

ABSTRACT
In many eukaryotes, the small GTPase Rheb functions as a switch to toggle activity of TOR complex 1 (TORC1) between anabolism and catabolism, thus controlling lifespan, development and autophagy. Our CRISPR-generated, fluorescently tagged endogenous Caenorhabditis elegans RHEB-1 and DAF-15/Raptor are expressed ubiquitously and localize to lysosomes. LET-363/TOR and DAF-15/Raptor are required for development beyond the third larval stage (L3). We observed that deletion of RHEB-1 similarly conferred L3 arrest. Unexpectedly, robust RNAi-mediated depletion of TORC1 components caused arrest at stages prior to L3. Accordingly, conditional depletion of endogenous DAF-15/Raptor in the soma revealed that TORC1 is required at each stage of the life cycle to progress to the next stage. Reversal of DAF-15 depletion permits arrested animals to recover to continue development. Our results are consistent with TORC1 functioning as a developmental checkpoint that governs the decision of the animal to progress through development.
KEY WORDS: Ral, RalGAP, TSC, Tuberous sclerosis complex, MTOR, MTORC1
Summary: RHEB-1 and DAF-15/Raptor, components of the nematode TORC1 complex, are necessary for animals to progress through each stage of post-embryonic development. Restoration of DAF-15 after depletion leads to resumed development.
INTRODUCTION
The serine/threonine protein kinase mTOR (mechanistic Target of Rapamycin) is a major signaling protein that is conserved from yeasts to humans (Blenis, 2017; Sabatini, 2017). The kinase exists in two discrete complexes, TORC1 and TORC2, that have distinct upstream inputs and downstream outputs. Raptor (Regulatory-associated protein of mTOR; DAF-15) and Rictor (Rapamycin-insensitive companion of mTOR) are scaffold proteins that define TORC1 and TORC2 substrate specificity, respectively, and hence the cellular roles of each complex. TORC1 functions as a central switch in metabolism, balancing opposing outputs of anabolism and catabolism. It promotes anabolic activities such as ribosome biogenesis and synthesis of proteins and other macromolecules, while conversely inhibiting a central facet of catabolism, autophagy. TORC1, a key regulator of cell growth, is the main focus of this study.
TORC1 integrates global systemic signals (e.g. insulin/IGF and secreted growth factors) with local cellular signals (including cellular nutrients such as amino acids, glucose, oxygen and ATP). Thus, TORC1 controls growth both at the level of individual cells and that of the whole animal. The small GTPase Rheb mediates the canonical growth factor input into TORC1. Insulin/IGF signaling (IIS) induces Rheb-mediated activation of TORC1 through an Akt-dependent negative regulatory cascade (Inoki et al., 2002, 2003; Long et al., 2005; Garami et al., 2003). The tuberous sclerosis complex, the TSC1/2 heterodimer, is a GAP (GTPase-activating protein) that specifically inhibits Rheb: TSC1/2 stimulates GTP hydrolysis, thus converting active Rheb-GTP into inactive Rheb-GDP. Activated Akt downstream of IIS, phosphorylates and inhibits TSC1/2, thereby derepressing Rheb and activating TORC1.
In addition to extensive analyses in mammalian cell culture that revealed important molecular mechanisms of TOR signaling, studies using model invertebrates have contributed significantly to our understanding of TORC1 function in vivo. TOR work was pioneered in yeasts but with certain mechanistic differences compared with metazoans (Eltschinger and Loewith, 2016; van Dam et al., 2011). Drosophila TORC1 controls normal cell growth and proliferation by regulating translational effectors during larval development (Oldham et al., 2000; Zhang et al., 2000). Overexpression of Drosophila dRheb confers eye and head overgrowth, whereas dRheb mutations suspend larval growth and prevent progression from first to second instar (Patel et al., 2003; Saucedo et al., 2003).
In Caenorhabditis elegans, mutation of the genes encoding TOR and Raptor coordinately suspends development at the third larval (L3) stage and causes intestinal atrophy (Hara et al., 2002; Jia et al., 2004; Long et al., 2002). Remarkably, C. elegans and related nematodes do not encode orthologs of the TSC1/2 Rheb GAP (Ashrafi, 2007; van Dam et al., 2011), raising the issue of how nematodes mediate global energy inputs into TORC1. One model proposes that TSC is functionally substituted by the structurally similar heterodimeric RalGAP, which inactivates Ral, a close relative of Rheb. We have previously shown that mammalian Ral binds and activates TORC1 (Martin et al., 2014), and mammalian TSC/RhebGAP and RalGAP are both repressed by upstream Akt activity (Chen et al., 2011, 2014; Inoki et al., 2002; Leto et al., 2013; Potter et al., 2002). TSC is well-known as a tumor suppressor, and a similar role has been suggested for RalGAP (Oeckinghaus et al., 2014; Saito et al., 2013). Additionally, C. elegans RalGAP and RAL-1 regulate lifespan (Martin et al., 2014). So, perhaps Rheb and Ral represent parallel and paralogous signaling inputs into TORC1. GAPs that are functionally distinct in other metazoans may be united in the single heterodimeric nematode GAP, the HGAP-1/2 RalGAP, to coregulate both RHEB-1 and RAL-1. Fittingly, the C. elegans RHEB-1 sequence has converged towards that of RAL-1 relative to Drosophila and mammalian orthologs (Reiner and Lundquist, 2018).
In a parallel noncanonical regulatory input, local cellular supply of amino acids and other metabolites adjusts local TORC1 activity by activating the heterodimer of RagA/B and RagC/D. This active Rag complex contributes to the targeting of TORC1 to the lysosome, where it encounters, and can be activated by, Rheb (Kim et al., 2008; Sancak et al., 2008). Single deletion mutants of raga-1(RagA) or ragc-1(RagC), or the raga-1/ragc-1 double mutant, extend lifespan but fail to suspend development (Schreiber et al., 2010; F.S.M. and D.J.R., unpublished), suggesting that the Rag heterodimer is not necessary for TORC1 activation in C. elegans. These results invite the obvious question of whether it is RHEB-1 or the RagA/C heterodimer that provides the TORC1 activation that is evidently necessary for developmental progression in vivo.
Here, we report that TORC1 is required at every larval stage for developmental progression. Deletion of either rheb-1 and daf-15 coordinately suspended development at the L3 stage but robust RNAi depletion or conditional protein degradation could arrest animals at all stages. Crucially, arrest conferred by conditional depletion was generally reversible. Tagged endogenous RHEB-1 and Raptor/DAF-15 colocalize at the lysosome. Our results are consistent with the model of the Rheb-TORC1 signaling axis functioning as a developmental checkpoint or licensing factor at each stage of development.
RESULTS
Ubiquitously expressed RHEB-1 and DAF-15 colocalize at lysosomes
Activated Rheb is predicted to conditionally associate with TORC1 at the lysosome (Groenewoud and Zwartkruis, 2013). The advent of CRISPR knock-in technology (Dickinson et al., 2013) permitted us to examine this association using endogenous protein. We used CRISPR to knock-in mKate2::3xFlag (mK2) into the 5′ end of rheb-1 (Fig. S1A) and mNeonGreen::2xHA (mNG) into the 3′ end of daf-15 (Fig. S1F), resulting in N-terminal and C-terminal tags of endogenous proteins, respectively. (LET-363/TOR itself was not analyzed because it also functions as a component of TORC2.) Validation of endogenous protein fusions was performed using western blot with antibodies against Flag or HA and α-Tubulin (control). Fusion proteins were of the expected size: 50 kDa for mK2::3xFlag::RHEB-1 (Fig. S1E) and 220 kDa for DAF-15::mNG::2xHA (Fig. S1G). The RHEB-1 tag was further validated by bacterially mediated RNAi depletion of rheb-1, which depleted both western blot and epifluorescence signal (Fig. S1B,E). Activity of the endogenous rheb-1 promoter, also visualized using our CRISPR tag, revealed the same spatiotemporal expression, with increased expression in the anteriormost intestinal cells (Fig. S1C,D). DAF-15 is expressed ubiquitously (Fig. S1H,I).
Both mK2::RHEB-1 and DAF-15::mNG were expressed throughout development and in all tissues (Fig. 1A; Fig. S1J). We analyzed tagged endogenous DAF-15 and RHEB-1 in live animals. Visualization of the epithelial syncytium (hypodermis) precluded concerns about strong autofluorescence signal observed in the intestine (see below). We observed colocalization of mK2::RHEB-1 and DAF-15::mNG in both vesicle-like and tubular structures in the lateral epithelium, consistent with colocalization of RHEB-1 and DAF-15 (Fig. 1A,C,E). mK2::RHEB-1 also colocalized with CTNS-1::GFP (Fig. 1B,D,E), which marks the lysosomal membrane in the hypodermis (Yu et al., 2008). These imaging techniques are limited by the low signal, a consequence of visualizing tagged endogenous proteins. Thus, we imaged rescuing transgenic high copy number mNG::RHEB-1 expressed in the intestine, where we observed numerous vesicle-shaped structures (Fig. S1K), consistent with lysosomal localization.
