

Abstract
DNA double-strand breaks (DSBs) are the most dangerous type of DNA damage because they can result in the loss of large chromosomal regions. In all mammalian cells, DSBs that occur throughout the cell cycle are repaired predominantly by the non-homologous DNA end joining (NHEJ) pathway. Defects in NHEJ result in sensitivity to ionizing radiation and the ablation of lymphocytes. The NHEJ pathway utilizes proteins that recognize, resect, polymerize and ligate the DNA ends in a flexible manner. This flexibility permits NHEJ to function on a wide range of DNA-end configurations, with the resulting repaired DNA junctions often containing mutations. In this Review, we discuss the most recent findings regarding the relative involvement of the different NHEJ proteins in the repair of various DNA-end configurations. We also discuss the shunting of DNA-end repair to the auxiliary pathways of alternative end joining (a-EJ) or single-strand annealing (SSA) and the relevance of these different pathways to human disease.
In dividing mammalian cells, there are an estimated ten DNA double-strand breaks (DSBs) per day per cell1–3. These pathological DSBs arise from ionizing radiation, reactive oxygen species, DNA replication errors and inadvertent cleavage by nuclear enzymes. Many of these pathological breaks, as well as the physiological (regulated) breaks that occur during V(D)J recombination and immunoglobulin heavy chain class switch recombination, require end processing by nucleases and DNA polymerases to repair the DNA (FIG. 1). In non-homologous DNA end joining (NHEJ), the DSB is first recognized by the Ku70–Ku80 hetero dimer (Ku), which acts as a ‘tool belt’ or loading protein to which other NHEJ proteins can be recruited as needed to promote the joining of DNA ends. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) has a high affinity for Ku–DNA ends and, together with Ku, forms the DNA-PK complex4 (FIG. 2a).
Figure 1 |. Overview of non-homologous end joining.
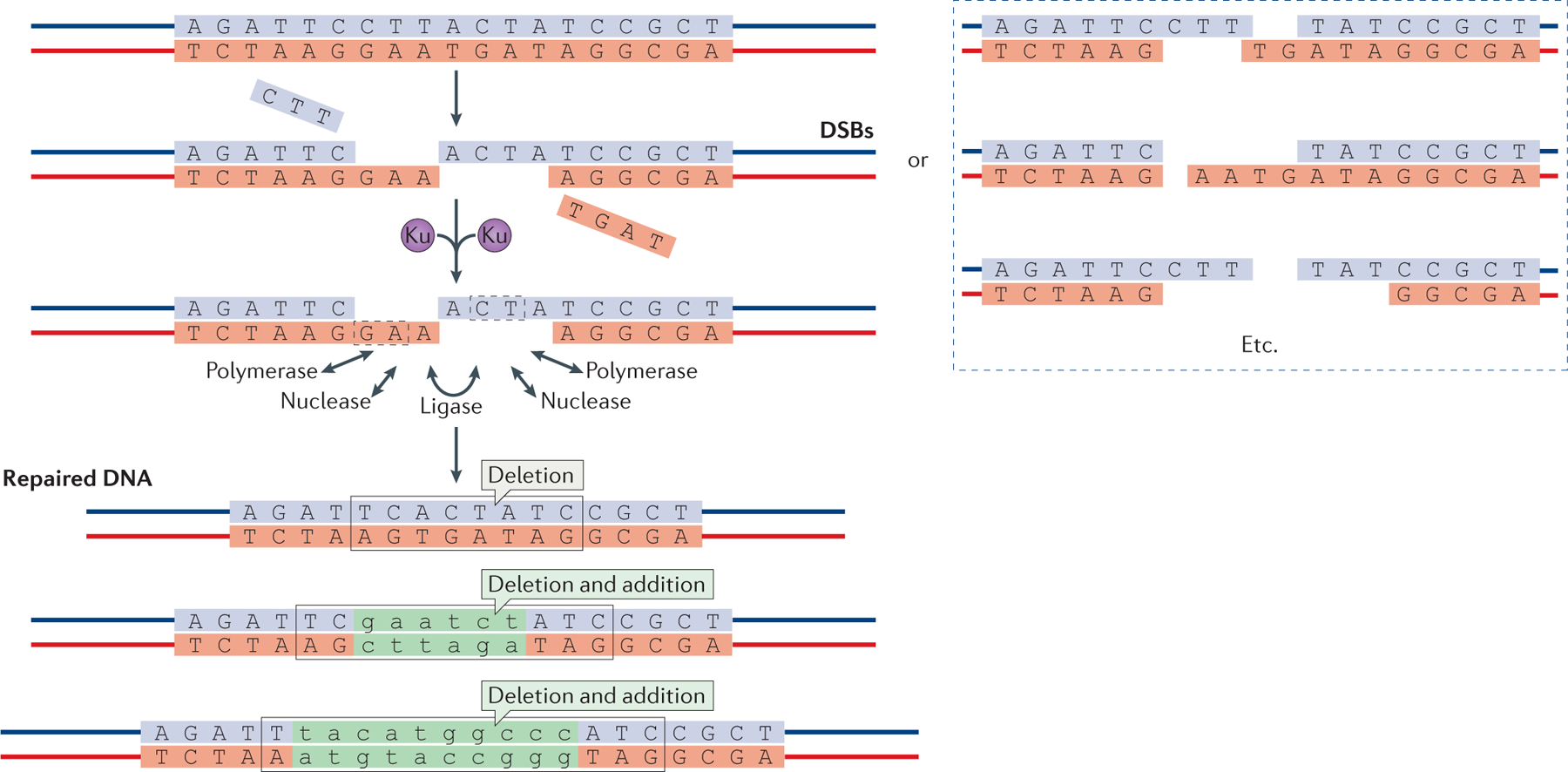
Schematic of DNA double-strand breaks (DSBs) and their repair by non-homologous end joining (NHEJ) (top). The Ku70–Ku80 heterodimer binds to DSBs and improves their subsequent binding by the NHEJ polymerase, nuclease and ligase complexes. These enzymes can act on DSBs in any order to resect and add nucleotides. Multiple rounds of resection and addition are possible, and nuclease and polymerase activities at each of the two DNA ends seem to be independent. Microhomology between the two DNA ends, which is either already present (dashed boxes) or newly created when the polymerases add nucleotides in a template-independent manner, is often used to guide end joining. The process is error-prone and can result in diverse DNA sequences at the repair junction (bottom). However, NHEJ is also capable of joining two DNA ends without nucleotide loss from either DNA end and without any addition. Nucleotide additions are depicted in green lower case.
Figure 2 |. Non-homologous end joining proteins and their known interactions.
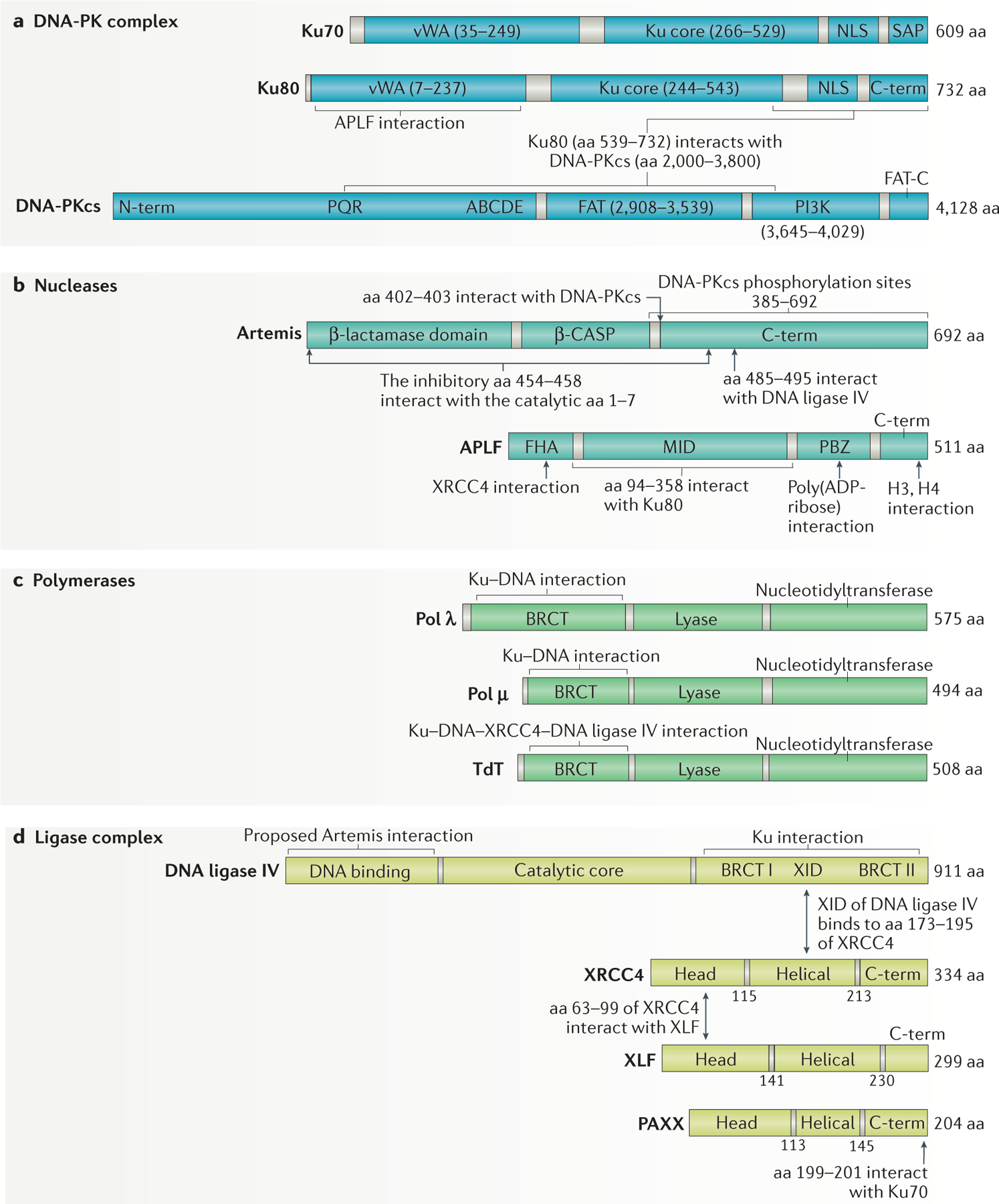
a | The non-homologous end joining (NHEJ) DNA protein kinase (DNA-PK) complex consists of a heterodimer of Ku70 and Ku80 plus DNA-PK catalytic subunit (DNA-PKcs). Ku70 and Ku80 consist of von Willebrand (vWA) domains, the Ku core and the nuclear localization sequence (NLS). Ku70 also contains a SAP (SAF-A/B, Acinus and PIAS) domain. DNA-PKcs consists of an amino-terminal domain with PQR and ABCDE autophosphorylation clusters implicated in its activation, a FAT (FRAP, ATM, TRRAP) domain, followed by the phosphatidylinositol 3-kinase (PI3K) domain, and the FAT-C (carboxy-terminal) domain. b | The NHEJ nucleases consist of Artemis and aprataxin and PNKP-like factor (APLF). Artemis has a catalytic β-lactamase domain, a cleavage and polyadenylation specificity factor (β-CASP) domain and a disordered C terminus. Amino acids 454–458 bind to aa 1–7 to auto-inhibit Artemis activity15. APLF consists of a forkhead-associated (FHA) domain, a middle (MID) domain and the poly(ADP-ribose)-binding zinc-finger (PBZ) domain151–154. c | The polymerases involved in NHEJ are Pol λ, Pol μ and terminal deoxynucleotidytransferase (TdT). They consist of a BRCA1 C terminus (BRCT) domain, a lyase domain and a nucleotidyltransferase domain. d | The DNA ligase complex consists of DNA ligase IV, X-ray repair cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF) and paralogue of XRCC4 and XLF (PAXX). DNA ligase IV consists of an N-terminal DNA binding domain, a catalytic core and an XRCC4 interaction domain (XID) flanked by the BRCT I and BRCT II domains. XRCC4, XLF and PAXX are structurally similar, with an N-terminal head domain, helical domain and a C terminus. Protein domains are shown in solid colour and linker regions in grey.
