

Abstract
Hereditary angioedema (HAE) is an uncommon genetic disorder characterized by recurrent episodes of edema involving subcutaneous tissue and submucosa. The pathogenesis of HAE reflects an intricate coordinated regulation of components of complement, kinin and hemostatic pathway. Till date, mutations in 4 different genes have been identified to cause HAE which includes serine protease inhibitor G1 (SERPING1), factor XII (F12), plasminogen (PLG) and angiopoietin 1 (ANGPT 1). These mutations lead to increased bradykinin 2 receptor mediated signalling via increased production of bradykinin except mutations in ANGPT1 gene that disturbs the cytoskeletal assembly of vascular endothelial cells. In this review we aim to summarize the recent advances in the pathogenesis and genetics of HAE. We also provide an overview of possible future prospects in the identification of new genetic defects in HAE.
Keywords: Angiopoietin 1, C1 inhibitor, Factor XII, Genetics, Hereditary angioedema, Plasminogen
Introduction
Hereditary angioedema (HAE) is a genetic disorder that predisposes an individual to develop vasogenic edema. Prevalence of HAE has been reported to be 1 in 10,000 to 1 in 150,000.1 The pathogenesis of HAE involves accumulation of extravascular fluid in various tissues via a non-inflammatory and non-allergic mechanism. Clinical manifestations include abrupt onset swelling around eyes, face and extremities; pain abdomen (as a result of bowel edema) and laryngeal edema leading to hoarseness of voice, breathing difficulty and occasionally death.2, 3 Awareness and recognition of this disease is important as HAE is often misdiagnosed as allergic angioedema or acute abdomen (especially acute appendicitis).4 This may often lead to inappropriate use of antihistamines, corticosteroids, adrenaline and sometimes even surgical interventions.4
Identification of defects in SERPING1 gene and several other recently identified genes have added significantly to the understanding of pathophysiology of HAE. This disease was initially believed to be a disorder of complement pathway caused by deficiency of C1-inhibitor (C1-INH). However, identification of novel genetic defects (other than SERPING1 gene) has expanded the spectrum of this disease beyond complement pathway. Genetic defects in some of the intermediate pathway components that may be possible cause HAE are yet to be identified. These advances may bring new therapeutic modalities for the management of HAE. In this review, we aim to update about current understanding in the pathogenesis and recent advances in the genetics of HAE.
Pathophysiology of HAE
Elevated serum levels of bradykinin in the blood are responsible for clinical manifestations seen in patients with HAE. Bradykinin is generated in the body via a complex interaction of various molecules involved in kinin, complement, hemostatic and fibrinolytic pathways (Fig. 1).5 Factor XII is activated at first by negatively charged surfaces. Activated FXII subsequently activates prekallikrein to kallikrein (prekallikrein may also be activated by enzyme prolylcarboxypeptidase). Kallikrein is capable of directly converting plasminogen to plasmin and also with the help of urokinase type plasminogen activator. Plasmin and kallikrein in turn augment Factor XIIa generation thereby creating an auto-activation loop. Kallikrein generates bradykinin, a nonapeptide, from kininogen (high molecular weight form (HMWK)).6 Kallidin, a decapeptide with an additional lysine residue at the amino-terminal end of bradykinin, is generated preferentially from low molecular weight kininogen (LMWK).7 Bradykinin can also be generated from kallidin by the action of aminopeptidases. Kininase I generates active metabolites from kallidin and bradykinin, whereas, angiotensin-converting enzyme, ACE (also known as Kininase II), and aminopeptidases both inactivate bradykinin and kallidin (Fig. 1).8
Figure 1.

A simplified summary of the kinin metabolism and the mutual interactions between the kinin, complement, hemostatic and fibrinolytic pathways. (The blue arrows represent the augmentation of the enzymatic reactions shown in brown arrows). The sites of action of C1 esterase inhibitor (C1-INH) are represented in green chevron shapes). Abbreviations: FXII – Factor XII, a as a suffix indicates the active form; pK – prekallikrien; K (in bold) – kallikrien; HMWK – high molecular weight kininogen; LMWK – low molecular weight kininogen; BK – bradykinin; KD – kallidin (lysine-bradykinin: K-BK), BK2-9 – bradykinin with its first amino acid cleaved off; BK1-7 – bradykinin with its last two amino acids cleaved off; Des-R BK – bradykinin with its ninth arginine residue cleaved off; meaning of subscripts in case of kallidin (K-BK) is the same as that for bradykinin derivatives; ACE (also called kininase II) – angiotensinogen converting enzyme, AP – aminopeptidase, K I – kininase I.