Fig. 1.

RHEB-1 and DAF-15 are expressed ubiquitously and colocalize at lysosomes. (A) DIC and confocal photomicrographs of endogenous DAF-15::mNG (green) and mK2::RHEB-1 (red) in a section through the hypodermis. Both mK2::RHEB-1 and DAF-15::mNG are expressed ubiquitously. Colocalization was observed at subcellular structures consistent with lysosomes, either in cross-section (arrows) or as a tubular side view (asterisks). The inset shows a vesicle-like shape, a putative lysosome, with a line scan in C (indicated by a yellow line) and quantification in E. Scale bar: 20 µm. (B) DIC and confocal photomicrographs of a hypodermal cross-section of lysosomal markers CNTS-1::GFP and mK2::RHEB-1, showing colocalization at subcellular structures, including cross-sections of vesicle-like shapes (arrows) and tubular lysosomes (asterisks). The inset shows a single vesicle-like shape, with a line scan in D (indicated by a yellow line) and quantification in E. Scale bar: 20 µm. (C) Measurement of green (DAF-15) and red (RHEB-1) signal intensity in a line scan through a putative lysosome, in arbitrary units (A.U.) of fluorescence. (D) Measurement of green (CTNS-1) and red (RHEB-1) signal intensity in a line scan through a putative lysosome, in arbitrary units (A.U.) of fluorescence. (E) Quantification of colocalization of total signal between RHEB-1 and DAF-15 (left) and CTNS-1 and RHEB-1 (right) was performed by counting total punctae from the magnified regions of A and B from a single animal (see Materials and Methods for numerical calculation).
Confocal images of large polyploid intestinal cells revealed that the expected intestinal autofluorescence, which is likely from the lipid storage lysosome-related organelles (LROs; Hermann et al., 2005), precluded delineation of subcellular localization of endogenously tagged mK2::RHEB-1 and DAF-15::mNG. To determine subcellular expression of endogenous mK2::RHEB-1 and DAF-15::mNG in the intestine, we compared raw confocal images of the intestine with spectrally unmixed images to remove autofluorescence (Fig. S1J, top versus bottom; see Materials and Methods). We observed mK2::RHEB-1 and DAF-15::mNG to be concentrated at the brush border of the apical (luminal) surface of intestinal cells, but bright punctae were removed. Thus, bright punctae in images without unmixing is intestinal autofluorescence.
rheb-1 and daf-15 mutants arrest at the 3rd larval stage
The rheb-1 gene encodes a small GTPase most closely related to yeast, Drosophila and mammalian Rheb proteins that function in concert with TORC1 (Reiner and Lundquist, 2018; Fig. 2A). To analyze the biological functions of RHEB-1, we examined the phenotypes conferred by the consortium-generated rheb-1 deletion alleles tm4931 and tm4642. Unexpectedly, tm4931 mutants arrested at the L2 stage, whereas tm4642 mutants died as embryos (Fig. S2A, Table S1). These observations were inconsistent with each other and with the published literature on C. elegans TORC1 function (Hara et al., 2002; Jia et al., 2004; Long et al., 2002).
Fig. 2.

Deletion mutations in rheb-1 and daf-15 confer L3 arrest. (A) Exon/intron gene model of rheb-1 with location of sequences deleted by mutations indicated by lateral lines. Key structural elements of small GTPase proteins are indicated: G boxes (G1-5), switch I and II, and HVR+CAAX (hypervariable region+Cys-A-A-X prenylation target, where A=aliphatic and X=any residue; Wennerberg et al., 2005). (B) Decreased pharyngeal pumping rate of rheb-1 and daf-15 arrested mutant animals versus wild type. Y-axis indicates pumps per minute. Number in columns indicate number of animals scored. ****P<0.001 (ANOVA test). (C) Merged DIC plus GFP images of rheb-1(tm4931), rheb-1(re242) and daf-15(ok1412) arrested animals with M-lineage reporter ayIs6 [hlh-8::GFP]. daf-15(ok1412) and rheb-1(re242) animals arrested at L3 (indicated by sex myoblasts located at the A-P midline), whereas rheb-1(tm4931) animals arrested at L2 stage (indicated by undifferentiated M-lineage blast cells located posteriorly). Scale bars: 50 µm. (D) Kaplan–Meier curves show rheb-1(re242), rheb-1(tm4931) and daf-15(ok1412) arrested animals have mean lifespan at 19, 12 and 13 days post-hatching, respectively. P-values were calculated using the Kaplan–Meier estimator with the OASIS website for statistical analysis (Yang et al., 2011) (****P<0.001). (E) Merged DIC and GFP epifluorescent images of knuSi221[FIB-1::GFP], an intestinal marker of the nucleolus, in rheb-1(re242)/+ (left) versus rheb-1(re242)/rheb-1(re242) animals. Scale bars: 25 µm. (F) Quantification of FIB-1::GFP nucleolar volume in heterozygous versus homozygous mutants. P-value was calculated by t-test (****P<0.001). (G) Bacterially mediated RNAi depletion of let-363 or rheb-1 induced nuclear translocation of HLH-30::GFP. Arrows indicate intestinal nuclei. Scale bar: 50 µm. (H) Quantification HLH-30::GFP nuclear localization relative to control. P-value was calculated by Fisher's exact test (****P<0.001).
To resolve this contradiction, we used CRISPR/Cas9 to isolate a series of deletion alleles in rheb-1. These alleles are divided into three groups: deletion of exon 6, mimicking tm4931 (re181, re182); deletion of the first five exons (re227, re228 and re229); and deletion of the entire rheb-1 gene (re242) (Fig. 2A). Animals mutant for each of these six rheb-1 deletion alleles confer coordinate arrest at the L3 larval stage, based on mutant animal size. By visual inspection using DIC microscopy, each tissue of arrested animals morphologically matched the apparent developmental stage. The stage of arrest of each mutant was corroborated independently of animal size by use of the transgenic M-lineage reporter line ayIs6 (Phlh-8::GFP; Harfe et al., 1998; Amin et al., 2007), which shows distinct expression patterns corresponding to each larval stage (Fig. 2C, Fig. S2B). To evaluate why tm4931 and tm4642 conferred phenotypes other than L3 arrest, we generated transheterozygotes of these alleles with the full-length deletion re242. tm4931/re242 and tm4642/re242 animals arrested at the L3 stage (Table S1), confirming that the rheb-1 null mutant phenotype is L3 arrest and suggesting that strains containing tm4931 and tm4642 harbor additional mutations that alter the final mutant phenotype. Thus, the observed rheb-1 deletion phenotypes are consistent with published phenotypes caused by mutations in daf-15/Raptor and let-363/TOR (Fig. 2C, Table S1).
Using the plasma membrane marker, mNG::RAP-1 (Rasmussen et al., 2018), we observed that the rheb-1(tm4931) germline cells are approximately comparable in size with their stage-matched siblings (Fig. S2H). Using the nuclear marker HIS-72::mNG (Dickinson et al., 2015), we observed that the polyploid intestinal nuclei of arrested mutant L2 and stage-matched siblings were approximately the same size (Fig. S2I). Given these observations of the similarities in body size between arrested and stage-matched siblings, we propose that the Rheb-TORC1 signaling axis does not control the size of somatic cells or endoreduplication of intestinal nuclei.
We additionally analyzed the phenotype of daf-15(ok1412) and daf-15(m81) mutants. Both mutants arrest at the L3 stage (Table S1). We observed small but significant differences in body length and width between rheb-1(re242) and daf-15(ok1412) mutants. However, using the M-lineage marker we conclude that both rheb-1 and daf-15 mutant animals arrested at the L3 stage (Fig. 2C, Fig. S2E,F,G).
Though rheb-1(tm4931) in trans to the re242 conferred L3 arrest, the L2 arrest phenotype of rheb-1(tm4931) homozygotes raised the possibility that Rheb-TORC1 activity is required at other stages of development. We depleted rheb-1 and daf-15 gene products by injecting dsRNA synthesized in vitro into the rrf-3(pk1426) RNAi-hypersensitive strain (Simmer et al., 2002). Depletion by daf-15(RNAi) caused variable arrest from L1 to L3 stages, as assayed using the M lineage reporter and animal size, whereas rheb-1(RNAi) caused arrest from L2 to L4 stages (Fig. S2C,D). Taken together, these observations are consistent with Rheb-TORC1 signaling being required at every stage throughout larval development. Perhaps maternal loading of RHEB-1, Raptor/DAF-15 and TOR/LET-363 gene products from heterozygotous mothers supports developmental progression until the L3 stage.