In this Review, we focus specifically on the differential requirements for the activity of the various NHEJ proteins depending on the configuration of the DNA ends, which can include blunt ends, 5′ overhangs and 3′ overhangs. Recent work has begun to systematically examine how various DNA-end configurations are processed differently5. Although a subset of this work concerns how specific DNA lesions are removed, the primary focus of this Review is on DNA ends with blunt or overhang configurations that do not have oxidative or other chemical damage. We also discuss how NHEJ relates to the other pathways of DSB repair, specifically alternative end joining (a-EJ) and single-strand annealing (SSA), and we discuss how NHEJ, a-EJ and SSA contribute to human disease.
Overview of NHEJ in humans
We provide a brief overview below of the types of proteins that are involved in NHEJ and their functions that applies to nearly all vertebrates.
The nucleases.
Most DSBs have two incompatible DNA ends that preclude direct ligation (FIG. 1). The ends are incompatible owing to chemical modifications or mismatching overhangs. Nuclease activity (also known as resection) is one method of ensuring that the two ends are compatible. In NHEJ, this end resection involves the degradation of short regions of the 5′ or 3′ overhangs by either exonuclease or endonuclease activity to expose or to generate small regions of microhomology (≤4 nucleotides) between the strands that can facilitate end joining. This differs from the extensive end resection (≥20 nucleotides) that occurs to initiate the homology-directed repair pathways. When DNA resection is required for NHEJ, DNA-PKcs is recruited in complex with the endonuclease Artemis. DNA-PKcs undergoes autophosphorylation and activates Artemis6,7, which then gains the ability to cut many DNA substrates at the boundaries between single-strand and double-strand DNA (ss–dsDNA)8,9.
Artemis belongs to the metallo-β-lactamase family of nucleases, which are characterized by conserved metallo-β-lactamase and β-CASP domains (FIG. 2a). This family of nucleases can hydrolyse DNA or RNA in various configurations10. Artemis has intrinsic 5′ exonuclease activity on ssDNA, even without DNA-PKcs11. At duplex DNA ends, Artemis, in complex with DNA-PKcs, has endonuclease activity on both the 5′ and the 3′ DNA overhangs (which are often created at pathological DNA breaks) and on DNA hairpins that are formed during V(D)J recombination. This DNA hairpin opening process during V(D)J recombination specifically requires Artemis, and thus patients lacking Artemis suffer from severe combined immunodeficiency (SCID) owing to a V(D)J recombination defect in antigen receptor gene assembly12,13. Amino acids 402–403 of Artemis interact with the FAT domain of DNA-PKcs (FIG. 2b), whereas the carboxy-terminal region of Artemis (aa 454–458) interacts with its own amino-terminal catalytic domain (aa 1–7) to inhibit the endonuclease activity14,15. Artemis (aa 485–495) also interacts with the N-terminal region of DNA ligase IV16,17.
Of ionizing radiation-induced DSBs, 20–50% require Artemis for repair18,19. It is unclear whether the remaining DSBs have DNA-end configurations that can be joined without the benefit of any nuclease. Other nucleases that might contribute to the repair of these DSBs include aprataxin and PNKP-like factor (APLF; also known as PALF)20–22, the MRN complex (MRE11–RAD50–NBS1 (Nijmegen breakage syndrome protein 1; also known as nibrin)), CtBP-interacting protein (CtIP; also known as RBBP8), Werner syndrome ATP-dependent helicase (WRN), flap endonuclease 1 (FEN1) and exonuclease 1 (EXO1)23. The abundance and localization of these nucleases at DSB sites may determine which nucleases are responsible for the most resection at DSBs (see Supplementary information S1 (table) for a list of the known cellular abundance of human NHEJ and auxiliary repair proteins). But for the limited resection that occurs during most NHEJ events, the Artemis–DNA-PKcs complex seems to be the primary nuclease8.
The polymerases.
DNA polymerase μ (Pol μ) and Pol λ are the two members of the Pol X family polymerases that are involved in NHEJ in humans24,25. These polymerases interact with Ku through their N-terminal BRCA1 C terminus (BRCT) domains26 (FIG. 2c). Primary cells derived from mice with genetic knockouts of both Pol μ and Pol λ exhibit little or no sensitivity to ionizing radiation, although knockouts in cell lines can have limited deficit in DSB repair in some assays27,28. Both Pol μ and Pol λ can incorporate either dNTPs or rNTPs24,25, and any ribonucleotides that are incorporated are likely to be subsequently removed by base excision repair29. Both polymerases can incorporate in a template-dependent or a template-independent manner27, although Pol μ does the latter more than Pol λ30,31.
Pol X family members also include Pol β and terminal deoxynucleotidyltransferase (TdT; also known as DNTT), but Pol β does not contain a BRCT domain to allow interaction with the Ku complex, and TdT is only expressed in early B lymphocytes and T lymphocytes during V(D)J recombination. DNA polymerases outside the Pol X family can incorporate nucleotides during NHEJ but only in a template-dependent manner32–36.
The ligase complex.
DNA ligase IV and X-ray repair cross-complementing protein 4 (XRCC4) (FIG. 2d) are the most central components of NHEJ in eukaryotes1. XRCC4 stimulates DNA ligase IV enzyme activity in biochemical assays37. XRCC4-like factor (XLF; also known as Cernunnos in humans or Nej1 in yeast), is a 33 kDa protein with weak sequence homology and structural similarity to XRCC4 (REFS 38–40). The N-terminal head domain of XLF interacts with the N-terminal head domain of XRCC4 (REF. 39), and the XRCC4–XLF complex forms a sleeve-like structure around a DNA duplex41. This proposed sleeve would presumably stabilize the positioning of the ends before covalent ligation, but this is still an area of active investigation. PAXX (paralogue of XRCC4 and XLF) is a recently discovered 22 kDa protein with structural similarity to XRCC4 and XLF42,43. The C terminus of PAXX (aa 199–201) interacts with Ku, and PAXX mutants are more sensitive to ionizing radiation and DSB-inducing agents42,44,45. It will be interesting to discover how PAXX participates in such a large assembly of other NHEJ proteins, especially in the context of chromatin.
Polynucleotide kinase, aprataxin and tyrosyl DNA phosphodiesterase 1.
Other proteins are involved in NHEJ if the chemistry of the DNA ends requires further proteins. For example, a 5′ end lacking a phosphate would require phosphorylation by polynucleotide kinase (PNK; also known as PNKP). Human PNK is also a phosphatase, which is important for removing 3′ phosphates that can arise from some types of oxidative damage46.
Ligase IV sometimes initiates but does not complete a covalent join, and this can result in the formation of an intermediate or an aborted ligation product in which an AMP group remains covalently bound to the 5′ end of one of the strands at the DSB. The enzyme aprataxin is required to remove the AMP group as part of a deadenylation reaction47. Both PNK and aprataxin bind to XRCC4 via their forkhead-associated domain (FHA), which is located near their N termini, but only after the kinase CK2 has phosphorylated XRCC4 (REF. 48).
Tyrosyl DNA phosphodiesterase 1 (TDP1) is the only identified enzyme that can specifically process 3′-phosphoglycolates (3′-PGs), which are by-products of ionizing radiation-induced DSBs at 3′ ends49. These 3′-PGs are unligatable ends that can account for 10% of radiation-induced DSBs50. However, TDP1 mutants in human cells show only marginal radiosensitivity, suggesting that another enzyme could be involved in processing 3′-PGs51.
End structure directs repair subpathway
Structural and biochemical studies support a model in which different sets of NHEJ proteins serve to align the two DNA ends in an end-to-end configuration (FIG. 3). One parameter that affects DNA end joining is how much transient base pairing can occur between the two DNA ends before joining or, in other words, the degree of microhomology between the ends. However, after several base pairs of DNA end breathing, any two DNA ends will share at least one nucleotide of homology that can be used for annealing, even if it is only by non-Watson–Crick base pairing52. Some DNA ends can be joined together using only the ligase complex, but other DNA ends require the action of polymerases or nucleases, which together form different NHEJ subpathways.
Figure 3 |. The various non-homologous end joining subpathways.
