-KO HAP1 cells, DIC microscopy. Left panels, objective magnification 63× (scale bar 10μm). Right panels, objective magnification 4× (scale bar 190μm). Blebs and filopodia-like processes are indicated by arrow and arrowhead, respectively. Dashed-bars highlight differences in colony sizes after 48 h of culturing. (B) Real-time viability assay. Two-way ANOVA with post hoc test (Sidak), ****p<0.0001, (n=3, 3 replicates). (C) Annexin V-FITC/PI flow cytometry demonstrated that TTC7A-KO have significantly increased Annexin-V staining. Data is quantified in Supplementary Figure 2B. (D) Western blot analysis of untreated and IFNγ (10 ng/ml)//TNFα-treated (30 ng/ml) WT and TTC7A-KO cells, 48 h (n=3). Refer to Supplementary Figure 2C for cleaved Caspase 3 (CC3) quantitation.
We found that HAP1 TTC7A-KO cells have reduced cell viability (Figure 1B) and increased apoptosis using several complementary methods. Flow cytometry revealed increased annexin V staining in TTC7A-KO cells, indicating that the cells were in the early stages of apoptosis (Figure 1C, Supplementary Figure 2B). Western blot analysis confirmed that TTC7A-KO cells have increased levels of baseline cleaved Caspase 3 (Figure 1D and Supplementary Figure 2C). Additionally, we determined that TTC7A-KO cells have a greater susceptibility for Caspase-dependent apoptosis following treatment with proinflammatory and proapoptotic stimuli (IFNγ/TNFα,) (Figure 1D). Cumulatively, the data indicate that there is increased apoptosis in TTC7A-KO cells, mirroring a primary histopathological feature found in TTC7A-deficiency patients1, 2.
To examine cytoskeletal features underlying the abnormal morphology observed in TTC7A-KO cells we examined cellular actin. Actin staining revealed abnormal cytoskeletal organization, with an abundance of filopodia-like processes in TTC7A-KO cells (Supplementary Figure 3A). Furthermore, when WT cells were treated with apoptotic stimuli (i.e. proinflammatory cytokines IFNγ/TNFα), we observed disorganization of F-actin that resembled the untreated TTC7A-KO cells, suggesting that the baseline loss of TTC7A induces cellular changes consistent with induction and progression of apoptosis (Supplementary Figure 3A). We also observed that TTC7A-KO cells underwent apoptosis prior to substrate detachment (Supplementary Figure 3B), suggesting that cell-death was not solely a result of anoikis (a form of apoptosis triggered by the loss of adhesion16). Taken together, we demonstrate that TTC7A-KO cells display phenotypes consistent with apoptosis.
Repurposing drug screen identifies hit compounds that reduce apoptosis in TTC7A-KO cells
Apoptosis is a pathologic feature of TTC7A loss observed both in vitro and clinically. We therefore designed a phenotypic drug screen to identify compounds that can reduce cellular apoptosis, without inducing uncontrolled cell proliferation. We used a highly specific and sensitive luciferase-based Caspase 3/7 activity assay (Caspase-Glo® 3/7 (Promega)17 for our primary screen. We developed the screen so that it could be multiplexed, where a single sample could provide readouts for both viability (using Calcein AM) and Caspase 3/7 activity. Assessing for viability in parallel was advantageous as it increased the sensitivity and specificity of the screen by reducing false positives and ensuring that low Caspase 3/7 readouts were not a result of compound toxicity and/or total cell annihilation. The Caspase-Glo® 3/7 assay revealed that TTC7A-KO cells consistently display elevated Caspase 3/7 cleavage relative to WT cells, which resulted in a Z’-factor score of 0.54 (Figure 2A), a value that is suitable for discerning hits among thousands of compounds in a high-throughput screen11.
Figure 2. High-throughput drug screening identifies approved-compounds that improve the apoptotic phenotype.

(A) Modeling the high-throughput apoptosis-phenotype assay. Caspase-Glo® 3/7 assay, z’=0.54, data are presented as the mean ±SD. Unpaired t test, ****p<0.0001, (n=3, 8 replicates). (B) Drug screen workflow. See Methods. (C) A sample of plate-results from the Prestwick Chemical Library screen showing mean Caspase 3/7 activity from drug-treated wells (green dots) and DMSO-treated control wells, WT (light blue dashes) and TTC7A-KO cells (dark blue dashes). Tiabendazole and tiaprofenic acid reduced Caspase 3/7 activity below the hit threshold (μWT-3σ), z’= 0.56. (D) Drug screen summary, 0.4% hit rate. Hit compounds represent the 10 drug classification families shown. (E) Comparison of hit compound inhibition of apoptosis in TTC7A-KO cells. Black borders represent FDA-approved drugs and bar colors correspond to Figure 2D. (F) Concentration-response curve for leflunomide inhibition of Caspase 3/7 activity in HAP1 TTC7A-KO cells, IC50=1.1 μM, (n=3). (G) Colony size formation in WT, TTC7A-KO, and drug-treated TTC7A-KO cells. Drug abbreviations: cyanocobalamin (CYANO), leflunomide (LEF), tiaprofenic acid (TIA), fenbufen (FEN), and fasudil (FAS). Plotted values represent individual cell colonies, error bars presented as mean ±SD. Statistical significance was relative to the KO/DMSO control. One-way ANOVA with post hoc test (Dunnett), ****p<0.0001, ***p<0.001, **p<0.01, (n=3, 6 replicates per condition). (H) Viability assay. Statistical significance relative to KO/DMSO (blue). WT (*p<0.05), KO/LEF (**p<0.01), KO/TIA (****p<0.0001), KO/FEN (***p<0.001), KO/FAS (****p<0.0001), two-way ANOVA with post hoc test (Dunnett), (n=3, 4 replicates per condition).