Most cases of hereditary angioedema result from mutations in the gene encoding for C1 esterase inhibitor (C1-INH) protein. C1-INH belongs to serine protease inhibitor (serpin) protein superfamily. This superfamily also includes other proteins such as α1-antitrypsin, antithrombin III, α1-antichymotrypsin, protein C inhibitor and heparin cofactor II. Genetic defects in these proteins have also been referred to as serpinopathies. As defects in these genes lead to conformational change in protein, its dimerization and loss of function, hence these defects may also be labeled as conformational disorders.9, 10 Mutation in SERPING1 gene leads to either decreased levels or functionally abnormal C1-INH protein. This results in loss of regulation of factor XIIa and kallikrein enzymes thereby leading to excess production of bradykinin. Bradykinin is the key molecule that mediates all clinical manifestations of HAE by acting on BR2 receptors leading to increased vascular permeability and edema.11 Other than C1-INH, defects in many other molecules involved in this pathway may lead to excess production of bradykinin and similar clinical manifestations.
Genetics of HAE
Mutations in the SERPING1 gene are responsible for majority of cases of hereditary angioedema. In addition, various genetic defects have also been found to result in HAE phenotype (Fig. 2).
Figure 2.
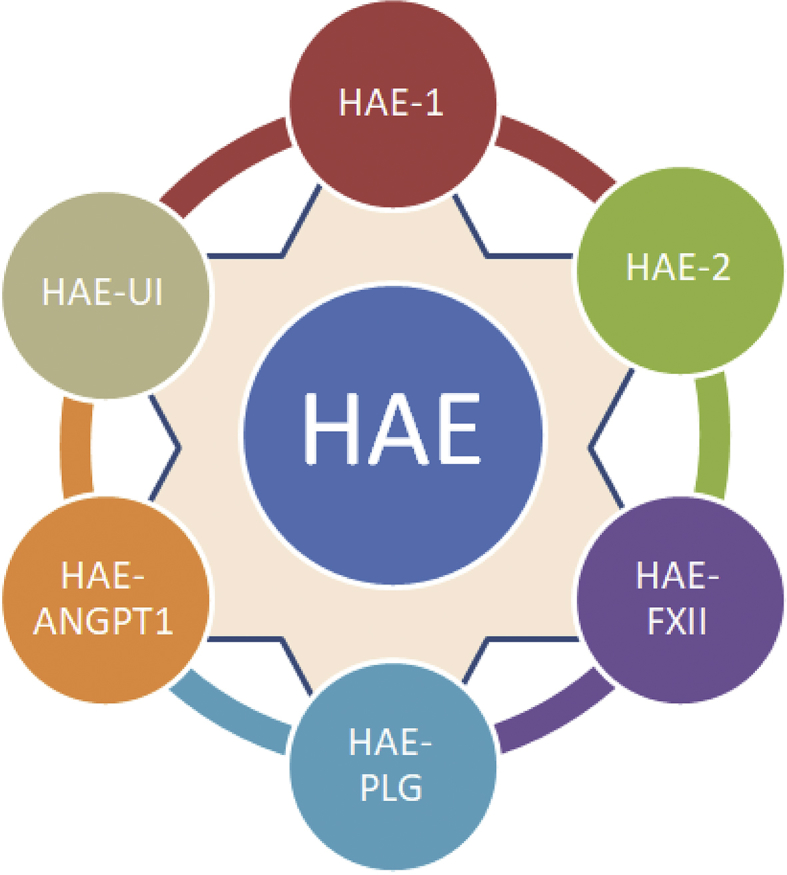
Summary of the genetic defects leading to the phenotype of hereditary angioedema. HAE-1 (Type 1) is due to mutations leading to a quantitative defect of C1 esterase, whereas HAE-2 (Type 2) represents qualitative defects of C1-INH function. HAE-FXII and HAE-PLG pertains to defects in factor XII and plasminogen respectively. HAE-ANGPT1 corresponds to the recently described defects in the angiopoietin-1 gene leading to a hereditary angioedema like phenotype, whereas HAE-UI represents still unidentified mutations.