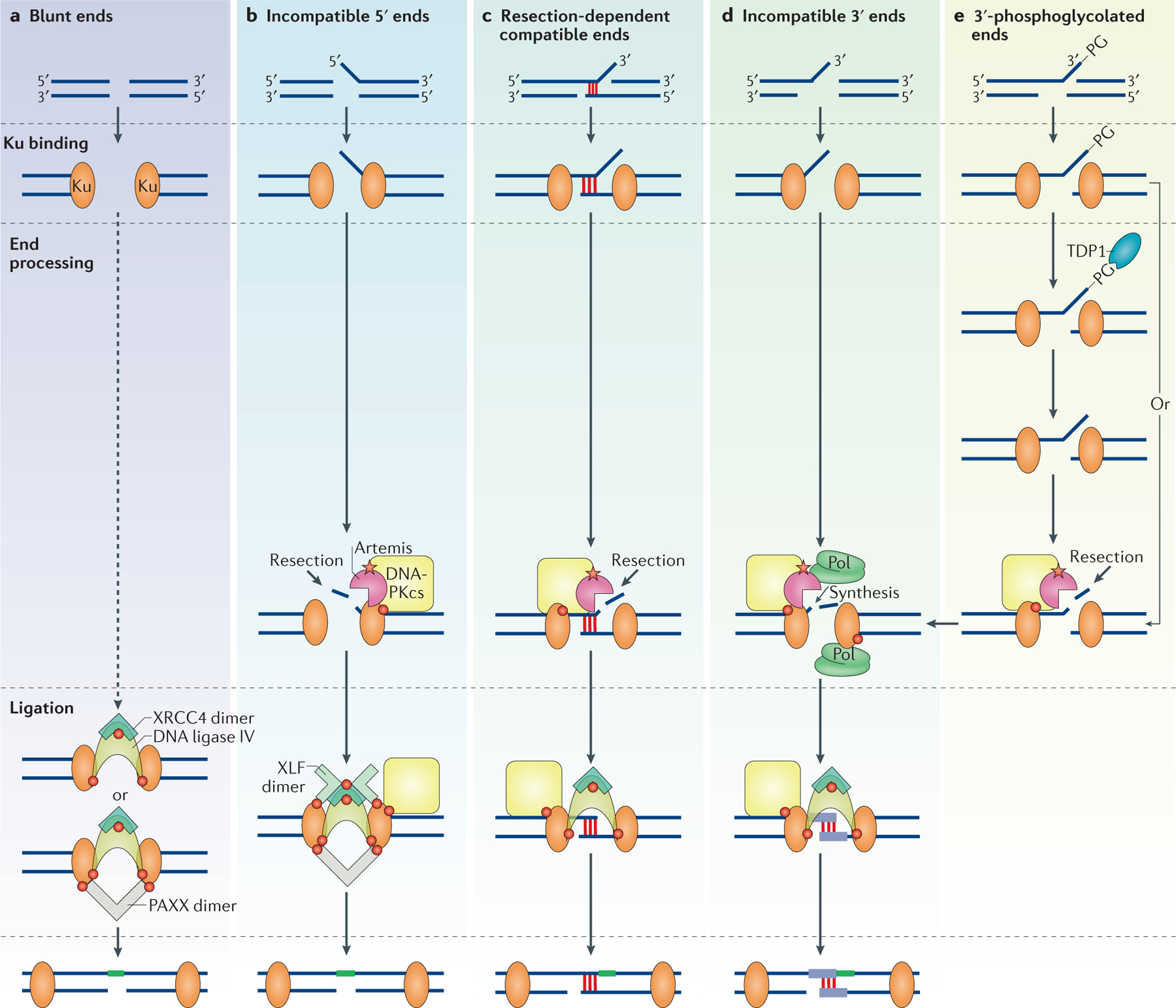
Various non-homologous end joining (NHEJ) proteins may associate at common NHEJ substrates. Red circles represent known protein–protein interactions depicted in FIG. 2. The red star represents the interaction between Artemis and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which results in the activation of the endonuclease activity of Artemis. The diagram depicts ligation (in green) only of the top strand, but this process will inevitably proceed to the bottom strand (Supplementary information S2 (figure)). a | Blunt DNA ends are preferentially repaired without end processing and their ligation can be stimulated by paralogue of XRCC4 and XLF (PAXX). b | Incompatible 5′ overhanging ends are preferentially processed with resection of the 5′ overhang by the Artemis–DNA-PKcs complex, followed by blunt-end ligation that is stimulated by XRCC4-like factor (XLF) and PAXX. c | Resection-dependent compatible ends that have a short stretch of microhomology (~4 nucleotides of base pairing) along with a non-base paired flap only require Artemis–DNA-PKcs to cleave off the flap for ligation to occur. d | Incompatible 3′ overhanging ends are processed by iterative events of end resection and nucleotide synthesis by DNA polymerases (Pol) to generate short regions of base pairing (purple) before ligation. e | 3′-phosphoglycolated (3′-PG) ends can form on recessed or blunt ends or on a DNA overhang, and can be processed by tyrosyl DNA phosphodiesterase 1 (TDP1). Alternatively, Artemis–DNA-PKcs can bypass the 3′-PG and probably other end modifications and can endonucleolytically resect the ends that contain the modifications. Ku, Ku70–Ku80; XRCC4, X-ray repair cross-complementing protein 4.
Blunt-end ligation by Ku–XRCC4–DNA ligase IV.
Biochemical studies have demonstrated that NHEJ of blunt DNA ends lacking microhomology rely on Ku for efficient joining (FIG. 3a). By contrast, DNA ends joined using microhomology do not require Ku, indicating that Ku becomes more important the less the ends are able to form terminal base pairs31. Ku has a high affinity for DNA ends (Kd = 6 × 10−10 M) and can promote the binding of XRCC4–DNA ligase IV to the DNA ends53. The C terminus of DNA ligase IV contains two BRCT domains, which bind Ku54, and the region between the two BRCT domains also binds to a homo-dimer of XRCC4 (FIG. 2d). Thus, XRCC4 associates with DNA ligase IV in a 2:1 ratio, which could contribute to the bridging between the two DNA ends55–57. This Ku–XRCC4–DNA ligase IV complex is required for the efficient reconstitution of the NHEJ pathway using human proteins58. The addition of DNA-PKcs, Artemis and Pol μ does not further stimulate ligation, suggesting that the direct ligation of blunt ends is preferred over their processing (FIG. 3a).
The relatively high efficiency of blunt-end ligation by human Ku and XRCC4–DNA ligase IV contrasts with in vivo observations in Saccharomyces cerevisiae, in which blunt-end joining is inefficient59,60. However, it is possible that such inefficiency in yeast could be the consequence of more aggressive DNA end resection that exposes long 3′ overhangs in preparation for homologous recombination (HR).
The 6.6 Å and the more recent 4.3 Å crystal structures of DNA-PKcs raise the possibility of dimerization of DNA-PKcs, and one could speculate that this contributes to bridging of the two DNA ends before ligation61,62. DNA is not present in these crystal structures, and thus one can only speculate about its location. The ligation of DNA ends with only Ku and XRCC4–DNA ligase IV provides biochemical evidence that DNA end bridging is not reliant on DNA-PKcs or on NHEJ factors other than Ku and XRCC4–DNA ligase IV5. It is clear that signal joint formation during V(D)J recombination also does not require any NHEJ proteins other than Ku and XRCC4–DNA ligase IV1, and this is consistent with the biochemistry of blunt end ligation (FIG. 3a).
Nuclease-dependent subpathways.
DNA-PKcs weakly interacts with DNA but its binding increases 100-fold when Ku is present63. The FAT domain of DNA-PKcs binds to the C terminus (aa 718–732) of Ku80 (REF. 64) (FIG. 2a). One of the major roles of DNA-PKcs is to interact with and activate the endonuclease activity of Artemis at DNA ends. DNA-PKcs autophosphorylation upon binding of the DNA end activates Artemis endonuclease activity6. DNA-PKcs also phosphorylates the C-terminal domain of Artemis65 (FIG. 2b). It is likely that autophosphorylated DNA-PKcs promotes the dissociation of the C-terminal inhibitory region of Artemis (aa 454–458) from the N-terminal catalytic domain (aa 1–7) of Artemis (FIG. 2b). The Artemis–DNA-PKcs complex endonucleolytically removes 5′ and 3′ DNA overhangs to create DNA end structures that can be ligated by the XRCC4–DNA ligase IV complex26,66. At 5′ overhangs, Artemis directly cuts at the ss–dsDNA boundary (FIG. 3b). However, when processing 3′ overhangs and DNA hairpins, Artemis preferentially leaves a 4-nucleotide 3′ overhang (FIG. 3c). DNA hairpins are structurally similar to DNA overhangs, because they have a sterically constrained hairpin tip that results in only transient base pairing of the terminal base pairs (4 nucleotides), thereby creating a ss–ds boundary67. From these observations, Artemis activity on duplex DNA can be explained using a model in which Artemis–DNA-PKcs binds to the ss–dsDNA boundary to occupy 4 nucleotides along the single-stranded segment at the boundary8. This binding is followed by nicking on the 3′ side of the 4 nucleotides.
In addition to stable ss–dsDNA boundaries, Artemis acts at blunt DNA ends that breathe to an open state, thereby forming transient ss–ds boundaries8. Such blunt DNA ends may be generated by chemotherapeutic agents, reactive oxygen species or ionizing radiation68. A more comprehensive version of this model, which can explain the essential structural features of all the DNA substrates at which Artemis functions, including blunt DNA ends (transient ss–ds boundaries), proposes that Artemis recognizes all ss–dsDNA boundaries through putative contact points in the duplex DNA that are either adjacent to the 5′ or 3′ overhang, or adjacent to the hairpin9. To achieve hydrolysis of the phosphodiester backbone, the Artemis active site would then act within the single-stranded portion of the overhang or the hairpin. Although this model must await the elucidation of a DNA–Artemis co-crystal, it does explain all the known cutting patterns of Artemis.
Although the role of Artemis in V(D)J recombination is well characterized, its role in NHEJ is less clear. One role of Artemis in NHEJ is when ionizing radiation-induced DSBs have a 3′-PG terminus69–71. These DNA ends are unable to undergo ligation because this step requires a 3′ hydroxyl on one end and a 5′ phosphate on the other. As discussed above, TDP1 can remove these 3′ modifications. However, TDP1-mutant cells are only marginally radiosensitive; Artemis mutants, however, are sensitive to ionizing radiation, and therefore it is likely that Artemis is involved in removing the damaged strand (FIG. 3e). Indeed, it has been shown biochemically that the Artemis–DNA-PKcs complex is able to process these ends72,73. More recently, the C-terminal region of Artemis (aa 485–495) has been shown to interact with the N-terminal head domain of ligase IV16,17,74 (FIG. 2b). This interaction may promote Artemis activity by recruiting Artemis to 3′ overhangs through the DNA ligase IV binding5.