A workflow schematic of our drug screen is provided in Figure 2B. With drug-repurposing as the primary aim of our study, we selected 3 libraries (see Methods) containing FDA-approved drugs as well as other compounds with known biological targets. To ensure that hit identification was stringent, compounds that reduced Caspase activity below 3 standard deviations of the positive control (i.e. WT Caspase 3/7 activity), providing a confidence limit of 99.73%, were selected as hits (see Figure 2C for a sample plate readout). We screened 3760 compounds and identified 16 compounds (henceforth referred to as ‘hits’) resulting in a 0.4% hit rate (Figure 2D, Table 1). Nonsteroidal anti-inflammatory drugs (NSAIDs) and Metabotropic Glutamate Receptor Subtype 5 (mGluR5) antagonists were the most well-represented drug classes among the hits.
Table 1. Summary of hits able to reduce Caspase 3/7 activity in TTC7A-KO cells.
Grey boxes are FDA-approved drugs. Compound structures, formulas, and molecular weights were provided by Prestwick, LOPAC and TOCRIScreen and compound descriptions were obtained from PubChem.27
| Compound Name | Structure | Formula Structure | Molecular Weight (g/mol) | IC 50 (μM) | Decrease in caspase 3/7 activity | Compound description |
|---|---|---|---|---|---|---|
| Leflunomide | 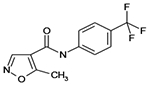 |
C12H9F3N2O2 | 270.2 | 1.1 | 96% | Disease Modifying Anti Rheumatic Drug (DMARD) - Pyrimidine synthesis inhibitor |
| Tiaprofenic acid | 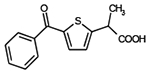 |
C14H12O3S | 260.3 | 2.5 | 87% | NSAID, arthritic pain |
| Tiabendazole | 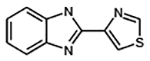 |
C10H7N3S | 201.3 | 11 | 60% | Fungicide and anthelmintic |
| Cyanocobalamin | 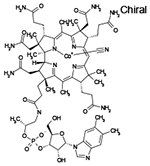 |
C63H88CoN14O14P | 1355 | 11 | 64% | Vitamin B12 |
| Flurbiprofen | 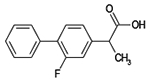 |
C15H13FO2 | 244.2 | 39 | 31% | NSAID, arthritic pain |
| Indoprofen | 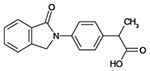 |
C17H15NO3 | 281.3 | 6.1 | 73% | NSAID, withdrawn due to carcinogenicity |
| Fenbufen | 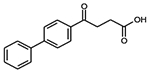 |
C16H14O3 | 254.3 | 30 | 56% | NSAID, osteoarthritis, ankylosing spondylitis, and tendinitis |
| Acacetin | 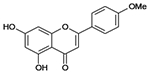 |
C16H12O5 | 284.3 | N/A | 47% | Flavone, anticonvulsant and plant metabolite |
| Resveratrol | 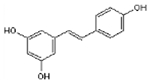 |
C14H12O3 | 228.3 | 4.9 | 45% | Stilbenoid, anti-inflammatory, polyphenolic phytoalexin |
| Isoliquiritigenin | 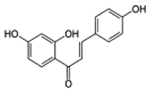 |
C15H12O4 | 256.3 | 9.7 | 89% | Soluble guanylyl cyclase activator |
| SB-366791 | 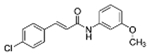 |
C16H14NO2Cl | 287.7 | 7.6 | 58% | Vanilloid antagonist |
| SIB 1893 | 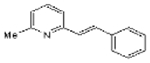 |
C14H13N | 195.3 | 9.1 | 58% | Selective antagonist of metabotropic glutamate receptor (mGlu5R) |
| SIB 1757 | 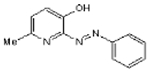 |
C12H11N3O | 213.2 | 18 | 50% | Selective antagonist of mGlu5R |
| Ketorolac tris salt | 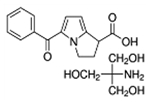 |
C15H12NO3•C4H12NO3 | 376.4 | 156 | 48% | NSAID |
| MPEP hydrochloride | 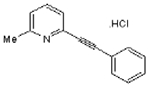 |
C14H11N.HCL | 229.7 | 6.3 | 33% | Selective antagonist of mGlu5R |
| Pifithrin-α hydrobromide | 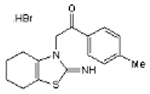 |
C16H18N2OS.HBr | 367.3 | 8.3 | 31% | p53 inhibitor |
All hits were confirmed via concentration-response curves in HAP1 TTC7A-KO cells (Supplementary Table S1). To ensure that hit compound efficacy was not an artefact specific to TTC7A-KO cells, we further demonstrated that our hit compounds could reduce Caspase 3/7 activity in a HeLa cell line stably expressing TTC7A point-mutations found in previously reported patients (p. E71K and A832T)1 (Supplementary Table S1). Leflunomide was identified as the most efficacious compound based on its ability to inhibit Caspase 3/7 activity (96% reduction; Figure 2E) with half-maximal inhibitory (IC50) concentration of 1.1 μM (Figure 2F).
Hit compounds rescue concomitant phenotypes related to TTC7A-deficiency
From the 16 hit compounds identified in the screen, we selected a group of candidate drugs with pharmacological class diversity and FDA-approved status for further validation. Our initial hits (Table 1) were narrowed to four candidate drugs for orthogonal validation and included cyanocobalamin (vitamin B12), leflunomide (DMARD), tiaprofenic acid (NSAID), and fenbufen (NSAID). We also compared the performance of our candidate drugs with small molecule ROCK-inhibitors (fasudil and Y27632) to determine whether aberrant TTC7A phenotypes might be driven by the constitutive activation of ROCK as previously suggested2.