An important consideration is whether loss of Rheb-TORC1 activity confers lethality or developmental arrest. rheb-1 mutant animals continued to move and pump their pharynges in feeding behavior, although at a significantly lower rate than stage-matched wild-type animals (Fig. 2B). rheb-1 and daf-15 mutants also showed lifespans of several days with active behaviors, as assayed by response to light touch: rheb-1(re242), rheb-1(tm4931) and daf-15(ok1412) arrested animals show 50% survival at 19, 12 and 13 days post-hatching, respectively (Fig. 2D). Importantly, the lifespan of arrested rheb-1(tm4931) animals was subject to activity of IIS: daf-2(e1370)/InsR extends, whereas daf-16(mu86)/FoxO shortens, lifespan (Fig. S2J).
Rheb-TORC1 signaling is thought to regulate general biosynthetic processes. Using the FIB-1::GFP marker of the nucleolus (Allen et al., 2014), we determined that nucleoli are significantly smaller in the rheb-1(re242)/re242 homozygote relative to the rheb-1(re242)/+ heterozygote (Fig. 2E,F), consistent with decreased ribosomal biogenesis in the rheb-1 mutant. TORC1 represses autophagy in diverse systems, in part through phosphorylation of the TFEB transcription factor, a master regulator of CLEAR genes that encode components of autophagy (Settembre et al., 2011). We used the nuclear translocation of HLH-30::GFP, the C. elegans ortholog of TFEB (Lapierre et al., 2013; Martina et al., 2012), as a readout of Rheb-TORC1 inhibition of autophagy. RNAi depletion of rheb-1 and let-363 promoted translocation of HLH-30::GFP into the nucleus (Fig. 2G,H), consistent with the C. elegans Rheb-TORC1 signaling axis also repressing TFEB activation.
Intestinal RHEB-1 expression is sufficient to support developmental progression
To determine in which tissue(s) RHEB-1 functions to promote developmental progression, we performed a series of transgenic experiments to rescue developmental arrest conferred by the rheb-1(re227) or rheb-1(re242) null mutation. L3 arrest of rheb-1(re227) homozygotes was rescued by three out of four transgenes harboring two overlapping cosmid clones covering the entire rheb-1 gene and its potential upstream and downstream regulatory sequences (Fig. S3A,B; Table S2). We then used a set of heterologous promoter constructs to transgenically express mNG::RHEB-1 in various tissues to rescue L3 arrest caused by the rheb-1(re242) full-length deletion: Plet858 (ubiquitous, positive control), Pges-1 (intestine), Pcol-10 (hypodermis, i.e. epithelium), Prgef-1 (neurons) and Punc-54 (muscle) (Fig. S3C). Ubiquitous expression of mNG::RHEB-1 (three out of three transgenes) fully rescued rheb-1(re242) mutant developmental progression (91% fertile and 9% sterile adults; Fig. 3A,B). Importantly, expression of mNG::RHEB-1 only in the intestine produced substantial rescue of L3 arrest in re242 mutants (two out of two transgenes) (23% fertile and 66% sterile adults, 11% L4 arrest; Fig. 3A,B, Table S2). Thus, intestinal expression of RHEB-1 rescues developmental progression but only partially rescues fertility.
Fig. 3.

Intestinal expression of mNG::RHEB-1 rescues the deletion mutant. (A) rheb-1(re242) homozygous mutants harbor transgenic arrays with mNeonGreen::RHEB-1 (green) expression under control of different promoters: Plet-858 (ubiquitous), Pges-1 (intestine), Pcol-10 (hypodermis), Prgef-1 (neuron) and Punc-54 (muscle). Pmyo-2::mCherry, which resulted in red pharynges, was used as a co-injection marker. Strains were propagated as balanced rheb-1(re242)/qC1nIs106 (green pharynges)+transgene heterozygotes by selecting for both red and green pharynges for each generation. Scale bars: 100 µm. (B) Percentage of array-bearing rheb-1(re242) homozygotes progressing to different stages was scored by evaluating animals with red but non-green pharynges at day 3 post-egg lay. Homozygous mutant animals harboring the Pges-1::mNG::rheb-1 transgene expressed in intestine were significantly different from mutants harboring transgenes expressed in other tissues (e.g. intestine versus hypodermis, P<0.0001, χ2 test). Expression of RHEB-1 in the hypodermis and neuron can partially rescue rheb-1 L3-arrested mutant to L4 stage (62% and 51%, respectively). Expression of RHEB-1 in muscle produced no rescue. Hypodermal and neuronal expression permit the rheb-1 L3 arrested mutant to proceed to one stage further. Control animals were rheb-1(re242) homozygous siblings from the Plet-858 group without the extrachromosomal array. The numbers in the rows indicate the number of animals scored for each genotype.
Intestinal DAF-15 expression is necessary for normal lipid storage and intestinal health but not for developmental progression
We used the auxin-inducible degron (AID) system (Zhang et al., 2015) to perform conditional depletion of the DAF-15/Raptor component of TORC1. We used CRISPR to knock-in mNG::AID into the 3′ end of the daf-15 gene (Fig. S4A). AID requires exogenous expression of the plant co-factor TIR1 for conditional degron to respond to auxin (Zhang et al., 2015). Thus, we generated a strain harboring daf-15(re257[daf-15::mNG::AID]) and ieSi57(Peft-3::TIR1::mRuby) for depletion in all somatic cells. To validate the efficacy of DAF-15::mNG::AID tag system, we observed that expression of tagged DAF-15 was abolished after 1 h at all three auxin concentrations (100 µM, 300 µM and 1 mM; Fig. S4B).
To test whether intestinal expression of DAF-15 is necessary for developmental progression, we generated a strain harboring daf-15(re257[daf-15::mNG::AID]) and ieSi61[Pges-1::TIR1::mRuby], where TIR1 is expressed only in the intestine. Auxin treatment of L1s depleted mNG fluorescent signal specifically where TIR1 was expressed, in the intestine (Fig. 4A). Animals with DAF-15 depletion in the intestine still grew to adulthood, but with a smaller, atrophied intestine (Fig. 4A; Fig. S4E) and a clear body phenotype (Fig. 4B, Fig. S4F). We stained with Oil Red O to measure lipid storage in the intestine (Wählby et al., 2014). We detected significantly lower Oil Red O staining in auxin- versus vehicle-treated animals (Fig. 4C,D; Fig. S4G), indicating a significant reduction in lipid storage in animals with intestinal knockdown of DAF-15.
Fig. 4.

Conditional TORC1 depletion reveals a functional requirement at every stage. (A) CRISPR-generated daf-15::mNG::AID (green) and intestinal Pges-1::TIR1::mRuby (red) are shown, processed by spectral unmixing to minimize signal from autofluorescence (see Materials and Methods). Vehicle (left) and auxin-treated animals (right) were mounted on the same slide for comparison of fluorescent signal intensity. Germline (green arrows) and intestine (red arrows) positions are indicated. mNG fluorescent signal in intestine was greatly reduced in the auxin-treated animal when compared with vehicle control. The intestine of the auxin-treated animal is smaller than that of the vehicle control (red arrows). Merged images of red and green channels with higher magnification images showing a clear reduction in DAF-15::mNG expression in the intestine of the treated animal. Scale bar: 25 µm. (B) Bright field image of daf-15::mNG::AID+Pges-1::TIR1::mRuby with vehicle or auxin (1 mM) treatment. Scale bar: 200 µm. (C) Oil Red O images of lipid staining in the 1 mM auxin-treated group compared with vehicle control (4% Ethanol). Scale bars: 50 µm. (D) Quantification of Oil Red O staining intensity using ImageJ. A.U.=Arbitrary Units (digitized gray levels, 0 to 255 per RGB channel). ****P<0.001 (t-test). (E) daf-15::mNG::AID+Peft-3::TIR1::mRuby animals were treated with vehicle (4% Ethanol) or 1 mM auxin at embryo, L1, L2, L3 or L4 stage. Stage-matched animals are shown for each condition. Scoring of animal stage was performed at day 2 post-egg lay (for embryo or L1 treatment group) or at day 3 post-egg lay (for L2, L3 and L4 treatment group). Scale bars: 100 µm. (F) Quantification of arrest stage for each treatment group is shown compared with stage-matched animals of the same genotype treated with vehicle. Treatment at each stage resulted in coordinated arrest at the following stage. (G) Brood size quantification on L4 treatment group compared with vehicle control. ****P <0.001 (t-test). (H-K) daf-15::mNG::AID+Peft-3::TIR1::mRuby animals were exposed to 100 µM or 1 mM auxin at either L1 stage (H,I) or L2 stage (J,K). Animals were removed from auxin after 24 h of initial treatment, and reverse of arrest phenotype was scored at day 4 post-hatching (∼48 h post-removal).
Changes in gut appearance and Oil Red O staining are consistent with grossly perturbed intestinal metabolism upon TORC1 disruption. We therefore examined intestinal autofluorescence as a surrogate for LRO size in the rheb-1(re242) homozygote versus the re242/+ heterozygote. We observed that the putative LROs were significantly larger in the homozygote (Fig. S4H,I). Conversely, cytosolic DAF-15::mNG signal was signficantly lower in the homozygote (Fig. S4H,J). These observations are consistent with large scale reorganization of metabolism in the absence of TORC1.