SERPING1 gene mutations (that lead to type 1 and type 2 HAE), constitute more than 95% of all cases of HAE. Clinical manifestations of CI-INH deficiency HAE are highly variable and depends upon the location of mutation in SERPING1 gene. This gene is characterized by a remarkable allelic heterogeneity and approximately 450 different mutations have been reported as per the Human Gene Mutation Database (HGMD)12 and a similar database specifically dedicated for HAE (HAEdb, hae.enzim.hu).13 Mutations in SERPING1 gene may be classified as shown in Fig. 3. This gene is located on chromosome 11 band position 11q12-q13.1 and has an unusual promoter devoid of TATA box. It rather contains TdT-like initiator and poly purine pyrimindine tract.14 SERPING1 gene consists of 8 exons and 7 introns. Various exonic mutations and mutations at intron/exon junctions have been identified in this gene and these mutations are transmitted with an autosomal dominant mode of inheritance. However, approximately a quarter of all C1-INH deficiency HAE patients are sporadic in nature (i.e. they are caused by de-novo mutations in SERPING1 gene).15 Since the availability of various mutations reported in SERPING1 gene, it has become easier to predict the structural and functional attributes of this gene thereby explaining the phenomena of allelic heterogeneity.16, 17 SERPING1 gene is an example of mutagenic liability and various theories have been postulated for the same: 1) SERPING1 gene is located near centromeric region; 2) Presence of 17 Alu repeats on intron 7 of the gene makes it a hotspot for non-homologous recombination and causes partial deletions or duplication in the gene,18 characterized by absence of protein product accounting for approximately 15–20% of all cases of CI-INH deficiency HAE; 3) Presence of CpG sites in the coding part of the gene predisposes it for mutations due to spontaneous deamination. The 466th residue of gene encodes for the arginine amino acid (CGC) that forms the reactive center of CI-INH and CpG is a natural target for recurrent amino acid substitution.19
Figure 3.

Various mutations reported in SERPING1 gene.
Missense mutations constitute approximately 30–40% of all mutations in SERPING1 gene.20, 21 Various nonsense and indel mutations lead to addition of premature stop codon and frame-shift mutations that eliminate protein synthesis via nonsense mediated mRNA decay.22
Single Nucleotide Polymorphisms (SNPs) in SERPING1 gene have also been found to be associated with some non-HAE disease conditions; e.g. rs4926 [c.1438G>A, p.V480M] polymorphism is a missense variant in the coding region and has been found to be associated with staphylococcal carriage of the anterior nares.23 In Caucasian populations, presence of intron 6 SNP (rs2511989) in SERPING1 gene has been found to be associated with age related macular degeneration.24
Factor XII gene
In the year 2000, Bork et al described another type of HAE with a quantitatively and qualitatively normal C1-INH (labeled as nlC1-INH-HAE). In this rare type of HAE, clinical manifestations were found to be similar to classical type 1 and 2 HAE but no mutation was detected in the SERPING1 gene. The genetic defect for this type of HAE was discovered in the year 2006 when two missense mutations (p.Thr309Lys and p.Thr309Arg) in F12 gene were detected for the first time in a German family (Table 1).25, 26
Table 1.
Summary of the mutations described in F12, plasminogen (PLG) and angiopoietin (ANGPT1) genes resulting in HAE.
| S no | Gene name | Variant observed in HAE |
|---|---|---|
| 1. | F12 | Exon 9 missense variants (p.Thr328Lys) and (p.Thr328Arg) Exon 9/intron 9 boundary large deletion of 72 bp (c.971_1018 + 24del72) duplication of 18 bp (c.892_909dup) |
| 2. | Plasminogen (PLG) | Exon 9 missense variant (c.988 A>G), (p.Lys330Glu) |
| 3. | Angiopoietin (ANGPT1) | Missense mutation c.807G>T, p.Ala119Ser |
Over the next few years after discovery of mutations in F12 gene, researchers found out that these mutations lead to increased production of activated Factor XII (Factor XIIa) via plasmin.27, 28 This also provides an insight into the effect of anti-fibrinolytic medications in this type of HAE. Around 300 patients with F12 gene mutations have been reported till date (OMIM#610619). F12 gene has a size of 12 kb and comprises of 14 exons.29 The promoter of the F12 gene is similar but not identical to the promoter of the estrogen responsive elements (EREs) gene. Because of this similarity, estrogen also enhances the concentration of FXII in the plasma and may produce manifestations similar to this type of HAE.30 F12 gene mutation associated with HAE is a gain of function mutation and is transmitted in an autosomal dominant mode of inheritance.31 Four most common pathogenic mutations in this gene have been identified so far. These include 2 missense variants in exon 9 (p.Thr328Lys and p.Thr328Arg); 72 bp deletion (c.971_1018 + 24del72) in two unrelated families from Turkey; mutation at the exon 9/intron 9 junction22, 32 and 18 bp duplication (c.892_909dup) in a Hungarian family causing repetition of 6 amino acids (p.298–303) at the same locus.33 The clinical phenotype of HAE caused by mutations in F12 gene is similar to C1INH deficiency HAE except that there is higher female predominance, more aggravation during pregnancy and more estrogen dependency.34 Complement C4 and C1-INH activity is often normal except occasionally during the attack. Assessment of activity of coagulation factor XII is not reliable for diagnosis.