Biochemical reconstitution of NHEJ with purified proteins has shown that Artemis resects 5′ and 3′ overhangs to generate regions of microhomology for NHEJ to occur5. In instances in which the overhangs have the potential for microhomology after partial resection of the overhang, the endonuclease activity of Artemis exposes the nucleotides within a stretch of ssDNA (FIG. 3c). However, in instances in which there are no regions of substantial microhomology in the overhangs, the Artemis–DNA-PKcs complex often resects into the duplex to generate overhangs that expose microhomology5. Interestingly, Artemis–DNA-PKcs does not strongly stimulate the ligation of blunt-ended DNA. This suggests that, even though Artemis–DNA-PKcs is able to resect at blunt ends, these ends are usually joined directly without resection5,8 (FIG. 3a). By contrast, the ligation of incompatible overhangs is strongly stimulated by the presence of the Artemis–DNA-PKcs complex, which is probably recruited to the DNA end only when resection is required.
Polymerase-dependent subpathways.
Pol μ and Pol λ are recruited to the DNA end by interaction of their N-terminal BRCT domain with the Ku–DNA complex26 (FIG. 2c). Pol μ primarily has template-independent polymerase activity, whereas Pol λ primarily has template-dependent polymerase activity (fill-in synthesis)30. This difference in activity is due to structural variations of these polymerases in a region known as loop 1 (REF. 75). This loop is structurally flexible and provides hydrogen bonding with the DNA template strand, allowing Pol μ to add nucleotides without an actual template.
In reactions that involve only the Ku–XRCC4–DNA ligase IV complex, Pol μ strongly promotes the ligation of incompatible 3′ overhangs31. At these overhangs, Pol μ can add nucleotides in a template-independent manner, generating regions of microhomology for subsequent base pairing and ligation31. Pol μ is also required for the joining of two DNA substrates with short (1 nucleotide or 2 nucleotides) incompatible 3′ overhangs28. In biochemical reactions involving DNA-PKcs and Artemis, Pol μ strongly stimulates the joining of two mismatched 3′ overhangs by promoting the formation of terminal microhomology5 (FIG. 3d). Sequences at the resulting junctions reveal nucleotides that represent template-independent nucleotide addition by Pol μ, as well as an absence of nucleotide resection by Artemis–DNA-PKcs5.
Pol λ primarily promotes the ligation of terminally compatible overhangs that require fill-in synthesis28,31. These situations arise when opposing DNA ends terminally base pair but leave a gap that needs to be filled in before ligation. Unsurprisingly, Pol λ has little effect on NHEJ of completely mismatched 3′ overhangs because these overhangs do not provide a template strand5.
Ligation by the XLF and PAXX subpathways.
XLF and PAXX are the most recently characterized NHEJ factors that have been shown to support ligation by the ligase IV complex. Both XLF and PAXX have structural similarity to XRCC4 (REFS 39,42). XLF forms homo-dimers and its head domain binds to XRCC4 (REF. 76). The XLF head domain also interacts with the Ku–DNA complex77. PAXX also forms homodimers, and its C terminus has been found to associate with Ku42,43 (FIG. 2d). XLF and PAXX promote NHEJ of a subset of DNA ends that require maximal stabilization by the ligase complex. In biochemical reactions involving only Ku and the XRCC4–DNA ligase IV complex, XLF was shown to only stimulate the ligation of short incompatible 3′ overhangs31. However, in another study involving Ku, DNA-PKcs and XRCC4–DNA ligase IV, XLF was shown to promote the ligation of all mismatched and noncohesive overhangs78. This difference may be partly due to the dependence of XLF on the length of the dsDNA present, as ~70 bp fragments were used in the first study mentioned above compared with >3 kb linearized plasmids in the second study. Alternatively, DNA-PKcs could be interfering with XLF interactions.
PAXX was shown to promote the ligation of two blunt ends in reactions involving only Ku and the XRCC4–DNA ligase IV complex42 (FIG. 3a). PAXX may also promote the ligation of a blunt end to a 3′ overhang in reactions involving Ku, XLF and the XRCC4–DNA ligase IV complex43. However, a more recent biochemical study that also included Artemis and Pol μ failed to demonstrate this effect, but did show that XLF and PAXX stimulate the NHEJ of 5′ incompatible overhangs5 (FIG. 3b). These data suggest that the role of XLF and PAXX may be to help to stabilize Ku along with other NHEJ proteins at a DNA end under conditions in which terminal microhomology is not available.
Inactivation of XLF and PAXX together is synthetic lethal in mice and reduces V(D)J recombination in human B lymphocytes79–82. These data suggest a possible redundant role of XLF and PAXX; however, additional roles may become apparent when DNA substrates can be studied in the context of chromatin.
Shunting to auxiliary pathways
When NHEJ is compromised owing to the lack of one or more of its key protein components, the activity of the other end joining pathways becomes apparent, which typically involve much more extensive resection of the DNA ends to reveal sequence homology, the annealing of which stabilizes the two ends of a break to allow for more efficient joining and ligation1. The a-EJ pathway83 (FIG. 4) (also known as microhomology-mediated end joining and Pol θ-mediated end joining) requires microhomology that ranges between 2 bp and 20 bp. At the low end of this range, NHEJ overlaps with a-EJ and requires usually ≤4 bp of microhomology; non-conservative homology-directed repair pathways (which involve the loss of nucleotides), such as SSA, require >20 bp of homology33,34,84,85 (FIG. 5). The conservative HR pathway (in which no nucleotides are lost) generally requires lengths of homology longer than 100 bp (HR is beyond the scope of this Review and is discussed in detail elsewhere86–88).
Figure 4 |. Double-strand break repair pathway choice.
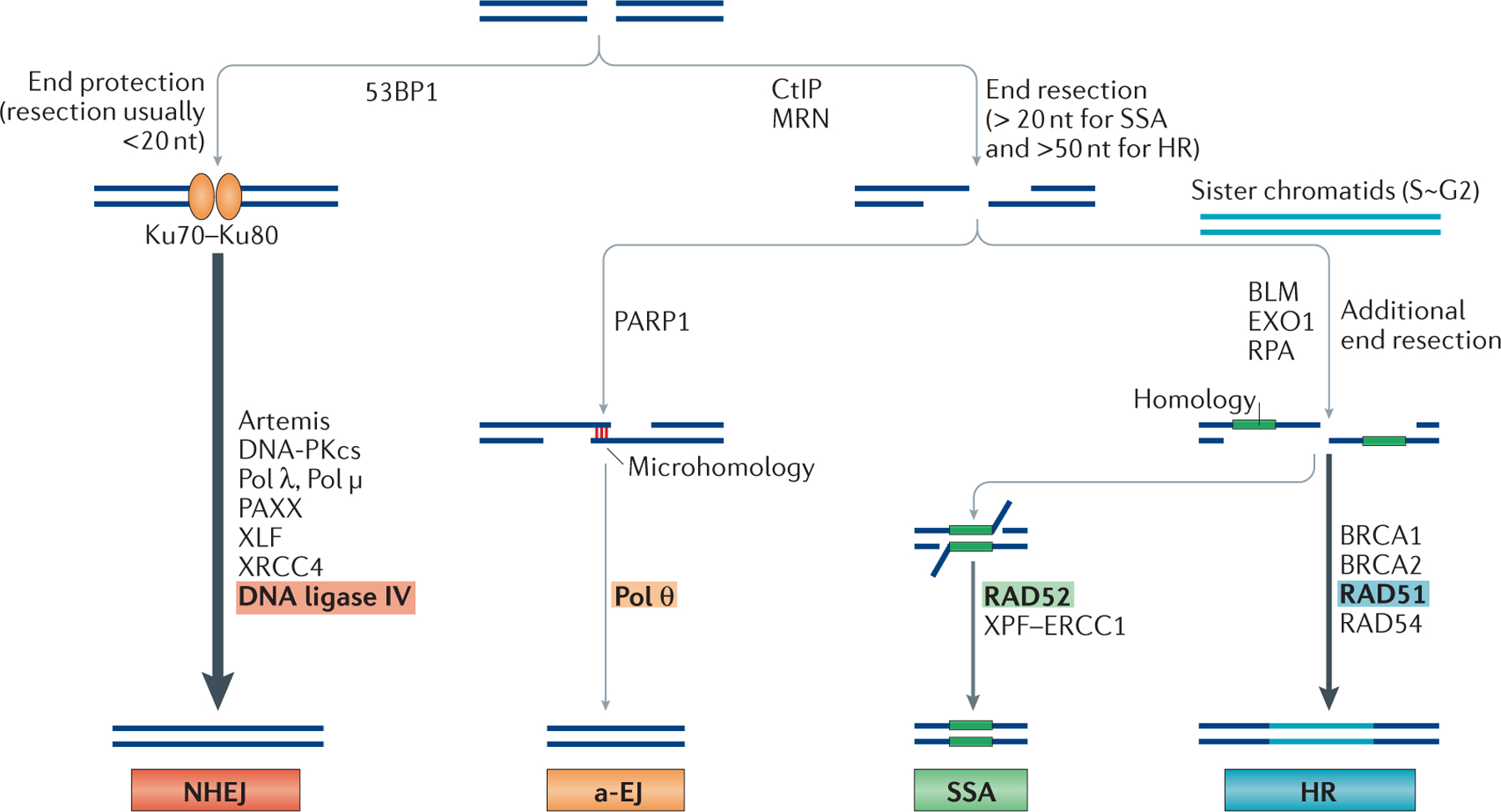
DNA double-strand breaks (DSBs) can be repaired by the classical non-homologous end joining (NHEJ) pathway, the alternative end joining (a-EJ) pathway, the single-strand annealing (SSA) pathway or by homologous recombination (HR). The major differences in pathway choice are the requirement for substantial DNA end resection. p53-binding protein 1 (53BP1) is a chromatin remodeller and a positive regulator of NHEJ. Although the complex of Artemis and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) can carry out some resection (typically <20 nucleotides), the NHEJ pathway does not require extensive end resection and the ends are mostly protected by the binding of Ku70–Ku80. By contrast, carboxy-terminal binding protein interacting protein (CtIP) and the MRN (MRE11–RAD50–NBS1 (Nijmegen breakage syndrome protein 1)) complex are involved in extensive 5′ to 3′ resection of regions of the duplex to generate stretches of single-strand DNA (ssDNA) at DNA ends for a-EJ, SSA and HR. SSA typically requires >20 bp of microhomology, whereas the requirement for a-EJ is <25 bp. Poly(ADP-ribose) polymerase 1 (PARP1) and DNA polymerase θ (Pol θ) are important for a-EJ. Bloom syndrome RecQ-like helicase (BLM) and exonuclease 1 (EXO1) provide additional resection, and replication protein A (RPA) binds to ssDNA to promote the SSA and the HR pathways. RAD52-mediated annealing of large regions of homology is key for the SSA pathway.