Following treatment with the candidate drugs, the morphology of TTC7A-KO cells resembled that of WT cells with a reduction in membrane blebbing (Supplementary Figure 4). Cell colony size, a proxy for both cell adhesion and viability, was diminished in untreated TTC7A-KO cells; however, we observed the formation of larger-adherent colonies after cyanocobalamin, leflunomide, tiaprofenic acid, fasudil, and Y27632 drug treatments (Figure 2G, Supplementary Figure 4). In addition to reducing levels of apoptosis to that of the control, we found that candidate drug treatments improved the viability of TTC7A-KO cells over the course of 48-hours (Figure 2H), consistent with the observed increase in colony sizes. Importantly, as inhibition of apoptosis may result in excessive cell-proliferation, we found that leflunomide improved viability to that of control levels, in contrast to the ROCK-inhibitor fasudil that increased viability above WT levels and induced excessive cell proliferation.
Leflunomide rescues abnormal intestinal features in ttc7a −/− zebrafish
To date, a major obstacle in TTC7A research is the lack of an appropriate disease model that recapitulates the intestinal abnormalities observed in TTC7A-deficiency patients. For example, Ttc7a-mutant mice display a prominent flaky skin phenotype but do not manifest the atretic and inflamed intestinal phenotypes seen in patients.18 We therefore sought to develop an animal model able to phenocopy the intestinal abnormalities seen in humans. Using CRISPR-Cas9 genome editing, we developed and validated a ttc7a zebrafish line (ttc7a−/−) with an 11-basepair deletion in exon 14 predicted to result in a frameshift mutation causing an early stop codon (p.T548LfsX41) (Supplementary Figure 5).
ttc7a−/− zebrafish (ttc7a-mutant) have normal survival but display a range of abnormal intestinal phenotypes (Figure 3). Histopathological zebrafish sections were taken along the sagittal plane, to assess a continuous stretch of the intestinal tract, including the widest and most anterior intestine (the intestinal bulb - IB) to the anal pore (Figure 3A). Sections were assessed for an open IB, villus structure, a discernable epithelial monolayer, intestinal epithelial cell integrity, and the presence of mature goblet cells with large secretory vesicles. Histological examination revealed that ttc7a−/− zebrafish had a combination of the following intestinal defects: stratification and crowding of intestinal epithelial cells, blunted villi, breaches in the intestinal mucosal layer, signs of apoptosis, and atresia along the intestinal tract (Figure 3A). The intestinal defects observed from zebrafish histology were consistent with patient histopathological findings,1, 2, 4 suggesting ttc7a-mutant fish appropriately model the intestinal atresia and loss of intestinal epithelial cell integrity observed in TTC7A patients.
Figure 3. Leflunomide rescues abnormal intestinal features in ttc7a −/− zebrafish.

(A) Histology from ttc7a −/− zebrafish (7 dpf) reveals pathological intestinal phenotypes. Control (ttc7a +/−) zebrafish display open luminal spaces with discernible villi projections (grey arrowhead), clefts (black arrowhead), monolayer epithelium (dotted-outlined area), and mature goblet cells with large vesicles (black arrows). ttc7a −/− zebrafish display narrowing of the intestinal bulb, stratified epithelium (yellow dotted-outlined area), signs of apoptosis (yellow arrows), and goblet cells with numerous small vesicles (yellow boxes). Objective magnification 10× and 40× for insets (scale bar 100 μm, inset scale bar 50 μm), (ttc7a +/− n=14, ttc7a −/− n=11). (B) Incidence of the narrow gut phenotype in DMSO and LEF treated fish. One-way ANOVA with post hoc test (Fisher’s LSD), ****p<0.0001, ttc7a +/− (DMSO n=49, LEF n=12), ttc7a −/− (DMSO n= 36, LEF n=26). (C) Intestinal histology from treated (see Methods) ttc7a−/− zebrafish. Leflunomide, cyanocobalamin and tiaprofenic acid suppressed the narrow-gut phenotype with minimal enterocyte crowding. Objective magnification 10×, (scale bar 100 μm). ttc7a+/− (DMSO n=49) ttc7a−/− (DMSO n= 36, CYANO n=10, LEF n=26, TIA n= 13, FEN n=13. (D) Assessment of apoptosis. DMSO or leflunomide treatment. ttc7a−/− zebrafish display fragmented, condensed or engulfed nuclei in the epithelium (arrows). Leflunomide treatment resulted in diminished signs of apoptosis, reduced intestinal epithelial cell crowding, and overall improved epithelium architecture in ttc7a−/− zebrafish. Refer to Supplementary Figure 7A for the quantitation of apoptotic cells/sample. Objective magnification 70× (scale bar 20 μm), (n=6 per group, across 3 experimental clutches). (E) Staining of the intestinal lumen in control (ttc7a+/−) and ttc7a-mutant (ttc7a−/−) zebrafish. Images are from peristalsis assays (Supplementary Video 2): intestinal lumen marked green fluorescent stain (DCFH-DA). ttc7a+/− fish have discernable villi (yellow arrow) and large continuous intestinal bulbs (double-headed white arrow). Representative ttc7a −/− intestinal phenotypes (i) atresia point (white arrow heads) (ii) narrow intestinal lumen and (iii) obstruction interrupting the intestinal bulb (white arrow). Objective magnification 4×, (scale bar 100 μm). (F) Incidence of ttc7a-mutant phenotypes. ttc7a−/− fish have significantly larger populations with motility and narrow lumen pathological phenotypes. Data are presented as the mean ±SD, one-way ANOVA with post hoc test (Fisher’s LSD), **p<0.0054, *p<0.0196, (n=50 total for each group, across 3 experimental clutches). (G) Phenotype summary from drug treated fish. ttc7a −/− fish with aberrant motility and narrow lumen phenotypes were significantly reduced with cyanocobalamin, leflunomide, tiaprofenic acid, and fenbufen treatment (3–7 dpf). Data are presented as the mean ±SD. Statistical significance was relative to ttc7a −/− DMSO control, and determined by two-way ANOVA with post hoc test (Dunnett), *p<0.05, **p<0.01, ***p<0.001, ns=not significant (DMSO n=18, CYANO n=21, LEF n=21, TIA n=17, FEN n=18, FAS n=13 for each group across 3 experimental clutches).