TORC1 is required at every larval stage for developmental progression
To evaluate the requirement for TORC1 throughout development, synchronized daf-15(re257[daf-15::mNG::AID]) animals with and without ieSi57(Peft-3::TIR1::mRuby) were subjected to vehicle or 1 mM auxin treatment at specific time points: embryo, L1, L2, L3 and L4 stage. Treatment at each stage resulted in the depletion of mNG-tagged DAF-15 signal (Fig. S4C,D) and coordinated arrest at the following stage (Fig. 4E). Quantification of arrest stage for each treatment group is shown compared with stage-matched animals of the same genotype treated with vehicle (Fig. 4F). Depletion at the L4 stage did not cause arrest, but treated animals displayed significant reduction of brood size (Fig. 4F,G). Notably, in this experiment DAF-15::mNG::AID was not depleted in the germline, as TIR-1 was expressed only in somatic cells. Consequently, we conclude that TORC1 is required somatically for proper fertility.
TORC1 depletion dependent arrest is reversible
We expect that arrest should be reversible, while a developmental defect should not be. We took advantage of the reversibility of AID-dependent depletion (Zhang et al., 2015) to determine whether DAF-15::mNG::AID-dependent arrest by auxin treatment was reversible. We treated with auxin at two different stages (L1 and L2) and at two concentrations (100 µM or 1 mM). Auxin was removed after 24 h of initial treatment and reverse of arrest phenotype was scored ∼48 h post-removal. Auxin treatment (1 mM) at L1 caused complete acute arrest with no recovery after auxin removal (Fig. 4H). In contrast, treatment with 100 µM auxin also conferred acute arrest but after removal from auxin these animals were able to recover to become fertile adults (89%), sterile adults (7%) and infrequent non-reversible L4 arrest (5%; Fig. 4I). Treatment at L2 was acute and reversible at both auxin treatment concentrations (Fig. 4J,K).
Importantly, with prolonged observation we observed that after initial arrest, auxin-arrested animals slowly continued larval development over the subsequent 2 days, to the L4 stage but not beyond. Retrospectively, we observed that arrest in previous experiments was not absolute, but was followed by very slow developmental progression that was rarely completed. These results are in marked contrast to those with mutants for rheb-1 or daf-15, the arrest of which was absolute. Therefore, auxin-dependent depletion has qualitatively different consequences from mutation of the gene. Yet we speculate that depletion is more likely to reflect strong starvation conditions, unlike gene deletion, and is thus more likely to be physiologically relevant.
TORC1 depletion dependent arrest is partially bypassed by excess DAF-9
Starvation dependent developmental checkpoint arrest has previously been shown to be bypassed by expression of functional DAF-9::GFP, which mediates developmental progression by producing steroid hormone (Schindler et al., 2014; Gerisch and Antebi, 2004; Mak and Ruvkun, 2004). We tested the ability of the dhIs64[DAF-9::GFP] transgene to bypass the arrest of daf-15 mutant animals (we did not test bypass of rheb-1 mutants because dhIs64 is linked to rheb-1 on chromosome III). Upon scrutinizing arrested rheb-1(re242) homozygotes for extended periods, we found that although no animals initiated vulval morphogenesis by day 5, many animals progressed to early vulval morphogenesis by day 7, accompanied by M-lineage proliferation to generate the vulval muscles (Fig. 5A). We observed a similar phenomenon in daf-15(ok1412) mutants, where some but not all animals at day 4, but not earlier, had begun vulval morphogenesis (Fig. 5B). As with the rheb-1 mutants, daf-15 mutants never progressed past early vulval morphogenesis. However, daf-15(ok1412) homozygotes harboring dhIs64[DAF-9::GFP] had frequently progressed to late vulval morphogenesis by day 3, and typically ruptured at the vulva at day 4 (Fig. 5C). Thus, continuous expression of DAF-9::GFP drove earlier developmental progression of daf-15 mutants, and animals progressed to a later stage of development. This observation is consistent with ectopic DAF-9 partially bypassing the arrest conferred by disrupted TORC1.
Fig. 5.
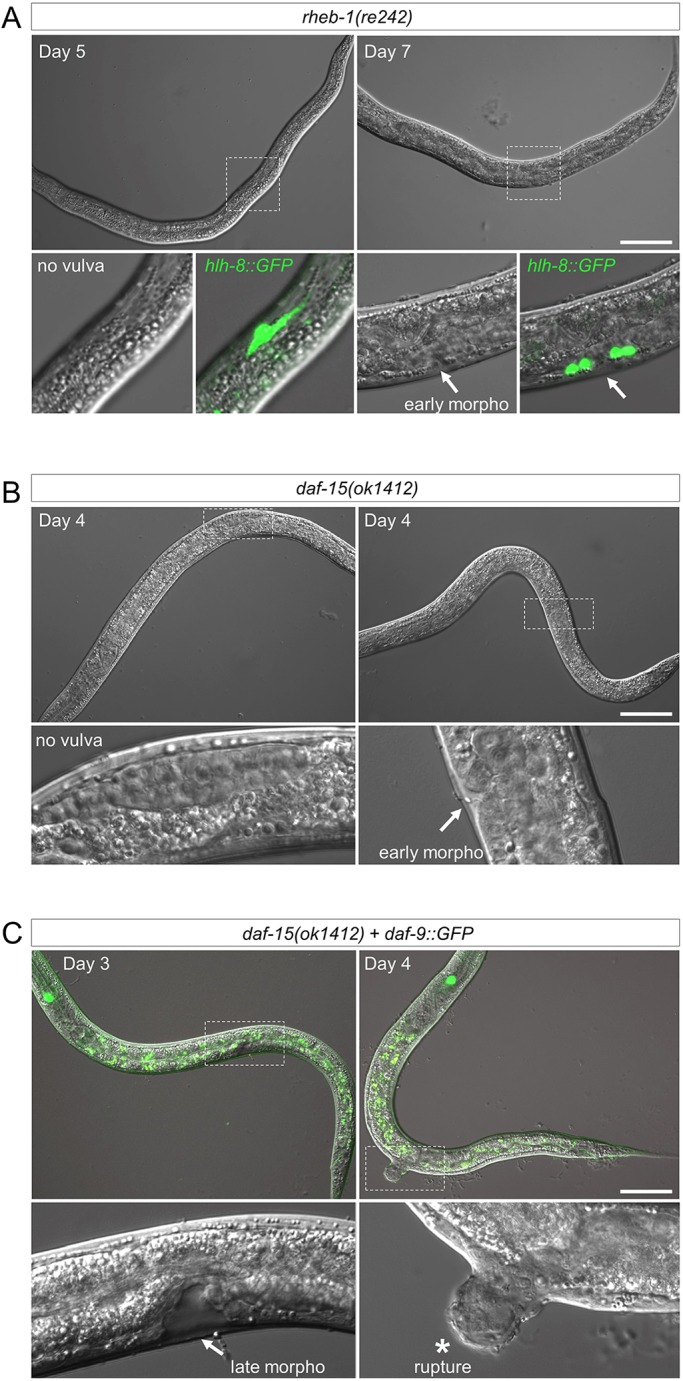
Ectopic DAF-9::GFP drives development beyond the stage of TORC1 mutant arrest. (A) rheb-1(re242) mutant animals have not formed vulval structures by day 5 after arrest, but show early vulval morphogenesis (‘early morpho’) at day 7. (Vulval morphogenesis in the wild type at 20°C occurs less than 1 day after the L2-L3 molt.) For each, the M-lineage reporter line ayIs6 (Phlh-8::GFP) reveals stage-specific development that is retarded for longer than the life cycle of the animal. Vulval development never progresses beyond this stage. (B) daf-15(o1412) mutants similarly show greatly retarded progression of vulval development in a subset of animals. (C) The dhIs64[DAF-9::GFP] transgene confers faster and farther developmental progression of daf-15(o1412) animals, which typically rupture at the vulva on day 4. Scale bars: 50 µm.
DISCUSSION
Previous studies found that disruption of let-363/TOR and daf-15/Raptor conferred arrest at the third larval stage (Hara et al., 2002; Jia et al., 2004; Long et al., 2002). In this study, we observed that RHEB-1 functions similarly. In contrast, the Rag heterodimer is not necessary for developmental progression. Thus, despite the absence of TSC/RhebGAP in C. elegans, RHEB-1 likely functions as the principal input into the TORC1 signaling axis. In contrast to other metazoans, we found no evidence that C. elegans TORC1 controls cell size. Unexpectedly, we showed that components of TORC1 signaling are required throughout post-embryonic development, not only at the third larval stage, as previously thought. Furthermore, arrested animals can resume development upon restoration of TORC1 signaling, and triggering of ectopic steroid hormone synthesis with ectopic DAF-9::GFP can partially bypass arrest. In consideration of other details of C. elegans TORC1 function, we hypothesize that TORC1 functions as a developmental checkpoint (Fig. 6).