The xeroderma pigmentosum group F (XPF)–ERCC1 complex cuts the remaining 3′ overhangs before ligation. By contrast, RAD51-mediated strand exchange and its association with BRCA1, BRCA2 and RAD54 are essential for promoting the HR pathway. PAXX, paralogue of XRCC4 and XLF; XLF, XRCC4-like factor; XRCC4, X-ray repair cross-complementing 4.
Figure 5 |. Microhomology length requirement of DNA-end joining pathways.
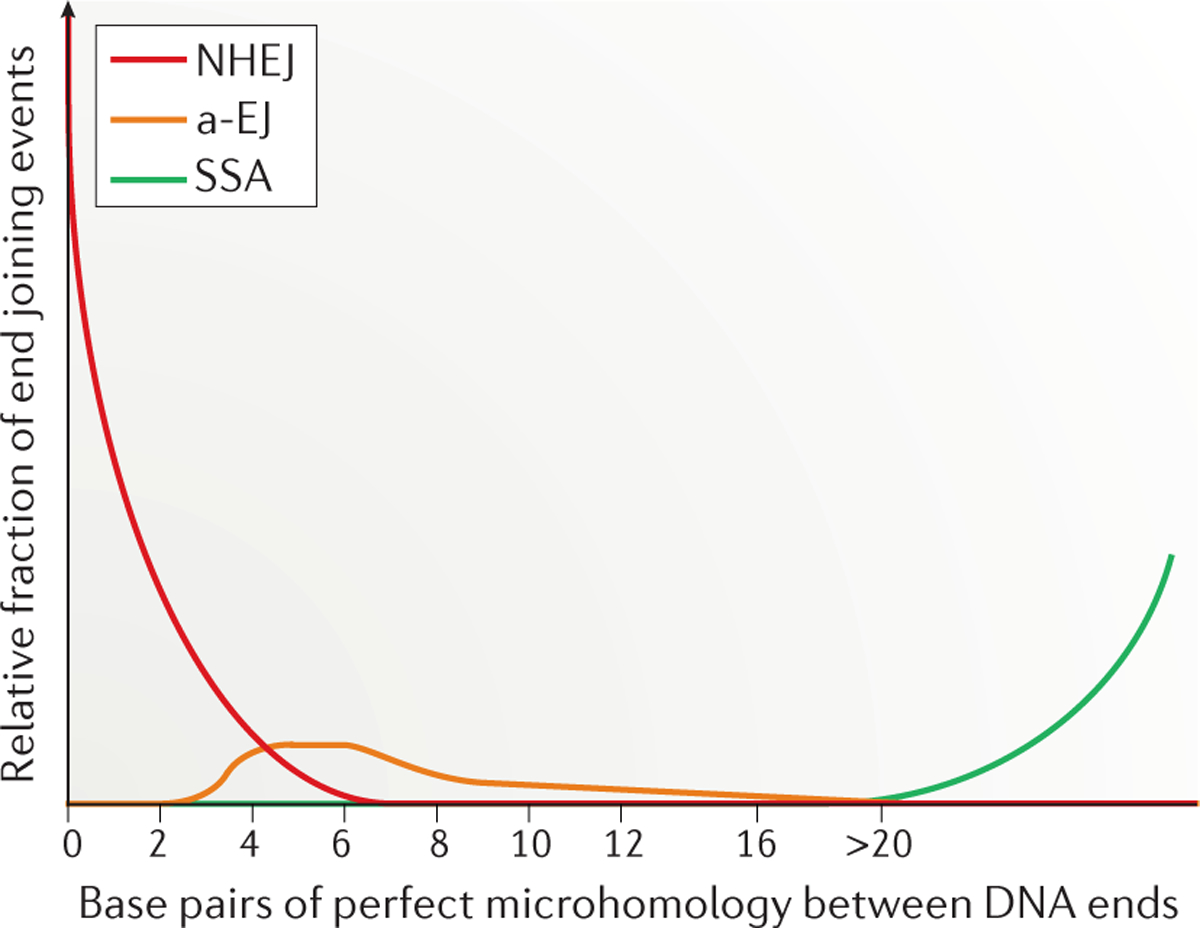
Non-homologous end joining (NHEJ) uses short stretches of microhomology (from 0 to 4 bp), whereas alternative end joining (a-EJ) uses <20 bp of microhomology (most commonly 4–6 bp). The single-strand annealing (SSA) pathway uses >20 bp of homology (for SSA, homology is a more appropriate term than microhomology); the relative use of SSA depends on the organism and the length of homology.
Extensive DNA-end resection is prevented by Ku89. The high abundance of Ku in cells (Supplementary information S1 (table)) increases the likelihood that Ku is the first protein to bind to a broken DNA end and, therefore, that repair is carried out through NHEJ (FIG. 4). There is evidence that the DNA damage response protein p53-binding protein 1 (53BP1; Rad9 in S. cerevisiae) acts as an antagonist to end resection, along with replication timing regulatory factor 1 (RIF1)90. 53BP1 and mediator of DNA damage checkpoint protein 1 (MDC1) are recruited to DSBs through several modified histone residues and seem to have distinct roles in DSB repair86,91,92. Further work is required to elucidate specific ally how 53BP1 recruitment inhibits extensive end resection. Overcoming this barrier to resection, however, is the first step to enable either a-EJ or SSA.
Alternative end joining.
Given the rarity of humans with NHEJ mutations, it is unclear whether a-EJ represents a standing pathway or whether the components of the pathway usually serve other functions in dsDNA processing, such as in replication, recombination or repair, and only become involved in end joining when NHEJ is compromised. Importantly, a-EJ requires Pol θ93–99 and may also include poly(ADP-ribose) polymerase 1 (PARP1), CtIP and the MRN complex100–103. The endonuclease function of MRN, which is stimulated by phosphorylated CtIP, seems to initiate a-EJ by processing DNA ends to generate 15–100-nucleotide 3′ overhangs (FIG. 4). MRN proteins are considerably less abundant than Ku (Supplementary information S1 (table)) and are therefore less likely to bind to dsDNA ends, suggesting a limited role for MRN in end joining when Ku is present104. However, in vitro studies demonstrated that the endonuclease function of MRN may remove protein adducts from the ends of DNA, suggesting a possible role for MRN and CtIP in a subset of reactions in NHEJ105,106. PARP1 is an ADP-ribosylating enzyme that is involved in sensing DNA damage and promoting the a-EJ pathway107.
Cells with mutations in both NHEJ proteins (to allow detection of a-EJ) and Pol θ have a marked reduction in a-EJ to nearly undetectable levels93–99. Pol θ is encoded by the POLQ gene and belongs to the A family of DNA polymerases. It has a C-terminal polymerase domain and, uniquely among DNA polymerases, an N-terminal helicase-like domain94,108,109. Pol θ has been shown to stabilize the annealing of two long 3′ ssDNA overhangs (often known as 3′ tails) with as little as 2 bp of homology, extending one 3′ DNA end by using the annealing partner as a template95. This creates a more stable annealed intermediate that can be sealed by either DNA ligase I or DNA ligase III. The polymerase activity of Pol θ probably prevents further extensive resection of ends, thereby minimizing the potential formation of large deletions by SSA. Pol θ also has terminal transferase activity and thus can add nucleotides to provide microhomology that is not already present97.
A subset of Pol θ-mediated end joining products includes templated insertions95,99,109. Some NHEJ templated insertions also arise owing to the activity of the error-prone polymerases, Pol μ and Pol λ26, but it seems that Pol θ creates longer (>10 nucleotides) templated insertions, which initiate from a short length (often 2 or more bp) of microhomology95,99,109. Short templated insertions (usually <10 nucleotides, and not necessarily associated with microhomology) are also seen in some normal murine lymphoid V(D)J recombination junctions110 and in a substantial proportion (20–50%) of human lymphoid translocations. These are likely to be mediated by Pol μ or Pol λ111–114. The junctional sequences in a large majority of human lymphoid translocations are most consistent with NHEJ, and the hairpin opening at the D or J coding ends are clearly mediated by Artemis–DNA-PKcs–Ku111. If Pol θ rather than Pol μ or Pol λ is responsible for the longest (>10 nucleotides) templated insertions, it is possible that Pol θ could modify some of these DNA ends in the context of NHEJ, but this possibility must be investigated. Experiments in Drosophila melanogaster predicted much of what is now being discovered in mammalian systems regarding Pol θ96,109. It is important to note that D. melanogaster does not have Pol μ or Pol λ, and it will be interesting to determine which templated insertions are carried out by which polymerase in mammalian cells.
Pol θ can also function when the annealed micro-homologies are embedded within the long 3′ ssDNA tails that are generated by extensive resection94–98. This would create non-homologous 3′ ssDNA tails that would need to be removed before extension by Pol θ. Therefore, nuclease activities from other repair pathways may be utilized during a-EJ. For example, the xeroderma pigmento sum group F (XPF)–ERCC1 nuclease complex, APLF or Artemis–DNA-PKcs could conceivably be used.
It is possible that a-EJ is slower than NHEJ. For example, in immunoglobulin class switch recombination, when DNA ligase IV is missing, DNA ligase I or DNA ligase III can substitute for DNA ligase IV, but with approximately tenfold slower kinetics101,102. This illustrates that even the most central NHEJ proteins such as ligase IV have back-up components, but that these back-up enzymes function with slower kinetics and lower repair efficiency. The slower end joining repair kinetics could be due to a requirement for more resection to reveal additional microhomology to stabilize the junction before the final ligation step. a-EJ junctional sequences in humans have microhomology lengths that are usually >4 bp, and often >10 bp (REF. 115) (FIG. 5). This observation suggests that Pol θ is active after the resection by a nuclease.