Intestinal atresia is a distinctive phenotype among TTC7A-deficiency patients. Similarly, intestinal narrowing was the most striking abnormality in ttc7a-mutant fish (Figure 3B, Supplementary Figure 6). There was a greater than 50% incidence of intestinal narrowing in the ttc7a-mutant fish population, whereas this phenotype was minimal (<5%) in control fish. We demonstrated that leflunomide treatment of ttc7a-mutant fish resulted in a reduced incidence of intestinal narrowing, suggesting that the drug ameliorated intestinal atresia formation (Figure 3B).
ttc7a−/− fish showed additional histological improvement, including increases in the size of intestinal luminal spaces, and discernible villi and goblet cell morphology following treatment with tiaprofenic acid, leflunomide, or cyanocobalamin (Figure 3C). Intact monolayers with normal columnar intestinal cell architecture and fewer apoptotic cells were observed in leflunomide treated ttc7a−/− fish (Figure 3D, Supplementary Figure 7A). Previous studies have suggested that the chaperone drug 4-phenylbutyrate (4-PBA, Ucyclyd Pharma), and ROCK-inhibitors (fasudil and Y27632) may be effective in improving defects related to TTC7A-deficiency.2, 9 Our data suggests that 4-PBA treatment improves intestinal defects seen in ttc7a −/− fish, in contrast to the ROCK-inhibitors that did not result in any histological improvements (Supplementary Figure 8).
The histopathological findings were further validated by real-time in vivo imaging of intestinal peristalsis, which demonstrated differences in gut architecture and contractile motility (Figure 3E, Supplementary Video 2). Normal motility is integral to intestinal health and loss of intestinal contractility is correlated with increased IBD severity.19 Live-labeling of the intestinal lumen was achieved by treating the fish with a water soluble non-fluorescent stain, 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA), that becomes oxidized to a fluorescent compound once ingested by the fish.13 Still-frames from peristalsis imaging are shown in Figure 3E and corresponding footage can be found in Supplementary Video 2. Normal intestinal architecture, including the presence of distinct villi, can be appreciated with light transmission (DIC) imaging, while delineation of a distinct luminal space can be seen by epifluorescence imaging. Control heterozygous fish, ttc7a+/−, consistently displayed coordinated peristaltic contractions and larger intestinal luminal spaces with discernable villi in the IB (Figure 3E–F, Supplementary Video 2). Peristalsis assays revealed that ttc7a−/− fish have narrow regions in the anterior IB resulting in bottlenecked contractions, severely narrow luminal spaces with absent peristalsis, and masses obstructing the IB resulting in uncoordinated gut motility (Figure 3E–F). In sum, we demonstrate that ttc7a-mutant fish have intestinal atresia resembling TTC7A-deficiency patients1, 2, 4 with severe atretic phenotypes.
Candidate drug treatment of ttc7a-mutant fish showed that leflunomide reduced the proportion of mutant fish with motility defects, rescued the atresia phenotype and increased intestinal luminal volumes (Figure 3G, Supplementary Figure 7B). Leflunomide treatment also resulted in the largest improvement in motility observed in ttc7a−/− fish (Figure 3G). Except for fenbufen, all candidate drugs increased intestinal luminal volume. (Supplementary Figure 7B). Taken together, we demonstrate that leflunomide is effective in improving gut motility, intestinal epithelium integrity, and preventing intestinal atresia in ttc7a−/− fish.
The role of AKT in TTC7A-deficiency
AKT is a crucial survival kinase and its activation (via phosphorylation) prevents apoptosis, promotes proliferation, increases protein expression, and regulates cell metabolism.7 Given that reduced viability and increased apoptosis were conspicuous TTC7A-deficiency phenotypes, we sought to determine whether TTC7A dysfunction might alter AKT activation. We observed that TTC7A-deficiency patients had loss of phosphorylated-AKT (p-AKT) in the intestinal epithelium, whereas changes in total AKT levels were less striking, suggesting that AKT activation (i.e. p-AKT levels) may be tied to the pathobiology of TTC7A-deficiency (Figure 4A–B). We examined AKT signaling using the HAP1 cell line. As shown in Figure 4C, WT cells lack cleaved Caspase 3 and have measurable levels of both p-AKT and X-linked inhibitor of apoptosis (XIAP), in contrast to TTC7A-KO cells which exhibit the presence of cleaved caspase 3 and reduction in p-AKT and XIAP levels. Treatment with a phosphatidylinositol 3-kinase (PI3K)-inhibitor (LY294002) blocks the formation of PI(3,4,5)P3 required for AKT activation and triggers Caspase 3 cleavage.7 WT cells treated with PI3K-inhibitor show a similar immunoblot pattern to TTC7A-KO cells treated with DMSO, suggesting that the apoptotic phenotype observed in TTC7A-deficiency may be mediated by AKT (Figure 4C). Importantly, TTC7A-KO cells treated with leflunomide resulted in an increase in p-AKT (Thr 308) and XIAP, and a reduction in cleaved Caspase 3 levels (Figure 4D). These data suggest that leflunomide may improve cell viability and reduces apoptosis by altering AKT activation. Furthermore, treatment with the PI3K-inhibitor resulted in undetectable AKT phosphorylation at both Threonine 308 and Serine 473 sites in WT and leflunomide-treated TTC7A-KO cells, suggesting that leflunomide is acting upstream of PI3K (Supplementary Figure 9).