Fig. 6.

TORC1 functions as a developmental checkpoint. A model for TORC1 function in C. elegans development. Outer circle (dark green): homozygous deletion mutants in rheb-1/Rheb, daf-15/Raptor or let-363/TOR born of heterozygous mothers experience decreasing maternal product during development, as indicated by progressively paler shades of green. At a certain low threshold, development arrests. Conversely, by precociously depleting maternal product, robust RNAi-mediated depletion could cause arrest prior to L3. Middle circle (C. elegans life cycle): TORC1 is required for developmental progression at all post-embryonic stages, as well as nonautonomously regulating brood size in the adult. Inner circle (pink): key or ‘bellweather’ nutrients (e.g. ceramide49) are hypothesized to be necessary to activate TORC1 and hence support developmental transitions at the L1/L2, L2/L3 and L3/L4 transitions.
Our results reinforce the correlation of L3 arrest of mutants for TORC1 components with dauer diapause, an alternate third developmental stage induced under adverse environmental conditions (Hu, 2007). Yet we propose that this correlation is coincidental. Mutants for rheb-1 and daf-15 only superficially resemble dauers morphologically and not at all behaviorally. Moreover, TORC1 activity is required throughout post-embryonic development. Perhaps L3 is the stage at which maternal product from heterozygous mothers is insufficient to support development in animals mutant for components of the TORC1 signaling axis.
Intestinal-specific expression of RHEB-1 is sufficient to support development in the rheb-1 mutant, consistent with the role of the intestine as the major metabolic and energy storage tissue in C. elegans (Ashrafi, 2007). Yet the requirements for TORC1 function might not be so simple. Intestinal-specific degradation of TORC1 still permitted developmental progression, suggesting potential roles of TORC1 elsewhere in the animal. Somatic depletion of TORC1 dramatically decreased brood size, suggestive of a nonautonomous role for TORC1 in reproduction. Such nonautonomous control of metabolism has a precedent in C. elegans (Dowen et al., 2016). However, intestinal TORC1 depletion drastically reduced intestinal lipid storage, indicating that TORC1 can also function autonomously in the intestine. Given the role in other systems of TORC1 promoting anabolism, a compelling hypothesis is that TORC1-depleted intestines are compromised in their ability to synthesize complex lipids and perhaps other energy storage molecules. This anabolic activity of TORC1 may function independently of the requirement of TORC1 for developmental progression.
Mutational disruption of LET-363/TOR and DAF-15/Raptor have previously been described as causing increased storage of lipids in the intestine (Jia et al., 2004), whereas we describe decreased storage of lipids upon depletion of DAF-15. Yet these results are not necessarily contradictory. First, we used Oil Red O staining rather than the Sudan Black used in the earlier study. These dyes may stain different complements of the lipid metabolome. Additionally, Sudan Black staining has been shown to be ‘highly variable’, whereas Oil Red O is much more consistent (O'Rourke et al., 2009). Second, the Sudan Black staining was conducted on arrested L3 animals, while our Oil Red O staining was conducted on animals of reproductive age. The extensive production of nutrients in the intestine for export to oocytes in the germline could result in greater depletion of lipid stores when TORC1 function is perturbed and anabolic metabolism reduced.
RHEB-1 has been loosely associated with the effects of dietary restriction, mutation of Ataxin/ATX-2 and animal size (Bar et al., 2016). RHEB-1 additionally plays a complex role in intermittent fasting-induced longevity and dietary restriction (Honjoh et al., 2009). On the one hand, RNAi depletion of rheb-1 extended lifespan. On the other hand, RNAi depletion of rheb-1 or let-363/tor blocked the effects of lifespan extension via intermittent fasting (Honjoh et al., 2009). When added to our observations about the requirement of TORC1 throughout development, we conclude that RHEB-1 and TORC1 play complex but central roles in multiple facets of metabolism, lifespan and development.
Our results using a null mutation in rheb-1, suggest that IIS functions in parallel to TORC1 to control lifespan. Deletion of RSKS-1, the C. elegans ortholog of Ribosomal S6 kinase, a major TORC1 output, was shown to be redundant with a reduced function mutation in DAF-2/InsR, extending lifespan fivefold relative to the wild type; this effect was blocked by mutation of the downstream DAF-16/FoxO (Chen et al., 2013). Under starvation conditions, germline overproliferation induced by mutation of DAF-18/PTEN, a negative regulator of IIS, was suppressed by RNAi depletion of rheb-1 or raga-1, consistent with TORC1 function downstream or in parallel to IIS (Fukuyama et al., 2012). Overexpression of RHEB-1 failed to rescue starvation-induced cell quiescence, while activated RAGA-1/RagA rescued this phenotype, suggesting that under certain circumstances RAGA-1 input into TORC1 is more important than that of RHEB-1 (Fukuyama et al., 2015). Removal from food at precise times induces coordinated and stereotyped arrest of development of the vulva, consistent with single developmental checkpoints in each larval stage (Schindler et al., 2014). Yet daf-16/FoxO was required for this arrest, while mutation of daf-16 failed to alter arrest due to the deletion of rheb-1.
Conditional depletion of tagged endogenous DAF-15/Raptor at every stage confers developmental arrest. These arrest points are mostly reversible upon restoration of DAF-15 expression. Reversible arrest under adverse conditions, as well as bypass of arrest by activation of a downstream protein, are hallmarks of checkpoints, defined originally for the yeast cell cycle (Hartwell and Weinert, 1989). An L1 arrest in C. elegans is conferred by deficiency of monomethyl-branched chain fatty acid (mmBCFA; (Zhu et al., 2013). This mmBCFA is further processed to generate a ceramide-like sphingolipid, which is required for survival of starvation, suggesting that it is a key nutrient (Cui et al., 2017). Intriguingly, activation of TORC1 by various means bypasses the L1 arrest conferred by ceramide deficit (Zhu et al., 2013). These observations, in conjunction with our results, induce us to propose that RHEB-1-TORC1 comprises a crucial checkpoint (Fig. 6) in C. elegans development that may also mediate starvation-based arrest: key nutrients for survival under adverse conditions – such as ceramide – are likely required for activation of TORC1 during development.
MATERIALS AND METHODS
C. elegans handling and genetics
Strains were grown with OP50 bacteria on NGM agar plates at 20°C except where otherwise noted. Strains used in this study are listed in Table S6. To obtain synchronized progeny, adults were transferred onto OP50 seeded NGM plates and allowed to lay eggs for 1 h. Primers used in this study are listed in Table S3. Genotyping conditions for each mutant are described in Table S5.
Plasmids, generation of transgenic lines, rescue
Plasmids used are listed in Table S6. The rheb-1 cDNA sequence was amplified from F54C8.5 ORF (Dharmacon). pTD8 [Plet-858::rheb-1::let-858 3′UTR] was generated by cloning amplified rheb-1 cDNA into the NheI cut site of pPD118.25 [Plet-858::GFP::let-858 3′UTR]. pTD63 [Plet-858::mNG::rheb-1::let-858 3′UTR] was generated by Gibson Assembly reaction using part of pTD8 to N-terminally fuse mNG to rheb-1. Both pTD64 [Prgef-1::mNG::rheb-1::let-858 3′UTR] and pTD66 [Pges-1::mNG::rheb-1::let-858 3′UTR] were generated by a Gibson Assembly reaction using part of pTD63 to make different promoters fused to mNG::rheb-1::let-858 3′UTR. For Punc-54::mNG::rheb-1::let-858 3′UTR and Pcol-10::mNG::rheb-1::let-858 3′UTR, we generated them by using PCR stitching approach by designing primers to combine the two fragments of promoter and mNG::rheb-1::let-858 3′UTR.
To perform tissue-specific rescue experiments, PCR fragments of all five promoters (Plet-858, Pges-1, Pcol-10, Prgef-1 and Punc-54) with mNG::rheb-1::let-858 3′UTR constructs were injected into the rheb-1(re242)/qC1[dpy-19(e1259) glp-1(q339) nIs189(Pmyo-2::GFP)]; ayIs6[Phlh-8::GFP] strain at 0.25 ng/µl, together with N2 genomic DNA/EcoRV (30 ng/µl) (filler DNA) and Pmyo-2::mCherry/ScaI 0.25 ng/µl (co-injection marker). Transgenic lines were scored for transmission rate based on percentage of red pharynges in the population. For rescue using cosmids, overlapping cosmids F54C8 (23 kb insert) and B0464 (41 kb insert) were injected into rheb-1(re227)/qC1[dpy-19(e1259) glp-1(q339) nIs189(Pmyo-2::GFP)]; ayIs6[Phlh8::GFP] at 5 ng/µl each, together with N2 genomic DNA/EcoRV 30 ng/µl (filler DNA) and Pmyo-2::mCherry/ScaI (0.25 ng/µl) (co-injection marker). A summary of transgenic lines made for rescue experiments is listed in Table S2.