Plasminogen gene
Plasminogen is inactive precursor protein of the enzyme plasmin. Plasmin plays its role in bradykinin production via activation of factor XII. A newly identified missense mutation in exon 9 of plasminogen (PLG) gene, c.988A>G (or named as c.1100A>G depending upon the assembly used), describes glutamic acid replacing lysine at position 330 (p.Lys330Glu) (or named as p.Lys311Glu depending on the numbering scheme used).35, 36 Interestingly, the same mutation has been described by various groups in about 25 families from different countries with affected individuals numbering more than 100 (Table 1).37 The mutation leads to change in the kringle 3 domain which alters the structure of the wild-type protein. The mutant protein leads to the increased production of the bradykinin thereby resulting in HAE. Clinical profile of patients with plasminogen mutation is different from C1INH deficiency HAE. Patients with plasminogen gene mutation present usually in adulthood as compared to pediatric onset in majority of patients with C1-INH deficiency HAE. In addition, these patients tend to have higher chances of head and neck swellings (tongue, face, laryngeal edema) and relatively less chances of extremity edema or swelling of the genitals. ACE inhibitors and Angiotensin 1 receptor blockers tend to precipitate the attacks. Icatibant, tranexamic acid seem to be the most effective drugs.35, 38, 39, 40
Angiopoietin 1 gene
A novel mutation was identified in angiopoietin 1 (ANGPT1) gene using whole exome sequencing (WES) in patients with normal C1-INH HAE (Table 1).41 This was an unusual mutation as this gene has not been shown to be associated with hemostatic system or kinin pathway. It has also been shown that p.Ala119Ser mutation reduces the plasma levels of ANGPT1 protein and further hampers its ability to form multimers. Deficiency of ANGPT1 protein leads to reduced ability of mutant protein to bind to its cognate receptor tunica interna endothelial cell kinase 2 (TIE2). This signaling cascade is involved instabilizing vasculature and diminishing vascular permeability. ANGPT2 protein, on the other hand, antagonizes ANGPT1 protein leading to enhanced vascular permeability. The ratio of ANGPT1/ANGPT2 was found to be decreased in patients with p.Ala119Ser mutation.42 A simplified summary of the pathogenic mechanism involved in HAE caused by ANGPT1 gene mutation is shown in Fig. 4.43 Discovery of novel pathways in the pathogenesis of HAE have remarkably opened new frontiers in understanding and managing this disease.
Figure 4.

Counteracting influences of angiopoietin 1 and bradykinin on vascular endothelial cells. Angiopoietin 1 acting via TIE 2 receptor reinforces the cell cytoskeletal arrangements and decreases the vascular permeability. While, bradykinin disrupts this orderly arrangement leading to increased vascular permeability.
Laboratory evaluation of patients with a clinical suspicion of HAE
The initial laboratory workup for hereditary angioedema is shown in Fig. 5. For genetic testing of HAE, molecular analysis is to be carried out for the genes associated with HAE. In the classical form of type 1 and type II HAE SERPING1 gene is the prominent target. Initially all the eight exons of SERPING1 gene with intronic/exonic junctions are amplified using oligonucleotides. Using MLPA kit, probe amplification depending on Multiplex Ligation can be done44, 45 when all the eight exons data of SERPING1 gene is clear than one might expect some large gene rearrangements like insertion/deletions. For other genes like F12, plasminogen and angiopoietin diseases associated exons are amplified with oligonucleotides and checked for the variants using Sanger sequencing. In addition, next generation sequencing (NGS) panel including all genes known to cause HAE and several other genes that may potentially lead to HAE may be utilized for diagnosis with an additional advantage of identifying newer genetic defects.
Figure 5.

Proposed diagnostic algorithm for evaluation of a suspected case of hereditary angioedema (HAE with normal C1 inhibitor function requires genetic analysis for diagnosis).