Figure 4. p-AKT is reduced in TTC7A-deficiency.

(A) Histopathology analysis of p-AKT in human colon tissue. Co-staining of cytokeratin 20 (CK20), a marker for the intestinal epithelium, and p-AKT is present in the normal and IBD control, while p-AKT is diminished in TTC7A-deficiency patients. Patient 1 is the colonoid donor and Patient 2 is unpublished with confirmed biallelic mutations. RedDot 2 nuclear counterstain (blue). Sections were magnified at 20× objective. (B) Histopathology analysis of pan-AKT (total AKT) in human colon tissue. Biopsies are the same as described in Figure 5A. Pan-AKT is present in all samples albeit with reduced intensity in the TTC7A-deficiency patient samples. Sections were magnified at 20× objective. (C) Immunoblot for p-AKT, XIAP, and cleaved Caspase 3 in WT and TTC7A-KO cells. After 3 h leflunomide treatment in TTC7A-KO cells, p-AKT and XIAP protein levels are detectable, while cleaved Caspase 3 is diminished. DMSO (*) (n=3). (D) Densitometric analysis of p-AKT, XIAP, and cleaved Caspase 3 from WT and TTC7A-KO cells. One-way ANOVA with post hoc test (Tukey), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, (n=3). (E) ttc7a+/− and ttc7a−/− whole mount zebrafish staining with p-AKT (red), Synaptic vesicle protein 2 (SV2) (green), and RedDot 2 nuclear counterstain (blue). SV2 staining, indicating neuromuscular junctions, is absent from the intestinal epithelial monolayer and aids in differentiating epithelial cells from other nearby cell types. In the DMSO treated panel, p-AKT staining (Ser473) in ttc7a+/− fish is evident along the gastrointestinal tract, while absent in ttc7a−/− fish. Leflunomide treatment (3–7 dpf) restores p-AKT staining in the intestinal epithelium of ttc7a−/− fish. Fish were magnified at 5× objective, (n=4).
The inhibition of pyrimidine synthesis via dihydroorotate dehydrogenase (DHODH) is the putative target of leflunomide.10 Immunoblot analysis using Vidofludimus, another DHODH inhibitor, did not increase p-AKT or reduce cleaved Caspase 3, suggesting that leflunomide increases p-AKT via an uncharacterized mechanism (Supplementary Figure 10). Treatment of TTC7A-KO cells with leflunomide results in a shift from an apoptotic to a survival phenotype; however, leflunomide’s mechanism for altering p-AKT, XIAP and cleaved Caspase 3 remains to be elucidated.
We also performed whole mount zebrafish staining that confirmed the loss of p-AKT (Figure 4E). Leflunomide was able to increase p-AKT levels in the gastrointestinal tract of ttc7a−/− fish, thus, providing in vivo drug validation in the zebrafish model (Figure 4E). These data point to AKT as a possible therapeutic target for restoring intestinal health in TTC7A-deficiency.
Leflunomide treatment enhances survival and function of TTC7A-deficiency patient-derived colonoids
To further validate the effect of leflunomide, we tested the drug’s efficacy in colonoids derived from TTC7A-deficiency patients. Biopsies were obtained from a 6-month old female with confirmed biallelic TTC7A mutations (termed TTC7Amut), c.211 G>A (p.E71K) and c.911delT (p.L304R) (Figure 5A). TTC7A immunofluorescence staining of a colonic tissue sample demonstrated that the TTC7Amut patient had loss of TTC7A staining compared to healthy and IBD control samples (Supplementary Figure 11A). Histological sections obtained from TTC7Amut patient intestinal biopsy specimens also showed features consistent with TTC7A-deficiency including villous blunting in the duodenum and crypt apoptosis and eosinophilic inflammation in the colon (Supplementary Figure 11B).
Figure 5. Leflunomide treatment enhances survival and function of TTC7A-deficiency patient-colonoids.

(A) Personalized medicine approach to evaluate drug efficacy in human TTC7A-deficiency using patient-derived colonoids. Endoscopic biopsies were obtained from healthy control and TTC7A-deficiency (E71K and L304R compound heterozygous, TTC7Amut) patients and cultured into colonoids, which were then tested with leflunomide. (B) Leflunomide improves the survival of human colonoids derived from TTC7A-deficiency patients (TTC7Amut). Example images of control (TTC7A+/+) and TTC7A-deficient (TTC7Amut) colonoids with and without leflunomide (10 μM) treatment at 2 and 6 days and grown in the absence of ROCK inhibitor (Y27632). Red arrows indicate examples of dead colonoids. (C) Summary graph showing the percentage of dead TTC7Amut colonoids +/− leflunomide treatment. TTC7Amut and TTC7A+/+ colonoids were cultured +/− ROCK inhibitor (Y27632) or with leflunomide and viability was assessed at day 2 and 6. TTC7Amut colonoids grown without ROCK inhibitor have significantly increased death, which is reversed following leflunomide treatment. One-way ANOVA with post-hoc test (Tukey-Kramer), *p<0.05. **p<0.01. (D) Immunofluorescence staining of TTC7A+/+ and TTC7Amut colonoids with villin (green) marking the apical brush border and cytokeratin 20 (CK20, red) marking basal epithelial structure and DAPI (blue, nuclei). TTC7A+/+ colonoids show normal polarity with defined basal CK20 staining. TTC7Amut colonoids treated with ROCK inhibitor show grossly normal staining with mild cytological atypia. TTC7Amut colonoids without ROCK inhibitor show normal sidedness but abnormal polarity with cytological atypia and basal structural abnormalities which is improved in leflunomide treated colonoids. Quantification of abnormal polarity as assessed by the presence of multiple lumens in colonoids. Percentage of colonoids with multiple lumens in TTC7A-deficient colonoids with and without leflunomide (10 μM). Healthy control colonoids did not have multiple lumens (data not shown). Two tailed t-test, *p<0.05. (E-F) TTC7A-deficient colonoids (TTC7Amut) have a reduced swelling response to forskolin stimulation. Example images showing reduced swelling in TTC7Amut compared to TTC7Amut treated with leflunomide (10 μM). Inset panel show magnified images with red arrows indicating example colonoids pre and post swelling. Summary data showing significantly increased swelling in leflunomide (10 μM) treated TTC7Amut colonoids. Two tailed t-test, **p<0.01.