Future work will help to identify all the components of a-EJ and explain how a-EJ is distinct from NHEJ116. The kinetics of repair by these various pathways is also an important factor to consider. In addition, the ataxia telangiectasia mutated (ATM)-mediated DNA damage response may be important for the balance of NHEJ and a-EJ, given that the absence of ATM favours NHEJ117. It is also important to note that humans who do not have major NHEJ components are exceedingly rare. Therefore, the a-EJ proteins and enzymes may have functions other than merely as substitutes for an absence of NHEJ that is too lethal to be usually found in mammals.
Single-strand annealing.
a-EJ has more in common with SSA than it does with NHEJ as both a-EJ and SSA require extensive resection to reveal microhomology. By contrast, NHEJ often uses 1–4 bp of microhomology but this is not a requirement5. Neither the a-EJ pathway nor the SSA pathway is reliant on Ku, and the binding of Ku to DNA ends may need to be attenuated for a-EJ and SSA to proceed. These pathways also rely on the initiation of extensive resection by the MRN complex and CtIP, which generate 15–100-nucleotide 3′ ssDNA tails. It is at this point that the a-EJ and SSA pathways diverge. In a-EJ, annealing of microhomology seems to be sufficient for Pol θ to extend one of the DNA strands to stabilize the intermediate for ligation.
SSA requires the exposure of more sequence homology; therefore, more extensive resection is required118. The 3′ ssDNA tails created by MRN and CtIP are further extended by the action of the nuclease EXO1 or Bloom syndrome RecQ-like helicase (BLM) or DNA replication helicase/nuclease 2 (DNA2) (acting as part of a complex) to generate longer 3′ ssDNA tails119,120 (FIG. 4). The long 3′ ssDNA tails do not remain exposed, but are bound by multiple copies of the replication protein A (RPA) complex, the components of which are abundant within the cell (Supplementary information S1 (table)). RPA forms a filament on the ssDNA to prevent the formation of secondary structures. During HR, the RecA homologue RAD51 replaces RPA to allow for homology search and strand invasion121 (FIG. 4). BRCA1, BRCA2 and RAD54 may have a role in promoting HR, but this is beyond the scope of this Review. SSA, however, is a RAD51-independent mechanism that generally depends on the presence of 3′ ssDNA tails that share suitable sequence homology to form a stable annealing intermediate. The promiscuity of joining partners is what makes SSA non-conservative and prone to generating deletions and translocations. The annealing of complementary ssDNA tails is mediated by the strand annealing protein RAD52 (FIG. 4). Before ligation, the unannealed, non-homologous portions of the 3′ ssDNA tails must be processed and removed. In this case, SSA uses the nucleotide excision repair complex XPF–ERCC1 (FIG. 4) and the mismatch repair complex MSH2–MSH3, further highlighting the overlap among repair pathways85,122,123.
The influence of the cell cycle.
Although binding of DNA ends by Ku inhibits extensive resection by MRN and CtIP, and favours repair by NHEJ, extensive resection is also dependent on the cell cycle owing to the action of cyclin-dependent kinases (CDKs)124,125. Factors that promote extensive end resection are more active during S and G2 phases, favouring HR when a sister chromatid is present. This is another reason why repair by NHEJ is dominant throughout the cell cycle, whereas repair by HR and SSA is favoured in S and G2 phases. Targets of CDKs include the DNA damage response checkpoint proteins ATM and ataxia telangiectasia and Rad3-related (ATR), as well as enzymes that promote extensive resection125. For example, CDK2 phosphorylates CtIP at Thr847 (REF. 126), and phosphorylated CtIP may form a complex with BRCA1 and MRN in a process that is important for removing the inhibitory 53BP1 from histones near the DNA ends, thereby allowing for longer resection90,91,127,128. In addition, CDK control over Dna2 in yeast and EXO1 in humans further limits the extent of end resection that can occur outside of S and G2 (REFS 118,129,130). In a recent study, DNA-PKcs phosphorylation of ATM was shown to contribute to the regulation of pathway choice between NHEJ and HR131.
Therefore, in G1 phase, NHEJ is favoured by more than 50-fold for the repair of DSBs owing to both the level of Ku and the suppression of extensive end resection by CtIP and MRN. Even in S and G2 phases, when extensive end resection can take place, the resection machinery must still overcome the presence of Ku at DNA ends either by outcompeting Ku for DNA-end binding or by processing the DNA ends to the point at which Ku binding is less favoured. The ratio of NHEJ to HR in wild-type mammalian somatic cells, even during S phase and G2 phase, is estimated to be 4:1 (REF. 132).
If Ku is absent (which is exceedingly rare in normal human tissues, as well as in neoplastic human tissues), a-EJ may be favoured over SSA in G1 phase, owing to the limited amount of resection that a-EJ involves. It remains to be determined what dictates the use of a-EJ versus SSA in S and G2 phases. However, time is likely to be a key determinant because the longer a DSB remains unrepaired, the more end processing can occur to generate longer 3′ ssDNA tails to favour SSA. Finally, quantification of the relative ratio of various pathways is complicated because the absence of one pathway results in the accumulation of substrate for other pathways to a level that may not reflect the situation in wild-type cells.
A recent study examined how resection differences in G1 versus G2 phase relate to NHEJ and end joining pathway choice but this study used doses of ionizing radiation that were sufficient to induce non-physiological levels of ~200 DSBs per cell133. The authors also fused S/G2-arrested cells to asynchronous cells, examined the G1 subset and inferred that several HR enzymes might also be involved in NHEJ. It will be interesting to discover how these findings relate to the more typical situation in cells in which far fewer DSBs are present. In any event, it highlights the complex relationship between end resection and pathway choice and reiterates that many key questions have yet to be fully answered.
Conclusions and future perspectives
Recent biochemical and genetic studies have provided clearer mechanistic insights into which NHEJ proteins are used depending on the DNA end configuration. For the joining of two blunt DNA ends, Ku and XRCC4–DNA ligase IV are sufficient, and the addition of other NHEJ proteins does not substantially improve the joining. By contrast, the joining of DSBs that require nuclease or polymerase activity is more dependent on Artemis–DNA-PKcs and the Pol X polymerases. When NHEJ is missing key components, as is the case in rare human genetic disorders (BOX 1) or in experimental animal models, a-EJ becomes increasingly important in somatic cells. The potential roles of a-EJ in meiosis, in the rare chromothripsis events in tumour cells, or in other DSB transactions in somatic cells (such as random integration of exogenous DNA), are areas for future study.
Box 1 |. Non-homologous end joining and human diseases.
Non-homologous end joining (NHEJ) is not the cause of DNA double-strand breaks (DSBs). Rather, DNA breakage occurs owing to a variety of causes, and NHEJ simply restores chromosomal structure, usually with the loss of a few nucleotides from one DNA end or both ends1. The role of NHEJ in repairing DSB sites during the formation of human chromosomal translocations has recently been discussed elsewhere111. NHEJ is the dominant pathway for the joining phase during chromosomal translocations in human cells111,135, although this may be different in murine cells136–138. The contribution of alternative end joining (a-EJ) to disease, including chromosomal translocations has not yet been proved or, at least, fully evaluated, except in cases in which there is an existing mutation in another major DSB repair pathway116.
Spontaneous mutations in NHEJ proteins are exceedingly rare in humans139. Mutations in Artemis arise in cases of consanguinity, especially in Athabascan-speaking Native Americans140. Artemis mutations can have a range of phenotypes from deficiency in antibody production to severe combined immunodeficiency (SCID) owing to deficiency in V(D)J recombination, as discussed in detail elsewhere139,141. The same range of Artemis mutations also has a corresponding range of effects on patient responses to therapeutic ionizing radiation141,142.
Rare mutations in DNA ligase IV also have a range of phenotypic severity142. Mutations in other components of the ligase IV complex, such as in X-ray repair cross-complementing protein 4 (XRCC4) and XRCC4-like factor (XLF), also have widely varying degrees of severity, including immunodeficiency, progeria-like features, microcephaly, growth retardation, autoimmunity and infections83,139,143,144. A subset of human mutations in XRCC4 can cause dwarfism without causing immunodeficiency83. Mutations in DNA-dependent protein kinase catalytic subunit (DNA-PKcs) have been described in only a few patients, and cause SCID, radiosensitivity and varying related abnormalities145–147.
Within an individual, repair in somatic cells is relevant to both cancer and ageing. But it is also interesting to consider the role of DSBs on a broader scale. On a population level and in regard to inherited genomic changes (such as in inherited disorders), meiotic changes are the relevant cell type in which DSB repair may lead to chromosomal changes. In somatic cells, NHEJ occurs 50-fold more frequently than DNA polymerase θ (Pol θ)-mediated a-EJ95. By contrast, in meiotic cells (based on the sequences at repaired breaks in inherited human disorders), Pol θ-mediated a-EJ and single-strand annealing may account for nearly as many joining events as NHEJ148,149.
Pol θ activity may be important in tumours that are deficient in homologous recombination116. Based on the template switching that has been observed in some studies150, we speculate that Pol θ activity may also be responsible for end joining in a subset of chromothripsis events that occur in a minority of genomes in human neoplasms, whereas NHEJ may be responsible for end joining of another subset of chromothripsis junctions.
Many questions remain. The first set of questions relates to DNA repair pathway choice. How much variation exists between different somatic cells or between mitotic versus meiotic cells in the use of NHEJ or in pathway choice? Ku is abundant and can thread onto dsDNA ends despite ssDNA overhangs that are longer than 20 nucleotides134. What other proteins affect repair pathway choice? Could pathway choice simply be stochastic? Could it be determined purely by the relative abundance of the proteins in the competing pathways? Could it be determined by the number of DSBs present in the cell? Which pathways contribute to chromothripsis in tumour cells and the formation of heritable chromosomal rearrangements in meiotic cells?
The second set of questions involves evaluating the contribution of Pol θ to NHEJ. Can Pol θ participate in a small subset of NHEJ events, in addition to its involvement in a-EJ? Many lymphoid chromosomal translocation junctions have templated insertions, which have been thought to be generated by Pol μ or Pol λ. Could some of these insertions actually be generated by Pol θ? Ku does not bind (or recruit) Pol θ, but does Ku obstruct Pol θ? How are non-homologous 3′ DNA tails removed before extension by Pol θ? Without such removal, the a-EJ pathway cannot proceed. Conversely, are all templated insertions generated by Pol θ, or are some generated by Pol μ or by Pol λ? The answer to this final question will determine whether some junctions form with the participation of both NHEJ proteins and this key protein of a-EJ.