CRISPR knockouts and knock-ins
CRISPR/Cas9-dependent rheb-1 deletions were generated using plasmid-based delivery or RNA/protein delivery. The rheb-1 full-length deletion mutation, re242, was generated by using a plasmid-based CRISPR/Cas9 strategy with dpy-10 co-CRISPR (Paix et al., 2014; Ward, 2015) by microinjection of pJA58 (25 ng/µl), rheb-1 sgRNA-Cas9#1 pTD15 (50 ng/µl), rheb-1 sgRNA-Cas9#4 pTD46 (50 ng/µl), dpy-10(cn64) ssODN repair template (500 nM) and co-injection marker pPD118.33 (10 ng/µl) into N2 wild-type animals. rheb-1 deletion mutations re181 and re182 were generated by using plasmid-based CRISPR/Cas9 and co-CRISPR strategy (Paix et al., 2014) by microinjection of pJA58 (25 ng/µl), rheb-1 sgRNA-Cas9#3 pTD45 (50 ng/µl), rheb-1 sgRNA-Cas9#4 pTD46 (50 ng/µl), dpy-10(cn64) ssODN repair template (500 nM) and co-injection marker pPD118.33 (10 ng/µl) into rheb-1(re64[mKate2^3xFlag::rheb-1]) animals (the guide RNAs used for these experiments are listed in Table S4). Additional rheb-1 deletion mutations re227, re228 and re229 were generated by using protein-based CRISPR/Cas9 and co-CRISPR strategy by microinjection of dpy-10 cRNA, rheb-1 crRNA#1, rheb-1 crRNA#2, tracer RNA, dpy-10(cn64) ssODN repair template (500 nM), rheb-1 ssODN repair template (500 nM), and Cas9 protein (50 ng/µl) into wild-type animals.
Endogenous rheb-1 and daf-15 were tagged with fluorescent protein::epitope tags, mKate2::3xFlag and mNeonGreen::2xHA, respectively, using CRISPR/Cas9 knock-in with a Self-Excising Cassette (SEC) approach (Dickinson et al., 2015). Schematics of the SEC strategy for rheb-1 and daf-15 knock-in are described in Fig. S1A,F. daf-15 was also tagged with mNG::AID using the same approach (Fig. S4A). Repair templates for CRISPR knock-ins using the SEC approach were made as described previously (Dickinson et al., 2015). However, for the AID knock-in into daf-15, the repair template was derived from pTD55(mNG^SEC^AID). This vector was modified from pDD268(mNG^SEC^3xFlag) to replace the 3xFlag with the Degron/AID sequence. The sgRNA targeting sequences were inserted into pJW1236, the Cas9+sgRNA (F+E) plasmid (Ward, 2015), by Q5 site-directed mutagenesis (NEB).
mKate2^3xFlag::rheb-1 was generated by microinjection of rheb-1 sgRNA-Cas9#1 pTD15 (50 ng/µl), rheb-1 sgRNA-Cas9#2 pTD16 (50 ng/µl), repair template pTD14 (10 ng/µl) and co-injection marker pPD118.33 (10 ng/µl) into wild-type animals. daf-15::mNG^2xHA was generated by microinjection of daf-15 sgRNA-Cas9#1 pTD34 (50 ng/µl), daf-15 sgRNA-Cas9#2 pTD35 (50 ng/µl), repair template pTD32 (10 ng/µl) and co-injection marker pPD118.33 (10 ng/µl) into wild-type animals. daf-15::mNG^AID was generated by microinjection of daf-15 sgRNA-Cas9#1 pTD34 (50 ng/µl), daf-15 sgRNA-Cas9#2 pTD35 (50 ng/µl), repair template pTD62 (10 ng/µl) and co-injection marker pPD118.33 (10 ng/µl) into wild-type animals.
AID and auxin treatment
AID (auxin-inducible degron) sequence (135 bp) (Zhang et al., 2015) was knocked into the 3′ end of the daf-15 gene together with the mNeonGreen gene (Fig. S4A). CRISPR knock-in daf-15(re257[daf-15::mNG::AID]) was crossed into the ieSi57[Peft-3::TIR1::mRuby::unc-54 3′UTR] strain to evaluate the effect of conditional knockdown of daf-15. With auxin (Alpha Aesar, A10556), we made auxin stock solution (400 mM in ethanol) that was subsequently stored at 4°C for up to 1 month. To obtain 16 mM auxin, the stock was diluted 1:25 in filtered water with 4% ethanol (final concentration). Synchronized animals were cultured on OP50 seeded on 60 mm NGM plates to the required stage and 500 µl of 16 mM auxin was added to each plate at a final concentration of 1 mM (plates contained ∼8 ml of agar). Ethanol (4%) was used as vehicle control. For treatment with auxin at 100 µM or 300 µM, 500 µl of 1.6 mM or 5.3 mM auxin was added to the plate, respectively. For recovery experiments, auxin (100 µM or 1 mM) was applied at either the L1 or L2 stage. Twenty-four hours after the initial auxin treatment timepoint, treated animals were then moved to either a freshly made vehicle control plate or an auxin plate at the desired concentration. Scoring for reverse phenotype was performed at day 4 post-hatching (∼48 h post-removal).
Imaging
Live animals were mounted in 5 µl of 2 mg/ml tetramisole in M9 buffer on slides with 3% agar pads. Fluorescent micrographs were captured using a Nikon Eclipse Ni and A1si Confocal Laser Microscope. Bright field images were captured using a Nikon SMZ18 stereo fluorescence microscope and a DS-Fi2 camera. DIC images of animal size were captured using a Nikon Eclipse Ni microscope and DS-Fi2 camera. NIS Elements Advanced Research, Version 4.40 software (Nikon) was used. For Fig. 1, line scans were performed using the ‘plot profile analyze’ plug-in in ImageJ, and quantification of total colocalization was performed using the ‘analyze counter’ plug-in in ImageJ. To measure the percentage of colocalization (Fig. 1E), we counted the total number of green punctae (CTNS/1-lysosomes or DAF-15::mNG) and the number of punctae that are both green+ and red+. The percentage of colocalization of RHEB-1 on lysosomes or DAF-15::mNG=(green+ and red+ puncta)/green+ puncta. The size of FIB-1::GFP punctae was measured with the measure tool in ImageJ. Spectral unmixing of signals from green and red channels to remove autofluorescence signal was performed using the ‘Spectral Unmixing’ plug-in in ImageJ.
RNA interference
RNAi plasmids used are listed in Table S6, including the previously published pREW2 (luciferase RNAi; Shin et al., 2018). Insert sequences were confirmed by sequencing using the DJR627 primer (Table S3). RNAi experiments were performed at 20°C on NGM agar plates supplemented with 1 mM IPTG and 50 µg/ml carbenicillin. Plates were seeded with 80 µl dsRNA-producing bacteria, grown for 6-8 h at 37°C in the shaker. Late L4 animals (sqIs17[Phlh-30::hlh-30::GFP]) were moved to freshly made RNAi-seeded plates, then transferred to another RNAi plate the next day. This latter plate was used for scoring HLH-30::GFP nuclear translocation (Fig. 2G,H).
rheb-1 and daf-15 double-stranded RNAs were designed following a previously published protocol (Dudley and Goldstein, 2005). Briefly, a fragment of target gene including exon regions was amplified using primers with a tail containing T7 sequence plus six extra nucleotides (for T7 polymerase activity) at the 5′ end. Purified PCR products were then used as template for transcription (T7 RiboMAX Express RNAi system, Promega, P1700). For daf-15 dsRNA, primers (TD92, TD93) were designed to amplify exons three and four of the daf-15 gene. For rheb-1 dsRNA, primers (TD88, TD89) were designed to amplify a region from exon 2 to exon 5 of the rheb-1 gene. For luciferase dsRNA, universal primers (TD217, TD218) were used to amplify a luciferase fragment from pREW2 (derived from L4440/pPD129.36).
Purified dsRNA was injected into the rrf-3 RNAi hypersensitive strain (Simmer et al., 2002) with M-lineage reporter GFP (rrf-3(pk1426); ayIs6[Phlh-8::GFP]) at 500 ng/µl (diluted in RNAse-free water). Injected animals were transferred to a new plate at the end of injection day (day 1) (first transfer). The second transfer was performed early on day 2 and the third transfer was carried out at the end of day 2. We observed that second and third transferred batches had the strongest knockdown effect from dsRNA injection, therefore these groups were used for phenotype scoring.
Assays: pumping, autophagy, survival/arrest lifespan, brood size, body length and width
The pumping assay was performed by counting the number of pharyngeal pumps of homozygous arrested animals or wild-type N2 control (L3 stage) in 60 s under the stereomicroscope.