Previous studies have shown that ROCK inhibition is able to improve survival of TTC7A deficient organoids2. Colonoids derived from both a healthy subject (TTC7A+/+) and our TTC7Amut patient were generated from endoscopic biopsies and treated with a ROCK-inhibitor (5 μM Y27632) and/or leflunomide. Compared to control TTC7A+/+ colonoids, a higher percentage of TTC7Amut colonoids died in the absence of ROCK inhibitor treatment (Figures 5B–C). Treatment of TTC7Amut colonoids with leflunomide (with and without ROCK-inhibitor) promoted colonoid survival in a dose-dependent manner.
Apicobasal polarity is essential to intestinal epithelial cell function and previous studies has suggested aberrant cell polarity in TTC7A-deficient epithelial cells.2 Intact TTC7A+/+ colonoids showed well-demarcated polarization of apical and basolateral membranes delineated by villin and CK20 staining, respectively (Figure 5D). Without ROCK-inhibitor treatment, epithelial cells were polarized in the correct orientation in TTC7Amut colonoids, yet showed other defects including disorganized cytoskeletal staining and the presence of abnormal cytoplasmic inclusions and the presence of multiple lumens. In conjunction with previous reports2 where treatment with a ROCK-inhibitor restored epithelial structure and polarity in TTC7A-deficient colonoids, TTC7Amut colonoids treated with a ROCK-inhibitor partially corrected polarity defects as assessed by the number of colonoids with multiple lumens (Figure 5D). Similar to the ROCK-inhibitor treatment, leflunomide (10 μM) also partially rescued cytoskeletal and polarity defects in TTC7Amut colonoids.
Transepithelial fluid and electrolyte homeostasis are essential functions of intestinal epithelial cells. Therefore, to assess whether leflunomide can restore epithelial function in TTC7A-deficiency, we measured agonist-stimulated fluid secretion in intact colonoids. Fluid secretion was stimulated by addition of the cAMP-elevating agonist forskolin, which activates cystic fibrosis transmembrane conductance regulator (CFTR)-mediated anion-and fluid-transport into the lumen of the colonoid. Fluid secretion can then be measured by the extent of colonoid swelling.15 TTC7Amut colonoids have impaired agonist-stimulated fluid secretion compared to control TTC7A+/+ colonoids (Supplementary Figure 11C–D). However, in TTC7Amut colonoids leflunomide was able to restore the ability of the epithelium to secrete fluid, suggesting recovery of epithelial transport function (Figures 5E–F). These data using TTC7A-deficient patient cells suggest that leflunomide treatment can improve intestinal epithelial survival, structure, and ion-transport function in humans.
Discussion
TTC7A-deficiency is a rare and often fatal disease that disrupts intestinal epithelial homeostasis and the immune system.1, 2 Challenged by the unmet need for therapies and reports that immune reconstitution via HSCT do not improve the intestinal disease,5 our goal was to identify candidate drugs that improve intestinal abnormalities. Given that apoptosis is a key clinical and experimental feature of TTC7A-deficiency, we performed a phenotypic high-throughput drug screen to identify compounds that could reduce the increased apoptosis seen in TTC7A-deficiency. We selected candidate drugs having class diversity, known safety profiles, and repurposing potential and tested their efficacy in various disease models. Based on our experimental data, safety profiles, FDA-approved status and its prior use for inflammatory conditions such as juvenile rheumatoid arthritis,20 leflunomide was selected as our lead candidate therapeutic. Leflunomide’s known mechanism of action is through inhibition of DHODH, an enzyme responsible for the de novo synthesis of pyrimidines important in T-cell activation.10 However, Vidofludimus, a selective DHODH inhibitor21 did not increase p-AKT or inhibit cleaved Caspase 3 in TTC7A-KO cells, indicating an alternative mechanism-of-action in rescuing TTC7A-deficient cells. We demonstrated that leflunomide is involved in regulating AKT, providing a potential mechanism for the increase in cell viability and reduction in apoptosis. These findings corroborate the research from Leger et al. showing that low dose leflunomide (10 μM) activates PI3K/AKT signaling in HEL and K562 leukemia cells.22 We propose that loss of AKT activation in TTC7A-deficiency results in a greater susceptibility for Caspase 3 cleavage, thereby altering cellular fitness and disrupting epithelial homeostasis (Figure 6).
Figure 6. Model of leflunomide’s mechanism of action in TTC7A-deficiency.

(A) TTC7A-competent. 1, TTC7A binds and recruits PI4KIIIα to the plasma membrane. 2, PI4KIIIα phosphorylates PI lipids to create PI4P, precursor to PI (4,5)P2/ PI(3,4,5)P3. 3, PI3K phosphorylates PI (4,5)P2, required for AKT phosphorylation. p-AKT activates multiple downstream substrates that promote cell survival. For example, XIAP polyubiquitylates Pro Caspases 3,7, and 9 for proteasomal degradation, thereby reducing the susceptibility for Caspase-dependent cell death. (B) TTC7A-deficiency + leflunomide. 1, PI4KIIIα trafficking to (or kinase activity at) the plasma membrane is compromised in TTC7A-deficiency resulting in reduced PI4P. 2, Reduced p-AKT provides a rationale for increases in apoptosis. 3, Caspase-dependent apoptosis is frequently associated with TTC7A-deficiency. 4, Leflunomide treatment increases p-AKT and XIAP levels. 5, The apoptotic phenotype is ameliorated when TTC7A-deficient HAP1 cells are treated with leflunomide, suggesting a shift toward a survival phenotype. 6, It has yet to be established how leflunomide mediates p-AKT activation, however, PI3K-inhibition with LY294002 hinders leflunomide’s effect on AKT, XIAP, and Caspase 3 cleavage.