Treatment
Treatment of HAE has evolved with advancement in understanding the pathogenesis of this disease and is either directed towards replacing the defective protein or blocking various molecules involved in the production of bradykinin or the bradykinin receptor (Fig. 6). In addition to various preparations of C1-INH analogues, other therapeutic options available include icatibant, a parenterally administered bradykinin receptor type 2 antagonist; ecallantide, a parenterally administered kallikrein inhibitor46, 47; Landelumab, a monoclonal antibody targeting kallikrein48 and a novel molecule BCX7353, an oral kallikrein inhibitor.49 Other management options include fresh frozen plasma (FFP)/single donor plasma (SDP) [aimed at replacing the defective or insufficient C1-INH]; attenuated androgens and tranexamic acid. Androgens act by augmenting bradykinin inactivation via aminopeptidase enzyme and by increasing the production of C1-INH.50, 51 Tranexamic acid limits the production of plasmin by blocking the cleavage of its proenzyme plasminogen (Table 2).52, 53
Figure 6.

Therapeutic modalities employed in the management of hereditary angioedema. (The blue arrows represent the augmentation of the enzymatic reactions shown in brown arrows; the green arrows with a plus sign indicate therapies for replacement or augmentation of the target product, while the red arrows with a minus sign indicate inhibitions. The sites of action of C1 esterase inhibitor (C1-INH) are represented in green chevron shapes). Abbreviations: FXII – Factor XII, a as a suffix indicates the active form; pK – prekallikrien; K (in bold) – kallikrien; HMWK – high molecular weight kininogen; LMWK – low molecular weight kininogen; BK – bradykinin; KD – kallidin (lysine-bradykinin: K-BK), BK2-9 – bradykinin with its first amino acid cleaved off; BK1-7 – bradykinin with its last two amino acids cleaved off; ACE (also called kininase II) – angiotensinogen converting enzyme, AP – aminopeptidase, K I – kininase I, TA – tranexamic acid, EACA – epsilon amino caproic acid.
Table 2.
Summary of effective treatment options for hereditary angioedema.
| Therapeutic options | Uses | Suggested doses | Remarks |
|---|---|---|---|
| Danazol Stanozolol Oxandrolone (Weak androgens) |
Short term & Long term prophylaxis |
Danazol - STP: 2.5–10 mg/kg/day (maximum 600 mg/day). LTP: 2.5–5 mg/kg/day (maximum 200 mg/day). | Easily available. Undesirable virilising effect, weight gain, acne, headaches, menstrual irregularities, depression. |
| Tranexamic acid Epsilon aminocaproic acid | Short term & Long term prophylaxis |
Tranexamic acid: 20–50 mg/kg/day in 2 or 3 divided doses (maximum 3–6 g/day). | Can be used in pregnancy if benefits outweigh risk (category B) |
| FFP (fresh frozen plasma)/ SDP (solvent detergent plasma) |
Short term prophylaxis Acute treatment |
10 ml/kg/dose | Easily available, inexpensive. Concern of infections with regular use. |
| C1 esterase inhibitors | pdC1-INHBe for short term prophylaxis, acute treatment | pdC1-INHBe 15–30 units/kg/dose IV. | Expensive, currently unavailable in many Asian countries. One unit parallels the amount of C1INH in 1 ml of plasma of a normal individual. |
| pdC1-INHCi for long term prophylaxis, acute treatment | pdC1-INHCi 10–20 units/kg/dose once or twice weekly (initial maximum dose 1000 units). | ||
| rhC1-INH for acute treatment, long term prophylaxis | rhC1-INH 50 units/kg/dose and is given by intravenous injection. | ||
| Subcutaneous preparations are also available. | |||
| Ecallantide | Acute treatment | 30 mg/dose subcutaneous injection | Expensive, risk of anaphylaxis. Not available in many Asian countries. |
| Icatibant | Acute treatment | 30 mg/dose subcutaneous injection (maximum three doses/day) | Expensive. Not available in many Asian countries. Local reactions. |
| Lanadelumab | Long term prophylaxis | Dose range 150–300 mg every 2–4 weeks. | Pediatric dosing yet to be established. |
| BCX7353 | Long term prophylaxis | Minimum effective dose used: 125 mg/day |
Future perspectives
HAE is a heterogeneous disease with complex pathophysiology that has been studied extensively. Despite detailed research and identification of novel defects, a proportion of patients with HAE are still labeled as HAE-Unidentified (HAE-UI) wherein the genetic defect has not yet been identified. There are several molecules involved in the pathway for production of bradykinin and mutations in genes encoding these molecules may be potential genetic etiology for HAE-UI. These could include activating mutations in Kallikrein gene; activating mutations in Bradykinin gene or its receptor; activation mutations in Kininase 1 gene and loss of function mutations in gene encoding aminopeptidase and ACE enzymes. There is a possibility that mutations in these and other related genes could be potential modifiers in the clinical phenotype of patients with known genetic causes of HAE. A recent study analyzed polymorphisms in 15 such genes and their effect on the clinical phenotype of patients with C1-INH HAE/FXII-HAE. Amongst several variants that were identified in this study, 5 were classified as probably pathogenic. These included p.Y244C, p.G354R, p.T916M in the ACE gene; p.C548Y in KLKB1 (kallikrein) gene and p.D287N in NOS3 (nitric oxide synthase) gene. However, the effect of these mutations on the final clinical phenotype was found to be rather complex. In patients with c.-22-2A>G mutations in C1-INH gene, p.Y244C resulted in infrequent attacks as compared to p.T916M. However, in patients with c.889G>A mutation in C1-INH gene, the clinical phenotype was very different even with the same variant p.G354R in the ACE gene.54 Identification of these novel polymorphisms in several genes leading to modification in the clinical phenotype of HAE is a relatively recent phenomenon. This needs more research work and needs to be studied in several other populations.