Supplementary Material
V(D)J recombination.
DNA recombination process during B or T lymphocyte activation in which the antigen receptors variable domain exons are assembled from sub-exonic segments called V, D and J to ultimately generate an immunoglobulin gene or T cell receptor, respectively.
Immunoglobulin heavy chain class switch recombination.
The DNA recombination process by which the immunoglobulin heavy chain isotype is changed from producing IgM to producing IgG, IgA or IgE.
Microhomology.
One or more base pairs of complementarity at the two DNA ends of a break.
FAT domain.
FRAP (FKBP12-rapamycin-associated protein), ATM (ataxia telangiectasia mutated), TRRAP (transformation/transcription domain-associated protein) domain. A structural domain found in phosphatidylinositol 3-kinase-like kinase family members.
Pol X family polymerases.
Subfamily of DNA polymerases; based on homology it includes Pol β, Pol μ, Pol λ and terminal deoxynucleotidyltransferase (TdT).
BRCA1 C terminus.
(BRCT). Protein domain of approximately 100 aa that binds to phosphoproteins that are often involved in the DNA damage response.
DNA end breathing.
Break of the hydrogen bonds between one or more base pairs in the anti-parallel strands of the DNA duplex break.
Templated insertions.
Nucleotide additions at a double-strand break repair junction that seem to be direct or inverted repeat copies derived from either strand of either of the two DNA ends.
Chromothripsis.
Shattering of chromosomal regions followed by random repair of the DNA fragments in some human neoplasms and inherited disorders.
Acknowledgements
The authors thank R. Mosteller for comments on the manuscript. Work in the authors’ laboratory is supported by the US National Institutes of Health (NIH) (M.R.L.) and by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (15H04323 to N.A.).
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lieber MR The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lieber MR & Karanjawala ZE Ageing, repetitive genomes and DNA damage. Nat. Rev. Mol. Cell Biol 5, 69–75 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Martin GM, Smith AC, Ketterer DJ, Ogburn CE & Disteche CM Increased chromosomal aberrations in first metaphases of cells isolated from the kidneys of aged mice. Isr. J. Med. Sci 21, 296–301 (1985). [PubMed] [Google Scholar]
- 4.Meek K, Dang V & Lees-Miller SP DNA-PK: the means to justify the ends? Adv. Immunol 99, 33–58 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Chang HHY et al. Different DNA end configurations dictate which NHEJ components are most important for joining efficiency. J. Biol. Chem 291, 24377–24389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a biochemical reconstitution of NHEJ using a direct gel assay (without PCR) and all of the major NHEJ components.
- 6.Goodarzi AA et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 25, 3880–3889 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu J et al. DNA-PKcs regulates a single-stranded DNA endonuclease activity of Artemis. DNA Repair (Amst.) 9, 429–437 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang HH, Watanabe G & Lieber MR Unifying the DNA end-processing roles of the artemis nuclease: Ku-dependent Artemis resection at blunt DNA ends. J. Biol. Chem 290, 24036–24050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang HH & Lieber MR Structure-specific nuclease activities of Artemis and the Artemis: DNA-PKcs complex. Nucleic Acids Res. 44, 4991–4997 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominski Z Nucleases of the metallo-β-lactamase family and their role in DNA and RNA metabolism. Crit. Rev. Biochem. Mol. Biol 42, 67–93 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Li S et al. Evidence that the DNA endonuclease ARTEMIS also has intrinsic 5′-exonuclease activity. J. Biol. Chem 289, 7825–7834 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moshous D et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105, 177–186 (2001). [DOI] [PubMed] [Google Scholar]; This paper reports that Artemis is mutated in human SCID.
- 13.Ma Y, Pannicke U, Schwarz K & Lieber MR Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108, 781–794 (2002). [DOI] [PubMed] [Google Scholar]; This paper reports the discovery that Artemis is a ss–ds endonuclease (and a 5′ exonuclease).
- 14.Niewolik D et al. DNA-PKcs dependence of artemis endonucleolytic activity: differences between hairpins and 5′ or 3′ overhangs. J. Biol. Chem 281, 33900–33909 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Niewolik D, Peter I, Butscher C & Schwarz K Autoinhibition of the nuclease ARTEMIS is mediated by a physical interaction between its catalytic and C-terminal domains. J. Biol. Chem 292, 3351–3365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malu S et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J. Exp. Med 209, 955–963 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Ioannes P, Malu S, Cortes P & Aggarwal AK Structural basis of DNA ligase IV–Artemis interaction in nonhomologous end-joining. Cell Rep. 2, 1505–1512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riballo E et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol. Cell 16, 715–724 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Kurosawa A et al. The requirement of Artemis in double-strand break repair depends on the type of DNA damage. DNA Cell Biol. 27, 55–61 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Kanno S et al. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 26, 2094–2103 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S et al. Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3′ exonuclease and can participate in DNA end joining in a biochemical system. J. Biol. Chem 286, 36368–36377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grundy GJ et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 32, 112–125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pannunzio NR, Li S, Watanabe G & Lieber MR Nonhomologous end joining often uses microhomology: implications for alternative end joining. DNA Repair (Amst.) 17, 74–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bebenek K, Pedersen LC & Kunkel TA Structure–function studies of DNA polymerase lambda. Biochemistry 53, 2781–2792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon AF et al. Sustained active site rigidity during synthesis by human DNA polymerase μ. Nat. Struct. Mol. Biol 21, 253–260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y et al. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 16, 701–713 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Bertocci B, DeSmet A, Weill J-C & Reynaud CA Non-overlapping functions of polX family DNA polymerases, pol m, pol l, and TdT, during immunoglobulin V(D)J recombination in vivo. Immunity 25, 31–41 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Pryor JM et al. Essential role for polymerase specialization in cellular nonhomologous end joining. Proc. Natl Acad. Sci. USA 112, E4537–E4545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.NickMcElhinny SA & Ramsden DA Polymerase μ is a DNA-directed DNA/RNA polymerase. Mol. Cell. Biol 23, 2309–2315 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.NickMcElhinny SA et al. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell 19, 357–366 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Gu J et al. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 26, 1010–1023 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieber MR The polymerases for V(D)J recombination. Immunity 25, 7–9 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Daley JM, Laan RLV, Suresh A & Wilson TE DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J. Biol. Chem 280, 29030–29037 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Daley JM, Palmbos PL, Wu D & Wilson TE Nonhomologous end joining in yeast. Ann. Rev. Genet 39, 431–451 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Daley JM & Wilson TE Rejoining of DNA double-strand breaks as a function of overhang length. Mol. Cell. Biol 25, 896–906 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daley JM & Wilson TE Evidence that base stacking potential in annealed 3′ overhangs determines polymerase utilization in yeast nonhomologous end joining. DNA Repair (Amst.) 7, 67–76 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grawunder U et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388, 492–495 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Dai Y et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc. Natl Acad. Sci. USA 100, 2462–2467 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahnesorg P, Smith P & Jackson SP XLF interacts with the XRCC4–DNA ligase IV complex to promote nonhomologous end-joining. Cell 124, 301–313 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Buck D et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–299 (2006). [DOI] [PubMed] [Google Scholar]; References 39 and 40 describe the discovery of Cernunnos (also known as NEJ1 and XLF).
- 41.Brouwer I et al. Sliding sleeves of XRCC4–XLF bridge DNA and connect fragments of broken DNA. Nature 535, 566–569 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Ochi T et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347, 185–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xing M et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun 6, 6233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 42 and 43 describe the discovery of PAXX.
- 44.Roy S et al. XRCC4/XLF interaction is variably required for DNA repair and is not required for ligase IV stimulation. Mol. Cell. Biol 35, 3017–3028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tadi SK et al. PAXX is an accessory c-NHEJ factor that associates with Ku70 and has overlapping functions with XLF. Cell Rep. 17, 541–555 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Bernstein NK et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell 17, 657–670 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Ahel I et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443, 713–716 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Koch CA et al. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 23, 3874–3885 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inamdar KV et al. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem 277, 27162–27168 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Chen B et al. GC/MS methods to quantify the 2-deoxypentos-4-ulose and 3′-phosphoglycolate pathways of 4’ oxidation of 2-deoxyribose in DNA: application to DNA damage produced by gamma radiation and bleomycin. Chem. Res. Toxicol 20, 1701–1708 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou T et al. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 33, 289–297 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinden RR & Wells RD DNA structure, mutations and human genetic diseases. Curr. Opin. Biotechnol 3, 612–622 (1992). [DOI] [PubMed] [Google Scholar]
- 53.Mimori T & Hardin JA Mechanism of interaction between Ku protein and DNA. J. Biol. Chem 261, 10375–10379 (1986). [PubMed] [Google Scholar]
- 54.Costantini S, Woodbine L, Andreoli L, Jeggo PA & Vindigni A Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst.) 6, 712–722 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Sibanda BL et al. Crystal structure of an Xrcc4–DNA ligase IV complex. Nat. Struct. Biol 8, 1015–1019 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Grawunder U, Zimmer D, Kulesza P & Lieber MR Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J. Biol. Chem 273, 24708–24714 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Grawunder U, Zimmer D & Lieber MR DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr. Biol 8, 873–876 (1998). [DOI] [PubMed] [Google Scholar]
- 58.NickMcElhinny SA, Snowden CM, McCarville J & Ramsden DA Ku recruits the XRCC4–ligase IV complex to DNA ends. Mol. Cell. Biol 20, 2996–3003 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrmann G, Lindahl T & Schar PS cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J. 17, 4188–4198 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Westmoreland JW, Summers JA, Holland CL, Resnick MA & Lewis LK Blunt-ended DNA double-strand breaks induced by endonucleases PvuII and EcoRV are poor substrates for repair in Saccharomyces cerevisiae. DNA Repair (Amst.) 9, 617–626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sibanda BL, Chirgadze DY & Blundell TL Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463, 118–121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the crystal structure of DNA-PKcs at 6.6 Å.
- 62.Sibanda BL, Chirgadze DY, Ascher DB & Blundell TL DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355, 520–524 (2017). [DOI] [PubMed] [Google Scholar]; This paper describes the crystal structure of DNA-PKcs at 4.3 Å. Although no DNA is present, the 20 kDa portion of the C terminus of Ku80 is present.