A phase 1 clinical study and uncontrolled pilot study suggested that leflunomide may be a safe treatment option for Crohn’s disease (CD), although not commonly used.23 Besides leflunomide, there were other potential candidate drugs. While tiaprofenic acid was able to rescue many TTC7A-related phenotypes, its repurposing should be approached with caution as it is typically not prescribed for children and NSAIDs are associated with gastric bleeding.24 Cyanocobalamin, the synthetic form of Vitamin B12, was less effective in rescuing the apoptotic phenotype compared with leflunomide; however, its relatively safe clinical profile makes it an appealing therapeutic to consider. Additionally, there is some limited evidence that high-dose Vitamin B12 may be beneficial in IBD patients.25 Interestingly, three hits (non-FDA-approved drugs) were antagonists of mGluR5, a type of G-protein coupled receptor linked to the inositol trisphosphate/DAG pathway. Previous research shows mGluR5 antagonists to be important in the gastrointestinal tract by improving epithelial barrier function.26 The drug screen herein identified a diverse range of biologically active compounds and illuminated potential pathways implicated in TTC7A-deficiency.
We demonstrated that ttc7a-mutant zebrafish have intestinal atresia, loss of villi, discordant intestinal epithelial cell integrity, and poor gut motility. The zebrafish model recapitulates the intestinal phenotypes seen in patients, and to our knowledge, establishes the only animal disease-model system for TTC7A-deficiency. Consistent with the mechanism of our model, Thakur and colleagues developed a zebrafish model (cdipt hi559) lacking PI synthesis, and interestingly also observed an intestinal atresia phenotype, thus, providing a precedent for linking PI deficiency with IBD and intestinal narrowing.9 Taken together, these findings further suggest that PI regulation could be an important target for improving intestinal health.
The GI tracts of zebrafish and humans have homologous structures, functions, tissues, and cell types, and their transparency during the larval stage makes for an advantageous model for intestinal research.9 However, fundamental differences in zebrafish and humans (e.g. microbiome, environmental) could possibly contribute to the discrepancies we see in disease severity and overall survival. Ultimately, differences in the microbiome, embryonic development, and drug dosing increase the complexity in translating these findings to patients. A thorough characterization of intestinal apoptosis in ttc7a-mutant fish is additionally required and may aid elucidating the extent of morbidity that is driven by cell death. Also, given that the fish were treated with leflunomide from 3 to 7 dpf, and that the gut is fully formed by 5 dpf, it remains to be elucidated whether leflunomide prevents or reverses the intestinal pathologies. In the future, the development of transgenic ttc7a−/− reporters that aid in visualizing intestinal infiltrate may allow for more thorough assessment of histopathological features in ttc7a-deficient fish.
Despite leflunomide’s merits of approved status and previous use in pediatrics, several limitations exist. Clinically, existing data pertaining to leflunomide’s pharmacokinetics, IC50 values, and mechanism of action are specific to its use in rheumatoid arthritis and inhibition of pyrimidine synthesis via DHODH. In our study, leflunomide’s precise mechanism of action remains to be elucidated, and previously reported IC50 values differ because they are for the inhibition of DHODH rather than caspase activity. Identifying leflunomide’s primary cellular target may therefore be critical to fully understanding how TTC7A functions in intestinal cells. Our study identified leflunomide’s IC50 for the inhibition of Caspase 3/7 activity in vitro but potency remains to be established for the in vivo situation, with alterations likely due to drug pharmacokinetics. Current juvenile rheumatoid arthritis dosing for leflunomide may differ for TTC7A-deficiency patients who are likely to be very young (<6 years). Therefore, establishing an effective and safe dose for various pediatric thresholds (i.e., <5 kg, <10 kg) will be necessary. Lastly, evaluating leflunomide’s efficacy in TTC7A-deficiency patients may be confounded by the effects of concomitant immunodeficiency.
In this study, we have identified FDA-approved drugs that rescue TTC7A-deficiency phenotypes and orthogonally validated the potential therapies in zebrafish and patient-derived colonoids. We identified AKT activation as part of the pathobiology in TTC7A-deficiency, and propose an alternative mechanism-of-action where leflunomide increases activated AKT and improves cellular fitness. The data presented here suggest that leflunomide is a repurposing candidate with potential to benefit patients with monogenic defects in TTC7A as well as in those in the broader IBD population suffering with apoptotic and/or stricturing intestinal disease. Based on these pre-clinical findings, leflunomide is a promising candidate to advance to clinical testing. Future investigation of the hits identified here could provide alternative therapeutic targets for TTC7A-deficiency and offer insight into the cellular functions of TTC7A.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
Mutations in the tetratricopeptide repeat domain 7A gene (TTC7A) cause intestinal epithelial and immune defects, apoptotic enterocolitis, multiple intestinal atresia, and recurrent intestinal stenosis.
NEW FINDINGS:
In a drug screen, the authors identified leflunomide as an agent that reduces apoptosis and levels of active caspase 3 in TTC7A-knockout cells. In zebrafish with disruption of ttc7a, leflunomide restored gut motility, reduced intestinal tract narrowing, and increased intestinal cell survival.
LIMITATIONS:
Further studies are needed in humans.
IMPACT:
Leflunomide might be repurposed for treatment of TTC7A deficiency.