In addition, the recent discovery of ANGPT1 gene mutation in an Italian family with HAE-UI phenotype has expanded our understanding about this disease beyond the complement, kinin cascade. This discovery has also raised several questions for researchers that in the process of finding out novel genes for HAE, they not only need to work on above mentioned potential genes involved in pathway of bradykinin synthesis but several other pathways as well. Increasing number of novel mutations in the ANGPT1 gene are now being reported to be associated with the HAE phenotype [c.23C>T (p.A8V) and c.1110G>C (p.Q370H)].55
Apart from this, several novel mutations are likely to be discovered in populations wherein the genetic etiology of HAE has not been evaluated till date. In our experience, we analyzed our cohort of pediatric HAE patients and found novel mutations in SERPING1 gene from 3 families (unreported data). Discovery of new genetic etiologies is likely to bring new therapeutic options in the management of patients with HAE.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Peer review under responsibility of Chongqing Medical University.
References
- 1.Ghazi A., Grant J.A. Hereditary angioedema: epidemiology, management, and role of icatibant. Biologics. 2013;7:103–113. doi: 10.2147/BTT.S27566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jalaj S., Scolapio J.S. Gastrointestinal manifestations, diagnosis, and management of hereditary angioedema. J Clin Gastroenterol. 2013;47:817–823. doi: 10.1097/MCG.0b013e31829e7edf. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y.Y., Zhi Y.X., Liu R.L. Upper airway edema in 43 patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(6):539–544. doi: 10.1016/j.anai.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Zanichelli A., Longhurst H.J., Maurer M. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016;117(4):394–398. doi: 10.1016/j.anai.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Zuraw B.L., Christiansen S.C. HAE pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol. 2016;51(2):216–229. doi: 10.1007/s12016-016-8561-8. [DOI] [PubMed] [Google Scholar]
- 6.Levi M., Cohn D.M., Zeerleder S. Hereditary angioedema: linking complement regulation to the coagulation system. Res Pract Thromb Haemost. 2019;3:38–43. doi: 10.1002/rth2.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pathak M., Wong S.S., Dreveny I. Structure of plasma and tissue kallikreins. Thromb Haemost. 2013;110:423–433. doi: 10.1160/TH12-11-0840. [DOI] [PubMed] [Google Scholar]
- 8.Maurer M., Bader M., Bas M. New topics in bradykinin research. Allergy. 2011;66:1397–1406. doi: 10.1111/j.1398-9995.2011.02686.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y., Swulius M.T., Moremen K.W. Elucidation of the molecular logic by which misfolded alpha-1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci U S A. 2003;100:8229–8234. doi: 10.1073/pnas.1430537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haslund D., Ryø L.B., Majidi S.S. Dominant-negative SERPING1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J Clin Invest. 2019;129(1):388–405. doi: 10.1172/JCI98869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan A.P., Joseph K. Complement, kinins, and hereditary angioedema: mechanisms of plasma instability when C1 Inhibitor is absent. Clin Rev Allergy Immunol. 2016;51(2):207–215. doi: 10.1007/s12016-016-8555-6. [DOI] [PubMed] [Google Scholar]
- 12.Stenson P.D., Mort M., Ball E.V. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalmár L., Hegedüs T., Farkas H. HAEdb: a novel interactive, locus-specific mutation database for the C1 inhibitor gene. Hum Mutat. 2005;25:1–5. doi: 10.1002/humu.20112. [DOI] [PubMed] [Google Scholar]
- 14.Davis A.E., Whitehead A.S., Harrison R.A. Human inhibitor of the first component of complement, C1: characterization of cDNA clones and localization of the gene to chromosome 11. Proc Natl Acad Sci USA. 1986;83:3161–3165. doi: 10.1073/pnas.83.10.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappalardo E., Cicardi M., Duponchel C. Frequent de novo mutations and exon deletions in the C1 inhibitor gene of patients with angioedema. J Allergy Clin Immunol. 2000;106:1147–1154. doi: 10.1067/mai.2000.110471. [DOI] [PubMed] [Google Scholar]