The TFEB/autophagy assay was performed by using a previously published reporter line, sqIs17[Phlh-30::hlh-30::GFP] (Lapierre et al., 2013). The assay is based on nuclear translocation of the HLH-30::GFP transcription factor upon reduction of TORC1 activity. We observed that sqIs17[Phlh-30::hlh-30::GFP] is highly sensitive to temperature change. Therefore, animals grown on different RNAi-producing HT115 bacteria were kept in the same box and moved together to the microscopy room, where animals were mounted on slides and scored 3-5 min after mounting.
Oil Red O staining was performed as described previously (Wählby et al., 2014). Staining intensity was quantified by imaging the posterior intestinal region using a Nikon Eclipse Ni, with images captured using a DS-Fi2 camera and NIS-Elements AR 4.20.00 software.
For survival assays, arrested animals were collected from a synchronized population (from 2 h after egg laying by heterozygote hermaphrodites). Arrested animals were checked for survival every other day. Kaplan–Meier curves were used to display survival of arrested mutants. P-values were calculated using the OASIS website for statistical analysis (sbi.postech.ac.kr/oasis/) (Yang et al., 2011).
To score total progeny (brood size), L4 hermaphrodites were picked from individual plates with or without auxin, and transferred to new plates every day over 4 days (until no more eggs were laid). The eggs or hatched larvae on each plate were counted after removing parents. Body length and width of arrested animals were measured using ImageJ, as previously published (Mörck and Pilon, 2006).
Western blotting
Animals were washed off unstarved plates using M9 buffer, lysed in 4% SDS loading buffer, then boiled at 90°C for 2 min. Protein samples were run on 4-15% SDS gel (Bio-Rad, 5671084) and blotted on Immobilon-P Membrane, PVDF (EMD Millipore, IPVH00010). Mouse monoclonal anti-Flag antibody (Sigma-Aldrich, F1804), mouse monoclonal anti-HA antibody (Sigma-Aldrich, 05-904) and mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich, T6199) were diluted 1:2000 in blocking solution (5% w/v nonfat dry milk in 1× PBS+0.1% Tween). Rabbit polyclonal mNeonGreen Antibody (Cell Signaling Technology, 53061) was diluted 1:1000 in blocking solution (5% w/v BSA in 1× PBS+0.1% Tween). HRP-linked secondary antibodies: goat anti-mouse (Sigma-Aldrich, 12-349) and goat anti-rabbit (Cell Signaling Technology, 7074) were diluted 1:5000 in blocking solutions (either nonfat dry milk or BSA). Chemiluminescent western blot reaction was performed using Pierce ECL Western Blotting Substrate (Thermo Fisher, 32106) and signals were detected using the film processor SRX-101A (Konica Minolta) on x-ray film (Phenix).
Quantification and statistical analysis
In every bar graph panel, animals were scored concurrently to avoid variability, using scoring standards described in the Materials and Methods. n equals the animals scored, and is indicated as a white number on each bar. To avoid bias, n was determined randomly, with all prepared animals scored and statistical tests only performed post hoc. General statistical methods are described in each figure legend. Briefly, data were analyzed using a pairwise t-test and a one-way ANOVA. Analysis of transgenic rescue counts was performed by pairwise χ2 test. P-value is shown in each panel. For statistical analysis of survival data we used GraphPad Prism 5. For survival assays, P-values were calculated using the Kaplan–Meier estimator and the OASIS website for statistical analysis (Yang et al., 2011). Statistical significance was defined as P<0.05.
Supplementary Material
Acknowledgements
We thank the B. Barstead, B. Grant, M. Hansen, K. Kornfeld, S. Mitani and D. Moerman labs for strains; wormbase.org for information; L. Vergara in the IBT Center for Advanced Imaging (CAI) for advice on microscopy and ImageJ quantification; Z. Zhou for helpful advice; and the labs of S. Arur and D. Reiner for helpful discussions. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by National Institutes of Health Office of Research Infrastructure Programs.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization, T.D. and D.J.R.; Methodology, T.D., N.R.R. and D.J.R.; Investigation, T.D., N.R.R., E.B., F.S.M.; Visualization, T.D., N.R.R., D.J.R.; Writing – original draft, T.D., D.J.R.; Project Administration, D.J.R.; Funding acquisition, D.J.R.; Resources, D.J.R.; Supervision, D.J.R. All authors read and approved the manuscript.
Funding
This work was supported by an American Cancer Society PF-16-083-01 post-doctoral fellowship to N.R.R. and National Institutes of Health grants R01GM121625 and R21HD090707 to D.J.R. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.181727.supplemental
References
- Allen A. K., Nesmith J. E. and Golden A. (2014). An RNAi-based suppressor screen identifies interactors of the Myt1 ortholog of Caenorhabditis elegans. G3 (Bethesda) 4, 2329-2343. 10.1534/g3.114.013649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin N. M., Hu K., Pruyne D., Terzic D., Bretscher A. and Liu J. (2007). A Zn-finger/FH2-domain containing protein, FOZI-1, acts redundantly with CeMyoD to specify striated body wall muscle fates in the Caenorhabditis elegans postembryonic mesoderm. Development 134, 19-29. 10.1242/dev.02709 [DOI] [PubMed] [Google Scholar]
- Ashrafi K. (2007). Obesity and the regulation of fat metabolism. WormBook, 1-20. 10.1895/wormbook.1.130.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar D. Z., Charar C., Dorfman J., Yadid T., Tafforeau L., Lafontaine D. L. J. and Gruenbaum Y. (2016). Cell size and fat content of dietary-restricted Caenorhabditis elegans are regulated by ATX-2, an mTOR repressor. Proc. Natl. Acad. Sci. USA 113, E4620-E4629. 10.1073/pnas.1512156113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenis J. (2017). TOR, the gateway to cellular metabolism, cell growth, and disease. Cell 171, 10-13. 10.1016/j.cell.2017.08.019 [DOI] [PubMed] [Google Scholar]
- Chen X.-W., Leto D., Xiong T., Yu G., Cheng A., Decker S. and Saltiel A. R. (2011). A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol. Biol. Cell 22, 141-152. 10.1091/mbc.e10-08-0665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Li P. W.-L., Goldstein B. A., Cai W., Thomas E. L., Chen F., Hubbard A. E., Melov S. and Kapahi P. (2013). Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans. Cell Rep. 5, 1600-1610. 10.1016/j.celrep.2013.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Quan C., Xie B., Chen L., Zhou S., Toth R., Campbell D. G., Lu S., Shirakawa R., Horiuchi H. et al. (2014). GARNL1, a major RalGAP alpha subunit in skeletal muscle, regulates insulin-stimulated RalA activation and GLUT4 trafficking via interaction with 14-3-3 proteins. Cell. Signal. 26, 1636-1648. 10.1016/j.cellsig.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Cui M., Wang Y., Cavaleri J., Kelson T., Teng Y. and Han M. (2017). Starvation-induced stress response is critically impacted by ceramide levels in Caenorhabditis elegans. Genetics 205, 775-785. 10.1534/genetics.116.194282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D. J., Ward J. D., Reiner D. J. and Goldstein B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028-1034. 10.1038/nmeth.2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D. J., Pani A. M., Heppert J. K., Higgins C. D. and Goldstein B. (2015). Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035-1049. 10.1534/genetics.115.178335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen R. H., Breen P. C., Tullius T., Conery A. L. and Ruvkun G. (2016). A microRNA program in the C. elegans hypodermis couples to intestinal mTORC2/PQM-1 signaling to modulate fat transport. Genes Dev. 30, 1515-1528. 10.1101/gad.283895.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley N. R. and Goldstein B. (2005). RNA interference in Caenorhabditis elegans. Methods Mol. Biol. 309, 29-38. 10.1385/1-59259-935-4:029 [DOI] [PubMed] [Google Scholar]
- Eltschinger S. and Loewith R. (2016). TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 26, 148-159. 10.1016/j.tcb.2015.10.003 [DOI] [PubMed] [Google Scholar]
- Fukuyama M., Sakuma K., Park R., Kasuga H., Nagaya R., Atsumi Y., Shimomura Y., Takahashi S., Kajiho H., Rougvie A. et al. (2012). C. elegans AMPKs promote survival and arrest germline development during nutrient stress . Biol. Open 1, 929-936. 10.1242/bio.2012836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama M., Kontani K., Katada T. and Rougvie A. E. (2015). The C. elegans hypodermis couples progenitor cell quiescence to the dietary state. Curr. Biol. 25, 1241-1248. 10.1016/j.cub.2015.03.016 [DOI] [PubMed] [Google Scholar]
- Garami A., Zwartkruis F. J. T., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L. and Thomas G. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457-1466. 10.1016/S1097-2765(03)00220-X [DOI] [PubMed] [Google Scholar]
- Gerisch B. and Antebi A. (2004). Hormonal signals produced by DAF-9/cytochrome P450 regulate C. elegans dauer diapause in response to environmental cues. Development 131, 1765-1776. 10.1242/dev.01068 [DOI] [PubMed] [Google Scholar]
- Groenewoud M. J. and Zwartkruis F. J. T. (2013). Rheb and Rags come together at the lysosome to activate mTORC1. Biochem. Soc. Trans. 41, 951-955. 10.1042/BST20130037 [DOI] [PubMed] [Google Scholar]