Acknowledgments & Grant Support:
The authors thank all the SickKids and Boston Children’s Hospital patients and their families who have consented and participated in this study as part of the NEOPICS (www.NEOPICS.org) and Helmsley VEOIBD (www.VEOIBD.org) consortia, as well as the health care professionals at The Hospital for Sick Children and Boston Children’s Hospital who care for these IBD patients. REB #1000024905, IRB- P00000529. They also thank Ramil Noche and the SickKids Zebrafish Genetics Core facility and Mark Jen for technical assistance. All zebrafish studies were performed via an IACUC approved protocol.
AMM, SBS, CK, and DK, are supported by the Leona M. and Harry B. Helmsley Charitable Trust to study Very Early Onset Inflammatory Bowel Disease. AMM is funded by a Canada Research Chair (Tier 1) in Pediatric IBD, and a CIHR Foundation Grant. AMM and JRT are supported by the NIDDK (RC2DK118640). CK and DK are supported by the Collaborative Research Consortium SFB1054 project A05. JJD is supported by CIHR Operating Grants. JRT is funded by the NIDDK (K08DK113106), American Gastroenterological Association (Research Scholar Award) and Boston Children’s Hospital (OFD Career Development Award).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests.
The authors declare no competing interests.
References
Author names in bold designate shared co-first authorship
- 1.Avitzur Y, Guo C, Mastropaolo LA, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 2014;146:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bigorgne AE, Farin HF, Lemoine R, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest 2014;124:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jardine S, Dhingani N, Muise AM. TTC7A: Steward of Intestinal Health. Cell Mol Gastroenterol Hepatol 2019;7:555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lien R, Lin YF, Lai MW, et al. Novel Mutations of the Tetratricopeptide Repeat Domain 7A Gene and Phenotype/Genotype Comparison. Front Immunol 2017;8:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kammermeier J, Lucchini G, Pai SY, et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood 2016;128:1306–8. [DOI] [PubMed] [Google Scholar]
- 6.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 1999;21:932–9. [DOI] [PubMed] [Google Scholar]
- 7.Tan J, Brill JA. Cinderella story: PI4P goes from precursor to key signaling molecule. Crit Rev Biochem Mol Biol 2014;49:33–58. [DOI] [PubMed] [Google Scholar]
- 8.Vaillancourt FH, Brault M, Pilote L, et al. Evaluation of phosphatidylinositol-4-kinase IIIalpha as a hepatitis C virus drug target. J Virol 2012;86:11595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thakur PC, Davison JM, Stuckenholz C, et al. Dysregulated phosphatidylinositol signaling promotes endoplasmic-reticulum-stress-mediated intestinal mucosal injury and inflammation in zebrafish. Dis Model Mech 2014;7:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breedveld F, Dayer J. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis 2000;59:841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999;4:67–73. [DOI] [PubMed] [Google Scholar]
- 12.Varshney GK, Carrington B, Pei W, et al. A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish. Nat Protoc 2016;11:2357–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Zhang Y, Zhao F, et al. Acetylcholine serves as a derepressor in Loperamide-induced Opioid-Induced Bowel Dysfunction (OIBD) in zebrafish. Sci Rep 2014;4:5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141:1762–72. [DOI] [PubMed] [Google Scholar]
- 15.Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013;19:939–45. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Hou Z, Gulbranson DR, et al. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell 2010;7:240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith CE, Soti S, Jones TA, et al. Non-steroidal Anti-inflammatory Drugs Are Caspase Inhibitors. Cell Chem Biol 2017;24:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helms C, Pelsue S, Cao L, et al. The Tetratricopeptide repeat domain 7 gene is mutated in flaky skin mice: a model for psoriasis, autoimmunity, and anemia. Exp Biol Med (Maywood) 2005;230:659–67. [DOI] [PubMed] [Google Scholar]
- 19.Bassotti G, Antonelli E, Villanacci V, et al. Gastrointestinal motility disorders in inflammatory bowel diseases. World J Gastroenterol 2014;20:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silverman E, Mouy R, Spiegel L, et al. Leflunomide or methotrexate for juvenile rheumatoid arthritis. N Engl J Med 2005;352:1655–66. [DOI] [PubMed] [Google Scholar]
- 21.Herrlinger KR, Diculescu M, Fellermann K, et al. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: the ENTRANCE study. J Crohns Colitis 2013;7:636–43. [DOI] [PubMed] [Google Scholar]
- 22.Leger DY, Liagre B, Beneytout JL. Low dose leflunomide activates PI3K/Akt signalling in erythroleukemia cells and reduces apoptosis induced by anticancer agents. Apoptosis 2006;11:1747–60. [DOI] [PubMed] [Google Scholar]
- 23.Holtmann MH, Gerts AL, Weinman A, et al. Treatment of Crohn’s disease with leflunomide as second-line immunosuppression : a phase 1 open-label trial on efficacy, tolerability and safety. Dig Dis Sci 2008;53:1025–32. [DOI] [PubMed] [Google Scholar]
- 24.Autret-Leca E, Bensouda-Grimaldi L, Maurage C, et al. Upper gastrointestinal complications associated with NSAIDs in children. Therapie 2007;62:173–6. [DOI] [PubMed] [Google Scholar]
- 25.Mortimore M, Florin TH. A role for B(1)(2) in inflammatory bowel disease patients with suppurative dermatoses? An experience with high dose vitamin B(1)(2) therapy. J Crohns Colitis 2010;4:466–70. [DOI] [PubMed] [Google Scholar]
- 26.Ferrigno A, Berardo C, Di Pasqua LG, et al. Localization and role of metabotropic glutamate receptors subtype 5 in the gastrointestinal tract. World J Gastroenterol 2017;23:4500–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SCJ, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. Volume 47(D1) 2019:D1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.