- 16.Pappalardo E., Zingale L.C., Terlizzi A. Mechanisms of C1-inhibitor deficiency. Immunobiology. 2002;205:542–551. doi: 10.1078/0171-2985-00153. [DOI] [PubMed] [Google Scholar]
- 17.Cicardi M., Zingale L., Zanichelli A. C1 inhibitor: molecular and clinical aspects. Semin Immunopathol. 2005;27:286–298. doi: 10.1007/s00281-005-0001-4. [DOI] [PubMed] [Google Scholar]
- 18.Stoppa-Lyonnet D., Carter P.E., Meo T. Clusters of intragenic Alu repeats predispose the human C1 inhibitor locus to deleterious rearrangements. Proc Natl Acad Sci USA. 1990;87:1551–1555. doi: 10.1073/pnas.87.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skriver K., Radziejewska E., Silbermann J.A. CpG mutations in the reactive site of human C1 inhibitor. J Biol Chem. 1989;264:3066–3071. [PubMed] [Google Scholar]
- 20.Pappalardo E., Caccia S., Suffritti C. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol. 2008;45:3536–3544. doi: 10.1016/j.molimm.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Freiberger T., Kolárová L., Mejstrík P. Five novel mutations in the C1 inhibitor gene (C1NH) leading to a premature stop codon in patients with type I hereditary angioedema. Hum Mutat. 2002;19:461. doi: 10.1002/humu.9029. [DOI] [PubMed] [Google Scholar]
- 22.Amrani N., Sachs M.S., Jacobsen A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- 23.Emonts M., de Jongh C.E., Houwing-Duistermaat J.J. Association between nasal carriage of Staphylococcus aureus and the human complement cascade activator serine protease C1 inhibitor (C1INH) valine vs. methionine polymorphism at amino acid position 480. Immunol Med Microbiol. 2007;50:330–332. doi: 10.1111/j.1574-695X.2007.00250.x. [DOI] [PubMed] [Google Scholar]
- 24.Ennis S., Jomary C., Mullins R. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case-control study. Lancet. 2008;372:1828–1834. doi: 10.1016/S0140-6736(08)61348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dewald G., Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286–1289. doi: 10.1016/j.bbrc.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 26.Bork K., Gul D., Hardt J., Dewald G. Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. Am J Med. 2007;120:987–992. doi: 10.1016/j.amjmed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 27.Cichon S., Martin L., Hennies H.C. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. 2006;79:1098–1104. doi: 10.1086/509899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Maat S., Bjorkqvist J., Suffritti C. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. 2016;138 doi: 10.1016/j.jaci.2016.02.021. 1414–1412. [DOI] [PubMed] [Google Scholar]
- 29.Royle N.J., Nigli M., Cool D. Structural gene encoding human factor XII is located at 5q33-qter. Somat Cell Mol Genet. 1988;14:217–221. doi: 10.1007/BF01534407. [DOI] [PubMed] [Google Scholar]
- 30.Schloesser M., Zeerleder S., Lutze G. Mutations in the human factor XII gene. Blood. 1997;90:3967–3977. [PubMed] [Google Scholar]
- 31.Bork K., Wulff K., Meinke P. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor. Clin Immunol. 2011;141:31–35. doi: 10.1016/j.clim.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Bork K., Wulff K., Hardt J., Witzke G., Lohse P. Characterization of a partial exon 9/intron 9 deletion in the coagulation factor XII gene (F12) detected in two Turkish families with hereditary angioedema and normal C1 inhibitor. Haemophilia. 2014;20:e372–e375. doi: 10.1111/hae.12519. [DOI] [PubMed] [Google Scholar]
- 33.Aulak K.S., Davis A.E., Donaldson V.H. Chymotrypsin inhibitory activity of normal C1-inhibitor and a P1 Arg to His mutant: evidence for the presence of overlapping reactive centers. Protein Sci. 1993;2:727–732. doi: 10.1002/pro.5560020504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bork K., Wulff K., Witzke G. Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy. 2015;70(8):1004–1012. doi: 10.1111/all.12648. [DOI] [PubMed] [Google Scholar]
- 35.Bork K., Wulff K., Steinmüller-Magin L. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2017;00:1–9. doi: 10.1111/all.13270. [DOI] [PubMed] [Google Scholar]