- Hara K., Maruki Y., Long X., Yoshino K.-I., Oshiro N., Hidayat S., Tokunaga C., Avruch J. and Yonezawa K. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177-189. 10.1016/S0092-8674(02)00833-4 [DOI] [PubMed] [Google Scholar]
- Harfe B. D., Branda C. S., Krause M., Stern M. J. and Fire A. (1998). MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development 125, 2479-2488. [DOI] [PubMed] [Google Scholar]
- Hartwell L. H. and Weinert T. A. (1989). Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629-634. 10.1126/science.2683079 [DOI] [PubMed] [Google Scholar]
- Hermann G. J., Schroeder L. K., Hieb C. A., Kershner A. M., Rabbitts B. M., Fonarev P., Grant B. D. and Priess J. R. (2005). Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell 16, 3273-3288. 10.1091/mbc.e05-01-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjoh S., Yamamoto T., Uno M. and Nishida E. (2009). Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457, 726-730. 10.1038/nature07583 [DOI] [PubMed] [Google Scholar]
- Hu P. J. (2007). Dauer. WormBook, 1-19. 10.1895/wormbook.1.144.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Li Y., Zhu T., Wu J. and Guan K.-L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648-657. 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Xu T. and Guan K.-L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829-1834. 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia K., Chen D. and Riddle D. L. (2004). The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897-3906. 10.1242/dev.01255 [DOI] [PubMed] [Google Scholar]
- Kim E., Goraksha-Hicks P., Li L., Neufeld T. P. and Guan K.-L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935-945. 10.1038/ncb1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre L. R., De Magalhaes Filho C. D., Mcquary P. R., Chu C.-C., Visvikis O., Chang J. T., Gelino S., Ong B., Davis A. E., Irazoqui J. E. et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267 10.1038/ncomms3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D., Uhm M., Williams A., Chen X.-W. and Saltiel A. R. (2013). Negative regulation of the RalGAP complex by 14-3-3. J. Biol. Chem. 288, 9272-9283. 10.1074/jbc.M112.426106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X., Spycher C., Han Z. S., Rose A. M., Müller F. and Avruch J. (2002). TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr. Biol. 12, 1448-1461. 10.1016/S0960-9822(02)01091-6 [DOI] [PubMed] [Google Scholar]
- Long X., Ortiz-Vega S., Lin Y. and Avruch J. (2005). Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 280, 23433-23436. 10.1074/jbc.C500169200 [DOI] [PubMed] [Google Scholar]
- Mak H. Y. and Ruvkun G. (2004). Intercellular signaling of reproductive development by the C. elegans DAF-9 cytochrome P450. Development 131, 1777-1786. 10.1242/dev.01069 [DOI] [PubMed] [Google Scholar]
- Martin T. D., Chen X.-W., Kaplan R. E. W., Saltiel A. R., Walker C. L., Reiner D. J. and Der C. J. (2014). Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 53, 209-220. 10.1016/j.molcel.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina J. A., Chen Y., Gucek M. and Puertollano R. (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903-914. 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mörck C. and Pilon M. (2006). C. elegans feeding defective mutants have shorter body lengths and increased autophagy. BMC Dev. Biol. 6, 39 10.1186/1471-213X-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A., Postler T. S., Rao P., Schmitt H., Schmitt V., Grinberg-Bleyer Y., Kühn L. I., Gruber C. W., Lienhard G. E. and Ghosh S. (2014). kappaB-Ras proteins regulate both NF-kappaB-dependent inflammation and Ral-dependent proliferation. Cell Rep. 8, 1793-1807. 10.1016/j.celrep.2014.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham S., Montagne J., Radimerski T., Thomas G. and Hafen E. (2000). Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 14, 2689-2694. 10.1101/gad.845700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'rourke E. J., Soukas A. A., Carr C. E. and Ruvkun G. (2009). C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10, 430-435. 10.1016/j.cmet.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A., Wang Y., Smith H. E., Lee C.-Y. S., Calidas D., Lu T., Smith J., Schmidt H., Krause M. W. and Seydoux G. (2014). Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 198, 1347-1356. 10.1534/genetics.114.170423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P. H., Thapar N., Guo L., Martinez M., Maris J., Gau C.-L., Lengyel J. A. and Tamanoi F. (2003). Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J. Cell Sci. 116, 3601-3610. 10.1242/jcs.00661 [DOI] [PubMed] [Google Scholar]
- Potter C. J., Pedraza L. G. and Xu T. (2002). Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4, 658-665. 10.1038/ncb840 [DOI] [PubMed] [Google Scholar]
- Rasmussen N. R., Dickinson D. J. and Reiner D. J. (2018). Ras-dependent cell fate decisions are reinforced by the RAP-1 small GTPase in Caenorhabditis elegans. Genetics 210, 1339-1354. 10.1534/genetics.118.301601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner D. J. and Lundquist E. A. (2018). Small GTPases. WormBook 2018, 1-65. 10.1895/wormbook.1.67.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini D. M. (2017). Twenty-five years of mTOR: uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 114, 11818-11825. 10.1073/pnas.1716173114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R., Shirakawa R., Nishiyama H., Kobayashi T., Kawato M., Kanno T., Nishizawa K., Matsui Y., Ohbayashi T., Horiguchi M. et al. (2013). Downregulation of Ral GTPase-activating protein promotes tumor invasion and metastasis of bladder cancer. Oncogene 32, 894-902. 10.1038/onc.2012.101 [DOI] [PubMed] [Google Scholar]
- Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L. and Sabatini D. M. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496-1501. 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo L. J., Gao X., Chiarelli D. A., Li L., Pan D. and Edgar B. A. (2003). Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 5, 566-571. 10.1038/ncb996 [DOI] [PubMed] [Google Scholar]
- Schindler A. J., Baugh L. R. and Sherwood D. R. (2014). Identification of late larval stage developmental checkpoints in Caenorhabditis elegans regulated by insulin/IGF and steroid hormone signaling pathways. PLoS Genet. 10, e1004426 10.1371/journal.pgen.1004426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M. A., Pierce-Shimomura J. T., Chan S., Parry D. and Mcintire S. L. (2010). Manipulation of behavioral decline in Caenorhabditis elegans with the Rag GTPase raga-1. PLoS Genet. 6, e1000972 10.1371/journal.pgen.1000972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V. A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S. U., Huynh T., Medina D., Colella P. et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429-1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H., Kaplan R. E. W., Duong T., Fakieh R. and Reiner D. J. (2018). Ral signals through a MAP4 kinase-p38 MAP kinase cascade in C. elegans cell fate patterning. Cell Rep. 24, 2669-2681.e5. 10.1016/j.celrep.2018.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F., Tijsterman M., Parrish S., Koushika S. P., Nonet M. L., Fire A., Ahringer J. and Plasterk R. H. A. (2002). Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol. 12, 1317-1319. 10.1016/S0960-9822(02)01041-2 [DOI] [PubMed] [Google Scholar]
- Van Dam T. J. P., Zwartkruis F. J. T., Bos J. L. and Snel B. (2011). Evolution of the TOR pathway. J. Mol. Evol. 73, 209-220. 10.1007/s00239-011-9469-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wählby C., Conery A. L., Bray M.-A., Kamentsky L., Larkins-Ford J., Sokolnicki K. L., Veneskey M., Michaels K., Carpenter A. E. and O'rourke E. J. (2014). High- and low-throughput scoring of fat mass and body fat distribution in C. elegans. Methods 68, 492-499. 10.1016/j.ymeth.2014.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. D. (2015). Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363-377. 10.1534/genetics.114.172361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K., Rossman K. L. and Der C. J. (2005). The Ras superfamily at a glance. J. Cell Sci. 118, 843-846. 10.1242/jcs.01660 [DOI] [PubMed] [Google Scholar]
- Yang J.-S., Nam H.-J., Seo M., Han S. K., Choi Y., Nam H. G., Lee S.-J. and Kim S. (2011). OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS ONE 6, e23525 10.1371/journal.pone.0023525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Lu N. and Zhou Z. (2008). Phagocytic receptor CED-1 initiates a signaling pathway for degrading engulfed apoptotic cells. PLoS Biol. 6, e61 10.1371/journal.pbio.0060061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Stallock J. P., Ng J. C., Reinhard C. and Neufeld T. P. (2000). Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 14, 2712-2724. 10.1101/gad.835000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Ward J. D., Cheng Z. and Dernburg A. F. (2015). The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 142, 4374-4384. 10.1242/dev.129635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Shen H., Sewell A. K., Kniazeva M. and Han M. (2013). A novel sphingolipid-TORC1 pathway critically promotes postembryonic development in Caenorhabditis elegans. eLife 2, e00429 10.7554/eLife.00429 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.