- 63.West RB, Yaneva M & Lieber MR Productive and nonproductive complexes of Ku and DNA-PK at DNA termini. Mol. Cell. Biol 18, 5908–5920 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spagnolo L, Rivera-Calzada A, Pearl LH & Llorca O Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 22, 511–519 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Ma Y et al. The DNA-PKcs phosphorylation sites of human artemis. J. Biol. Chem 280, 33839–33846 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Lu H et al. A biochemically defined system for coding joint formation in human V(D)J recombination. Mol. Cell 31, 485–497 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blommers MJ et al. Effects of base sequence on the loop folding in DNA hairpins. Biochemistry 28, 7491–7498 (1989). [DOI] [PubMed] [Google Scholar]
- 68.Povirk LF Processing of damaged DNA ends for double-strand break repair in mammalian cells. ISRN Mol. Biol 2012, 345805 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henner WD, Grunberg SM & Haseltine WA Enzyme action at 3′ termini of ionizing radiation-induced DNA strand breaks. J. Biol. Chem 258, 15198–15205 (1983). [PubMed] [Google Scholar]
- 70.Henner WD, Rodriguez LO, Hecht SM & Haseltine WA γ ray induced deoxyribonucleic acid strand breaks. 3’ glycolate termini. J. Biol. Chem 258, 711–713 (1983). [PubMed] [Google Scholar]
- 71.Valerie K & Povirk LF Regulation and mechanisms of mammalian double-strand break repair. Oncogene 22, 5792–5812 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Povirk LF, Zhou T, Zhou R, Cowan MJ & Yannone SM Processing of 3′-phosphoglycolate-terminated DNA double strand breaks by Artemis nuclease. J. Biol. Chem 282, 3547–3558 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Yannone SM et al. Coordinate 5′ and 3′ endonucleolytic trimming of terminally blocked blunt DNA double-strand break ends by Artemis nuclease and DNA-dependent protein kinase. Nucleic Acids Res. 36, 3354–3365 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ochi T, Gu X & Blundell TL Structure of the catalytic region of DNA ligase IV in complex with an Artemis fragment sheds light on double-strand break repair. Structure 21, 672–679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 16, 17 and 74 describe the interaction of Artemis with DNA ligase IV.
- 75.Moon AF et al. Structural insight into the substrate specificity of DNA polymerase mu. Nat. Struc. Mol. Biol 14, 45–53 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Junop MS et al. Crystal structure of the XRCC4 DNA repair protein and implications for end joining. EMBO J. 19, 5962–5970 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andres SN, Modesti M, Tsai CJ, Chu G & Junop MS Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol. Cell 28, 1093–1101 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Tsai CJ, Kim SA & Chu G Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl Acad. Sci. USA 104, 7851–7856 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Shao Z, Jiang W, Lee BJ & Zha S PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun 8, 13816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balmus G et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 30, 2152–2157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lescale C et al. Specific roles of XRCC4 paralogs PAXX and XLF during V(D)J recombination. Cell Rep. 16, 2967–2979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar V, Alt FW & Frock RL PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc. Natl Acad. Sci. USA 113, 10619–10624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saito S, Kurosawa A & Adachi N Mutations in XRCC4 cause primordial dwarfism without causing immunodeficiency. J. Hum. Genet 61, 679–685 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Gauss GH & Lieber MR Mechanistic constraints on diversity in human V(D)J recombination. Mol. Cell. Biol 16, 258–269 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bhargava R, Onyango DO & Stark JM Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lisby M & Rothstein R Cell biology of mitotic recombination. Cold Spring Harb. Perspect. Biol 7, a016535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haber JE Mating-type gene switching in Saccharomyces cerevisiae. Annu. Rev. Genet 32, 561–599 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Moynahan ME & Jasin M Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol 11, 196–207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mimitou EP & Symington LS Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Escribano-Diaz C et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Daley JM, Niu H, Miller AS & Sung P Biochemical mechanism of DSB end resection and its regulation. DNA Repair (Amst.) 32, 66–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie A et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell 28, 1045–1057 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koole W et al. A polymerase theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun 5, 3216 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Wood RD & Doublie S DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair (Amst.) 44, 22–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper comprehensively summarizes the current information about Pol θ.
- 95.Wyatt DW et al. Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol. Cell 63, 662–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan SH, Yu AM & McVey M Dual roles for DNA polymerase θ in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 6, e1001005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kent T, Mateos-Gomez PA, Sfeir A & Pomerantz RT Polymerase θ is a robust terminal transferase that oscillates between three different mechanisms during end-joining. eLife 5, e13740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mateos-Gomez PA et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yousefzadeh MJ et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 10, e1004654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen X et al. Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 409, 39–52 (2006). [DOI] [PubMed] [Google Scholar]
- 101.Han L & Yu K Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV-deficient B cells. J. Exp. Med 205, 2745–2753 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the effect of DNA ligase IV knockout on immunoglobulin class switch recombination in a murine B cell line.
- 102.Masani S, Han L, Meek K & Yu K Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc. Natl Acad. Sci. USA 113, 1261–1266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sfeir A & Symington LS Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci 40, 701–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Makharashvili N et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell 54, 1022–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anand R, Ranjha L, Cannavo E & Cejka P Phosphorylated CtIP Functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell 64, 940–950 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Deshpande RA, Lee JH, Arora S & Paull TT Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol. Cell 64, 593–606 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Robert I, Dantzer F & Reina-San-Martin B Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med 206, 1047–1056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lange SS, Takata K & Wood RD DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu AM & McVey M Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 38, 5706–5717 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lieber MR, Hesse JE, Mizuuchi K & Gellert M Lymphoid V(D)J recombination: nucleotide insertion at signal joints as well as coding joints. Proc. Natl Acad. Sci. USA 85, 8588–8592 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lieber MR Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 16, 387–398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Murga Penas EM et al. The t(14;18)(q32;q21)/IGH-MALT1 translocation in MALT lymphomas contains templated nucleotide insertions and a major breakpoint region similar to follicular and mantle cell lymphoma. Blood 115, 2214–2219 (2010). [DOI] [PubMed] [Google Scholar]
- 113.Jaeger U et al. Follicular lymphomas BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood 95, 3520–3529 (2000). [PubMed] [Google Scholar]
- 114.Welzel N et al. Templated nucleotide addition and immunoglobulin JH-gene utilization in t(11;14) junctions: implications for the mechanism of translocation and the origin of mantle cell lymphoma. Cancer Res. 61, 1629–1636 (2001). [PubMed] [Google Scholar]; References 113 and 114 were the first to describe template insertions in lymphoid chromosomal translocations.
- 115.Pan-Hammarstrom Q et al. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J. Exp. Med 201, 189–194 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ceccaldi R, Rondinelli B & D’Andrea AD Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bhargava R, Carson CR, Lee G & Stark JM Contribution of canonical nonhomologous end joining to chromosomal rearrangements is enhanced by ATM kinase deficiency. Proc. Natl Acad. Sci. USA 114, 728–733 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Symington LS Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mimitou EP & Symington LS Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu Z, Chung WH, Shim EY, Lee SE & Ira G Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 119 and 120 describe resection mechanisms for a-EJ and SSA in yeast.
- 121.Sung P, Krejci L, Van Komen S & Sehorn MG Rad51 recombinase and recombination mediators. J. Biol. Chem 278, 42729–42732 (2003). [DOI] [PubMed] [Google Scholar]
- 122.Paques F & Haber JE Two pathways for removal of nonhomologous DNA ends during double-strand break repair in S. cerevisiae. Mol. Cell. Biol 17, 6765–6771 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pannunzio NR, Manthey GM & Bailis AM RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair (Amst.) 7, 788–800 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ira G et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jazayeri A et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol 8, 37–45 (2006). [DOI] [PubMed] [Google Scholar]
- 126.Huertas P & Jackson SP Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem 284, 9558–9565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen L, Nievera CJ, Lee AY & Wu X Cell cycle-dependent complex formation of BRCA1. CtIP. MRN is important for DNA double-strand break repair. J. Biol. Chem 283, 7713–7720 (2008). [DOI] [PubMed] [Google Scholar]
- 128.Cannavo E & Cejka P Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014). [DOI] [PubMed] [Google Scholar]
- 129.Chen X et al. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol 18, 1015–1019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tomimatsu N et al. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun 5, 3561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou Y et al. Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM. Mol. Cell 65, 91–104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Beucher A et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 28, 3413–3427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biehs R et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 65, 671–684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Falzon M, Fewell J & Kuff EL EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J. Biol. Chem 268, 10546–10552 (1993). [PubMed] [Google Scholar]
- 135.Ghezraoui H et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 55, 829–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boboila C et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl Acad. Sci. USA 107, 3034–3039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the clearest example of a-EJ in mice with the absence of Ku70 and ligase IV.
- 137.Gostissa M, Alt FW & Chiarle R Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu. Rev. Immunol 29, 319–350 (2011). [DOI] [PubMed] [Google Scholar]
- 138.Chiarle R et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Villartay JP Congenital defects in V(D)J recombination. Br. Med. Bull 114, 157–167 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Li L et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J. Immunol 168, 6323–6329 (2002). [DOI] [PubMed] [Google Scholar]
- 141.Volk T et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum. Mol. Genet 24, 7361–7372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Woodbine L, Gennery AR & Jeggo PA The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst.) 16, 84–96 (2014). [DOI] [PubMed] [Google Scholar]
- 143.de Villartay JP When natural mutants do not fit our expectations: the intriguing case of patients with XRCC4 mutations revealed by whole-exome sequencing. EMBO Mol. Med 7, 862–864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jspeert I, et al H. XLF deficiency results in reduced N-nucleotide addition during V(D)J recombination. Blood 128, 650–659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Burg M.v.d. et al. A DNA-PKcs mutation in a radiosensitive T-B− SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest 119, 91–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Woodbine L et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Invest 123, 2969–2980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mathieu AL et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J. Allergy Clin. Immunol 135, 1578–1588.e5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carvalho CM & Lupski JR Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet 17, 224–238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abyzov A et al. Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nat. Commun 6, 7256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stephens PJ et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iles N, Rulten S, El-Khamisy SF & Caldecott KW APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol 27, 3793–3803 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Eustermann S et al. Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose). Nat. Struct. Mol. Biol 17, 241–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li GY et al. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc. Natl Acad. Sci. USA 107, 9129–9134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mehrotra PV et al. DNA repair factor APLF is a histone chaperone. Mol. Cell 41, 46–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.