- 36.Dewald G. A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2018;498(1):193–198. doi: 10.1016/j.bbrc.2017.12.060. [DOI] [PubMed] [Google Scholar]
- 37.Recke A., Massalme E.G., Jappe U. Identification of the recently described plasminogen gene mutation p.Lys330Glu in a family from Northern Germany with hereditary angioedema. Clin Transl Allergy. 2019;9:9. doi: 10.1186/s13601-019-0247-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belbézier A., Hardy G., Marlu R. Plasminogen gene mutation with normal C1 inhibitor hereditary angioedema: three additional French families. Allergy. 2018;73:2237–2239. doi: 10.1111/all.13543. [DOI] [PubMed] [Google Scholar]
- 39.Yakushiji H., Hashimura C., Fukuoka K. A missense mutation of the plasminogen gene in hereditary angioedema with normal C1 inhibitor in Japan. Allergy. 2018;73:2244–2247. doi: 10.1111/all.13550. [DOI] [PubMed] [Google Scholar]
- 40.Germenis A.E., Loules G., Zamanakou M. On the pathogenicity of the plasminogen K330E mutation for hereditary angioedema. Allergy. 2018;73:1751–1753. doi: 10.1111/all.13324. [DOI] [PubMed] [Google Scholar]
- 41.Bafunno V., Firinu D., D'Apolito M. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141:1009–1117. doi: 10.1016/j.jaci.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 42.Zuraw B.L. Hereditary angioedema with normal C1 inhibitor: Four types and counting. J Allergy Clin Immunol. 2018;141(3):884–885. doi: 10.1016/j.jaci.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 43.d'Apolito M., Santacroce R., Colia A.L. Angiopoietin-1 haploinsufficiency affects the endothelial barrier and causes hereditary angioedema. Clin Exp Allergy. 2019;00:1–10. doi: 10.1111/cea.13349. [DOI] [PubMed] [Google Scholar]
- 44.Roche O., Blanch A., Caballero T. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94:498–503. doi: 10.1016/S1081-1206(10)61121-0. [DOI] [PubMed] [Google Scholar]
- 45.Schouten J.P., McElgunn C.J., Waaijer R. Relative quantifications of 40 nucleic acid sequences by multiplex ligation dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farkas H., Martinez-Saguer I., Bork K. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. 2017;72:300–313. doi: 10.1111/all.13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maurer M., Magerl M., Ansotegui I. The international WAO/EAACI guideline for the management of hereditary angioedema – the 2017 revision and update. Allergy. 2018;73(8):1575–1596. doi: 10.1111/all.13384. [DOI] [PubMed] [Google Scholar]
- 48.Barmettler S., Li Y., Banerji A. New and evolving therapies for hereditary angioedema. Allergy Asthma Proc. 2019;40:7–13. doi: 10.2500/aap.2019.40.4195. [DOI] [PubMed] [Google Scholar]
- 49.Aygören-Pürsün E., Bygum A., Grivcheva-Panovska V. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N Engl J Med. 2018;379:352–362. doi: 10.1056/NEJMoa1716995. [DOI] [PubMed] [Google Scholar]
- 50.Drouet C., Desormeaux A., Robillard J. Metallopeptidase activities in hereditary angioedema: effect of androgen prophylaxis on plasma aminopeptidase P. J Allergy Clin Immunol. 2008;121(2):429–433. doi: 10.1016/j.jaci.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pappalardo E., Zingale L.C., Cicardi M. Increased expression of C1-inhibitor mRNA in patients with hereditary angioedema treated with Danazol. Immunol Lett. 2003;86(3):271–276. doi: 10.1016/s0165-2478(03)00029-4. [DOI] [PubMed] [Google Scholar]
- 52.Mannucci P.M., Levi M. Prevention and treatment of major blood loss. N Engl J Med. 2007;356(22):2301–2311. doi: 10.1056/NEJMra067742. [DOI] [PubMed] [Google Scholar]
- 53.Zuraw B.L. Clinical practice. Hereditary angioedema. N Engl J Med. 2008;359(10):1027–1036. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 54.Veronez C.L., Aabom A., Martin R.P. Genetic variation of kallikrein-kinin system and related genes in patients with hereditary angioedema. Front Med (Lausanne) 2019;6:28. doi: 10.3389/fmed.2019.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cagini N., Veronez C.L., Azevedo B.F. In silico analysis of alterations in ANGPT1 gene supports a new pathway responsible to mediate hereditary angioedema in Brazilian patients with no mutations in SERPING1 and F12 genes. J Allergy Clin Immunol. 2018;141(2):AB46. [Google Scholar]